Finding Haplotype Block Boundaries by Using the Minimum-Description-Length Principle (original) (raw)
Abstract
We present a method for detecting haplotype blocks that simultaneously uses information about linkage-disequilibrium decay between the blocks and the diversity of haplotypes within the blocks. By use of phased single-nucleotide polymorphism data, our method partitions a chromosome into a series of adjacent, nonoverlapping blocks. The partition is made by choosing among a family of Markov models for block structure in a chromosomal region. Specifically, in the model, the occurrence of haplotypes within blocks follows a time-inhomogeneous Markov process along the chromosome, and we choose among possible partitions by using the two-stage minimum-description-length criterion. When applied to data simulated from the coalescent with recombination hotspots, our method reliably situates block boundaries at the hotspots and infrequently places block boundaries at sites with background levels of recombination. We apply three previously published block-finding methods to the same data, showing that they either are relatively insensitive to recombination hotspots or fail to discriminate between background sites of recombination and hotspots. When applied to the 5q31 data of Daly et al., our method identifies more block boundaries in agreement with those found by Daly et al. than do other methods. These results suggest that our method may be useful for designing association-based mapping studies that exploit haplotype blocks.
Introduction
In the past 2 years, a series of surveys of SNPs has documented linkage disequilibrium (LD) extending across long distances in the human genome (Daly et al. 2001; Reich et al. 2001 ; Gabriel et al. 2002). This long-range LD is not confined to occasional, distantly separated marker pairs in LD; rather, these studies report a blocklike LD structure. The blocks of LD are regions, typically <100 kb, in which LD decreases very little with distance between markers. Between these blocks, however, LD is observed to decay rapidly with physical distance. Concomitant with the blocks of LD, these studies also find low haplotype diversity within blocks. Often, >90% of the chromosomes in a sample possess one of only two to five haplotypes within a block (Daly et al. 2001; Patil et al. 2001; Gabriel et al. 2002).
The existence of haplotype block structure has serious implications for association-based methods for the mapping of disease genes. On the one hand, if a causative allele occurs within a long block of LD, then it may be difficult to localize that causative gene at a fine scale by association mapping. On the other hand, if the diversity of haplotypes within blocks is low, then common disease genes may be mapped, at least to within a haplotype block, by using far fewer markers than previously imagined (Goldstein 2001). Accordingly, a variety of methods for the identification of haplotype blocks from SNP data have been proposed (Daly et al. 2001; Patil et al. 2001; Gabriel et al. 2002; Wang et al. 2002; Zhang et al. 2002_b_). These methods can be classified into two categories: those that divide strings of SNPs into blocks on the basis of the decay of LD across block boundaries and those that delineate blocks on the basis of some haplotype-diversity measure within the blocks.
Daly et al. (2001), Gabriel et al. (2002), and Wang et al. (2002) define haplotype blocks on the basis of LD decay between blocks. Within a 500-kb region on 5q31, Daly et al. (2001) use a hidden Markov model in which the transition probabilities between states at adjacent markers are related to the decay of LD, as measured by the statistic _D_′. Unfortunately, their method appears to require the ability to identify a small set of “ancestral” haplotypes spanning the region of interest, so their method is not generally useful for new data sets. Wang et al. (2002) define haplotype blocks by using the four-gamete test (FGT) of Hudson and Kaplan (1985). Gabriel et al. (2002) define a block to be a region in which a small proportion of marker pairs show evidence for historical recombination. Hence, Gabriel et al.’s (2002) method is similar to Wang et al.’s (2002) method, except that the evidence for historical recombination is pairwise _D_′<1 in the latter, whereas, in the former, the evidence for historical recombination relies on whether the upper and lower confidence limits on estimates of pairwise _D_′ fall within certain threshold values. The threshold values for these confidence limits are subjectively determined for the populations under study, making them somewhat arbitrary and not easily applied to data sets that are from different populations or that have different sample sizes.
Patil et al. (2001) and Zhang et al. (2002_b_) partition sequences of SNPs into blocks by using methods that are based on haplotype diversity within blocks, instead of the decay of LD across block boundaries. They locate blocks so that the most common haplotypes within blocks in a data set can be identified using the smallest number of SNPs, called, following Johnson et al. (2001), “haplotype tagging SNPs” (htSNPs). Patil et al. (2001) find block boundaries by using a greedy algorithm, whereas Zhang et al. (2002_b_) present a dynamic programming algorithm to find the block partition corresponding to a globally minimal number of htSNPs. These methods are appealing because they suggest that, by defining blocks in an appropriate fashion, the number of SNPs that must be typed for association studies may be reduced (Zhang et al. 2002_a_). Zhang et al. (2002_b_) also show that other measures of block quality can be used in their dynamic programming algorithm, so long as the measures of block quality are functions of the SNPs within blocks, rather than between blocks. For example, they illustrate the use of their algorithm in conjunction with Clayton’s haplotype-diversity measure (Johnson et al. 2001).
Like LD, patterns of haplotype diversity are influenced by recombination, and, therefore, block boundaries based on measures of within-block haplotype diversity should coincide with drops in LD. However, these within-block methods are not specifically designed to define blocks in terms of the drop in LD across block boundaries, and both Patil et al. (2001) and Zhang et al. (2002_b_) have been careful not to attach specific biological meaning to the position of block boundaries found by their methods.
Here, we present a method for the detection of blocks in phased SNP data by simultaneously using information about LD decay between blocks and about the diversity of haplotypes within blocks. We develop our method by formalizing the task of finding block boundaries as a problem in statistical-model selection, and we then apply the minimum-description-length (MDL) criterion (Rissanen 1978, 1989) to select the block designations that best capture the structure within the data. The MDL criterion is an application of information theory to statistical-model selection. The “description length” of a data set is a penalized negative log-likelihood, which is a function of the number and position of block boundaries. The best set of block boundaries, by the MDL criterion, is the set that achieves the shortest description length for the data. (An introduction to the MDL principle may be found in Hansen and Yu 2001.) Lanterman (2001) provides a discussion of the relationship between MDL and other model-selection procedures. The MDL criterion has been successfully applied to a wide variety of model-selection problems in the fields of computer science, electrical engineering, and database mining.
Computing a description length for SNP data requires a probability model for the data. Within the MDL framework, this model is intended to provide a statistical description of the data and is not necessarily designed to mimic the genetic processes generating the data (Hansen and Yu 2001). Such a descriptive framework is appropriate for inference of haplotype blocks, because, even though the etiology of the blocks is unclear, the patterns of block structure observed in empirical studies are detailed enough to be useful in forming a model. We developed a variety of different probability models for haplotype blocks in SNP data, and we report here the simplest of those models, which captures both LD decay between blocks and low haplotype diversity within blocks. In this model, the haplotypes possessed by a chromosome at adjacent blocks occur as a Markov process, with varying degrees of dependence between different types. This variable dependence is motivated by the observation of Daly et al. (2001) that the blocks in 5q31 occur at locations where there is a reduction of LD, yet considerable association between a small number of common haplotypes at adjacent blocks may still occur. The Markov chain of our model is different from that of Daly et al. (2001), in that the “time steps” in their chain were individual SNPs, whereas, in ours, the “time steps” are different blocks. Consequently, the MDL method does not rely on LD between pairs of markers; rather, it exploits LD between whole haplotypes carried at adjacent blocks. Such a block-based measure of LD should be primarily affected by recombination between blocks and less affected by multiple mutation, gene conversion, and genotyping error than are LD measures based on pairwise comparisons between loci.
In the past half-year, two other block-partitioning methods using the MDL criterion have been independently developed and presented at symposia. Although these two other methods also use the MDL criterion, they are implemented differently. Koivisto et al. (2003) report a method that uses an underlying probability model, which is different from the Markov model that we employ. In their method, haplotypes within blocks are clustered using _k-_means clustering, and the description length depends on the number of clusters within a block and how closely haplotypes within blocks cluster together (as well as on the number and position of blocks). Their method is thus based on a measure of within-block haplotype diversity. Unlike our method, Koivisto et al.’s (2003) model does not account for the association between common haplotypes at adjacent blocks, as observed by Daly et al. (2001). Greenspan and Geiger (2003) present an MDL-based haplotype block–partitioning method that has the attractive feature in that it accepts both phased haplotype data and unphased genotype data. When using unphased genotype data, their program is capable of estimating the underlying haplotypes. They base their description length on a Markov model similar to the one that we use, but their model underlying the distribution of types within a block is more complex, allowing for the specification of a limited number of ancestral haplotypes from which the observed haplotypes are derived by way of (possibly recurrent) mutation. This extra complexity makes minimizing the description length over the different parameters of their model a difficult task, which they tackle with a heuristic search strategy. We employ a more efficient optimization via a dynamic programming algorithm.
In the second part of the present article, we assess how our method and other methods behave when applied to simulated data. We undertake a series of simulations using the coalescent with recombination hotspots to induce LD decay at defined locations and produce a known block structure in the data. Recombination hotspots have been shown to be associated with blocks of LD in single-sperm–typing studies (Jeffreys et al. 2001; Cullen et al. 2002), and population-level data are consistent with the occurrence of recombination hotspots at block boundaries (Gabriel et al. 2002; Kauppi et al. 2003). Thus, our simulations provide an approximation to one of the possible processes giving rise to haplotype blocks. Regardless of how well these simulations correspond to the actual process that produces haplotype blocks, they do reveal useful results about the behavior of different block-finding methods. The main conclusions from these simulations are that (1) in the presence of recombination hotspots, our MDL-based method locates block boundaries at the hotspot sites and rarely places block boundaries at sites that recombine with lower intensity; (2) methods based on haplotype diversity are relatively insensitive to LD decay across block boundaries; and (3) the FGT-based method of Wang et al. (2002) locates block boundaries not only at the sites of recombination hotspots but also at numerous other, nonhotspot locations.
