The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD) (original) (raw)
Abstract
Nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes mellitus (T2DM) remain as one of the most global problematic metabolic diseases with rapidly increasing prevalence and incidence. Epidemiological studies noted that T2DM patients have by two-fold increase to develop NAFLD, and vice versa. This complex and intricate association is supported and mediated by insulin resistance (IR). In this review, we discuss the NAFLD immunopathogenesis, connection with IR and T2DM, the role of screening and noninvasive tools, and mostly the impact of the current antidiabetic drugs on steatosis liver and new potential therapeutic targets.
1. Introduction
The liver is one of the main houses that control the metabolic homeostasis. Metabolic diseases such as obesity, IR, T2DM, dyslipidaemia, and NAFLD are connected through molecular-biochemical, and complex immune mechanism [1, 2]. Both diabetes and NAFLD are chronic diseases that usually portray nonalarming changes that can lead to disability and many other metabolic complications. They are all independently mortality and morbidity risk promoters, and overall global financial consumer disorders [3–5].
Currently, NAFLD remains one of the most frequent liver diseases, affecting up to 25% of the general adult population [6–8], and with reported increased incidence in children [9]. Soon it may become the most common indication for liver transplant [10]. This multifactorial condition can derive from an unhealthy lifestyle, obesity, dyslipidaemia, type2 diabetes mellitus, and/or other metabolic syndromes [11, 12]. It is characterized by a wide spectrum of liver diseases that vary from simple fat accumulation (benign steatosis), to inflammation (nonalcoholic steatohepatitis (NASH)), fibrosis, cirrhosis, liver failure, and finally to hepatocellular carcinoma (HCC), in the absence of excessive alcohol consumption, medications, or viral aetiology [13–16]. Researchers found that individuals with diagnosed NAFLD have a two-fold increased risk of T2DM [17], and higher risk to develop oncologic [18], cardiovascular [19, 20], and renal disease [21] especially when is associated with T2DM [22].
By now, TD2M is reported to affect 1 in 11 adults and up to 463 million people worldwide [23]. Since the 90s, there is an increased incidence and prevalence of prediabetes and T2DM among the paediatric population which is linked to a high-fat diet, sedentarism, obesity, and liver-related diseases [24, 25]. As T2DM is defined by high serum glucose levels, IR, and damaged islet cell function, it is possible that patients with NAFLD have a higher risk of developing diabetes as they usually express abnormal glucose metabolism [26]. Interestingly, recent evidence shows that T2DM is an independent risk factor for NAFLD [27], women with a history of Gestational Diabetes Mellitus (GDM) have a higher risk of NAFLD, and vice versa [28, 29], and that hepatic steatosis resolution can prevent T2DM onsets [30, 31].
Over the years, substantial efforts were made in order to elucidate the immunopathogenic mechanism behind NAFLD and its connections with T2DM. Even if they are still insufficiently known data, IR seems to be one of the key events that appear in both disorders [32, 33]. The interplay between T2DM, NAFLD, and IR could be considered a two-way street. It is difficult to establish if IR is the cause or the consequence of NAFLD and T2DM, and what is the full relationship with other metabolic syndromes [34, 35]. Nonetheless, it is very important to understand as much as possible this codependency relationship.
In this review, we aim at describing the immunopathogenesis behind NAFLD, how IR is the hallmark that coexist in booth diseases, how we can prevent and assert hepatic changes through noninvasive methods, what are the best therapeutic approaches in T2DM subjects that have NAFLD, and the new desirable possible therapeutic options, and foremost we hope to raise awareness among clinicians about how we should look beyond one disease and the importance of screening for better prevention, management, and outcomes.
2. Immunopathogenesis
The link between T2DM and NAFLD can be described by a spectrum of metabolic changes represented by IR, defective hepatic lipidic profile, and triglyceride (TG) metabolism which lead to fat accumulation, immune responses, and/or subsequently hyperinsulinemia determined by the _β_-cell dysfunction in T2DM [36]. Normally, there is a balanced scale between lipid uptake (free fatty acids (FFAs) or “de novo lipogenesis” (DNL), and esterification) and lipid disposal (metabolism or _β_-oxidation, and elimination as very-low-density lipoproteins (VLDL)). In NAFLD, VLDL removal cannot keep up with the increased rate of TG uptake and intrahepatic production [37]. Thus, NAFLD immunopathogenesis can be described by two hypotheses. One that includes increased intake of dietary fats that lead to free fatty acids (FFAs) surplus, increased DNL, and decreased hepatic TG excretion, and one that encompasses oxidative stress, lipid peroxidation, mitochondrial dysfunction, and release of inflammatory mediators [29, 38, 39].
2.1. Lipotoxicity
To maintain strict control of the hepatic lipid homeostasis, compound interactions are made between hormones, nuclear receptors, and transcription factors [40]. As known, carbohydrate excess contributes to steatosis via DNL that produces lipogenic molecules such as acetyl-CoA carboxylase (ACC), fatty acid synthesis (FAS), and stearoyl CoA-desaturase-1 (SCD-1). DNL plays a very substantial role in the development of NAFLD and is characterized by a series of enzymatic transformations. First, glucose is converted to acetyl CoA by glycolysis and pyruvate oxidation. Acetyl-CoA is then converted to malonyl-CoA by ACC. FAS catalyse the formation of palmitic acid from malonyl-CoA and acetyl-CoA. Palmitic acid is then desaturated by long-chain fatty acid elongase 6 and SCD1 to generate saturated fatty monoacids, which are the main constituents of triglyceride fatty acids. Glycerol-3-phosphate acyltransferase (GPAT) then catalyses the esterification of glycerol-3-phosphate from glycolysis with newly synthesized fatty acid to phosphatidic acids. The phosphatidic acids are then processed into diacylglycerols (DAG) by lipin, followed by the formation of triglycerides by acyl-CoA: diacylglycerol acyltransferase [41–43]. Glucose and insulin promote lipogenesis through activation of the carbohydrate response element-binding protein (ChREBP) and the sterol regulating element-binding protein 1c (SREBP1c) [38, 44]. Fructose increases the expression and induction of CD36 lipogenic pathways, including uniquely regulation of ChREBP and SREBP1c, increased steatosis, and reduced hepatic insulin signalling [45, 46]. SREBPs are the most skilled regulators of lipid uptake and cholesterol biosynthesis, and can induce liver steatosis by enhancing TG expression. Deletion of SREBP-1a promotes toll-like receptor-4 (TLR4) stimulation, increased inflammatory gene expression via reprogramming the fatty acid production [47], systemic IR, and reduction of the glucose transport [48]. Transcriptional regulation of DNL is primarily orchestrated by SREBP1c. This process connects DNL to cholesterol metabolism and indirectly to IR, mainly because SREBP1c can enhance the generation of harmful lipid molecules such as DAG and ceramides which further enhance IR, resulting in a positive feedback loop, in which hepatic DNL help IR and IR stimulate hepatic DNL [49]. Ceramides are an additional class that combines cellular toxicity with proinflammatory actions. Ceramides contribute to inflammation through interaction with TNF_α_, are involved in oxidative stress, and cell death [50]. They damage the mitochondria and the endoplasmic reticulum (ER) function, elicit oxidative stress, promote apoptosis, and histologic dysmorphic liver lesions like ballooning and Mallory-Denk bodies [45, 51]. In addition, lipid overload in the pancreatic β_-cells impels insulin secretion and changes the expression of peroxisome proliferator-activated receptor- (PPAR-) α, glucokinase, glucose transporter 2, preproinsulin, and of pancreatic duodenal homeobox, which enhance IR as a result of apoptosis [52]. The PPAR-γ is a transcriptional regulator of adipose metabolism that binds to SREBPs. Experimental data shows that in obese models PPAR-γ and SREBP1 expression are elevated [53] and that SREBP-1c/PPAR_α ratio can be used as an index of hepatic steatosis [54].
2.2. Oxidative Stress
As seen, lipid excess leads to fat storage accumulation, abnormal lipid peroxidation, the release of the proinflammatory cytokine, high reactive oxygen species (ROS), and reactive nitrogen species (RNS). Lipid peroxidation then promotes stellate cell proliferation, which contributes to fibrogenesis. The nuclear erythroid 2-related factor 2/antioxidant response element (NRF2/ARE) pathway which modulates the antioxidant effect of ROS and RNS is flawed in subjects with obesity and IR [55]. ROS induce the release of cytokines from hepatocytes, trigger TLR-4 synthesis, and promotes inflammatory liver macrophage activation [56].
2.3. Hepatic Cell Activation
In major metabolic diseases, the onset is characterized by alteration of peripheral macrophage number and functional phenotype, especially in the hepatic and obese tissue [57]. In increased adipose tissue, researchers found high levels of macrophages, mainly resident adipose tissue macrophages (ATMs), which were detected in clusters named crown-like structures (CLS) [58, 59]. These ATMs are fundamental for tissue homeostasis, are involved in tissue remodelling, clearance of cellular debris, inflammation, and fibrosis [60–62]. In human lean adipose tissue, ATMs are represented by CD14+/CD16− and express markers like CD11c+ and CD206 that are associated with IR [63]. The elements that activate ATMs into metabolically activated macrophage are the lipoproteins, FFAs, glucose, and insulin. Liver macrophages (LM) are transformed from an anti-inflammatory (M2) to a proinflammatory cell function (M1) [64, 65]. M1 enhances the production of chemokines such as chemokine ligand 2 (CCL2) that induces the synthesis of tumor necrosis factor-alpha (TNF-α), IL-1 β, and interleukin-6 (IL-6) which can alter the insulin sensitivity in the adipose tissue [66, 67]. Importantly, TNF-α activates two major proinflammatory signaling pathways: the c-Jun N terminal kinase (JNK) a mitogen-activated protein kinase family member and the nuclear-kappa B (NF-_κ_B) pathway, both linked to IR and NAFLD. Under normal conditions, NF-_κ_B is sequestered in the cytoplasm and binds to the inhibitor of kappa B (I_κ_B) proteins, which then inhibit the nuclear localization of NF-_κ_B. The NF-_κ_B kinase of nuclear factor kappa-B kinase (IKK-β) inhibitor plays an important role in the activation of NF-_κ_B. It seems that the deletion of IKK-β improves glucose tolerance and insulin sensitivity and suppresses the NF-_κ_B pathway which can limit the lipogenesis and inflammation processes [64, 68]. Also, TNF-α reduces AMP-activated protein kinase (AMPK) activity which has a role in NAFLD development [69]. The JNK pathways constant activation is maintained by stimuli-like oxidative stress or various drug via a feedback loop mechanism. Subsequently, JNK phosphorylates insulin receptor substrate (IRS) which instigates the inhibition of the insulin signaling [70, 71].
Kupffer cells (KCs) are the most flourishing population of resident macrophages that inhabit the liver. Among the immune homeostasis regulation, KCs coordinate the metabolism of bilirubin, cholesterol, iron [72, 73], and can recruit neutrophils and natural killer T cells (NKT-cells) into the liver [74]. This recruitment is modulated by chemotactic factors, such as monocyte chemotactic protein-1 (MCP-1). MCP-1 production is initiated by hepatocytes during simple steatosis and is supported by the infiltrating macrophages. Blocking or absence of MCP-1 or C-C Motif Chemokine Receptor2 (CCR2) the receptor for MCP-1 reduces the influx of monocytes and macrophages into the liver, effectively stopping the development and evolution of inflammation and fibrosis [75–77]. Studies show that CCR2+ macrophage is a pioneer in the hepatic monocyte uptake; it can induce lipolysis through regulation of epinephrine and norepinephrine levels and promote liver injury [78]. In T2DM-NAFLD subjects, authors found increased levels of Fatty acid-binding protein 1 (FABP1), a protein that facilitates the storage of FFAs and urges liver damage [79, 80]. Some suggested that this protein may be used as a biomarker to detect liver injury [81]. Another key protein is the Fatty acid transport protein 1 (FATP1) which along with FATP4/5 contributes to the inflammatory macrophages function. Deletion of FATP1 causes glucose intolerance, whereas inhibition/deletion of FATP4/FATP5 has beneficial metabolic effects [82]. An indispensable component of the amino acid metabolism that provides macrophage activation and polarization is glutamine. Glutamine attenuates inflammasome activation, macrophage cell death and has overall beneficial effects. In M1 macrophages, glutamine intensify their proinflammatory effects via succinate in response to LPS, enhances lipotoxicity which promotes inflammation in adipose tissue and trigger IR. Subjects with diabetes or obesity usually have decreased glutamine and increased succinate concentrations [83, 84]. Notably, oral supplementation with glutamine ameliorated diet-induced NASH progression in C57BL/6J mice models [85]. This vicious loop of inflammatory events that are connected and influenced by IR may provide new tools for detection, or new therapeutic targets that can stop the onset and progression of NAFLD.
