Болезнь Крейтцфельдта-Якоба | это... Что такое Болезнь Крейтцфельдта-Якоба? (original) (raw)
| Болезнь Крейтцфельдта — Якоба | |
|---|---|
| МКБ-10 | A81.0 F02.1 |
| МКБ-9 | 046.1 |
| OMIM | 123400 |
Болезнь Кройцфе́льдта — Я́коба, (более распространена транскрипция Крейтцфельдта — Якоба, названо по именам немецких врачей Hans Gerhard Creutzfeldt, Alfons Maria Jakob; синонимы: псевдосклероз спастический, синдром кортико-стриоспинальной дегенерации,трансмиссивная спонгиоформная энцефалопатия, коровье бешенство) — прогрессирующее дистрофическое заболевание коры большого мозга, базальных ганглиев и спинного мозга. Считается основным проявлением губчатой энцефалопатии (прионная болезнь).
Содержание
- 1 История
- 2 Эпидемиология
- 3 Этиология, патогенез и пути заражения
- 4 Формы болезни Кройцфельдта — Якоба
- 5 Клиника
- 6 Диагностика
- 7 Лечение
- 8 Источники
История
Заболевание было впервые описано в 1920 г. Гансом Кройцфельдтом. Альфонс Якоб в 1921 году отметил сочетание при этом заболевании психических нарушений с симптомами поражения передних рогов спинного мозга, экстрапирамидной и пирамидной системы, и определил заболевание как спастический псевдосклероз или энцефалопатию с рассеяными очагами поражения ткани мозга. Шпильмейер предложил называть болезнь по имени авторов, впервые описавших её.[1]
Эпидемиология
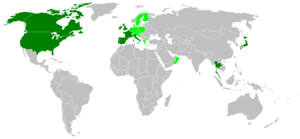
Темно-зеленым отмечены страны, где были случаи заболевания людей, светло-зеленым — случаи коровьего бешенства.
Болезнь Крейцфельда — Якоба составляет около 85 % всех прионовых энцефалопатий человека, поражает людей всех национальностей и рас, мужчин и женщин, взрослых и детей. Отмечается некоторое преобладание частоты случаев болезни куру у женщин, что связывают с особенностями национальных традиций (ритуального каннибализма) аборигенов острова Новой Гвинеи, когда женщины поедают мозг умерших и получают высокую дозу РrРSс. Начало заболевания наступает, как правило, в среднем или позднем возрасте, в типичных случаях на пятом десятке жизни, но может возникнуть в любом возрасте. Так, возраст дебюта классической формы варьирует от 17 до 87 лет (средний возраст — 64 года), в то время как средний возраст новой формы значительно меньше и составляет 29 лет.
Этиология, патогенез и пути заражения
См. Прионная болезнь
Попадая в организм, прион оседает на поверхности клетки, взаимодействуя с нормальными белками и изменяя их структуру на патологическую.
Накапливающиеся на поверхности клетки патологические белки блокируют процессы происходящие на мембране, что приводит к гибели клетки.
Накапливающиеся на поверхности клетки белки запускают апоптоз (запрограммированный процесс смерти клетки).
Клетка, стараясь избавиться от белков на поверхности, начинает производить активные кислородные соединения, однако белок на поверхности не даёт им выйти за пределы клетки. Активные вещества разрушают саму клетку.
Вокруг поражённых клеток начинаются воспалительные процессы с участием высокоактивных ферментов, поражающих соседние здоровые клетки.
Формы болезни Кройцфельдта — Якоба
Спонтанная — классическая (sCJD)
Согласно современным представлениям (прионной теории), прионы при этой форме заболевания возникают в мозге спонтанно, без какой-либо видимой внешней причины. Болезнь обычно поражает людей в возрасте более 50 лет и проявляется с вероятностью 1-2 случая на миллион жителей. Вначале проявляется в форме кратких потерь памяти, изменениями настроения, потерей интереса к происходящему вокруг. Больной постепенно перестает осиливать и текущие действия, связанные с каждодневной жизнью. В конечной фазе наступает расстройство зрения, галлюцинации и расстройство речи (особенная медленная речь).
Основные характеристики:
- около 40 % больных со спорадической формой имеют подострое течение с прогрессирующими когнитивными нарушениями, в 40 % случаев встречаются мозжечковые нарушения, в 20 % — их комбинация.