Finally, we apply various block-finding methods to the 5q31 data of Daly et al. (2001) and to the biallelic polymorphisms found in 86 complete human mtDNA sequences. Daly et al. (2001) originally discovered and assessed haplotype blocks in 5q31, with a significant amount of human interaction with the data. The blocks delineated by Daly et al. (2001) provide a compelling picture of block structure, which one would hope could be recovered comparably with “unsupervised” (i.e., not requiring considerable human input) block-finding methods. The blocks found by the MDL method are nearly in agreement with the original blocks found by Daly et al. (2001). The htSNP method of Zhang et al. (2002_b_), although largely insensitive to LD decay in the simulated data, also locates blocks in the 5q31 data, close to the blocks described by Daly et al. (2001). The FGT-based method, however, finds no fewer than 75 blocks in the 5q31 data, each with an average length of only 1.37 SNPs, suggesting that the FGT method’s utility may be reduced in the presence of the gene conversion, multiple mutation, and genotyping errors that are expected in real data. This finding is corroborated by analysis of the mitochondrial data.
Methods
In the present section, we formulate our statistical model and elaborate on the theory behind the MDL criterion. A detailed account of the computation of the description length and its optimization over possible block assignments is given in appendixes A–C. The method is implemented in the program MDBlocks, available on the World Wide Web (see MDBlocks Home).
We use the MDL criterion (Rissanen 1989) to infer block boundaries from SNP-marker data typed on a sample of chromosomes. The MDL criterion relies on the relationship between the probability of a random variable and the amount of information, measured in binary digits, or “bits,” required in order to encode the values of that random variable in a binary prefix code (see Cover and Thomas 1991, chap. 5). In brief, for a random variable W taking values in a set 𝒲, a prefix code is a code that establishes an association between each value _w_∈𝒲 and a unique code word, consisting of a series of 0s and 1s, such that no code word is the prefix of any other code word. We denote by 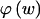 the length, in bits, of the code word associated with the value w. This quantity is called the “code length” of w. An efficient code—one that requires, on average, few bits in order to transmit a series of instances of _W_—will assign short code words to the frequent values of W and longer code words to the less frequent values of W. This is a fundamental principle of data compression. Two laws from the field of information theory, Kraft’s inequality and Shannon’s source-coding theorem, together give the result that the prefix code with the smallest expected code length for transmitting a sequence of values of W is achieved by assignment of code words to values of w, such that
the length, in bits, of the code word associated with the value w. This quantity is called the “code length” of w. An efficient code—one that requires, on average, few bits in order to transmit a series of instances of _W_—will assign short code words to the frequent values of W and longer code words to the less frequent values of W. This is a fundamental principle of data compression. Two laws from the field of information theory, Kraft’s inequality and Shannon’s source-coding theorem, together give the result that the prefix code with the smallest expected code length for transmitting a sequence of values of W is achieved by assignment of code words to values of w, such that 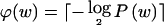 for all _w_∈𝒲, where
for all _w_∈𝒲, where 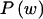 is the probability mass function for W and
is the probability mass function for W and 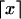 denotes the smallest integer equal to or larger than x. In other words, the optimal coding scheme is one that assigns a code word of length
denotes the smallest integer equal to or larger than x. In other words, the optimal coding scheme is one that assigns a code word of length 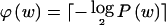 to every _w_∈𝒲.
to every _w_∈𝒲.
The MDL criterion exploits this connection between probability and information in the case in which the random variable in question is a data set with random features that can be described by a parameterized probability model for the data. In such a case, the code length (dispensing with the “ ” function and allowing for fractional bits, as we will do for the remainder of the present article) of the data set, conditional on the model and all its parameters, is the negative log-likelihood function with the log taken to base 2. Under this interpretation, the familiar maximum-likelihood estimate is identical to the value of the parameter that minimizes the code length of the observed data. The MDL framework extends more readily, however, than the maximum-likelihood framework to problems of model selection between nonnested models, because models and their parameters may also be described in terms of the code length required in order to encode them, providing a natural way of penalizing more-complex models for the addition of parameters. One way of formalizing this model-selection framework is by choosing the model that minimizes the description length for a two-stage coding of the model and the data (Rissanen 1989; Hansen and Yu 2001; Lee 2001). In a two-stage coding scheme, the description length is the sum of the code length required in order to encode the estimated values of the parameters in the model and the code length of the data set under the chosen model and parameter values.
” function and allowing for fractional bits, as we will do for the remainder of the present article) of the data set, conditional on the model and all its parameters, is the negative log-likelihood function with the log taken to base 2. Under this interpretation, the familiar maximum-likelihood estimate is identical to the value of the parameter that minimizes the code length of the observed data. The MDL framework extends more readily, however, than the maximum-likelihood framework to problems of model selection between nonnested models, because models and their parameters may also be described in terms of the code length required in order to encode them, providing a natural way of penalizing more-complex models for the addition of parameters. One way of formalizing this model-selection framework is by choosing the model that minimizes the description length for a two-stage coding of the model and the data (Rissanen 1989; Hansen and Yu 2001; Lee 2001). In a two-stage coding scheme, the description length is the sum of the code length required in order to encode the estimated values of the parameters in the model and the code length of the data set under the chosen model and parameter values.
In the next subsection (“Probability Model for Haplotypes and Blocks”), we introduce a descriptive probability model for the data. This probability model provides the basis for computing the code length of the data. The subsequent subsection (“Two-Stage Coding”) describes in more detail the two-stage coding. Appendixes A, B, and C respectively describe how we compute the description length of the parameters, how we optimize the description length over possible choices of block boundaries by using a dynamic programming algorithm, and how we treat missing data. Appendix D summarizes the notation used in the present article.
Probability Model for Haplotypes and Blocks
The data consist of N homologous chromosomes sampled from a population and typed at M SNPs each. The map location of the SNPs is used to order the SNPs from 1 to M along each chromosome, but our model does not otherwise consider the distance between SNPs. The two alleles at each SNP may be coded as 0 or 1, and we denote the allele carried on the _i_th chromosome at the _j_th SNP by 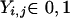 , for _i_=1,…,N and _j_=1,…,M. The data set can be thought of as a matrix Y of 0s and 1s with N rows and M columns. We use the term “block” to refer to a collection of consecutive SNPs, and we describe a haplotype block structure in the data set by the specification of R adjacent, nonoverlapping blocks, indexed by k (_k_=1,…,R). The index of the last SNP in the _k_th block is denoted by
, for _i_=1,…,N and _j_=1,…,M. The data set can be thought of as a matrix Y of 0s and 1s with N rows and M columns. We use the term “block” to refer to a collection of consecutive SNPs, and we describe a haplotype block structure in the data set by the specification of R adjacent, nonoverlapping blocks, indexed by k (_k_=1,…,R). The index of the last SNP in the _k_th block is denoted by 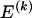 , and the _k_th block contains
, and the _k_th block contains 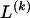 SNPs. Since the blocks are adjacent, the _k_th block begins at SNP index
SNPs. Since the blocks are adjacent, the _k_th block begins at SNP index 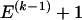 and ends at
and ends at 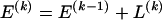 . Thus, the right endpoints of the blocks
. Thus, the right endpoints of the blocks 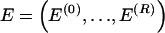 , where
, where 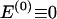 , specify the block structure. Our notation follows the convention that variables superscripted by “(k)” pertain to the _k_th block.
, specify the block structure. Our notation follows the convention that variables superscripted by “(k)” pertain to the _k_th block.
We use the words “type” or “haplotype” only in reference to a particular block of SNPs: a haplotype is characterized by a sequence of 0s and 1s within a block. At the _k_th block, the N chromosomes in the data set will exhibit a number 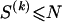 of distinct haplotypes. Across all blocks, we use S to denote the vector
of distinct haplotypes. Across all blocks, we use S to denote the vector 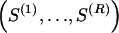 . We denote the set of distinct haplotypes observed in the data set at block k by
. We denote the set of distinct haplotypes observed in the data set at block k by 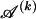 .
.
We model the occurrence of haplotypes along a chromosome, given R and E , by a simple Markov model with blocks playing the role of “time” and with time-inhomogeneous transition probabilities. In other words, the probability that a chromosome carries a certain haplotype at block k, conditional on the haplotypes carried on the chromosome at all blocks to the left of k, depends only on the haplotype carried by the chromosome at block _k_-1. The parameters of the Markov model are the unknown population frequencies of the types at the first block, 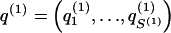 , and the unknown population frequencies,
, and the unknown population frequencies, 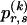 , of haplotype pairs in adjacent blocks. The parameter
, of haplotype pairs in adjacent blocks. The parameter 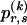 is the probability that a chromosome sampled from the population and carrying type r at block _k_-1 carries type s at block k. We let
is the probability that a chromosome sampled from the population and carrying type r at block _k_-1 carries type s at block k. We let 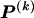 be an
be an 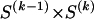 transition-probability matrix with entry
transition-probability matrix with entry 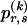 at the _r_th row and the _s_th column, and
at the _r_th row and the _s_th column, and 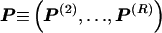 . The statistics of the data under the Markov model are the observed counts,
. The statistics of the data under the Markov model are the observed counts, 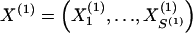 , of the
, of the 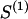 different haplotypes at block 1 and, for each _k_=2,…,R , an
different haplotypes at block 1 and, for each _k_=2,…,R , an 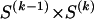 matrix
matrix 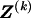 whose entry at the _r_th row and the _s_th column is
whose entry at the _r_th row and the _s_th column is 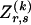 , the number of chromosomes in the data set having haplotype r at block _k_-1 and type s at block k. Given R, E , S , P, and
, the number of chromosomes in the data set having haplotype r at block _k_-1 and type s at block k. Given R, E , S , P, and 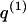 , the probability of the occurrence of types along every chromosome in the data set can be expressed as a function of
, the probability of the occurrence of types along every chromosome in the data set can be expressed as a function of 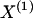 and
and 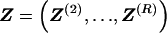 . The log of the probability of the types on all the chromosomes, with the order of chromosomes within the data set considered fixed, is hence
. The log of the probability of the types on all the chromosomes, with the order of chromosomes within the data set considered fixed, is hence
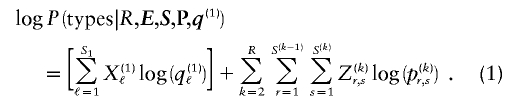
This probability distribution does not attempt to follow the stochastic process of genealogical inheritance, mutation, and sampling in a population that gives rise to the sample. Considerable work has been done on such models of coalescence with recombination that try to capture the actual process generating the data. However, inference based on the coalescent with recombination is difficult in this particular application, both because of computational considerations and because the specific demographic or recombinational processes generating the block structure are not well known. Instead, we propose our model as a computationally tractable statistical description of the dominant features of interest in the data.