2.4. Adipokines
Obesity is a major risk factor for diseases like T2DM, hyperlipidemia, and NAFLD. This metabolic disease emerges from an imbalance in energy input, energy consumption, and fat accumulation [86]. Adipose tissue is a well endocrine organ that secretes hormones and cytokines known as adipokines. The development of IR in NAFLD is also likely related to the imbalance between proinsulin (adiponectin, leptin) and anti-insulin (i.e., TNF_α_) cytokines [87–89]. Adiponectin is a specific secretory adipokine that regulates of fatty acid oxidation (FAO), inhibits the accumulation of FFAs, maintains the glucose homeostasis throughout the body, and the sensitivity to hepatic insulin. Hypoadiponectinemia affects the metabolism of fatty acids and promotes a chronic state of inflammation in the liver [90]. On the other hand, leptin can impel the hepatic stellate cell activation and liver fibrosis, control the energy balance, and suppress appetite [91]. Increased levels of leptin were found in subjects with increased body fat and cardiometabolic disorders [92]. Authors noted in a cross-sectional study that NAFLD patients have lower adiponectin levels, higher serum leptin levels, and higher leptin-to-adiponectin (L/A) ratio [93]. Adiponectin and leptin can independently predict the onset of NAFLD that is why they may be taken into consideration as potential predictive biomarkers for NAFLD [94]. Recently, researchers found that novel adipokine Gremlin 1 can antagonize insulin signaling, is positively correlated with the percentage of body fat and IR in T2DM and NAFLD/NASH subjects, and also could represent a potential biomarker or therapeutic target [95].
2.5. Gut Microbiota
Another theory suggest that gut microbiome alteration and dietary habits are another mechanism that induce and maintain T2DM and/or NAFLD [96, 97]. Indeed, data shows that gut dysbiosis enhances bacteria production that can regulate KCs inflammatory activation, promotes short-chain fatty acids (SCFAs) production, changes the enterohepatic circulation of bile acid, and can lead to inflammation and finally hepatic steatosis [98, 99]. Gut-derived metabolites such as tryptophan modulate inflammatory responses in the macrophages and in hepatocytes [100]. Inflammasome key protein complexes when activated induce cell apoptosis and proinflammatory cells release and are essential in host defences mechanism. NLRP3 inflammasome contributes through gut microbiota to control the NAFLD/obesity progression via overproduction of leptin, downregulation of adiponectin generation, and promotion of fibrosis [101]. Circulating microbiota-derived metabolites could be used for NAFLD diagnosis [102].
2.6. Insulin Resistance
Insulin is an anabolic hormone that can mediate the fluid homeostasis, ionic transport, storage of TG in the adipose tissue, can promote esterification and storage of fatty acids in lipid droplets, and can inhibit the lipolysis. Under normal conditions, the pancreatic _β_-cells are secreting insulin after a meal or after hormone release (i.e., catecholamine, glucagon). Insulin suppresses the production of hepatic glucose and stimulates peripheral glucose uptake, while hormones such as glucagon-like peptide-1(GLP-1) stimulates gluconeogenesis, glycogenolysis, and hepatic glucose production. Insulin mediates the glucose metabolism not only by promoting glucose uptake by the adipose tissue and by the hepatic tissue but also by suppressing the hepatic glucose production [103]. Hepatic insulin clearance is dampened in T2DM subjects and is correlated with the metabolic syndrome severity. In fact, insulin exhibits both anti-inflammatory and proinflammatory properties [104]. The term “insulin resistance” is generally used to describe insulin-mediated glucose uptake in the skeletal muscle. IR is defined by suboptimal cellular response to physiological levels of insulin in diverse tissues. This is the pioneer that in critical conditions, increases glycolysis, and release of FFAS for peripheral needs, as most of the glucose, is directed to the brain [40, 105]. Subsequently, hyperinsulinemia results from the beta cells effort to overcome IR by enhancing insulin release. High caloric intake damages the insulin receptor signaling resulting in a flawed suppression of FFAs release from the adipose cells and also flawed nitric oxide (NO) release [106]. Hence, IR and inflammation form a vicious circle, each condition promoting the other and accelerating the development of NAFLD and other metabolic disorders in the presence of lipotoxicity [107]. In both obese and lean subjects, high IR was found to be the most significant predictive factor for NAFLD [108], and research showed that serum insulin levels are firmly associated with ballooning and hepatic lobular inflammation [109]. The intricate relationship between IR, NAFLD, and T2DM is based on a vicious circle. Obesity induced by a high-fat diet is the main precursors that trigger the lipotoxicity and the glucotoxicity pathways which are both mediated by insulin through IR (Figure 1.).
Figure 1.
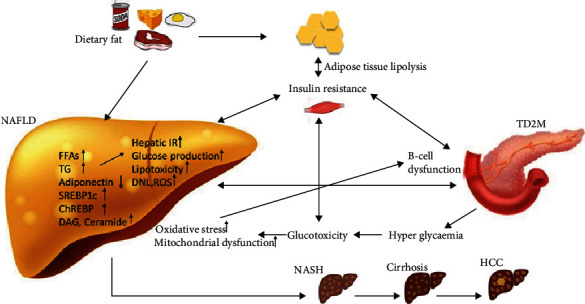
NAFLD, IR, and T2DM complex immunopathogenesis. T2DM: type 2 diabetes mellitus; NAFLD: nonalcoholic fatty liver disease; NASH: nonalcoholic steatohepatitis; HCC: hepatocellular carcinoma; IR: insulin resistance; FFAs: free fatty acids; TG: triglyceride; ChREBP: carbohydrate response element-binding protein; SREBP1c: sterol regulating element-binding protein 1c; DAG: diacylglycerols; ROS: high reactive oxygen species;
3. Screening and Assessment
Steatosis liver is defined when liver fat exceeds 5% of hepatocytes in absence of other secondary causes for lipid hepatic accumulation or by >5.6% of proton density fat fraction measured by MRI/spectroscopy [110]. Even if usually NAFLD is detected by chance through noninvasive imaging, the gold standard for NAFLD diagnosis remains the liver biopsy. However, this procedure is invasive has many serious side effects and is expensive. Thus, it is imperative to develop new systems and guidelines that include, screening, serological, and noninvasive imaging methods to help prevent and diagnose NAFLD in TD2M patients. Recently Bertot et al. [111] reported that noninvasive scoring systems are less accurate at liver outcome prediction in individuals with NAFLD and diabetes. Further data is needed so that we can underline a robust and firm conclusion regarding the practical use of scoring systems. As seen, individuals with T2DM have an increased risk of developing moderate-severe liver damage and have a higher chance to develop HCC [112]. In patients with prediabetes or T2DM serial liver biopsies revealed progressive fibrosis [113]. Scientific research found that Liver Index (FLI) and GGT correlate with peripheral IR and the risk of prediabetes, diabetes, and hypertension development [114, 115]. IR remains one of the key pathogenic tools that onset and maintains the progression of NAFLD to NASH. Homeostasis Model Assessment of Insulin Resistance (HOMA-IR) and liver stiffness measurement (LSM) using acoustic radiation force impulse (ARFI) elastography correlated with the liver fibrosis grade in obese-NAFLD patients [116]. These results can be attributed to the fact that obese subjects have higher peripheral insulin and lower hepatic insulin clearance. Histological alterations are positively correlated with parameters of IR and authors suggest that in the near future 2-hour oral glucose tolerance test (OGTT) may be used to assess NAFLD severity [117]. Along with the OGTT, authors recommend that Impaired Fasting Glycemia (IFG) should be used in the young population for screening at-risk of metabolic syndrome development [118]. Also, the Hepatic steatosis index (HSI) may be a useful tool in the primary screening of NAFLD [119]. So far, a proper screening for NAFLD in TD2M patients is currently unavailable because of the limits of the noninvasive diagnostic tools and the lack of therapeutic options. Nonetheless, every T2DM subject has one or more risk factors to develop liver damage and should be periodically checked [120]. The newly ADA (American Diabetes Association) guidelines updates recommend that individuals with prediabetes or T2DM that have elevated hepatic enzymes or steatosis liver should be investigated for fibrosis/NASH through noninvasive techniques [121].
The most known techniques approved by the European Association for the Study of Diabetes (EASD) [122] include NAFLD liver fat score (NLFS) and the fatty liver index (FLI) for NAFLD diagnosis [123], and fibrosis-4 index (FIB-4) and NAFLD fibrosis score (NFS) for fibrosis assessment [124]. Also, known the SteatoTest, NashTest, ActiTest, FibroTest, and FibroScan are handy tools in quantifying liver impairment [125]. FibroScan® is a reliable instrument in detecting and staging fibrosis in NAFLD/NASH that can also identify macrovascular and microvascular complications of diabetes [126, 127]. Experts propose as the gold standard for detection and grading of the hepatic steatosis, using the intrahepatic TG measurement with the magnetic resonance imaging derived proton density fat fraction (MRI-PDFF) [128, 129]. Subjects with intermediate or high risk of severe fibrosis may benefit from FibroMeter that performs better than the simple FIB4 and NFS tests, and can measure the hepatic extracellular matrix components [130]. Also, Ampuero et al. [131] developed and validated recently the Hepamet fibrosis scoring system, a noninvasive scoring test with 97.2% specificity and 74% sensitivity that uses clinical and laboratory information from NAFLD subjects and can identify fibrosis stage with better accuracy than NFS and FIB-4.
4. Therapeutic Options
Since NAFLD has a complex immunopathogenesis, is dependable by numerous exogenous and endogenous factors, and has tight associations with other metabolic disease, a few therapeutic strategies are available. Efforts are currently in progress to find new promising treatment options to combat NAFLD [122]. In lack of a concrete pharmacotherapy, the current guidelines recommendation for NAFLD mainly emphasize revisions of lifestyle [108, 132].
4.1. Dietary and Lifestyle Revisions
We know that genetics, heritability, and gender type are major factors that raise the susceptibility to develop T2DM, IR, NAFLD, obesity, and/or other metabolic syndromes [133, 134]. While these cannot be changed, factors such as the circadian rhythm that interestingly promotes metabolic disruptions [135, 136], lifestyle (exercise, weight loss), and dietary changes can improve clinical and paraclinical outcomes of NAFLD and T2DM on long term [137, 138]. More than that, continuous exposure to environmental factors like endocrine-disrupting chemicals such as the ubiquitous phthalates and heavy metals adversely affect human health. These synthetic phthalate esters are found anywhere around us from air, to industrial products, and industrial food [139]. They act like hormones and interfere with different receptors such as PPAR-α, as well as androgen receptors (AR), thyroid hormone receptors (TR_α_, TR_β_) which interrupt the normal lipid and glucose homeostasis. There is sufficient evidence that links metabolic disorders development through phthalates exposure [140]. This shows that not only personal habits are important but also the overall living condition. Recently, The Diabetes Remission Clinical Trial (DiRECT) noted that weight loss in TDM patients led to liver fat loss and recovery of the _β_-cell function [141]. Also, in a recent systematic review and meta-analysis, authors observed that caloric deficient diet and periodic exercise ameliorated hepatic functions [142]. Low intake of fibres, vitamins, and mineral nutrients support NAFLD progression [143], whereas dietary habits rich in fruits and vegetables have antioxidant, anti-inflammatory effects, and can improve IR [144]. Protein diets in subjects with T2DM and NAFLD promotes loss of hepatic fat associated with better IR and decreased hepatic cytolytic profile [145], and can improve the glycated haemoglobin A1c (HbA1c) levels [146]. Contrary, others showed that a high protein diet may have negative effects on insulin sensitivity, and its beneficial effects are linked to the amount and quality of the products [147]. Additionally, fructose is commonly added into artificially-sweetened beverages and other sweet solid products. High fructose intake is associated with increased risk of steatosis, liver fibrosis, obesity, and IR [148]. Long-term sucrose ingestion led to increased fat accumulation, glucose intolerance and hyperinsulinemia, and histological damage like increased hepatocyte size and ballooning [149]. Dietary intake of monosaturated FAs found in foods like olive oil and avocado has been shown to improve insulin sensitivity and hepatic fat in prediabetic [150] and in paediatric patients with NAFLD [151]. Those who followed a Mediterranean diet showed a better decrease of liver transaminase, body mass index (BMI) changes, and improvement in IR [152, 153]. Enormous scientific data shows that diet and exercise are first and highly beneficial in metabolic syndromes, and any physician should recommend to their patients additionally to therapeutic medication.