- клиническая картина включает расстройства поведения, нарушения высших корковых функций, корковые нарушения зрения (вплоть до корковой слепоты), мозжечковую дисфункцию, сочетание пирамидной и экстрапирамидной симптоматики
- эпилептические припадки — Практически у всех больных развиваются фокальные, в том числе миоклонус века, миоклонус губы и/или вторично-генерализованные миоклонические припадки, которые могут провоцироваться фоно- и фотостимуляцией, тактильным раздражением (прикосновением). У большинства больных во время ЭЭГ-исследования выявляются характерные периодические или псевдопериодические пароксизмы острых волн и/или спайков на общем замедленном низкоамплитудном фоне биоэлектрической активности головного мозга.
в терминальной стадии — глобальные выраженные когнитивные нарушения,
летальный исход через 8 месяцев от дебюта заболевания.
Наследственная (fCJD)
Карта хромосомы с указанием места мутации.
Болезнь возникает в семьях, где наследуется повреждение гена для прионового протеина. Дефектный прионовый протеин является намного более подверженным спонтанному превращению в прион. Признаки и ход болезни подобны классической форме.
Ятрогенная (1CJD)
Подробнее см. «Словарь медицинских терминов»
Болезнь возникает непреднамеренным внесением прионов в тело пациента при медицинском вмешательстве. Источником прионов ранее были некоторые лекарства, инструменты или мозговые оболочки, которые забирались у мертвых людей и использовались для закрытия раны при операциях мозга. Сегодня этот источник заражения полностью устранен. Признаки и ход болезни подобен классической форме.
Новый вариант (nvCJD)
Болезнь появилась впервые в 1995 г. в Великобритании и от того момента от нее умерло не более 100 человек. Вероятнее всего, что они заразились мясными продуктами, содержащими бычьи прионы из мозга «бешеных коров». В отличие от «классической» формы болезнь поражает и молодых людей в возрасте около 20 лет. В первую очередь она проявляется в изменении личности. Люди теряют интерес к своему хобби, сторонятся и своих самых близких, поддаются депрессиям. Далее следует похудание, нарушение координации движений. Пациент неспособен сам о себе позаботиться, не соблюдает основные правила гигиены, не может без чужой помощи поесть. Повреждение мозга все больше и больше нарушает основные жизненные функции, и, наконец, пациент умирает. В отличие от классической формы при новом варианте болезни Кройцфельдта — Якоба отдалено наступление слабоумия, и пациент очень долго осознает свое ухудшающееся состояние.
Основные характеристики:
- психические расстройства и сенсорные нарушения,
- характерны глобальные когнитивные нарушения и атаксия.
- описано несколько случаев заболевания, дебютировавшего с корковой слепоты (вариант Heidenhain).
- эписиндром представлен так же миоклоническими припадками.
- мозжечковая симптоматика выявляется в 100 %
Клиника
Клиническими критериями деменции при БКЯ являются:
- быстро прогрессирующая — в течение 2 лет — («опустошающая») деменция с дезинтеграцией всех высших корковых функций; пирамидные нарушения (спастические парезы);
- экстрапирамидные нарушения (хореоатетоз);
- миоклонус;
- атаксия, акинетический мутизм;
- дизартрия;
- эпилептические припадки;
- зрительные нарушения (диплопия)
Стадии заболевания:
- Продромальный период — симптомы неспецифичны и возникают примерно у 30 % больных. Они появляются за недели и месяцы до возникновения первых признаков деменции и включают астению, нарушения сна и аппетита, внимания, памяти и мышления, снижение массы тела, потерю либидо, изменение поведения.
- Инициальный период — для первых признаков заболевания обычно характерны зрительные нарушения, головные боли, головокружение, неустойчивость и парестезии. У основной части больных постепенно развивается, реже — острый или подострый дебют. В некоторых случаях, как при так называемых амиотрофических формах, неврологические знаки могут предшествовать началу деменции.
- Развернутый период — обычно отмечается прогрессирующий спастический паралич конечностей с сопутствующими экстрапирамидными знаками, тремором, ригидностью и характерными движениями. В других случаях может отмечаться атаксия, падение зрения или мышечная фибрилляция и атрофия верхнего двигательного нейрона.
Для спорадической БКЯ характерны:
- Терминальный период — тяжелая деменция вплоть до маразма. Все прионозы — быстро прогрессирующие заболевания. Течение может быть подострым, но обычно приводит к смерти не более чем через 1-2 года от момента четко очерченной клинической манифестации. Средняя длительность спорадической — около 8 месяцев. Длительность новой формы больше и достигает в среднем 16 месяцев. Средняя длительность наследственной формы — 26 месяцев, после чего наступает смерть.
Диагностика
БКЯ должна предполагаться во всех случаях деменций, которые прогрессируют быстро в течение месяцев или 1-2 лет и сопровождаются множественными неврологическими симптомами.