Although the pattern of largely independent haplotypes across block boundaries, with occasional association between a few types, can be reasonably modeled using a Markov chain, the above formulation demands the specification of more parameters than are typically necessary. Imagine, for example, that there is no association between haplotypes in blocks _k_-1 and k. It would be superfluous to specify every single 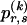 in this case, because
in this case, because 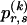 would be the same for all r and would simply equal the marginal frequency of the types at block k. Because the MDL criterion imposes a penalty for models with additional parameters, we consider parameterizing the Markov model with transition matrices
would be the same for all r and would simply equal the marginal frequency of the types at block k. Because the MDL criterion imposes a penalty for models with additional parameters, we consider parameterizing the Markov model with transition matrices 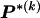 (as opposed to
(as opposed to 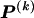 ), which have their entries determined in a way that requires fewer parameters than the
), which have their entries determined in a way that requires fewer parameters than the 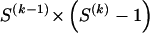 required in order to specify all the elements of each
required in order to specify all the elements of each 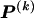 . We do this by setting each value
. We do this by setting each value 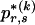 to the marginal frequency of the _s_th type in the _k_th block, unless there is a strong enough association between one or more types across the block boundary such that the overall description length (of the data and the parameters) may be reduced by explicitly modeling that dependence.
to the marginal frequency of the _s_th type in the _k_th block, unless there is a strong enough association between one or more types across the block boundary such that the overall description length (of the data and the parameters) may be reduced by explicitly modeling that dependence.
To specify the elements of 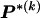 as above, we use the unknown marginal frequencies of the
as above, we use the unknown marginal frequencies of the 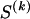 types at block k (_k_=2,…,R). Let the row vector
types at block k (_k_=2,…,R). Let the row vector 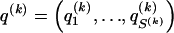 denote those unknown marginal frequencies. Also, let Δ be an
denote those unknown marginal frequencies. Also, let Δ be an 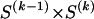 matrix of indicators
matrix of indicators 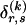 , which take the value 1 if the dependence between type r in block _k_-1 and type s in block k is to be explicitly modeled and the value 0 otherwise. We define the transition-probability matrix
, which take the value 1 if the dependence between type r in block _k_-1 and type s in block k is to be explicitly modeled and the value 0 otherwise. We define the transition-probability matrix 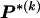 to have elements equal to those of
to have elements equal to those of 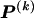 at all entries for which the corresponding elements in Δ are 1. For the remaining entries (corresponding to
at all entries for which the corresponding elements in Δ are 1. For the remaining entries (corresponding to 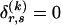 ), the values of
), the values of 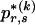 are set to be proportional to the corresponding components of
are set to be proportional to the corresponding components of 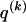 and are scaled so that the rows of
and are scaled so that the rows of 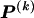 sum to unity. That is,
sum to unity. That is,
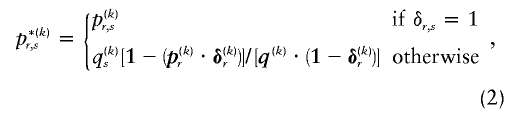
where 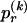 and
and 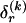 respectively denote the _r_th rows of
respectively denote the _r_th rows of 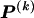 and
and 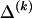 , 1 is a vector of 1s, and · is the vector dot product. This scheme allows the significant dependence between a small number of types between adjacent blocks to be modeled without having to invoke a very large number of parameters. In appendix A, we give the details of how we choose values for Δ. The log probability of
, 1 is a vector of 1s, and · is the vector dot product. This scheme allows the significant dependence between a small number of types between adjacent blocks to be modeled without having to invoke a very large number of parameters. In appendix A, we give the details of how we choose values for Δ. The log probability of 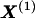 and Z, given
and Z, given 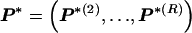 , is given by equation (1) but with
, is given by equation (1) but with 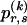 replaced by
replaced by 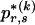 .
.
The above formulas describe a probability model for the occurrence of types along a chromosome. To apply the MDL criterion, however, we must compute the code length of the whole data set, which includes the sequences of the types at each block. Thus, we need a probability model for the sequence of 0s and 1s of each type in 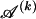 for _k_=1,…,R. Coalescent theory informs us of a reasonable form for the distribution of such sequences in a sample, but the actual calculation of that probability is computationally expensive because the number of possible coalescent topologies is so large. Some approximations have been developed (e.g., see Stephens and Donnelly 2000), but these too are difficult to implement and are also computationally demanding. Instead, we assume a simple Bernoulli model for sequences within each type in
for _k_=1,…,R. Coalescent theory informs us of a reasonable form for the distribution of such sequences in a sample, but the actual calculation of that probability is computationally expensive because the number of possible coalescent topologies is so large. Some approximations have been developed (e.g., see Stephens and Donnelly 2000), but these too are difficult to implement and are also computationally demanding. Instead, we assume a simple Bernoulli model for sequences within each type in 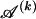 —the value of 0 or 1 at each site ℓ within a block k is drawn independently—with probability
—the value of 0 or 1 at each site ℓ within a block k is drawn independently—with probability 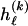 if the site is a 1 and with probability
if the site is a 1 and with probability 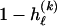 if it is a 0. We denote the vector of values of
if it is a 0. We denote the vector of values of  for all
for all  SNPs in block k by
SNPs in block k by 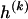 , and we define
, and we define 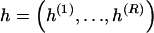 . When we let
. When we let 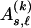 denote the type of the ℓth SNP of the _s_th type in the set
denote the type of the ℓth SNP of the _s_th type in the set 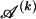 , the code length of the list of sequences in
, the code length of the list of sequences in 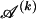 is computed as
is computed as
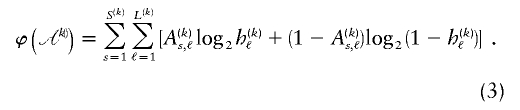
Two-Stage Coding
A useful way of thinking about two-stage coding is to imagine that you wish to transmit, in binary data format, the data set of SNPs, transmitting as few bits as possible. If you naively sent only one bit (a 0 or a 1) for each value of Y i,j , then you would have to send N_×_M bits to transmit the whole data set. This might be a reasonable coding scheme if every SNP in the data set were a 0 or a 1, with equal probability, independently of all other SNPs; however, that is not the case. Instead, there is a structure to the pattern of 0s and 1s in haplotype data, and this structure may be exploited to minimize the total number of bits required in order to transmit the data. Such data compression may be achieved by imagining that the data set is a realization from a suitable descriptive probability model, the family of which has been agreed on ahead of time by the transmitter and the receiver. By the relationship between probability and code length, the probability model and its parameter values determine the best prefix codes to be used to denote and transmit certain features of the data set. For the transmitted message to be properly received, the values of the parameters must be sent before the rest of the data features, so that the receiver will know how to properly decode the portion of the message that includes the data set. Hence, the total number of bits required is the description length of the parameters plus the description length needed to transmit the data by using the coding scheme implied by the probability model and its previously transmitted parameter values.
We have implemented a two-stage coding scheme in which the family of descriptive models is that given by equation (1) for the blocks and that implied by equation (3) for the sequences within each block. The overall description length 𝒟1 for the first stage is the sum of the code lengths for each of the parameters, R, E , S , P*, q , and h (see appendix A). For the second stage, the types carried by each chromosome must first be transmitted by sending a series of prefix code words for each type carried at each block on each chromosome. The number of bits required in order to transmit this information is given by equation (1), with the log taken to base 2. The final part of the second stage involves transmitting the sequences of the types within each block. For all the types at all the blocks, this requires the code length given by equation (3). The overall description length of the second stage is thus
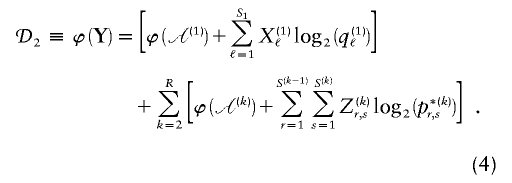
In the two-stage MDL framework, the best model is the one that minimizes the total description length, 𝒟1 + 𝒟2. Thus, there is a trade-off: increasing the complexity of the model requires more bits in order to describe the model and parameters, but, if the extra description length required for the model is more than offset by a reduction in the description length of the data themselves, then the more complex model is justified.
In appendix B, we present an algorithm for minimizing the total description length over models indexed by the number of blocks and the positions of the block boundaries. Because the method relies on successively performing a dynamic programming algorithm, we call the algorithm the “iterative dynamic programming (IDP) algorithm.” We also have developed a more computationally efficient, but approximate, algorithm that we call the “iterative approximate dynamic programming (IADP) algorithm.” The input to each algorithm is the N_×_M matrix of phased SNP data, and the output is the number of blocks and positions of the block boundaries corresponding to the MDL found by each algorithm. Appendix C describes how our program handles missing data by imputing the missing values in such a way as to minimize the entropy of the observed distribution of haplotypes within a block.
Applications
Coalescent Simulations with Recombination Hotspots
To evaluate our method’s ability to identify regions of rapid LD decay, we applied it to data simulated using the coalescent with recombination hotspots. Although these simulations do not recreate all of the patterns observed in haplotype data, they do create LD patterns, in the data, that are like those described in recent empirical studies. We also applied other block-finding algorithms to the same simulated data, to compare the results and behaviors of these different methods.