4.2. T2DM Medication and NAFLD
The current guidelines endorse that for the HbA1C and serum glycaemic control in T2DM, patients should use one of the seven drug classes approved by the American Diabetes Association (ADA). These classes include metformin, sulfonylureas, thiazolidinediones, dipeptidyl peptidase 4 inhibitors, glucagon-like peptide-1 receptor agonists, sodium-glucose cotransporter 2 inhibitors, and insulin [154]. Substantial research has been made over the last years in order to find among the existing antidiabetic drugs other potential tools that can be used in T2DM-NAFLD patients [155].
4.2.1. Metformin
Metformin also known as Glucophage is the first-line medication for the treatment of type 2 diabetes [156]. This drug lowers both basal and postprandial plasma glucose levels by suppressing the liver gluconeogenesis via phosphorylation of cAMP-response element-binding protein (CREBBP or CBP), decreases intestinal absorption of glucose, and improves insulin sensitivity by increasing peripheral glucose uptake and utilization [157]. It was thought that it could be beneficial along with hypocaloric diet and weight control in nondiabetic patients with NAFLD. However, in many clinical trials that included nondiabetic and diabetic patients, even if metformin administration improved at some degree the serum aminotransferase levels, histological outcomes on long-term failed to show significant differences [158–160]. Nonetheless, metformin has proven to impediment the risk of HCC development and cardiovascular complications related to NAFLD and T2DM [161].
4.2.2. Thiazolidinediones
Thiazolidinediones bind to a transcription factor identified as PPAR-γ that enhances the transcription of various genes in the adipose tissue; they induce preadipocyte differentiation into adipocytes, raise adiponectin levels, and help with insulin sensitivity [162]. In a 3-year clinical trial, patients with NAFLD who received Rosiglitazone had reduced liver enzymes and better insulin sensitivity after 1 year of treatment [163]. However, the two-year period following the FILTR-2 extension trial revealed no further improvements or changes in the fibrosis or liver ballooning [164]. Interestingly, a 13 years cross-sectional retrospective analysis noted that pioglitazone use in US patients decreased significantly over the years; however, NAFLD prevalence and incidence increased in T2DM patients which can imply that pioglitazone could have ameliorated this raise [165]. Using long term treatment with pioglitazone along with a hypocaloric diet in prediabetic and T2DM subjects with NAFLD resulted in histological and circulating liver enzymes improvement which persisted over 3 years [166]. Others showed that this drug can improve histologic features but not the mean fibrosis score [167]. In experimental NASH-induced models, pioglitazone administration reduced ceramides and DAG levels and improved the hepatic mitochondria function [168]. These results suggest that this therapy is effective and quite feasible for NAFLD [169]. Currently, there is the ongoing randomized open-label pilot (ToPiND) study that tries to investigate the effects of tofogliflozin 20 mg/day or/and pioglitazone 15–30 mg/day administration. At 6 months and at 1 year of therapy, the hepatic steatosis grade will be measured by MRI-PDFF and collective serum data would be assessed [170]. It remains to be seen whether monotherapy or combination therapy would have the most beneficial effects in NAFLD-T2DM. Many other robust studies are desired however, authors should take into consideration that pioglitazone has many serious side effects such as weight gain, worsening heart failure, osteoporosis, and raised risk of bladder cancer [171].
4.2.3. Glucagon-Like Peptide-1 (GLP-1) Analogues
GLP-1 analogue can promote glucose-mediated insulin secretion, decrease glucagon synthesis, and suppress appetite, which is why it can be a potential medication for NAFLD. Liraglutide administration in NASH subjects resulted in raised insulin sensitivity, decrease of DNL, reduced BMI, cholesterol-LDL, and suppression of lipolysis especially within the subcutaneous adipose tissue [172, 173]. An experimental in vivo and vitro study noted that Liraglutide administration promoted expression of autophagy markers via the AMPK/mTOR pathways leading to antilipotoxic effects [174]. Its ingestion for half a year reduces the subcutaneous body fat (from 361 ± 142 cm2 to 339 ± 131 cm2) but not visceral, hepatic, or epicardial fat [175]. Also, an open-label, active-controlled parallel-group, multicentre trial showed that liraglutide and sitagliptin added to metformin but not insulin glargine reduced body weight, visceral adipose tissue, and intrahepatic lipid levels in individuals with T2DM and NAFLD [176]. Evidence from clinical trials presents the quality of GLP-1 analogues to become disease-modifying tools in NAFLD [177–179]. The glucose-dependent insulinotropic polypeptide (GIP)/GLP-1 agonist could be used not only for glucose metabolism control but also for NAFLD treatment, as this combination has synergic effects, promotes lipogenesis and weight loss [180]. The physiological effects, the therapeutic implication of GIP antagonism, and agonism in T2DM-NAFLD patients need further exploration in larger human trials.
4.2.4. Sodium-Glucose Cotransporter-2 (SGLT2) Inhibitors
The newly T2DM therapy SGLT-2 inhibitors increase glucagon levels, diminish renal reabsorption of glucose, and increases its excretion. SGLT2 inhibitors could benefit the hepatic function because it promotes glucagon secretion, DNL, and urinary caloric losses with subsequently weight loss [181]. The main SGLT2 representants are composed by canagliflozin, dapagliflozin, and empagliflozin, used as second-line treatment in association with metformin as well as third-line treatment. Molecules such as luseogliflozin and tofogliflozin are only approved in Japan, while ipragliflozin was also approved last year in Russia [154]. A recent systematic review described 8 studies that evaluated the role of SGLT-2 inhibitors on NAFLD. The results illustrated that in most of these studies, patients with SGLT-2 therapy had AST and GGT levels decrease, 5 studies noted reduction in hepatic fat, and 2 studies found improvement in liver fibrosis [182]. Many trials involving canagliflozin [183–186] noted comprehensive results regarding SGLT-2 inhibitors in NAFLD. Also, empagliflozin [187, 188] and dapagliflozin [189, 190] administration showed similar beneficial results. A combination of exenatide once weekly plus dapagliflozin once a day reduced biomarkers of liver steatosis and fibrosis in patients with T2DM uncontrolled by metformin monotherapy [191]. The DURATION-8 (NCT02229396) phase 3 trial displayed similar effects when exenatide once weekly plus dapagliflozin once daily, improved glycemic control and body weight [192–194]. Also, long term use of luseogliflozins led to the improvement of steatosis, fibrosis, and histological activity score [195–197]. Recently, authors demonstrated that the use of the novel SGLT2 inhibitor, NGI001 in high fat diet-induced mice instigates suppression of lipid accumulation, inflammation, upregulation of _β_-oxidation, and they suggest that this inhibitor may be as a new therapeutic approach that can delay the onset of NAFLD [198]. Robust evidence supports the idea that antidiabetic drugs are quite feasible for NAFLD treatment (Table 1), and so far, pioglitazone and liraglutide noted the most promising results. We await further larger clinical trials that can prove better histological outcomes in T2DM-NAFLD patients.
Table 1.
Clinical trials which encompass the antidiabetic drug effects on NAFLD. BMI: body mass index; AST: aspartataminotransferaza, ALT: alanine aminotransferase, HbA1c: glycated haemoglobin, DNL: de novo lipogenesis.
| Agents | Study type | Dose | Time | Outcomes | Ref. |
|---|---|---|---|---|---|
| Rosiglitazone | Randomized placebo-controlled (FLIRT) Trial | 4 mg/day 1 month and after 8 mg/day | 1 year and 4 months | Improvement of steatosis correlated with reduction of transaminase level improvement in insulin sensitivity. | [163] |
| Randomized placebo-controlled (FLIRT 2) extension trial | 4 mg/day 1 month and after 8 mg/day | 3 years | Improvement in ALT and liver steatosis but no results on ballooning and fibrosis. | [164] | |
| Pioglitazone | Randomized, double-blind, placebo-controlled trial | 30 mg/day | 2 years | Improvement in individual histologic scores, adipose tissue, hepatic, and muscle insulin sensitivity. | [166] |
| Pioglitazone vs. ipragliflozin | Open-label, randomized, active-controlled trial | 15-30 mg/day vs. 50 mg/day | 6 months | AST and ALT, HbA1c, and fasting plasma glucose were similarly reduced in the two-treatment group. | [169] |
| Liraglutide | Randomised, placebo-controlled phase 2 study (LEAN) study | 1.8 mg/day | 1 year | Improvements in histological steatosis and hepatocyte ballooning. | [172] |
| Double-blind, randomised, placebo-controlled trial | 1.8 mg/day | 3 months | Reduced BMI, DNL, IR, and hepatic steatosis. | [173] | |
| Open-label, active-controlled parallel-group, multicentre trial | 3 mg/day | 6 months | Reduced BMI, hepatic steatosis, and hepatocellular apoptosis. | [176] | |
| Prospective, single-center study (LEAN-J). | 1.2 mg/day | 2 years | 31% reduction of hepatic steatosis | [179] | |
| Canagliflozin | A prospective study | 100 mg/day | 6 months | Improvement in histopathologic features and markers of liver dysfunction | [183] |
| A prospective study | 100 mg/day | 6 months | Histological improvement, improvement of IR. | [184] | |
| Prospective study | 100 mg/day | 6 months | Improvement in ALT and liver steatosis | [186] | |
| Empagliflozin | Randomised controlled trials (E-LIFT Trial) | 10 mg/day | 5 months | Improvement in liver steatosis and serum ALT level. | [187] |
| Single-arm, open-label, pilot study | 25 mg/day | 6 months | Reduction in BMI, cholesterol, GGT, ballooning, and fibrosis. | [188] | |
| Dapagliflozin | Randomised controlled trial | 10 mg/day | 4 months | Reduction in BMI, AST, ALT, and liver steatosis. | [189] |
| An open-label, randomized trial | 5 mg/day | 6 months | Improvements in hepatic steatosis, along with attenuation of fibrosis in a subset of patients with significant fibrosis. | [190] | |
| Luseogliflozin | A prospective, single-arm trial (LEAD trial) | 2.5 mg/day | 6 months | Improvement of steatosis correlated with reduction of transaminase level | [195] |
4.3. New Potential Therapeutic Targets
New promising treatment options emerged over the years to combat NAFLD [199]. Interestingly, micronutrients like choline and polyphenols also interfere with the liver-gut axis and contribute to several pathways that are crucial for diabetes or the NAFLD development. For example, low choline intake has been associated with worsening liver fibrosis and a higher risk of NAFLD development [200]. The polyphenol family encompasses a wide spectrum of molecules such as curcumin, resveratrol, or quercetin that can be found in vegetables, fruit, and coffee. Recently, researches showed that polyphenol can reduce TG accumulation through antioxidant, and anti-inflammatory effects, and by blockage of lipogenesis via SREBP1c downregulation [201]. Resveratrol administration can regulate PPAR expression, IL-1_β_, and TNF-α, and have antisteatosis effects with secondary increase of body weight [202]. Histopathological improvement in NASH models was observed after the administration of a dual PPAR_α_/γ agonist named Saroglitazar [203]. Another dual agonist named Elafibranor (GFT505) had similar results in murine models of NAFLD and NASH [204]. (PEGylated) FGF21 analogue marked as Pegbelfermin is also under investigation to see if it can be a feasible tool for NAFLD/NASH treatment [205]. A new potential target that may be used in T2DM treatment is the protein tyrosine phosphatase 1B (PTPIB) that was shown to inhibit insulin signaling and can normalize plasma glucose levels [206]. Considering the rise incidence of NAFLD in men and postmenopausal woman, authors demonstrated in vitro and in vivo study that 17_β_-estradiol (E2) therapy improves IR and fatty acid accumulation by interfering with the JNK activation pathway [207]. Recently, in an open-label prospective studies and trials administration of GS-0976 (Firsocostat), a small molecule inhibitor of acetyl-CoA carboxylase in NASH patients reduced de novo lipogenesis, intrahepatic TG levels, markers of liver injury and steatosis, and may soon be approved by FDA for NASH treatment [208, 209]. Another approach is the inhibition of certain inflammatory pathways involved in NAFLD. Animal studies showed that the inhibition of CCR2 or the ligand CCL2-IR with cenicriviroc may diminish fibrosis especially after 1-year treatment [210]. In a phase 2 randomized, placebo-controlled trial, phase II study, the use of Hepatic thyroid hormone receptor beta (THR-β) agonist VK2809 elicited dose dependent improvement on liver fat reduction and liver function [211]. Similar results were obtained when using a similar agent THR-β agonist named MGL-3196. However, more studies are needed to mark the side effects of these molecules and their interaction with metabolic systems [212]. A number of drugs that target steatosis and/or fibrosis are currently investigated in Phase II and Phase III clinical trials [213]. More than that, potential therapies that target the gut-liver axis could represent in the future a key strategy in the management of TD2M and NAFLD [214]. For example, the administration of probiotic and synbiotics may reduce histologically-confirmed liver fat and improve liver function [215, 216]. There are many therapeutic possibilities for NAFLD discovered by research teams that we did not discuss here. From the inflammatory, immune, metabolic, oxidative stress, hormonal, and gut-axis pathways to other T2DM therapeutic ways, NAFLD onset and progression should be modulated not just by one single therapeutic approach. Further trials and investigations in this matter are needed.