Определенная БКЯ:
- Характерная неврологическая и морфологическая в том числе патолого-анатомическая и нейрорадиологическая симптоматика.
- Протеазорезистентный РrР (по данным Western-блоттинга).
- Выявление скрепи-ассоциированных фибрилл.
Вероятная БКЯ:
- Прогрессирущая деменция.
- Характерный ЭЭГ-паттерн (для спорадической БКЯ).
- По крайней мере, 2 признака из нижеперечисленных:
- миоклонус;
- ухудшение зрения;
- мозжечковая симптоматика;
- пирамидные или экстрапирамидные симптомы;
- акинетический мутизм.
Возможная БКЯ:
- Прогрессирующая деменция.
- Нетипичные изменения на ЭЭГ (или ЭЭГ провести невозможно).
- По крайней мере, 2 признака из нижеперечисленных:
- миоклонус;
- ухудшение зрения;
- мозжечковая симптоматика;
- пирамидные или экстрапирамидные симптомы;
- акинетический мутизм.
- Продолжительность заболевания — менее 2 лет.
Из параклинических методов диагностики БКЯ наиболее информативными являются:
- характерные данные магнитно-резонансной томографии (МРТ) головного мозга (особенно на поздних стадиях развития заболевания) в виде билатеральных гиперинтенсивных сигналов на Т2-взвешенных изображениях (симптом «медовых сот»), преимущественно в области хвостатых ядер (в области головки), подушки главным образом таламуса; показано, что симптом «медовых сот» наиболее характерен для нвБКЯ; могут выявляться признаки атрофии коры больших полушарий и мозжечка;
- позитронно-эмиссионая томография (Позитронно-эмиссионная томография) менее информативна, выявляются множественные зоны гипометаболизма глюкозы на уровне подкорковых ядер и коры больших полушарий и полушарий мозжечка;
- для спорадической БКЯ характерно выявление на ЭЭГ трехфазной активности, ранняя пароксизмальная активность обычно диагностируется через 12 нед и более от дебюта спорадической БКЯ (у 80-88 % больных); фокальная, билатеральная и генерализованная миоклоническая пароксизмальная активность диагностируется в 15, 53 и 100 % случаев при продромальной, начальной и терминальной стадиях БКЯ соответственно; могут регистрироваться различные виды периодической пароксизмальной активности: двухфазные или трехфазные периодические комплексы, возникающие каждые 1-2 с; периодические комплексы с мультифазной конфигурацией; периодические полиспайковые разряды. ЭЭГ-паттерн «вспышка-подавление» характерен для терминальной стадии заболевания с явлениями декортикации;
- для новой формы выше описанные изменения ЭЭГ не характерны, ЭЭГ-паттерн может значимо не меняться по сравнению с возрастной нормой;
- результаты тестирования когнитивных функций (например, менее 24 баллов по краткой шкале ММSЕ;
- анализ ликвора (люмбальная пункция должна проводиться во всех случаях заболевания), учитываются давление ликвора, уровень сахара, цитоз, наличие бактериальных и вирусных культур, криптококкового антигена и др.; может быть небольшое повышение уровня белка (но не более 100 мг/дл); важным диагностическим критерием является уровень маркера в ликворе — чувствительность и специфичность этого теста превышает 90 %;
- если диагноз неясен, то возможно проведение прижизненной биопсии мозга (при наличии информированного согласия со стороны родственников или опекунов в случае недееспособности пациента);
- морфологическое и гистологическое исследование тканей головного мозга (коры, подкорковых ядер) при аутопсии (посмертная диагностика).
Лечение
Этиотропной терапии нет. Проводится симптоматическое лечение. При выявлении БКЯ необходимо отменить все лекарственные препараты, которые могут негативно влиять на мнестические функции и поведение пациента. Множество потенциальных терапевтических вмешательств при БКЯ остаются в настоящее время на уровне дискуссии. Традиционные противовирусные средства, такие как амантадин, интерфероны, пассивная иммунизация и вакцинация человека и животных, оказались неэффективными.
Среди лекарств положительно влияющих на процесс:
- Брефелдин А, разрушая аппарат Гольджи, препятствует синтезу PrPSc в инфицированной культуре клеток.
- Блокаторы кальциевых каналов, в частности NMDA-рецепторов, способствуют более длительному выживанию инфицированных нейрональных культур.
Источники
- ↑ Крейтцфельдта — Якоба болезнь // Большая медицинская энциклопедия, Т. 11. — 3-е изд. — М.: «Советская энциклопедия», 1979. — С. 519.
2. http://www.health-ua.com/articles/1736.html
Wikimedia Foundation.2010.