To simulate SNP data with recombination hotspots, we modified the program of Hudson (2002) to include variable recombination rates at sites of recombination. In the program, the scaled population recombination rate ρ for the entire simulated chromosomal segment was apportioned to equally spaced sites separating 200 chromosomal segments, and, among those sites, 10 equally spaced ones were chosen to be hotspots, having recombination rates R h times greater than the “background” rate of recombination. We performed sets of simulations for R h values equal to 50, 100, and 200 and ρ values equal to 200 and 500 for sample sizes of _N_=18 and _N_=100. The proportion of all recombinations that occurred within intermarker intervals containing hotspots was, on average, ∼75%, ∼85%, and ∼90% for R h values of 50, 100, and 200, respectively. For R _h_=200, the percentage of recombinations occurring at hotspots is on the order of the value of 95%, found by Jeffreys et al. (2001) in the major histocompatibility complex (MHC) region. The presence of hotspots leads to a bimodal distribution of the number of recombinations occurring between adjacent marker pairs: most adjacent marker pairs experience few recombination events in the ancestry of the sample; however, adjacent markers separated by hotspots experience many more recombinations. Figure 1 shows an example of this bimodal distribution. In addition, we simulated five hotspots equally spaced in the first half of the chromosome and no hotspots in the second half. The conclusions regarding the behavior of block-finding methods from these simulations with 5 hotspots were qualitatively similar to those with 10 hotspots (results not shown). All simulations assumed a population of constant size.
Figure 1.

Distribution of the number of recombination events between adjacent marker pairs. In 1,000 ancestries simulated from the coalescent with 10 recombination hotspots, we recorded the number of recombinations occurring in intervals between adjacent marker pairs. The plot shows a histogram of the number of intermarker intervals within which a given number of recombination events occurred. This histogram has two modes: the right mode, which represents recombinations occurring at simulated hotspots, accounts for only ∼9% of the intermarker intervals but ∼95% of all recombinations; the left mode represents recombinations that did not occur at hotspots. The distribution was obtained for the parameter settings of R _h_=200, ρ=200, and _N_=18. (Note the _Y-_axis break between 2,000 and 8,000.)
Although the recombination locations were equally spaced, the mutations were placed randomly within the entire segment according to the infinite-sites model. Only sites with a minor-allele frequency >0.05 in a sample of 100 were included in the sample, and the number of segregating sites was chosen so that a mean of ∼110 sites was obtained in each simulation replicate. Although genotyping technologies are constantly evolving, realistic marker densities are on the order of one marker every 2–5 kb, and suggested block lengths range from 5 to 100 kb (Daly et al. 2001; Patil et al. 2001; Gabriel et al. 2002). If we assume a recombination fraction of 1% per Mb and an effective size of 10,000 for the human population, then the simulations with ρ=200 are equivalent to a marker density of approximately one marker every 4.5 kb and block lengths of, on average, 45 kb. Under the same assumptions, a value of ρ=500 is approximately equivalent to one marker every 11 kb and average block lengths of 110 kb. We can also use the relationship suggested by Nordborg and Tavare (2002), wherein ρ=100 is equivalent to ∼100 kb. Under this assumption, in our simulations, ρ=200 corresponds to one marker every 1.8 kb with blocks of average length 18 kb, and ρ=500 corresponds to one marker every 4.5 kb with blocks of average length 45 kb.
We applied the following four block-finding algorithms to the simulated data and compared their behavior.
htSNP:
Patil et al.’s (2001) htSNP-based criterion as implemented by Zhang et al. (2002_b_) in the program Hapblock. We used the default settings of 80% coverage and ⩾80% of all haplotypes in a block represented more than once.
DB:
The diversity-based method suggested by Zhang et al. (2002_b_), using Clayton’s haplotype-diversity measure (Johnson et al. 1997). The method is implemented in Zhang et al.’s (2002_b_) Hapblock program, and we again used the default settings.
FGT:
The method presented by Wang et al. (2002), based on the FGT. We implemented the method in the C programming language.
MDB:
Our MDL-based method with the IADP algorithm, implemented in the computer package MDBlocks.
We applied all four methods to the same set of 1,000 simulated data sets with _N_=18. We also applied the FGT and MDB methods to 1,000 data sets of N = 100; the execution of the DB and htSNP methods was too slow to apply them to these data sets.
To describe the behavior of various methods, we calculated the proportion of all recombination hotspots that were identified as block boundaries, calling this statistic the “hotspot sensitivity.” Although none of the four methods are specifically designed to detect hotspots of recombination, the extent to which a method detects locations where LD drops sharply is relevant to SNP selection for association studies. In the simulations, LD dropped markedly at recombination hotspots, so the assessment of how well each method identifies simulated recombination hotspots is of practical interest. We also recorded the proportion of block boundaries that were not concordant with hotspots. We refer to this number as the hotspot nonconcordance (HNC) rate. Since relatively few recombinations occurred at sites that are not hotspots (fig. 1), the HNC rate measures how often methods declare block boundaries where the drop in LD is, on average, relatively small. “Sensitivity” and “nonconcordance” could be defined more loosely; for example, a block boundary could be considered concordant when it is separated from a hotspot by some small number of SNPs, instead of when it is exactly within the interval containing the hotspot. The conclusions from such a relaxed definition are similar to what is reported here for the more strict definition (results not shown).
For _N_=18, the hotspot sensitivity of the MDB method was comparable to that of the most sensitive methods, but the HNC rate of the MDB method was consistently lower than the HNC rates of the other methods (table 1). The differences in hotspot sensitivity between the MDB, FGT, and htSNP methods were never >15%. In contrast, the decrease in the HNC rate for the MDB method relative to the other methods is never <18% and is typically >22%. Overall, the results suggest that the MDB method performs well in the inference of block boundaries at locations with sharp drops in LD and that the MDB method also has the discriminating power to not identify as block boundaries those locations with comparatively small drops in LD. The DB method had the lowest hotspot sensitivity across all parameter sets, perhaps because our simulations did not create patterns of low haplotype diversity between hotspots. The results suggest that haplotype-diversity–based block-finding methods can be insensitive to sharp drops in LD.
Table 1.
Hotspot Sensitivity and Nonconcordance for Simulated Data with _N_=18[Note]
| Hotspot SensitivityWhen R ha Is | |||
|---|---|---|---|
| Method | 50 | 100 | 200 |
| MDB:b | |||
| ρ=200 | .52 (.18) | .62 (.17) | .67 (.16) |
| ρ=500 | .47 (.23) | .63 (.19) | .73 (.15) |
| FGT:c | |||
| ρ=200 | .58 (.16) | .60 (.16) | .62 (.15) |
| ρ=500 | .63 (.16) | .65 (.15) | .67 (.15) |
| DB:d | |||
| ρ=200 | .35 (.15) | .36 (.15) | .37 (.14) |
| ρ=500 | .39 (.15) | .42 (.16) | .44 (.16) |
| htSNP:e | |||
| ρ=200 | .51 (.16) | .54 (.16) | .55 (.15) |
| ρ=500 | .56 (.16) | .60 (.14) | .63 (.16) |
| Hotspot NonconcordanceWhen R ha Is | |||
| 50 | 100 | 200 | |
| MDB:b | |||
| ρ=200 | .47 (.17) | .38 (.16) | .32 (.14) |
| ρ=500 | .55 (.16) | .45 (.15) | .35 (.14) |
| FGT:c | |||
| ρ=200 | .70 (.09) | .63 (.11) | .55 (.12) |
| ρ=500 | .77 (.06) | .72 (.07) | .65 (.09) |
| DB:d | |||
| ρ=200 | .69 (.14) | .64 (.15) | .61 (.15) |
| ρ=500 | .73 (.11) | .68 (.13) | .62 (.14) |
| htSNP:e | |||
| ρ=200 | .74 (.10) | .70 (.11) | .68 (.11) |
| ρ=500 | .78 (.07) | .74 (.08) | .70 (.10) |
HNC rates were typically highest for the FGT and htSNP methods. Both of those methods identified block boundaries at many more locations than the 10 hotspots that were simulated. For instance, with ρ=500 and R _h_=200, the FGT and htSNP methods found, on average, ∼20 block boundaries, whereas the MDB method found, on average, only ∼10 block boundaries (table 2). This is not surprising for the FGT method, because it will declare a block boundary between any two adjacent sites with evidence of one or more recombinations in the history of the sample.
Table 2.
Average Number of Blocks Inferred across Various Simulation Settings[Note]
| Number of Blocks When R ha Is | ||||
|---|---|---|---|---|
| Method | 1 | 50 | 100 | 200 |
| _N_=18 | ||||
| MDB:b | ||||
| ρ=200 | 8.8 (3.0) | 10.8 (2.2) | 10.9 (1.8) | 10.9 (1.6) |
| ρ=500 | 3.9 (3.2) | 11.1 (4.0) | 12.4 (2.4) | 12.3 (1.7) |
| FGT:c | ||||
| ρ=200 | 27.1 (3.7) | 20.1 (2.8) | 17.4 (2.4) | 15.0 (1.9) |
| ρ=500 | 36.9 (4.0) | 28.0 (3.2) | 24.2 (3.1) | 20.5 (2.6) |
| DB:d | ||||
| ρ=200 | 14.6 (2.0) | 12.0 (1.6) | 11.2 (1.5) | 10.4 (1.4) |
| ρ=500 | 19.3 (2.0) | 15.8 (1.7) | 14.1 (1.6) | 12.67(1.4) |
| htSNP:e | ||||
| ρ=200 | 25.1 (5.4) | 21.5 (4.9) | 20.2 (5.2) | 19.2 (4.9) |
| ρ=500 | 32.2 (4.9) | 27.0 (4.6) | 24.7 (4.8) | 22.4 (4.7) |
| _N_=100 | ||||
| MDB:b | ||||
| ρ=200 | 11.8 (1.8) | 11.4 (1.1) | 11.1 (.9) | 10.9 (.7) |
| ρ=500 | 18.8 (2.5) | 14.1 (1.7) | 12.6 (1.3) | 11.7 (.8) |
| FGT:c | ||||
| ρ=200 | 38.3 (3.9) | 28.0 (3.0) | 23.6 (2.7) | 19.6 (2.3) |
| ρ=500 | 50.5 (4.5) | 38.7 (3.6) | 33.3 (3.3) | 27.9 (2.9) |
The difference in HNC between the MDB and FGT methods is far more pronounced for _N_=100 (table 3). With ρ=200 and R _h_=200, the mean HNC rate of the MDB method was 0.10, whereas, for the FGT method, it was 0.52. These rates reflect that, on average, the MDB method inferred 9 of the 10 hotspots as block boundaries and inferred only one additional block boundary where a hotspot did not exist. By contrast, the FGT method also typically identified as block boundaries 9 of the 10—or all 10—hotspots, but it also inferred block boundaries at an additional ⩾10 locations that were not at recombination hotspots. Relative to the simulations with _N_=18, when _N_=100, the hotspot sensitivity is higher for both the MDB and FGT methods, and the HNC rate is lower for the MDB method, suggesting that a large sample size is valuable for the accurate location of block boundaries where sharp drops in LD occur.