5. Conclusions
For many years, robust evidence tried to demonstrate the association between NAFLD, TD2M, IR, obesity, and other metabolic syndromes. Unfortunately, this is a vast and complex territory and so far, there is no clear evidence that links the glucose metabolism and IR with diabetes and NAFLD manifestations. Many countries lack exhaustive public health response to NAFLD, screening for NAFLD in diabetes patients, awareness campaigns, early validation of risk factors, large scale educational programs, guidelines/algorithms, and long-term strategies for these patients. With the high chance that a steatosis liver advances to NASH especially when multiple comorbidities are associated, early assessment and management of NAFLD in T2DM subjects are imperative. Therapeutic inertia continues to remain a general problem in diabetes; that is why it is crucial that a physician early recognises patients at risk. Clinicians must know what are the best noninvasive tools and therapeutic approaches to prevent and delay NAFLD, and also how to maintain an open collaboration with other specialities. Currently, there is no proper approved pharmaceutical treatment for NAFLD. Multitarget agents or combination of agents should have twice the beneficial effect compared to monotherapy. Numerous trials are currently under development that investigate new promising pharmacological agents for NAFLD and test the effect of antidiabetic drugs on liver function. We hope that in the future, larger clinical trials can assess and approve new therapeutic drugs for NAFLD that can be used safely in T2DM patients.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Evelina Maria Gosav, Minela Aida Maranduca, and Anca Ouatu contributed equally to this work.
References
- 1.Lindenmeyer C. C., McCullough A. J. The natural history of nonalcoholic fatty liver disease-an evolving view. Clinics in Liver Disease. 2018;22(1):11–21. doi: 10.1016/j.cld.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleinert M., Clemmensen C., Hofmann S. M., et al. Animal models of obesity and diabetes mellitus. Nature Reviews Endocrinology. 2018;14(3):140–162. doi: 10.1038/nrendo.2017.161. [DOI] [PubMed] [Google Scholar]
- 3.Younossi Z. M. The epidemiology of nonalcoholic steatohepatitis. Clinical Liver Disease. 2018;11(4):92–94. doi: 10.1002/cld.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Younossi Z., Anstee Q. M., Marietti M., et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nature Reviews Gastroenterology & Hepatology. 2018;15(1):11–20. doi: 10.1038/nrgastro.2017.109. [DOI] [PubMed] [Google Scholar]
- 5.Younossi Z. M., Golabi P., de Avila L., et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. Journal of Hepatology. 2019;71(4):793–801. doi: 10.1016/j.jhep.2019.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Li J., Zou B., Yeo Y. H., et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999-2019: a systematic review and meta-analysis. The Lancet Gastroenterology & Hepatology. 2019;4(5):389–398. doi: 10.1016/S2468-1253(19)30039-1. [DOI] [PubMed] [Google Scholar]
- 7.Bellentani S. The epidemiology of non-alcoholic fatty liver disease. Liver International. 2017;37(S1):81–84. doi: 10.1111/liv.13299. [DOI] [PubMed] [Google Scholar]
- 8.Pimpin L., Cortez-Pinto H., Negro F., et al. Burden of liver disease in Europe: epidemiology and analysis of risk factors to identify prevention policies. Journal of Hepatology. 2018;69(3):718–735. doi: 10.1016/j.jhep.2018.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Mann J. P., Valenti L., Scorletti E., Byrne C. D., Nobili V. Nonalcoholic fatty liver disease in children. Seminars in Liver Disease. 2018;38(1):001–013. doi: 10.1055/s-0038-1627456. [DOI] [PubMed] [Google Scholar]
- 10.Mikolasevic I., Filipec-Kanizaj T., Mijic M., et al. Nonalcoholic fatty liver disease and liver transplantation - where do we stand? World Journal of Gastroenterology. 2018;24(14):1491–1506. doi: 10.3748/wjg.v24.i14.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cusi K., Sanyal A. J., Zhang S., et al. Non-alcoholic fatty liver disease (NAFLD) prevalence and its metabolic associations in patients with type 1 diabetes and type 2 diabetes. Diabetes, Obesity and Metabolism. 2017;19(11):1630–1634. doi: 10.1111/dom.12973. [DOI] [PubMed] [Google Scholar]
- 12.Krishan S. Correlation between non-alcoholic fatty liver disease (NAFLD) and dyslipidemia in type 2 diabetes. Diabetes & Metabolic Syndrome: Clinical Research & Reviews. 2016;10(2):S77–S81. doi: 10.1016/j.dsx.2016.01.034. [DOI] [PubMed] [Google Scholar]
- 13.Machado M. V., Diehl A. M. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology. 2016;150(8):1769–1777. doi: 10.1053/j.gastro.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benedict M., Zhang X. Non-alcoholic fatty liver disease: an expanded review. World Journal of Hepatology. 2017;9(16):715–732. doi: 10.4254/wjh.v9.i16.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanwal F., Kramer J. R., Mapakshi S., et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology. 2018;155(6):1828–1837.e2. doi: 10.1053/j.gastro.2018.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Younossi Z. M., Otgonsuren M., Henry L., et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology. 2015;62(6):1723–1730. doi: 10.1002/hep.28123. [DOI] [PubMed] [Google Scholar]
- 17.Ballestri S., Zona S., Targher G., et al. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. Journal of Gastroenterology and Hepatology. 2016;31(5):936–944. doi: 10.1111/jgh.13264. [DOI] [PubMed] [Google Scholar]
- 18.Fujiwara N., Qian T., Koneru B., Hoshida Y. Omics-derived hepatocellular carcinoma risk biomarkers for precision care of chronic liver diseases. Hepatology Research. 2020;50(7):817–830. doi: 10.1111/hepr.13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brouwers M. C. G. J., Simons N., Stehouwer C. D. A., Isaacs A. Non-alcoholic fatty liver disease and cardiovascular disease: assessing the evidence for causality. Diabetologia. 2020;63(2):253–260. doi: 10.1007/s00125-019-05024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koo B. K., Allison M. A., Criqui M. H., Denenberg J. O., Wright C. M. The association between liver fat and systemic calcified atherosclerosis. Journal of Vascular Surgery. 2020;71(1):204–211.e4. doi: 10.1016/j.jvs.2019.03.044. [DOI] [PubMed] [Google Scholar]
- 21.Byrne C. D., Targher G. NAFLD as a driver of chronic kidney disease. Journal of Hepatology. 2020;72(4):785–801. doi: 10.1016/j.jhep.2020.01.013. [DOI] [PubMed] [Google Scholar]
- 22.Wild S. H., Walker J. J., Morling J. R., et al. Cardiovascular disease, cancer, and mortality among people with type 2 diabetes and alcoholic or nonalcoholic fatty liver disease hospital admission. Diabetes Care. 2018;41(2):341–347. doi: 10.2337/dc17-1590. [DOI] [PubMed] [Google Scholar]
- 23.International Diabetes Federation. IDF DIABETES ATLAS -9TH EDITION. April 2020, http://www.diabetesatlas.org. [PubMed]
- 24.Mayer-Davis E. J., Kahkoska A. R., Jefferies C., et al. ISPAD Clinical Practice Consensus Guidelines 2018: definition, epidemiology, and classification of diabetes in children and adolescents. Pediatric Diabetes. 2018;19(Suppl 27):7–19. doi: 10.1111/pedi.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koutny F., Weghuber D., Bollow E., et al. Prevalence of prediabetes and type 2 diabetes in children with obesity and increased transaminases in European German-speaking countries. Analysis of the APV initiative. Pediatric Obesity. 2020;15(4, article e12601) doi: 10.1111/ijpo.12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strey C. B. M., de Carli L. A., Pioner S. R., et al. Impact of diabetes mellitus and insulin on nonalcoholic fatty liver disease in the morbidly obese. Annals of Hepatology. 2018;17(4):585–591. doi: 10.5604/01.3001.0012.0922. [DOI] [PubMed] [Google Scholar]
- 27.American Diabetes Association. 2. Classification and Diagnosis of Diabetes:Standards of Medical Care in Diabetes—2018. Diabetes Care. 2017;41(Supplement 1):S13–S27. doi: 10.2337/dc18-S002. [DOI] [PubMed] [Google Scholar]
- 28.Sattari M., Bril F., Egerman R., Kalavalapalli S., Cusi K. Relationship between non-alcoholic fatty liver disease during pregnancy and abnormal glucose metabolism during and after pregnancy. Journal of Investigative Medicine. 2020;68(3):743–747. doi: 10.1136/jim-2019-001186. [DOI] [PubMed] [Google Scholar]
- 29.Akazawa Y., Nakao K. To die or not to die: death signaling in nonalcoholic fatty liver disease. Journal of Gastroenterology. 2018;53(8):893–906. doi: 10.1007/s00535-018-1451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sung K. C., Wild S. H., Byrne C. D. Resolution of fatty liver and risk of incident diabetes. The Journal of Clinical Endocrinology & Metabolism. 2013;98(9):3637–3643. doi: 10.1210/jc.2013-1519. [DOI] [PubMed] [Google Scholar]
- 31.Arab J. P., Arrese M., Trauner M. Recent Insights into the pathogenesis of nonalcoholic fatty liver disease. Annual Review of Pathology: Mechanisms of Disease. 2018;13(1):321–350. doi: 10.1146/annurev-pathol-020117-043617. [DOI] [PubMed] [Google Scholar]
- 32.Brar G., Tsukamoto H. Alcoholic and non-alcoholic steatohepatitis: global perspective and emerging science. Journal of Gastroenterology. 2019;54(3):218–225. doi: 10.1007/s00535-018-01542-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith G. I., Shankaran M., Yoshino M., et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. Journal of Clinical Investigation. 2020;130(3):1453–1460. doi: 10.1172/JCI134165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yi M., Chen R. P., Yang R., Chen H. Increased prevalence and risk of non-alcoholic fatty liver disease in overweight and obese patients with type 2 diabetes in South China. Diabetic Medicine. 2017;34(4):505–513. doi: 10.1111/dme.13174. [DOI] [PubMed] [Google Scholar]
- 35.Valenti L., Bugianesi E., Pajvani U., Targher G. Nonalcoholic fatty liver disease: cause or consequence of type 2 diabetes? Liver International. 2016;36(11):1563–1579. doi: 10.1111/liv.13185. [DOI] [PubMed] [Google Scholar]
- 36.Forlani G., Giorda C., Manti R., et al. The burden of NAFLD and its characteristics in a nationwide population with type 2 diabetes. Journal of Diabetes Research. 2016;2016:9. doi: 10.1155/2016/2931985.2931985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendez-Sanchez N., Cruz-Ramon V. C., Ramirez-Perez O. L., Hwang J. P., Barranco-Fragoso B., Cordova-Gallardo J. New aspects of lipotoxicity in nonalcoholic steatohepatitis. International Journal of Molecular Sciences. 2018;19(7):p. 2034. doi: 10.3390/ijms19072034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawano Y., Cohen D. E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. Journal of Gastroenterology. 2013;48(4):434–441. doi: 10.1007/s00535-013-0758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedman S. L., Neuschwander-Tetri B. A., Rinella M., Sanyal A. J. Mechanisms of NAFLD development and therapeutic strategies. Nature Medicine. 2018;24(7):908–922. doi: 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kitade H., Chen G., Ni Y., Ota T. Nonalcoholic fatty liver disease and insulin resistance: new insights and potential new treatments. Nutrients. 2017;9(4):p. 387. doi: 10.3390/nu9040387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ertunc M. E., Hotamisligil G. S. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. Journal of Lipid Research. 2016;57(12):2099–2114. doi: 10.1194/jlr.R066514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lefere S., Tacke F. Macrophages in obesity and non-alcoholic fatty liver disease: crosstalk with metabolism. JHEP Reports. 2019;1(1):30–43. doi: 10.1016/j.jhepr.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang W., Metlakunta A., Dedousis N., et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. 2010;59(2):347–357. doi: 10.2337/db09-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu W., Baker R. D., Bhatia T., Zhu L., Baker S. S. Pathogenesis of nonalcoholic steatohepatitis. Cellular and Molecular Life Sciences. 2016;73(10):1969–1987. doi: 10.1007/s00018-016-2161-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marra F., Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. Journal of Hepatology. 2018;68(2):280–295. doi: 10.1016/j.jhep.2017.11.014. [DOI] [PubMed] [Google Scholar]