Table 3.
Hotspot Sensitivity and Nonconcordance for Simulated Data with _N_=100[Note]
| Hotspot SensitivityWhen R ha Is | |||
|---|---|---|---|
| Method | 50 | 100 | 200 |
| MDB:b | |||
| ρ=200 | .92 (.09) | .95 (.07) | .96 (.06) |
| ρ=500 | .90 (.09) | .95 (.07) | .97 (.06) |
| FGT:c | |||
| ρ=200 | .97 (.06) | .97 (.05) | .98 (.05) |
| ρ=500 | .9 (.10) | .91 (.09) | .93 (.08) |
| Hotspot NonconcordanceWhen R ha Is | |||
| 50 | 100 | 200 | |
| MDB:b | |||
| ρ=200 | .21 (.13) | .14 (.11) | .09 (.09) |
| ρ=500 | .30 (.11) | .18 (.10) | .09 (.09) |
| FGT:c | |||
| ρ=200 | .69 (.06) | .62 (.07) | .52 (.08) |
| ρ=500 | .76 (.03) | .72 (.04) | .65 (.05) |
A boundary declared by the MDB method is more likely to have had a high number of recombinations than a boundary declared by the FGT method or the htSNP method. This can be seen in the distribution of the number of recombinations occurring at block boundaries inferred by the various methods (fig. 2), which shows that a higher proportion of block boundaries inferred by the MDB method have had a high number of recombinations when compared to block boundaries inferred by the FGT method or the htSNP method.
Figure 2.

Distribution of the number of recombinations within marker intervals identified as block boundaries, for the MDB (A), FGT (B), and htSNP (C) methods. Plus symbols (+) show the proportion of intermarker intervals, identified as block boundaries by each method, within which a given number of recombinations occurred in 1,000 simulated ancestries. Open circles (○) show the proportion of all intermarker intervals (irrespective of whether they were inferred to contain block boundaries) within which a given number of recombinations occurred (as in fig. 1). If block boundaries were inferred completely at random, the two distributions within each panel would be nearly identical. The relative height of the right mode of the plus symbols reflects the extent to which block-finding methods selectively infer intervals containing hotspots to be block boundaries. Simulation parameters were R _h_=200, ρ=200, and _N_=18 for 10 hotspots.
Coalescent Simulations with Uniform Recombination
We also investigated the behavior of block-finding algorithms on data simulated with uniformly distributed recombination. The simulation parameters were identical to those in the simulations with recombination hotspots, except that R _h_=1 for uniform recombination. Without hotspots, only 10%, on average, of all recombination events in the history of a sample of 100 chromosomes occurred at the 10 sites with the most recombination. The same figure for the simulations with hotspots was >75% for all values of R h. Furthermore, in the absence of hotspots, Phillips et al. (2003) and Wang et al. (2002) have shown that the distribution of allele frequencies, marker spacing, and recombination rates is an important factor influencing the observation of block lengths similar to those observed in human data. None of these factors were manipulated in a manner to produce block structure in our uniform-recombination simulations. For these reasons, there will be less-pronounced block structure in the simulations with uniform recombination than in the simulations with recombination hotspots.
The MDB method consistently identifies block boundaries in the absence of recombination hotspots. This finding makes it clear that the MDB method is not a test for the presence of recombination hotspots. For instance, with _N_=100 and ρ=500, the MDB method found, on average, 18 blocks in the simulated data. That the MDB method finds blocks even when there should not be strong block structure in the data may seem to be an undesirable property of the method; however, we note that the MDB method identifies fewer blocks in the uniform-recombination case than do all three of the other methods (table 2). On average, the FGT method inferred the highest number of blocks, followed by the htSNP method, with both methods inferring at least three times the number of blocks inferred by the MDB method for almost all values of ρ and N investigated. The DB method inferred fewer blocks than the htSNP and FGT methods, but it still inferred almost twice as many blocks, on average, as did the MDB method.
Without the inclusion of recombination hotspots in the simulations, it is difficult to interpret the significance, with respect to blocks of LD, of the blocks delineated by each of the different methods. The blocks inferred by each method will, in some way, reflect the history of recombination and mutation in the sample. However, further work will be necessary to understand the relationship between the blocks inferred by any method and the history of the sample, as well as the functional significance of that relationship.
Mitochondrial and Chromosome 5 Data
To test block-finding algorithms on real data, we applied our method to a data set of 822 biallelic sites in 86 complete human mtDNA sequences (Ingman et al. 2000; Maca-Meyer et al. 2001). Because there is little evidence for widespread recombination in human mtDNA (Posada et al. 2002), it would be surprising to find block structure in mtDNA data. Accordingly, the mtDNA data serve as a useful negative control for methods that detect block structure. We applied all four block-finding methods to the mtDNA data; however, we report results only for the MDB and FGT methods. The DB and htSNP methods made slow progress in analyzing the mtDNA data set; thus, their execution was terminated, since it appeared as though they would not complete the analysis within less than several weeks. Using the IDP algorithm, the MDB method found four blocks in the mitochondrial data. The block boundaries had endpoints 279, 304, and 306. The IADP algorithm did not find any block boundaries in the data. Moreover, the description length of 19,005.5 bits for the IDP result was not very different from the description length of 19,010.9 bits for the IADP result of no block boundaries. We can infer from the small difference in description length that the few blocks found by the IDP algorithm are not particularly strong block boundaries.
In contrast, the FGT method inferred the presence of 91 blocks in the mtDNA data. The inferred blocks likely occur where pairs of sites carry all four possible haplotypes owing to multiple mutations or genotyping error. This appears to be a limitation of using such a stringent definition for blocks as the FGT applied in a pairwise manner to SNPs. The contrast in the number of boundaries called by the MDB and FGT methods is still present when we exclude from the data set segregating sites in the D-loop (results not shown).
To analyze human data with known block structure, we applied the MDB, FGT, DB, and htSNP methods to the data of Daly et al. (2001). The data consist of 103 SNP markers from 5q31, haplotyped on 516 chromosomes from case and control trios in an association study of Crohn disease. The data set includes many missing values. Of the 103×516=53,148 SNPs typed in the sample, 12.7% are missing data, and 7.3% could not be unambiguously haplotyped. For the MDB method, we handled the missing data by using a heuristic approach of filling in missing data points in proportion to their associations among known subsequences of length 10 (i.e., _w_=10; see appendix C). Daly et al. established the existence of blocks in the 5q31 data by a variety of criteria and techniques. They first defined blocks on the basis of haplotype diversity by using an ad hoc criterion involving the ratio of observed to expected heterozygosity. They then confirmed that their block boundaries correspond to regions of high historical recombination by applying a hidden Markov model under the assumption that four classes of ancestral haplotypes across the whole region are known. Finally, they noted that jumps in the value of multiallelic _D_′ (calculated in a sliding window, over SNPs) occur at locations that they identified as block boundaries. Overall, the blocks designated by Daly et al. may be taken as a characterization of well-established block structure. Daly et al. reported the following set of block boundaries for their data set: {8–9,14–15,24,35,40,45,76–77,84–85,91,98}, where dashes indicate block-region boundaries that are imprecise because Daly et al. occasionally allowed single markers to not be included in any block.
We analyzed the 5q31 data six different times by the MDB method using the IDP algorithm. Each time, different random-number seeds were used to impute the missing data (appendix C). Across these six different imputations of the missing data, the block boundaries found by the MDB method were unchanged except for the movement of one boundary position. The resulting block boundaries were {8,14,24,36,47,57,76,86,91,98}, with 57 being replaced by 61 in two of the six runs. The average description length over the six runs was 11,556 bits.
The MDB method using the IADP algorithm produced results similar to the IDP algorithm, but in much less running time. When the IADP algorithm was used, all of the boundaries were identical to those identified by the IDP algorithm, except that the boundary at 57 was never moved to 61, and the average description length was 11,579 bits. These block designations compare favorably to those of Daly et al. (2001). Both the block calls made by Daly et al. and those made by the MDB method recognize the common boundaries {8,14,24,35/36,45/47,76,85/86,91,98}, where slash marks denote corresponding but not identical boundaries. The three cases (35/36, 45/47, and 85/86) for which the MDB method calls a boundary that is close to but not exactly concordant with one of Daly et al.’s block boundaries correspond to the three boundaries with the lowest historical recombination rates estimated by Daly et al. Furthermore, as shown in their figure 2, there is little support in the data for a distinction between a boundary at 45 versus 47 and, likewise, for a boundary at 85 versus 86. Specifically, transitions between Daly et al.’s ancestral haplotype states observed at high frequency ( >2% of chromosomes) occurring at boundary 45 would be indistinguishable from transitions occurring at boundary 47; transitions would be similarly indistinguishable between boundaries 85 and 86. The only boundary not found by the MDB method but found by Daly et al. was 40. The only boundary found by the MDB method but not by Daly et al.’s method was the boundary identified as either 57 or 61. Although the Daly et al. and MDB boundaries are generally in agreement, the description length obtained using the Daly et al. block boundaries is >850 bits longer than that obtained using the MDB method (see table 4). The large difference in the description lengths indicates that the small differences between the two sets of block boundaries are in some sense significant; however, there is no formally established method of relating description-length differences to statistical certainty.