- 46.Van Herck M. A., Weyler J., Kwanten W. J., et al. The differential roles of T cells in non-alcoholic fatty liver disease and obesity. Frontiers in Immunology. 2019;10 doi: 10.3389/fimmu.2019.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Apostolopoulou M., Gordillo R., Koliaki C., et al. Specific hepatic sphingolipids relate to insulin resistance, oxidative stress, and inflammation in nonalcoholic steatohepatitis. Diabetes Care. 2018;41(6):1235–1243. doi: 10.2337/dc17-1318. [DOI] [PubMed] [Google Scholar]
- 48.Vijayakumar A., Aryal P., Wen J., et al. Absence of carbohydrate response element binding protein in adipocytes causes systemic insulin resistance and impairs glucose transport. Cell Reports. 2017;21(4):1021–1035. doi: 10.1016/j.celrep.2017.09.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xia J. Y., Holland W. L., Kusminski C. M., et al. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metabolism. 2015;22(2):266–278. doi: 10.1016/j.cmet.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Birkenfeld A. L., Shulman G. I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. 2014;59(2):713–723. doi: 10.1002/hep.26672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oishi Y., Spann N. J., Link V. M., et al. SREBP1 contributes to resolution of pro-inflammatory TLR4 signaling by reprogramming fatty acid metabolism. Cell Metabolism. 2017;25(2):412–427. doi: 10.1016/j.cmet.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hill D. A., Lim H. W., Kim Y. H., et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proceedings of the National Academy of Sciences. 2018;115(22):E5096–E5105. doi: 10.1073/pnas.1802611115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dubois V., Eeckhoute J., Lefebvre P., Staels B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. Journal of Clinical Investigation. 2017;127(4):1202–1214. doi: 10.1172/JCI88894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pettinelli P., del Pozo T., Araya J., et al. Enhancement in liver SREBP-1c/PPAR-alpha ratio and steatosis in obese patients: correlations with insulin resistance and n-3 long-chain polyunsaturated fatty acid depletion. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2009;1792(11):1080–1086. doi: 10.1016/j.bbadis.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 55.Morgantini C., Jager J., Li X., et al. Liver macrophages regulate systemic metabolism through non-inflammatory factors. Nature Metabolism. 2019;1(4):445–459. doi: 10.1038/s42255-019-0044-9. [DOI] [PubMed] [Google Scholar]
- 56.Masarone M., Rosato V., Dallio M., et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxidative Medicine and Cellular Longevity. 2018;2018:14. doi: 10.1155/2018/9547613.9547613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Daemen S., Schilling J. D. The interplay between tissue niche and macrophage cellular metabolism in obesity. Frontiers in Immunology. 2020;10 doi: 10.3389/fimmu.2019.03133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amano S. U., Cohen J. L., Vangala P., et al. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metabolism. 2014;19(1):162–171. doi: 10.1016/j.cmet.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng C., Yang Q., Cao J., et al. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death & Disease. 2016;7(3, article e2167) doi: 10.1038/cddis.2016.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sakai M., Troutman T. D., Seidman J. S., et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain Kupffer cell identity. Immunity. 2019;51(4):655–670.e8. doi: 10.1016/j.immuni.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alisi A., Carpino G., Oliveira F. L., Panera N., Nobili V., Gaudio E. The role of tissue macrophage-mediated inflammation on NAFLD pathogenesis and its clinical implications. Mediators of Inflammation. 2017;2017:15. doi: 10.1155/2017/8162421.8162421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coats B. R., Schoenfelt K. Q., Barbosa-Lorenzi V. C., et al. Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Reports. 2017;20(13):3149–3161. doi: 10.1016/j.celrep.2017.08.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wentworth J. M., Naselli G., Brown W. A., et al. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. 2010;59(7):1648–1656. doi: 10.2337/db09-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ipsen D. H., Lykkesfeldt J., Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cellular and Molecular Life Sciences. 2018;75(18):3313–3327. doi: 10.1007/s00018-018-2860-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tan H. Y., Wang N., Li S., Hong M., Wang X., Feng Y. The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxidative Medicine and Cellular Longevity. 2016;2016:16. doi: 10.1155/2016/2795090.2795090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Monteillet L., Gjorgjieva M., Silva M., et al. Intracellular lipids are an independent cause of liver injury and chronic kidney disease in non alcoholic fatty liver disease-like context. Molecular Metabolism. 2018;16:100–115. doi: 10.1016/j.molmet.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jager J., Aparicio-Vergara M., Aouadi M. Liver innate immune cells and insulin resistance: the multiple facets of Kupffer cells. Journal of Internal Medicine. 2016;280(2):209–220. doi: 10.1111/joim.12483. [DOI] [PubMed] [Google Scholar]
- 68.Hao Y. R., Tang F. J., Zhang X., Wang H. Suppression of NF-κB activation by PDLIM2 restrains hepatic lipogenesis and inflammation in high fat diet induced mice. Biochemical and Biophysical Research Communications. 2018;503(2):564–571. doi: 10.1016/j.bbrc.2018.05.187. [DOI] [PubMed] [Google Scholar]
- 69.Perry R. J., Samuel V. T., Petersen K. F., Shulman G. I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510(7503):84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Solinas G., Becattini B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Molecular Metabolism. 2017;6(2):174–184. doi: 10.1016/j.molmet.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Win S., Than T. A., Zhang J., Oo C., Min R. W. M., Kaplowitz N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology. 2018;67(5):2013–2024. doi: 10.1002/hep.29689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krenkel O., Tacke F. Liver macrophages in tissue homeostasis and disease. Nature Reviews Immunology. 2017;17(5):306–321. doi: 10.1038/nri.2017.11. [DOI] [PubMed] [Google Scholar]
- 73.Scott C. L., Guilliams M. The role of Kupffer cells in hepatic iron and lipid metabolism. Journal of Hepatology. 2018;69(5):1197–1199. doi: 10.1016/j.jhep.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heymann F., Peusquens J., Ludwig-Portugall I., et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 2015;62(1):279–291. doi: 10.1002/hep.27793. [DOI] [PubMed] [Google Scholar]
- 75.Parker R., Weston C. J., Miao Z., et al. CC chemokine receptor 2 promotes recruitment of myeloid cells associated with insulin resistance in nonalcoholic fatty liver disease. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2018;314(4):G483–G493. doi: 10.1152/ajpgi.00213.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krenkel O., Puengel T., Govaere O., et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. 2018;67(4):1270–1283. doi: 10.1002/hep.29544. [DOI] [PubMed] [Google Scholar]
- 77.Guillot A., Tacke F. Liver macrophages: old dogmas and new insights. Hepatology Communications. 2019;3(6):730–743. doi: 10.1002/hep4.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim S. J., Feng D., Guillot A., et al. Adipocyte death preferentially induces liver injury and inflammation through the activation of chemokine (C-C motif) receptor 2-positive macrophages and lipolysis. Hepatology. 2019;69(5):1965–1982. doi: 10.1002/hep.30525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu Y. C., Chang C. C., Wang C. P., et al. Circulating fatty acid-binding protein 1 (FABP1) and nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus. International Journal of Medical Sciences. 2020;17(2):182–190. doi: 10.7150/ijms.40417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li M., Xu C., Shi J., et al. Fatty acids promote fatty liver disease via the dysregulation of 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Gut. 2018;67(12):2169–2180. doi: 10.1136/gutjnl-2017-313778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akbal E., Koçak E., Akyürek Ö., Köklü S., Batgi H., Şenes M. Liver fatty acid-binding protein as a diagnostic marker for non-alcoholic fatty liver disease. Wiener klinische Wochenschrift. 2016;128(1-2):48–52. doi: 10.1007/s00508-014-0680-8. [DOI] [PubMed] [Google Scholar]
- 82.Johnson A. R., Qin Y., Cozzo A. J., et al. Metabolic reprogramming through fatty acid transport protein 1 (FATP1) regulates macrophage inflammatory potential and adipose inflammation. Mol Metab. 2016;5(7):506–526. doi: 10.1016/j.molmet.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He L., Weber K. J., Schilling J. D. Glutamine Modulates Macrophage Lipotoxicity. Nutrients. 2016;8(4):p. 215. doi: 10.3390/nu8040215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ren W., Xia Y., Chen S., et al. Glutamine metabolism in macrophages: a novel target for obesity/type 2 diabetes. Advances in Nutrition. 2019;10(2):321–330. doi: 10.1093/advances/nmy084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sellmann C., Baumann A., Brandt A., Jin C. J., Nier A., Bergheim I. Oral supplementation of glutamine attenuates the progression of nonalcoholic steatohepatitis in C57BL/6J mice. The Journal of Nutrition. 2017;147(11):2041–2049. doi: 10.3945/jn.117.253815. [DOI] [PubMed] [Google Scholar]
- 86.Verboven K., Wouters K., Gaens K., et al. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. ScientificReports. 2018;8(1):p. 4677. doi: 10.1038/s41598-018-22962-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gastaldelli A., Gaggini M., DeFronzo R. A. Role of adipose tissue insulin resistance in the natural history of type 2 diabetes: results from the San Antonio Metabolism Study. Diabetes. 2017;66(4):815–822. doi: 10.2337/db16-1167. [DOI] [PubMed] [Google Scholar]
- 88.González-Muniesa P., Mártinez-González M. A., Hu F. B., et al. Obesity. Nature Reviews Disease Primers. 2017;3(1) doi: 10.1038/nrdp.2017.34. [DOI] [PubMed] [Google Scholar]
- 89.Kazankov K., Jørgensen S. M. D., Thomsen K. L., et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nature Reviews Gastroenterology & Hepatology. 2019;16(3):145–159. doi: 10.1038/s41575-018-0082-x. [DOI] [PubMed] [Google Scholar]
- 90.Li G., Feng D., Qu X., et al. Role of adipokines FGF21, leptin and adiponectin in self-concept of youths with obesity. European Neuropsychopharmacology. 2018;28(8):892–902. doi: 10.1016/j.euroneuro.2018.05.015. [DOI] [PubMed] [Google Scholar]
- 91.Boutari C., Mantzoros C. S. Adiponectin and leptin in the diagnosis and therapy of NAFLD. Metabolism. 2020;103:p. 154028. doi: 10.1016/j.metabol.2019.154028. [DOI] [PubMed] [Google Scholar]
- 92.Li L. J., Rifas-Shiman S. L., Aris I. M., Mantzoros C., Hivert M. F., Oken E. Leptin trajectories from birth to mid-childhood and cardio-metabolic health in early adolescence. Metabolism. 2019;91:30–38. doi: 10.1016/j.metabol.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mikami K., Endo T., Sawada N., et al. Leptin/adiponectin ratio correlates with hepatic steatosis but not arterial stiffness in nonalcoholic fatty liver disease in Japanese population. Cytokine. 2020;126:p. 154927. doi: 10.1016/j.cyto.2019.154927. [DOI] [PubMed] [Google Scholar]
- 94.Kim Y. S., Lee S. H., Park S. G., et al. Low levels of total and high-molecular-weight adiponectin may predict non-alcoholic fatty liver in Korean adults. Metabolism. 2020;103:p. 154026. doi: 10.1016/j.metabol.2019.154026. [DOI] [PubMed] [Google Scholar]
- 95.Hedjazifar S., Khatib Shahidi R., Hammarstedt A., et al. The novel adipokine Gremlin 1 antagonizes insulin action and is increased in type 2 diabetes and NAFLD/NASH. Diabetes. 2020;69(3):331–341. doi: 10.2337/db19-0701. [DOI] [PubMed] [Google Scholar]
- 96.Campo L., Eiseler S., Apfel T., Pyrsopoulos N. Fatty liver disease and gut microbiota: a comprehensive update. Journal of Clinical and Translational Hepatology. 2018;7(1):56–60. doi: 10.14218/JCTH.2018.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Augustyn M., Grys I., Kukla M. Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease. Clinical and Experimental Hepatology. 2019;5(1):1–10. doi: 10.5114/ceh.2019.83151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chávez-Carbajal A., Nirmalkar K., Pérez-Lizaur A., et al. Gut microbiota and predicted metabolic pathways in a sample of Mexican women affected by obesity and obesity plus metabolic syndrome. International Journal of Molecular Sciences. 2019;20(2):p. 438. doi: 10.3390/ijms20020438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rowland I., Gibson G., Heinken A., et al. Gut microbiota functions: metabolism of nutrients and other food components. European Journal of Nutrition. 2018;57(1):1–24. doi: 10.1007/s00394-017-1445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Krishnan S., Ding Y., Saeidi N., et al. Gut microbiota-derived tryptophan metabolites modulate inflammatory response in hepatocytes and macrophages. Cell Reports. 2019;28(12):p. 3285. doi: 10.1016/j.celrep.2019.08.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aragonès G., Colom-Pellicer M., Aguilar C., et al. Circulating microbiota-derived metabolites: a liquid biopsy? International Journal of Obesity. 2020;44(4):875–885. doi: 10.1038/s41366-019-0430-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang X., Dai J., Li L., Chen H., Chai Y. NLRP3 inflammasome expression and signaling in human diabetic wounds and in high glucose induced macrophages. Journal of Diabetes Research. 2017;2017:7. doi: 10.1155/2017/5281358.5281358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hari Kumar K. V. S. The good, the bad, and the ugly facets of insulin resistance. Medical Journal Armed Forces India. 2020;76(1):4–7. doi: 10.1016/j.mjafi.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bergman R. N., Piccinini F., Kabir M., Kolka C. M., Ader M. Hypothesis: Role of Reduced Hepatic Insulin Clearance in the Pathogenesis of Type 2 Diabetes. Diabetes. 2019;68(9):1709–1716. doi: 10.2337/db19-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Akuta N., Kawamura Y., Fujiyama S., et al. Predictors of insulin secretion in Japanese patients with histopathologically-confirmed non-alcoholic fatty liver disease. Internal Medicine. 2020;59(3):329–338. doi: 10.2169/internalmedicine.3555-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gambino R., Bugianesi E., Rosso C., et al. Different serum free fatty acid profiles in NAFLD subjects and healthy controls after oral fat load. International Journal of Molecular Sciences. 2016;17(4):p. 479. doi: 10.3390/ijms17040479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hong S. H., Choi K. M. Sarcopenic Obesity, Insulin Resistance, and Their Implications in Cardiovascular and Metabolic Consequences. International Journal of Molecular Sciences. 2020;21(2):p. 494. doi: 10.3390/ijms21020494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huang J. F., Tsai P. C., Yeh M. L., et al. Risk stratification of non-alcoholic fatty liver disease across body mass index in a community basis. International Journal of Molecular Sciences. 2020;119(1):89–96. doi: 10.1016/j.jfma.2019.03.014. [DOI] [PubMed] [Google Scholar]
- 109.Enooku K., Kondo M., Fujiwara N., et al. Hepatic IRS1 and ß-catenin expression is associated with histological progression and overt diabetes emergence in NAFLD patients. Journal of Gastroenterology. 2018;53(12):1261–1275. doi: 10.1007/s00535-018-1472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dai W., Ye L., Liu A., et al. Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus. Medicine. 2017;96(39, article e8179) doi: 10.1097/MD.0000000000008179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bertot L. C., Jeffrey G. P., de Boer B., et al. Diabetes impacts prediction of cirrhosis and prognosis by non-invasive fibrosis models in non-alcoholic fatty liver disease. Liver International. 2018;38(10):1793–1802. doi: 10.1111/liv.13739. [DOI] [PubMed] [Google Scholar]
- 112.Chalasani N., Younossi Z., Lavine J. E., et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67(1):328–357. doi: 10.1002/hep.29367. [DOI] [PubMed] [Google Scholar]
- 113.Hazlehurst J. M., Woods C., Marjot T., Cobbold J. F., Tomlinson J. W. Non-alcoholic fatty liver disease and diabetes. Metabolism. 2016;65(8):1096–1108. doi: 10.1016/j.metabol.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bonnet F., Gastaldelli A., Pihan-le Bars F., et al. Gamma-glutamyltransferase, fatty liver index and hepatic insulin resistance are associated with incident hypertension in two longitudinal studies. Journal of Hypertension. 2017;35(3):493–500. doi: 10.1097/HJH.0000000000001204. [DOI] [PubMed] [Google Scholar]
- 115.Kwok R., Choi K. C., Wong G. L. H., et al. Screening diabetic patients for non-alcoholic fatty liver disease with controlled attenuation parameter and liver stiffness measurements: a prospective cohort study. Gut. 2016;65(8):1359–1368. doi: 10.1136/gutjnl-2015-309265. [DOI] [PubMed] [Google Scholar]
- 116.Isokuortti E., Zhou Y., Peltonen M., et al. Use of HOMA-IR to diagnose non-alcoholic fatty liver disease: a population-based and inter-laboratory study. Diabetologia. 2017;60(10):1873–1882. doi: 10.1007/s00125-017-4340-1. [DOI] [PubMed] [Google Scholar]
- 117.Coccia F., Testa M., Guarisco G., et al. Insulin resistance, but not insulin response, during oral glucose tolerance test (OGTT) is associated to worse histological outcome in obese NAFLD. Nutrition, Metabolism and Cardiovascular Diseases. 2020;30(1):106–113. doi: 10.1016/j.numecd.2019.08.001. [DOI] [PubMed] [Google Scholar]
- 118.Li Y., Feng D., Esangbedo I. C., et al. Insulin resistance, beta-cell function, adipokine profiles and cardiometabolic risk factors among Chinese youth with isolated impaired fasting glucose versus impaired glucose tolerance: the BCAMS study. BMJ Open Diabetes Research & Care. 2020;8(1, article e000724) doi: 10.1136/bmjdrc-2019-000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bugianesi E., Rosso C., Cortez-Pinto H. How to diagnose NAFLD in 2016. Journal of Hepatology. 2016;65(3):643–644. doi: 10.1016/j.jhep.2016.05.038. [DOI] [PubMed] [Google Scholar]
- 120.Wong V. W. S., Chalasani N. Not routine screening, but vigilance for chronic liver disease in patients with type 2 diabetes. Journal of Hepatology. 2016;64(6):1211–1213. doi: 10.1016/j.jhep.2016.02.032. [DOI] [PubMed] [Google Scholar]
- 121.American Diabetes Association. 4. Comprehensive Medical Evaluation and Assessment of Comorbidities: Standards of Medical Care in Diabetes—2020. Diabetes Care. 2019;43(Supplement 1):S37–S47. doi: 10.2337/dc20-S004. [DOI] [PubMed] [Google Scholar]
- 122.Marchesini G., Roden M., Vettor R. Response to: Comment to "EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease". Journal of Hepatology. 2017;66(2):466–467. doi: 10.1016/j.jhep.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 123.Bedogni G., Bellentani S., Miglioli L., et al. The Fatty Liver Index: a simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterology. 2006;6(1) doi: 10.1186/1471-230X-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Patel P. J., Cheng J. C. Y., Banh X., et al. Clinically significant fibrosis is associated with longitudinal increases in fibrosis-4 and nonalcoholic fatty liver disease fibrosis scores. Clinical Gastroenterology and Hepatology. 2020;18(3):710–718.e4. doi: 10.1016/j.cgh.2019.07.036. [DOI] [PubMed] [Google Scholar]
- 125.Poynard T., Lassailly G., Diaz E., et al. Performance of biomarkers FibroTest, ActiTest, SteatoTest, and NashTest in patients with severe obesity: meta analysis of individual patient data. PLoS One. 2012;7(3, article e30325) doi: 10.1371/journal.pone.0030325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Eddowes P. J., Sasso M., Allison M., et al. Accuracy of FibroScan controlled attenuation parameter and liver stiffness measurement in assessing steatosis and fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology. 2019;156(6):1717–1730. doi: 10.1053/j.gastro.2019.01.042. [DOI] [PubMed] [Google Scholar]
- 127.Lombardi R., Airaghi L., Targher G., et al. Liver fibrosis by FibroScan®independently of established cardiovascular risk parameters associates with macrovascular and microvascular complications in patients with type 2 diabetes. Liver International. 2019;40(2):347–354. doi: 10.1111/liv.14274. [DOI] [PubMed] [Google Scholar]
- 128.Caussy C., Alquiraish M. H., Nguyen P., et al. Optimal threshold of controlled attenuation parameter with MRI-PDFF as the gold standard for the detection of hepatic steatosis. Hepatology. 2018;67(4):1348–1359. doi: 10.1002/hep.29639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Runge J. H., Smits L. P., Verheij J., et al. MR Spectroscopy-derived proton density fat fraction is superior to controlled attenuation parameter for detecting and grading hepatic steatosis. Radiology. 2018;286(2):547–556. doi: 10.1148/radiol.2017162931. [DOI] [PubMed] [Google Scholar]
- 130.Guillaume M., Moal V., Delabaudiere C., et al. Direct comparison of the specialised blood fibrosis tests FibroMeterV2Gand enhanced liver fibrosis score in patients with non-alcoholic fatty liver disease from tertiary care centres. Alimentary Pharmacology & Therapeutics. 2019;50(11-12):1214–1222. doi: 10.1111/apt.15529. [DOI] [PubMed] [Google Scholar]
- 131.Ampuero J., Pais R., Aller R., et al. Development and validation of Hepamet fibrosis scoring system-a simple, noninvasive test to identify patients with nonalcoholic fatty liver disease with advanced fibrosis. Clinical Gastroenterology and Hepatology. 2020;18(1):216–225.e5. doi: 10.1016/j.cgh.2019.05.051. [DOI] [PubMed] [Google Scholar]
- 132.Dibba P., Li A. A., Perumpail B. J., et al. Emerging therapeutic targets and experimental drugs for the treatment of NAFLD. Diseases. 2018;6(3):p. 83. doi: 10.3390/diseases6030083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Eslam M., George J. Genetic contributions to NAFLD: leveraging shared genetics to uncover systems biology. Nature Reviews Gastroenterology & Hepatology. 2020;17(1):40–52. doi: 10.1038/s41575-019-0212-0. [DOI] [PubMed] [Google Scholar]