Table 4.
Results of Application of Each Method to the Daly et al. Data
| Method | No. of Blocks | Boundaries | Average 𝒟a | Computational Timeb |
|---|---|---|---|---|
| MDB: | ||||
| IDP algorithm | 11 | {8,14,24,36,47,57/61,76,86,91,98} | 11,556 | 45 min |
| IADP algorithm | 11 | {8,14,24,36,47,57,76,86,91,98} | 11,579 | 2 min 45 s |
| Daly et al.c | 11 | {8–9,14–15,24,35,40,45,76–77,84–85,91,98} | 12,462 | … |
| htSNPd | 10 | {8,9,11,24,40,63,80,82,91} | 13,094 | 65 min |
| DBe | 5 | {24,44,77,88} | 14,003 | 31 min |
| FGTf | 75 | {1,3,4,5,6,…,98,99,101} | 20,393 | <1 min |
The DB method found five blocks in the 5q31 data. The block boundaries were {24,44,77,88}. The first three boundaries correspond closely with boundaries identified by Daly et al. (2001).
The htSNP method found 10 blocks in the 5q31 data. The block boundaries were {8,9,11,24,40,63,80,82,91}. Most of these boundaries correspond with Daly et al.’s (2001) block boundaries; boundaries in the set {11,63,80} do not correspond. Relative to its behavior in simulations, the htSNP method appears to perform better with real data. This difference in performance may be because the simulations do not re-create observed patterns of within-block diversity.
The FGT method found 75 blocks in the Daly et al. data set. The numerous incompatibilities in this data set cause the FGT to declare a large number of block boundaries—so many, in fact, that, on the basis of these results, one might be led to conclude that there are no long blocks in the Daly et al. data. The FGT method likely performs poorly on the 5q31 data because, like the mtDNA data set, the Daly et al. data set probably has sites with multiple mutations and with genotyping errors that create incompatibilities that are not the result of recombination. In addition, nuclear sequences experience gene conversion that can cause additional incompatibilities that are not the result of reciprocal recombination.
Running on a 1.66-GHz AMD Athlon processor, to find blocks in the 5q31 data, the MDB method required 2 min 45 s when the IADP algorithm was used and 45 min when the full IDP algorithm was used. For comparison, the FGT method required <1 s, the DB method required 31 min, and the htSNP method required 65 min. In the analysis of the mtDNA data set, the IADP algorithm ran overnight, whereas the IDP algorithm took ∼1 wk. Since the IADP algorithm is much faster than the IDP algorithm, performs well on simulated data, and yields a similar result to the IDP algorithm in most cases, we recommend the IADP algorithm when computational efficiency is an issue.
Discussion
We have proposed a haplotype block–detection method with many advantages over existing methods. We have formulated our method as a problem in statistical-model selection by use of the MDL principle. This allows us to invoke a probability model that captures both of the empirically described, dominant features of haplotype blocks: the decay of LD across block boundaries and the low haplotype diversity often encountered within blocks. The method, which we have implemented in the computer package MDBlocks, does not depend on population-specific or arbitrarily set thresholds. Furthermore, in factoring LD into its block-boundary designations, our MDL-based method incorporates information from numerous sites within a haplotype block at once and, in doing so, avoids being overly sensitive to the patterns of variation observed in LD measures based on pairwise comparisons. If the decay of LD across block boundaries is an important determinant of the degree to which blocks are useful in gene mapping, then our MDL-based method might be preferable to methods based solely on within-block haplotype diversity.
Using computer simulations, we show that MDBlocks reliably locates the boundaries between blocks at regions of rapid LD decay. We generated data by coalescent simulations with recombination hotspots across a range of recombination rates and hotspot intensities. The method detected most hotspots, particularly when the sample size was large (100 individuals) and the intensity of the hotspots was high. Simulations show that the method discriminates between the hotspots and regions having lower, background rates of recombination. This is important because empirical studies in the MHC region (Jeffreys et al. 2001; Cullen et al. 2002) have noted that, even though recombination hotspots will strongly structure the LD in a region, recombination does not occur exclusively at hotspots. In the absence of recombination hotspots in our simulations, MDBlocks continues to find blocks, demonstrating that it does not provide a test for the existence of recombination hotspots. However, MDBlocks identifies fewer blocks than other block-finding methods in data simulated with uniform recombination.
Application of MDBlocks to two published data sets confirms that it effectively identifies haplotype block structure when it clearly exists. The MDL-based method found a set of block boundaries that were nearly in complete agreement with those identified by Daly et al. (2001) for their data on 5q31. In mtDNA data, MDBlocks finds few blocks, as one would hope, considering that there is little evidence for recombination in the mitochondrial genome.
We applied three other block-finding algorithms to the same simulated and real data, concluding that our method captures the underlying block structure more effectively than any of these methods. The FGT method of Wang et al. (2002), based on the FGT, divides both the simulated data and the real data into many small blocks. Block boundaries determined by the FGT method do not necessarily correspond to locations with large drops in LD. The consequences of such undiscerning block-finding behavior are clear when the FGT method is applied to the 5q31 data of Daly et al. (2001); despite clear evidence for the existence of ∼10–12 extended haplotype blocks in that data set, the FGT method finds 75 blocks, each one containing, on average, fewer than two markers. Application of the FGT method to mtDNA data further demonstrates that the stringent pairwise nature of the FGT method makes it particularly susceptible to mistaking as block boundaries the results of genetic and sampling processes that may have little to do with recombination.
The two haplotype-diversity–based block-finding methods implemented by Zhang et al. (2002_b_) are largely insensitive to recombination hotspots in the simulated data. Across all parameter sets in our simulations with recombination hotspots, the block boundaries declared by these two methods (htSNP and DB) were among the least likely to be at recombination hotspots. We emphasize that these methods based on within-block diversity are not specifically designed to locate block boundaries where LD decays. Nonetheless, it is interesting and important that even the LD breakdown simulated under the assumption of recombination hotspots does not induce block structure that these methods are capable of detecting in the data.
Although the performance of our MDL-based method on simulated and real data is encouraging, we suggest here several areas of research that may lead to better methods for the detection of haplotype blocks. First, like the htSNP and DB methods, MDBlocks finds a single set of block boundaries but does not provide a summary of other sets of block boundaries that fit the data well. The description length itself, of course, provides a measure by which to compare different sets of boundaries, but determination of CIs on the basis of description lengths is an area that awaits further theoretical research. Sampling-based approaches may be helpful, in the future, for assessing the degree of confidence that may be placed in any particular set of blocks. Second, as currently implemented, our MDL method is not invariant to the direction in which SNPs are read along the chromosome. It is possible to obtain a slightly different set of blocks from MDBlocks, for any particular data set, when the data are read from left to right, rather than from right to left, along each chromosome. It might be possible, by imposition of conditions on the form of the matrix P, to ensure that the same result is achieved regardless of the direction the data are read, and this remains an unsolved problem. In the meantime, even though it is inelegant to have a description length that depends on the direction in which the markers are read, we have demonstrated that the method performs well regardless of the direction in which the data are read. Third, our method, like the FGT, htSNP, and DB methods, does not use physical distance between markers in the characterization of haplotype blocks. This is partly because the occurrence of hotspots makes the map between physical and genetic distance highly variable at fine scales (Jeffreys et al. 2001), so physical distance may not be a reliable criterion for block structure. Finally, our method, as well as the FGT, htSNP, and DB methods, requires that the phase of the markers be known, so some method of haplotype reconstruction must be applied to the data before applying these methods to it. Including the inference of haplotype blocks or the inference of recombination hotspots into the actual machinery for haplotype inference could provide some advantages, as noted by Niu et al. (2002) and as demonstrated by Greenspan and Geiger (2003).
We have presented an MDL-based method with desirable properties relative to such methods as those based on the FGT or on minimization of the number of htSNPs. Our simulations suggest that the MDL criterion, in conjunction with an appropriate statistical model, can be used to discern blocklike LD structure in the genome. Further work will be required in order to assess the degree to which the quality of the block inference is affected by the underlying statistical model and, specifically, how the performance of our method compares with that of the other recently developed MDL-based approaches of Koivisto et al. (2003) and Greenspan and Geiger (2003). A fruitful area of research may be found in the combination of elements of the model and optimization methods used here with the other MDL-based methods. In particular, Koivisto et al.’s (2003) method might be modified to include Markov dependence between a small number of their haplotype clusters defined in adjacent blocks. Additionally, the IADP algorithm with 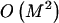 time complexity (eq. [B2], in appendix B) may provide a means to improve the optimization search strategy of Greenspan and Geiger (2003).
time complexity (eq. [B2], in appendix B) may provide a means to improve the optimization search strategy of Greenspan and Geiger (2003).
Although further development is possible, it is already clear from the results of the present study that the MDL approach is well suited to the problem of identifying haplotype blocks. In general, block-finding methods will be useful in exploring the population- and genome-level processes that give rise to observations of haplotype block structure. In particular, these methods may offer a better understanding of the relative roles that recombination-rate heterogeneity and population history play in shaping the patterns of LD in human populations. Finally, descriptions of haplotype blocks may be useful in improving the feasibility of large-scale gene-mapping studies and in aiding to their design.