- 134.Schernthaner-Reiter M. H., Stratakis C. A., Luger A. Genetics of diabetes insipidus. Endocrinology and Metabolism Clinics of North America. 2017;46(2):305–334. doi: 10.1016/j.ecl.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 135.Shi D., Chen J., Wang J., et al. Circadian clock genes in the metabolism of non-alcoholic fatty liver disease. Frontiers in Physiology. 2019;10:p. 423. doi: 10.3389/fphys.2019.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hudec M., Dankova P., Solc R., Bettazova N., Cerna M. Epigenetic regulation of circadian rhythm and its possible role in diabetes mellitus. International Journal of Molecular Sciences. 2020;21(8):p. 3005. doi: 10.3390/ijms21083005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Romero-Gómez M., Zelber-Sagi S., Trenell M. Treatment of NAFLD with diet, physical activity and exercise. Journal of Hepatology. 2017;67(4):829–846. doi: 10.1016/j.jhep.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 138.Patterson R. E., Sears D. D. Metabolic effects of intermittent fasting. Annual Review of Nutrition. 2017;37(1):371–393. doi: 10.1146/annurev-nutr-071816-064634. [DOI] [PubMed] [Google Scholar]
- 139.Milošević N., Milanović M., Sudji J., et al. Could phthalates exposure contribute to the development of metabolic syndrome and liver disease in humans? Environmental Science and Pollution Research. 2020;27(1):772–784. doi: 10.1007/s11356-019-06831-2. [DOI] [PubMed] [Google Scholar]
- 140.Taylor R., al-Mrabeh A., Zhyzhneuskaya S., et al. Remission of human type 2 diabetes requires decrease in liver and pancreas fat content but is dependent upon capacity for β cell Recovery. Cell Metabolism. 2018;28(4):547–556.e3. doi: 10.1016/j.cmet.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 141.Shoshtari-Yeganeh B., Zarean M., Mansourian M., et al. Systematic review and meta-analysis on the association between phthalates exposure and insulin resistance. Environmental Science and Pollution Research. 2019;26(10):9435–9442. doi: 10.1007/s11356-019-04373-1. [DOI] [PubMed] [Google Scholar]
- 142.Koutoukidis D. A., Astbury N. M., Tudor K. E., et al. Association of weight loss interventions with changes in biomarkers of nonalcoholic fatty liver Disease. JAMA Internal Medicine. 2019;179(9):1262–1271. doi: 10.1001/jamainternmed.2019.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hodson L., Rosqvist F., Parry S. A. The influence of dietary fatty acids on liver fat content and metabolism. Proceedings of the Nutrition Society. 2020;79(1):30–41. doi: 10.1017/S0029665119000569. [DOI] [PubMed] [Google Scholar]
- 144.Luukkonen P. K., Sädevirta S., Zhou Y., et al. Saturated fat is more metabolically harmful for the human liver than unsaturated fat or simple sugars. Diabetes Care. 2018;41(8):1732–1739. doi: 10.2337/dc18-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Drummen M., Dorenbos E., Vreugdenhil A. C. E., et al. Long-term effects of increased protein intake after weight loss on intrahepatic lipid content and implications for insulin sensitivity: a PREVIEW study. American Journal of Physiology-Endocrinology and Metabolism. 2018;315(5):E885–E891. doi: 10.1152/ajpendo.00162.2018. [DOI] [PubMed] [Google Scholar]
- 146.Skytte M. J., Samkani A., Petersen A. D., et al. A carbohydrate-reduced high-protein diet improves HbA1c and liver fat content in weight stable participants with type 2 diabetes: a randomised controlled trial. Diabetologia. 2019;62(11):2066–2078. doi: 10.1007/s00125-019-4956-4. [DOI] [PubMed] [Google Scholar]
- 147.Devan S. R. K., Arumugam S., Shankar G., Poosala S. Differential sensitivity of chronic high-fat-diet-induced obesity in Sprague-Dawley rats. Journal of Basic and Clinical Physiology and Pharmacology. 2018;29(5):553–563. doi: 10.1515/jbcpp-2017-0030. [DOI] [PubMed] [Google Scholar]
- 148.Cho Y. E., Kim D. K., Seo W., Gao B., Yoo S. H., Song B. J. Fructose promotes leaky gut, endotoxemia, and liver fibrosis through ethanol-inducible cytochrome P 450-2E1-mediated oxidative and nitrative stress. Hepatology. 2019;10 doi: 10.1002/hep.30652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Souza Cruz E. M., Bitencourt de Morais J. M., Dalto da Rosa C. V., et al. Long-term sucrose solution consumption causes metabolic alterations and affects hepatic oxidative stress in Wistar rats. Biology Open. 2020;9(3):p. bio047282. doi: 10.1242/bio.047282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Errazuriz I., Dube S., Slama M., et al. Randomized controlled trial of a MUFA or fiber-rich diet on hepatic fat in prediabetes. The Journal of Clinical Endocrinology & Metabolism. 2017;102(5):1765–1774. doi: 10.1210/jc.2016-3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Boyraz M., Pirgon Ö., Dündar B., Çekmez F., Hatipoğlu N. Long-term treatment with n-3 polyunsaturated fatty acids as a monotherapy in children with nonalcoholic fatty liver disease. Journal of Clinical Research in Pediatric Endocrinology. 2015;7(2):121–127. doi: 10.4274/jcrpe.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Anania C., Perla F. M., Olivero F., Pacifico L., Chiesa C. Mediterranean diet and nonalcoholic fatty liver disease. World Journal of Gastroenterology. 2018;24(19):2083–2094. doi: 10.3748/wjg.v24.i19.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bullón-Vela V., Abete I., Tur J. A., et al. Influence of lifestyle factors and staple foods from the Mediterranean diet on non-alcoholic fatty liver disease among older individuals with metabolic syndrome features. Nutrition. 2020;71:p. 110620. doi: 10.1016/j.nut.2019.110620. [DOI] [PubMed] [Google Scholar]
- 154.American Diabetes Association. 8. Pharmacologic Approaches to Glycemic Treatment:Standards of Medical Care in Diabetes—2018. Diabetes Care. 2017;41(Supplement 1):S73–S85. doi: 10.2337/dc18-S008. [DOI] [PubMed] [Google Scholar]
- 155.Iogna Prat L., Tsochatzis E. A. The effect of antidiabetic medications on non-alcoholic fatty liver disease (NAFLD) Hormones. 2018;17(2):219–229. doi: 10.1007/s42000-018-0021-9. [DOI] [PubMed] [Google Scholar]
- 156.Buse J. B., Wexler D. J., Tsapas A., et al. 2019 Update to: Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) Diabetes Care. 2020;43(2):487–493. doi: 10.2337/dci19-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.He L., Sabet A., Djedjos S., et al. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell. 2009;137(4):635–646. doi: 10.1016/j.cell.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Loomba R., Lutchman G., Kleiner D. E., et al. Clinical trial: pilot study of metformin for the treatment of non-alcoholic steatohepatitis. Alimentary Pharmacology & Therapeutics. 2009;29(2):172–182. doi: 10.1111/j.1365-2036.2008.03869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lavine J. E., Schwimmer J. B., van Natta M., et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and Adolescents. JAMA. 2011;305(16):1659–1668. doi: 10.1001/jama.2011.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bugianesi E., Gentilcore E., Manini R., et al. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. The American Journal of Gastroenterology. 2005;100(5):1082–1090. doi: 10.1111/j.1572-0241.2005.41583.x. [DOI] [PubMed] [Google Scholar]
- 161.Bhat A., Sebastiani G., Bhat M. Systematic review: preventive and therapeutic applications of metformin in liver disease. World Journal of Hepatology. 2015;7(12):1652–1659. doi: 10.4254/wjh.v7.i12.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kahn C. R., Chen L., Cohen S. E. Unraveling the mechanism of action of thiazolidinediones. Journal of Clinical Investigation. 2000;106(11):1305–1307. doi: 10.1172/JCI11705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ratziu V., Giral P., Jacqueminet S., et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135(1):100–110. doi: 10.1053/j.gastro.2008.03.078. [DOI] [PubMed] [Google Scholar]
- 164.Ratziu V., Charlotte F., Bernhardt C., et al. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51(2):445–453. doi: 10.1002/hep.23270. [DOI] [PubMed] [Google Scholar]
- 165.Le P., Chaitoff A., Rothberg M. B., McCullough A., Alkhouri N. Trends in pioglitazone use among U.S. adults with type 2 diabetes and suspected nonalcoholic fatty liver disease. Expert Opinion on Investigational Drugs. 2020;29(2):205–208. doi: 10.1080/13543784.2020.1704731. [DOI] [PubMed] [Google Scholar]
- 166.Cusi K., Orsak B., Bril F., et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Annals of Internal Medicine. 2016;165(5):305–315. doi: 10.7326/M15-1774. [DOI] [PubMed] [Google Scholar]
- 167.Sanyal A. J., Chalasani N., Kowdley K. V., et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. New England Journal of Medicine. 2010;362(18):1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kalavalapalli S., Bril F., Koelmel J. P., et al. Pioglitazone improves hepatic mitochondrial function in a mouse model of nonalcoholic steatohepatitis. American Journal of Physiology-Endocrinology and Metabolism. 2018;315(2):E163–E173. doi: 10.1152/ajpendo.00023.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ito D., Shimizu S., Inoue K., et al. Comparison of ipragliflozin and pioglitazone effects on nonalcoholic fatty liver disease in patients with type 2 diabetes: a randomized, 24-week, Open-Label, Active-Controlled Trial. Diabetes Care. 2017;40(10):1364–1372. doi: 10.2337/dc17-0518. [DOI] [PubMed] [Google Scholar]
- 170.Ozaki A., Yoneda M., Kessoku T., et al. Effect of tofogliflozin and pioglitazone on hepatic steatosis in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: a randomized, open-label pilot study (ToPiND study) Contemporary Clinical Trials Communications. 2020;17:p. 100516. doi: 10.1016/j.conctc.2019.100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tuccori M., Filion K. B., Yin H., Yu O. H., Platt R. W., Azoulay L. Pioglitazone use and risk of bladder cancer: population based cohort study. BMJ. 2016;352:p. i1541. doi: 10.1136/bmj.i1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Armstrong M. J., Gaunt P., Aithal G. P., et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387(10019):679–690. doi: 10.1016/S0140-6736(15)00803-X. [DOI] [PubMed] [Google Scholar]
- 173.Armstrong M. J., Hull D., Guo K., et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. Journal of Hepatology. 2016;64(2):399–408. doi: 10.1016/j.jhep.2015.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.He Y., Ao N., Yang J., Wang X., Jin S., Du J. The preventive effect of liraglutide on the lipotoxic liver injury via increasing autophagy. Annals of Hepatology. 2020;19(1):44–52. doi: 10.1016/j.aohep.2019.06.023. [DOI] [PubMed] [Google Scholar]
- 175.Bizino M. B., Jazet I. M., de Heer P., et al. Placebo-controlled randomised trial with liraglutide on magnetic resonance endpoints in individuals with type 2 diabetes: a pre-specified secondary study on ectopic fat accumulation. Diabetologia. 2020;63(1):65–74. doi: 10.1007/s00125-019-05021-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yan J., Yao B., Kuang H., et al. Liraglutide, sitagliptin, and insulin glargine added to metformin: the effect on body weight and intrahepatic lipid in patients with type 2 diabetes mellitus and nonalcoholic fatty liver disease. Hepatology. 2019;69(6):2414–2426. doi: 10.1002/hep.30320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Eguchi Y., Kitajima Y., Hyogo H., et al. Pilot study of liraglutide effects in non-alcoholic steatohepatitis and non-alcoholic fatty liver disease with glucose intolerance in Japanese patients (LEAN-J) Hepatology Research. 2015;45(3):269–278. doi: 10.1111/hepr.12351. [DOI] [PubMed] [Google Scholar]
- 178.Khoo J., Hsiang J. C., Taneja R., et al. Randomized trial comparing effects of weight loss by liraglutide with lifestyle modification in non-alcoholic fatty liver disease. Liver International. 2019;39(5):941–949. doi: 10.1111/liv.14065. [DOI] [PubMed] [Google Scholar]
- 179.Petit J. M., Cercueil J. P., Loffroy R., et al. Effect of liraglutide therapy on liver fat content in patients with inadequately controlled type 2 diabetes: the Lira-NAFLD study. The Journal of Clinical Endocrinology & Metabolism. 2016;102(2):407–415. doi: 10.1210/jc.2016-2775. [DOI] [PubMed] [Google Scholar]
- 180.Thondam S. K., Cuthbertson D. J., Wilding J. P. H. The influence of glucose-dependent insulinotropic polypeptide (GIP) on human adipose tissue and fat metabolism: implications for obesity, type 2 diabetes and non-alcoholic fatty liver disease (NAFLD) Peptides. 2020;125:p. 170208. doi: 10.1016/j.peptides.2019.170208. [DOI] [PubMed] [Google Scholar]
- 181.Fujita Y., Inagaki N. Renal sodium glucose cotransporter 2 inhibitors as a novel therapeutic approach to treatment of type 2 diabetes: clinical data and mechanism of action. Journal of Diabetes Investigation. 2014;5(3):265–275. doi: 10.1111/jdi.12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Raj H., Durgia H., Palui R., et al. SGLT-2 inhibitors in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: a systematic review. World Journal of Diabetes. 2019;10(2):114–132. doi: 10.4239/wjd.v10.i2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Akuta N., Watanabe C., Kawamura Y., et al. Effects of a sodium-glucose cotransporter 2 inhibitor in nonalcoholic fatty liver disease complicated by diabetes mellitus: preliminary prospective study based on serial liver biopsies. Hepatology Communications. 2017;1(1):46–52. doi: 10.1002/hep4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Akuta N., Kawamura Y., Watanabe C., et al. Impact of sodium glucose cotransporter 2 inhibitor on histological features and glucose metabolism of non-alcoholic fatty liver disease complicated by diabetes mellitus. Hepatology Research. 2019;49(5):531–539. doi: 10.1111/hepr.13304. [DOI] [PubMed] [Google Scholar]
- 185.Seko Y., Sumida Y., Tanaka S., et al. Effect of sodium glucose cotransporter 2 inhibitor on liver function tests in Japanese patients with non-alcoholic fatty liver disease and type 2 diabetes mellitus. Hepatology Research. 2017;47(10):1072–1078. doi: 10.1111/hepr.12834. [DOI] [PubMed] [Google Scholar]
- 186.Gautam A., Agrawal P. K., Doneria J., Nigam A. Effects of canagliflozin on abnormal liver function tests in patients of type 2 diabetes with non-alcoholic fatty liver disease. Journal of the Association of Physicians of India. 2018;66(8):62–66. [PubMed] [Google Scholar]
- 187.Kuchay M. S., Krishan S., Mishra S. K., et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (E-LIFT Trial) Diabetes Care. 2018;41(8):1801–1808. doi: 10.2337/dc18-0165. [DOI] [PubMed] [Google Scholar]
- 188.Lai L. L., Vethakkan S. R., Nik Mustapha N. R., Mahadeva S., Chan W. K. Empagliflozin for the treatment of nonalcoholic steatohepatitis in patients with type 2 diabetes mellitus. Digestive Diseases and Sciences. 2020;65(2):623–631. doi: 10.1007/s10620-019-5477-1. [DOI] [PubMed] [Google Scholar]
- 189.Eriksson J. W., Lundkvist P., Jansson P. A., et al. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: a double-blind randomised placebo-controlled study. Diabetologia. 2018;61(9):1923–1934. doi: 10.1007/s00125-018-4675-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shimizu M., Suzuki K., Kato K., et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes, Obesity and Metabolism. 2019;21(2):285–292. doi: 10.1111/dom.13520. [DOI] [PubMed] [Google Scholar]
- 191.Gastaldelli A., Repetto E., Guja C., et al. Exenatide and dapagliflozin combination improves markers of liver steatosis and fibrosis in patients with type 2 diabetes. Diabetes, Obesity and Metabolism. 2020;22(3):393–403. doi: 10.1111/dom.13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Frías J. P., Guja C., Hardy E., et al. Exenatide once weekly plus dapagliflozin once daily versus exenatide or dapagliflozin alone in patients with type 2 diabetes inadequately controlled with metformin monotherapy (DURATION-8): a 28 week, multicentre, double-blind, phase 3, randomised controlled trial. The Lancet Diabetes & Endocrinology. 2016;4(12):1004–1016. doi: 10.1016/S2213-8587(16)30267-4. [DOI] [PubMed] [Google Scholar]
- 193.Jabbour S. A., Frías J. P., Hardy E., et al. Safety and efficacy of exenatide once weekly plus dapagliflozin once daily versus exenatide or dapagliflozin alone in patients with type 2 diabetes inadequately controlled with metformin monotherapy: 52-week results of the DURATION-8 Randomized Controlled Trial. Diabetes Care. 2018;41(10):2136–2146. doi: 10.2337/dc18-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jabbour S. A., Guja C., Hardy E., Bhattacharya S., Ohman P. K., Frias J. P. DURATION-8 Randomized Controlled Trial 104-Week Results—Once-Weekly Exenatide (ExQW) plus Once-Daily Dapagliflozin (DAPA) vs. ExQW or DAPA Alone. Diabetes. 2018;67(Supplement 1):104–1LB. doi: 10.2337/db18-104-LB. [DOI] [Google Scholar]
- 195.Sumida Y., Murotani K., Saito M., et al. Effect of luseogliflozin on hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease: a prospective, single-arm trial (LEAD trial) Hepatology Research. 2019;49(1):64–71. doi: 10.1111/hepr.13236. [DOI] [PubMed] [Google Scholar]
- 196.Fujimori N., Tanaka N., Kimura T., et al. Long-term luseogliflozin therapy improves histological activity of non-alcoholic steatohepatitis accompanied by type 2 diabetes mellitus. Clinical Journal of Gastroenterology. 2020;13(1):83–89. doi: 10.1007/s12328-019-01018-1. [DOI] [PubMed] [Google Scholar]
- 197.Shibuya T., Fushimi N., Kawai M., et al. Luseogliflozin improves liver fat deposition compared to metformin in type 2 diabetes patients with non-alcoholic fatty liver disease: a prospective randomized controlled pilot study. Diabetes, Obesity and Metabolism. 2017;20(2):438–442. doi: 10.1111/dom.13061. [DOI] [PubMed] [Google Scholar]
- 198.Chiang H., Lee J. C., Huang H. C., Huang H., Liu H. K., Huang C. Delayed intervention with a novel SGLT2 inhibitor NGI001 suppresses diet-induced metabolic dysfunction and non-alcoholic fatty liver disease in mice. British Journal of Pharmacology. 2020;177(2):239–253. doi: 10.1111/bph.14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Reimer K. C., Wree A., Roderburg C., Tacke F. New drugs for NAFLD: lessons from basic models to the clinic. Hepatology International. 2020;14(1):8–23. doi: 10.1007/s12072-019-10001-4. [DOI] [PubMed] [Google Scholar]
- 200.Mazidi M., Katsiki N., Mikhailidis D. P., Banach M. Adiposity may moderate the link between choline intake and non-alcoholic fatty liver disease. Journal of the American College of Nutrition. 2019;38(7):633–639. doi: 10.1080/07315724.2018.1507011. [DOI] [PubMed] [Google Scholar]
- 201.Rodriguez-Ramiro I., Vauzour D., Minihane A. M. Polyphenols and non-alcoholic fatty liver disease: impact and mechanisms. Proceedings of the Nutrition Society. 2016;75(1):47–60. doi: 10.1017/S0029665115004218. [DOI] [PubMed] [Google Scholar]
- 202.Jimoh A., Tanko Y., Ahmed A., Mohammed A., Ayo J. O. Resveratrol prevents high-fat diet-induced obesity and oxidative stress in rabbits. Pathophysiology. 2018;25(4):359–364. doi: 10.1016/j.pathophys.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 203.Jain M. R., Giri S. R., Bhoi B., et al. Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver International. 2018;38(6):1084–1094. doi: 10.1111/liv.13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ratziu V., Harrison S. A., Francque S., et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150(5):1147–1159.e5. doi: 10.1053/j.gastro.2016.01.038. [DOI] [PubMed] [Google Scholar]
- 205.Sanyal A., Charles E. D., Neuschwander-Tetri B. A., et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2018;392(10165):2705–2717. doi: 10.1016/S0140-6736(18)31785-9. [DOI] [PubMed] [Google Scholar]
- 206.Verma M., Gupta S. J., Chaudhary A., Garg V. K. Protein tyrosine phosphatase 1B inhibitors as antidiabetic agents - a brief review. Bioorganic Chemistry. 2017;70:267–283. doi: 10.1016/j.bioorg.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 207.Galmés-Pascual B. M., Martínez-Cignoni M. R., Morán-Costoya A., et al. 17β-estradiol ameliorates lipotoxicity-induced hepatic mitochondrial oxidative stress and insulin resistance. Free Radical Biology and Medicine. 2020;150:148–160. doi: 10.1016/j.freeradbiomed.2020.02.016. [DOI] [PubMed] [Google Scholar]
- 208.Lawitz E. J., Coste A., Poordad F., et al. Acetyl-CoA carboxylase inhibitor GS-0976 for 12 weeks reduces hepatic de novo lipogenesis and steatosis in patients with nonalcoholic steatohepatitis. Clinical Gastroenterology and Hepatology. 2018;16(12):1983–1991.e3. doi: 10.1016/j.cgh.2018.04.042. [DOI] [PubMed] [Google Scholar]
- 209.Alkhouri N., Lawitz E., Noureddin M., DeFronzo R., Shulman G. I. GS-0976 (Firsocostat): an investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH) Expert Opinion on Investigational Drugs. 2020;29(2):135–141. doi: 10.1080/13543784.2020.1668374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Friedman S. L., Ratziu V., Harrison S. A., et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67(5):1754–1767. doi: 10.1002/hep.29477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Loomba R., Neutel J., Mohseni R., et al. LBP-20-VK2809, a novel liver-directed thyroid receptor beta agonist, significantly reduces liver fat with both low and high doses in patients with non-alcoholic fatty liver disease: a Phase 2 Randomized, Placebo-Controlled Trial. Journal of Hepatology. 2019;70(1):e150–e151. doi: 10.1016/S0618-8278(19)30266-X. [DOI] [Google Scholar]
- 212.Harrison S., Moussa S., Bashir M., et al. MGL-3196, a selective thyroid hormone receptor-beta agonist significantly decreases hepatic fat in NASH patients at 12 weeks, the primary endpoint in a 36 week serial liver biopsy study. Journal of Hepatology. 2018;68:p. S38. doi: 10.1016/s0168-8278(18)30292-7. [DOI] [Google Scholar]
- 213.Francque S., Vonghia L. Pharmacological treatment for non-alcoholic fatty liver disease. Advances in Therapy. 2019;36(5):1052–1074. doi: 10.1007/s12325-019-00898-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hu H., Lin A., Kong M., et al. Intestinal microbiome and NAFLD: molecular insights and therapeutic perspectives. Journal of Gastroenterology. 2020;55(2):142–158. doi: 10.1007/s00535-019-01649-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bomhof M. R., Parnell J. A., Ramay H. R., et al. Histological improvement of non-alcoholic steatohepatitis with a prebiotic: a pilot clinical trial. European Journal of Nutrition. 2019;58(4):1735–1745. doi: 10.1007/s00394-018-1721-2. [DOI] [PubMed] [Google Scholar]
- 216.Scorletti E., Afolabi P. R., Miles E. A., et al. Design and rationale of the INSYTE study: a randomised, placebo controlled study to test the efficacy of a synbiotic on liver fat, disease biomarkers and intestinal microbiota in non-alcoholic fatty liver disease. Contemporary Clinical Trials. 2018;71:113–123. doi: 10.1016/j.cct.2018.05.010. [DOI] [PubMed] [Google Scholar]