Acknowledgments
We thank Montgomery Slatkin, for discussions that led to the identification of the problem; Terry Speed, for suggesting the MDL approach and offering valuable advice; and Bin Yu and Michael Stumpf, for helpful discussions. We also gratefully acknowledge Mikko Koivisto, Gideon Greenspan, and two anonymous reviewers, for comments on our manuscript. This research was supported by National Institutes of Health grant GM-40282 (to M. Slatkin) and by a Howard Hughes Medical Institute Predoctoral Fellowship (to J.N.).
Appendix A: Code Lengths of the Parameters
By summing the code lengths for each parameter, we obtain the description length 𝒟1 for the first stage in our two-stage coding scheme:
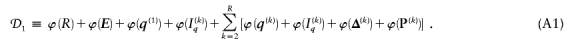
To compute the code length for a parameter, one must assume a prior distribution for it. This has the form of a Bayesian prior distribution but need not have a Bayesian interpretation (Lanterman 2001; Lee 2001). In most cases, we apply Laplace’s principle of indifference and assume that the unknown parameters are uniformly distributed throughout their range. Under the assumption that R is equally likely to take any value between 1 and M, it requires 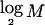 bits to encode R. Given R, there are
bits to encode R. Given R, there are 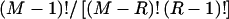 different values that the vector E may take. Under the assumption of a uniform distribution on E,
different values that the vector E may take. Under the assumption of a uniform distribution on E,
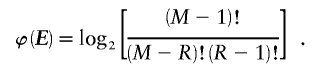
Since 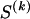 may take any value between 1 and N, we have
may take any value between 1 and N, we have 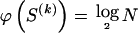 . Finally, each
. Finally, each  is a real number, but, given the
is a real number, but, given the  types in
types in 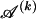 , it would be possible to estimate it only to the precision of
, it would be possible to estimate it only to the precision of 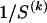 . Therefore, we calculate the code length for specification of
. Therefore, we calculate the code length for specification of 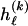 only to that precision. Following the method of Hansen and Yu (2001),
only to that precision. Following the method of Hansen and Yu (2001), 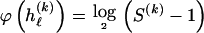 . We assume that the code lengths for
. We assume that the code lengths for 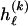 are additive within and across blocks, so that
are additive within and across blocks, so that 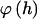 is the sum of
is the sum of 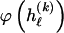 over all k and ℓ.
over all k and ℓ.
The 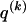 value that minimizes the code length of the data is the maximum-likelihood estimate of the frequencies of the
value that minimizes the code length of the data is the maximum-likelihood estimate of the frequencies of the 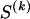 types. This is a function of
types. This is a function of 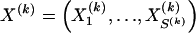 , the vector of observed counts of different types, taken to be in decreasing order (i.e.,
, the vector of observed counts of different types, taken to be in decreasing order (i.e., 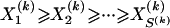 ). Therefore, the code length for
). Therefore, the code length for 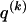 depends on
depends on 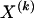 and a prior distribution for
and a prior distribution for 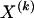 . We consider three different prior distributions that give the following three different code lengths:
. We consider three different prior distributions that give the following three different code lengths:
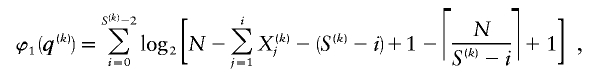
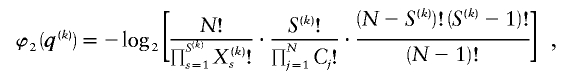
and
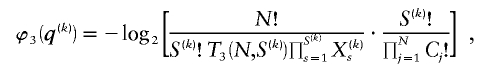
where C j is the number of types represented by j sequences in the sample and 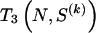 denotes a Stirling number of the third kind (Johnson et al. 1997). The code length
denotes a Stirling number of the third kind (Johnson et al. 1997). The code length 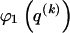 arises by assuming that each component of
arises by assuming that each component of 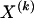 is uniformly distributed, conditional on the value of the preceding component; it favors haplotype frequencies in which a small number of types account for most of the chromosomes in the sample. The code length
is uniformly distributed, conditional on the value of the preceding component; it favors haplotype frequencies in which a small number of types account for most of the chromosomes in the sample. The code length 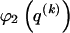 arises by assuming that every configuration of chromosomes into the
arises by assuming that every configuration of chromosomes into the 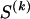 types is equally probable; it favors haplotype frequencies in which all the types within a block are at comparable frequencies. The code length
types is equally probable; it favors haplotype frequencies in which all the types within a block are at comparable frequencies. The code length 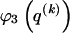 arises by assuming the multivariate Ewens distribution as the prior distribution for haplotype frequencies (i.e., by assuming the distribution of types within nonrecombining segments expected under neutral evolution). We choose the code length that best matches the data for each block k. To do so, we specify an indicator
arises by assuming the multivariate Ewens distribution as the prior distribution for haplotype frequencies (i.e., by assuming the distribution of types within nonrecombining segments expected under neutral evolution). We choose the code length that best matches the data for each block k. To do so, we specify an indicator 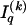 that we set to 1, 2, or 3, according to which prior distribution yields the shortest code length for
that we set to 1, 2, or 3, according to which prior distribution yields the shortest code length for 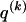 . Specification of
. Specification of 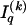 requires an additional
requires an additional 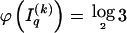 bits.
bits.
To encode the _r_th row of 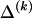 , we must first specify the number of 1s in the row. We denote this quantity by
, we must first specify the number of 1s in the row. We denote this quantity by 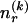 . Often, there will be no 1s in the row, so we specify
. Often, there will be no 1s in the row, so we specify 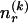 by first committing a single bit to indicate whether
by first committing a single bit to indicate whether 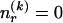 or
or 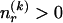 . Then, in the latter case, we require another
. Then, in the latter case, we require another 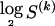 bits in order to specify
bits in order to specify 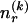 ; therefore,
; therefore,
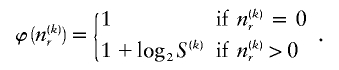
For 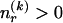 , there are
, there are 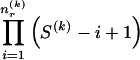 ways of arranging the
ways of arranging the 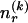 1s into the
1s into the 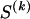 entries of the _r_th row of
entries of the _r_th row of 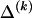 . If each of those arrangements is assumed to have equal prior probability, then
. If each of those arrangements is assumed to have equal prior probability, then
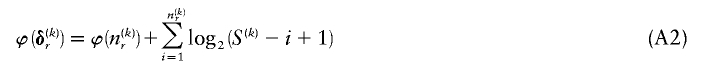
bits. The total code length for 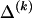 is a sum over rows:
is a sum over rows: 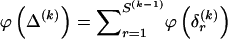 .
.
Determining 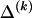 and Computing
and Computing 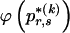
Although it is easy to compute 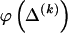 for any
for any 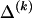 , we must still find
, we must still find 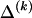 for each block, _k_=2,…,R , that minimizes the description length associated with the _k_th block:
for each block, _k_=2,…,R , that minimizes the description length associated with the _k_th block:
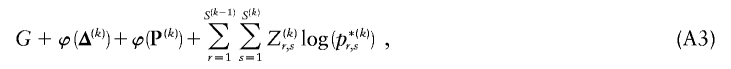
where 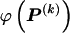 is the code length required in order to specify the values of
is the code length required in order to specify the values of 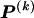 for which the corresponding elements in
for which the corresponding elements in 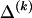 are equal to 1,
are equal to 1, 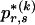 are elements of
are elements of 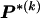 whose values are determined by
whose values are determined by 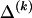 and
and 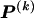 as shown in equation (2), and G is a sum of terms that are constant with respect to
as shown in equation (2), and G is a sum of terms that are constant with respect to 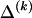 and hence do not factor into the optimization over possible values of
and hence do not factor into the optimization over possible values of 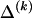 . Minimization of equation (A3) can be done by minimizing over
. Minimization of equation (A3) can be done by minimizing over 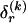 , for each r, the quantity
, for each r, the quantity
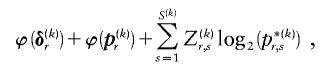
where, as in the main text, 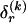 and
and 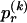 denote the _r_th rows of the matrices
denote the _r_th rows of the matrices 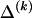 and
and 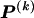 , respectively, and
, respectively, and 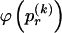 is the code length for those entries in the _r_th row of
is the code length for those entries in the _r_th row of 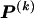 for which the corresponding elements in
for which the corresponding elements in 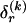 equal 1. The number of possible configurations of 1s and 0s in
equal 1. The number of possible configurations of 1s and 0s in 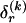 is
is 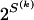 , making the problem of determining the optimal
, making the problem of determining the optimal 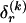 exponential in
exponential in 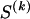 . As an alternative, we present a linear-time approximate algorithm for finding
. As an alternative, we present a linear-time approximate algorithm for finding 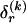 . In this algorithm, we order the haplotypes at the _k_th block in decreasing order of the distance between the observed conditional distribution of types given the haplotype in the previous block and the marginal distribution of types. We then cycle over the haplotypes in that order, and we set the corresponding values in
. In this algorithm, we order the haplotypes at the _k_th block in decreasing order of the distance between the observed conditional distribution of types given the haplotype in the previous block and the marginal distribution of types. We then cycle over the haplotypes in that order, and we set the corresponding values in 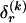 to 1 if doing so decreases the overall description length for the data and model parameters at the _k_th block.
to 1 if doing so decreases the overall description length for the data and model parameters at the _k_th block.
The approximate algorithm is defined more formally after the establishment of the following notation. As before, 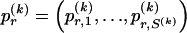 is a vector where
is a vector where 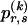 is the observed proportion of chromosomes having haplotype r in block _k_-1 and haplotype s in block k. We consider an ordering ν of the
is the observed proportion of chromosomes having haplotype r in block _k_-1 and haplotype s in block k. We consider an ordering ν of the 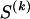 types in block k in decreasing order of their contribution to the cross-entropy between
types in block k in decreasing order of their contribution to the cross-entropy between 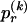 and
and 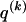 . The newly ordered vector is
. The newly ordered vector is 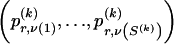 , and
, and 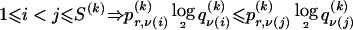 . Let
. Let 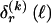 be the vector
be the vector 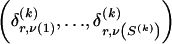 , in which the first ℓ components may be 0 or 1 and the remaining components,
, in which the first ℓ components may be 0 or 1 and the remaining components, 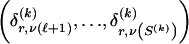 , are all 0. The code length of
, are all 0. The code length of 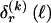 is
is
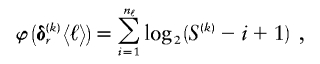
where _n_ℓ is the number of 1s in 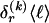 . This expression is consistent with equation (A2), because
. This expression is consistent with equation (A2), because 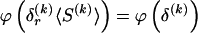 , as it should. Also, denote by
, as it should. Also, denote by 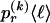 the values of
the values of 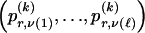 for which the corresponding entries in
for which the corresponding entries in 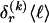 are 1. If
are 1. If 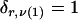 , then
, then 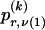 will be a maximum-likelihood estimate given by
will be a maximum-likelihood estimate given by 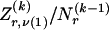 , where
, where 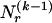 is the total number of chromosomes having type r at block _k_-1; thus,
is the total number of chromosomes having type r at block _k_-1; thus, 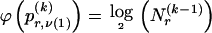 , and
, and
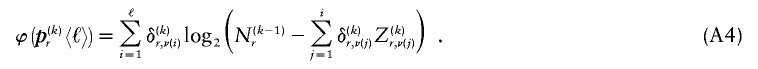
Finally, the code length of the haplotypes at the _k_th block on all chromosomes carrying type r at block _k_-1 is
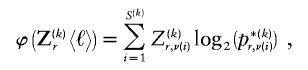
where the values for 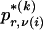 are obtained by application of equation (2), but with
are obtained by application of equation (2), but with  in place of
in place of  . Three more notational conventions are useful: given the vector
. Three more notational conventions are useful: given the vector 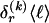 , we define
, we define 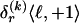 to be
to be 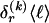 , with
, with 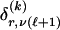 set to 1 instead of 0;
set to 1 instead of 0; 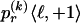 denotes the values of
denotes the values of 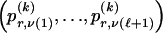 for which the corresponding entries in
for which the corresponding entries in 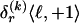 are 1; and
are 1; and 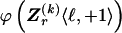 extends
extends 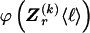 in the obvious way.
in the obvious way.
With the above definitions established, the algorithm may be described as follows.
The value 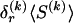 , found by the algorithm, is then used as
, found by the algorithm, is then used as 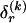 to compute
to compute 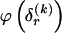 . Additionally,
. Additionally, 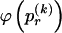 is computed as
is computed as 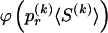 , using equation (A4). The above algorithm is applied to each
, using equation (A4). The above algorithm is applied to each 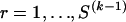 , and the overall code lengths used in the calculation of 𝒟1 in equation (A1) are
, and the overall code lengths used in the calculation of 𝒟1 in equation (A1) are 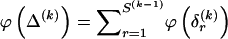 and
and 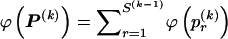 .
.
Appendix B: Optimization
We seek to minimize 𝒟=𝒟1+𝒟2 over all possible choices of R and E . To do so, it helps to express 𝒟 as a function of R and E . The form of the resulting function is
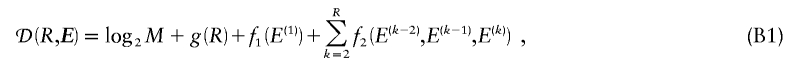
where
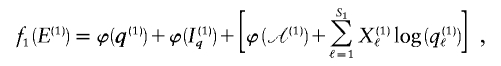
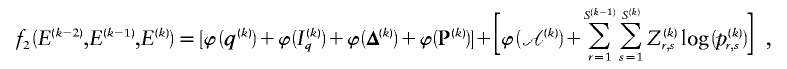
and
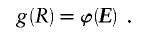
The last line above follows on the basis that 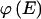 is a function of R. Minimization of equation (B1) with respect to R and E requires a different approach than that of Zhang et al. (2002_b_), because our model involves a sum over functions that are dependent on two blocks at a time. The Zhang et al. (2002_b_) solution is for minimization of objective functions with a simpler form. Because
is a function of R. Minimization of equation (B1) with respect to R and E requires a different approach than that of Zhang et al. (2002_b_), because our model involves a sum over functions that are dependent on two blocks at a time. The Zhang et al. (2002_b_) solution is for minimization of objective functions with a simpler form. Because 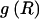 is not a linear function of R, it is not possible to efficiently minimize equation (B1), but, if we assume a value of
is not a linear function of R, it is not possible to efficiently minimize equation (B1), but, if we assume a value of  —for example,
—for example, 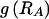 —then we can specify a dynamic programming algorithm to globally minimize the latter two terms of equation (B1). The algorithm can be described in terms of a dynamic programming matrix 𝔻 with the elements assigned as follows:
—then we can specify a dynamic programming algorithm to globally minimize the latter two terms of equation (B1). The algorithm can be described in terms of a dynamic programming matrix 𝔻 with the elements assigned as follows:
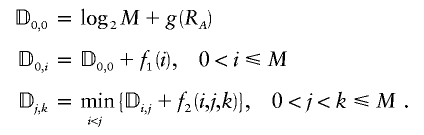
The minimal value of 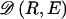 is
is 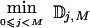 , and the values of R and E minimizing the description length can be obtained by a trace-back algorithm. The optimality of the algorithm for minimization of
, and the values of R and E minimizing the description length can be obtained by a trace-back algorithm. The optimality of the algorithm for minimization of 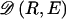 with
with 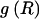 fixed at
fixed at 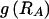 can be proved by induction. The time complexity of this algorithm is
can be proved by induction. The time complexity of this algorithm is 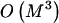 , and the space complexity is
, and the space complexity is 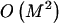 . Generally,
. Generally, 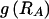 is small relative to the value of
is small relative to the value of 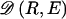 , so the assumed value has little effect on the solution; nonetheless, to explore a reasonable set of assumptions for R A, we take an iterative approach. We first minimize
, so the assumed value has little effect on the solution; nonetheless, to explore a reasonable set of assumptions for R A, we take an iterative approach. We first minimize 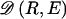 with R A set to 1, calling the number of blocks found
with R A set to 1, calling the number of blocks found 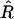 . We then set R A equal to
. We then set R A equal to 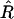 and repeat until R A converges to a single value. Very rarely, the value of R A begins cycling, and then we choose, from the cycle, the solution that has the lowest description length. We refer to the combination of a dynamic programming algorithm with iteration over the values of R A as the “IDP algorithm.”
and repeat until R A converges to a single value. Very rarely, the value of R A begins cycling, and then we choose, from the cycle, the solution that has the lowest description length. We refer to the combination of a dynamic programming algorithm with iteration over the values of R A as the “IDP algorithm.”
To improve computation speed, we have also developed an approximation to the dynamic programming algorithm that has time complexity 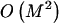 and space complexity
and space complexity 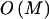 . This approximate algorithm, with
. This approximate algorithm, with 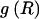 fixed at
fixed at 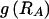 , is defined recursively as
, is defined recursively as
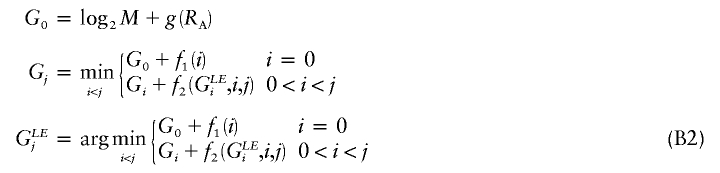
for 1⩽j<M. The resulting approximation to the MDL is G M , and the minimizing values of R and E are obtained by a trace back, through the assignments, to vector G. The algorithm will work well to the extent that G LE j is the same as the minimizing second-to-last endpoint (i.e., 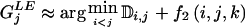 for 0<j<k_⩽_M). In the majority of simulations, the performance of the approximate algorithm is similar to that of the full dynamic programming algorithm. As above, we iterate this approximate algorithm over values of
for 0<j<k_⩽_M). In the majority of simulations, the performance of the approximate algorithm is similar to that of the full dynamic programming algorithm. As above, we iterate this approximate algorithm over values of 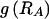 , and we refer to this algorithm as the “IADP algorithm.”
, and we refer to this algorithm as the “IADP algorithm.”
Appendix C: Missing Data
It is not typical to obtain perfect haplotype data from autosomal regions, so most data sets will have a number of holes in them. Little work has been done on the formal treatment of missing data from within the MDL framework. To apply our method with missing data, we employ a simple, iterative method for the imputation of missing values according to their frequency of occurrence in the rest of the data set, given a window of neighboring sites. These imputed values are used to generate a set of candidate sequences at each block, and then haplotypes with data missing in each block k are assigned to these candidate sequences in a manner such that the entropy of the distribution of types at block k in the data set is minimized. First, the imputation method is described below, and then the method for assigning haplotypes to the candidate sequences is described.
Imagine that the _i_th chromosome is missing data at the _j_th SNP. We fill in the missing data at Y i,j on the basis of a “window” of size w, as follows:
- 1.
Identify all sets of w SNPs not missing in chromosome i such that each member of the set is separated from the _j_th SNP by no more than _w_-1 sites not missing in chromosome i. There will be, at most, w+1 such sets (in cases in which j is near one of the ends of the string of SNPs in the data set there could be fewer than w+1). Let the indexes of the SNPs in such a set be denoted 𝒥+. These are like “windows” of nonmissing sites around j. - 2.
Let 𝒦0 be the set of all chromosomes having nonmissing data of allelic value 0, at site j, and for which no SNP at a site in 𝒥+ carries an allele different from that on chromosome i. Let 𝒦1 be defined similarly for chromosomes with the 1 allele at position j. For any chromosome k, let p k denote the number of nonmissing sites it has in 𝒥+, and define _P_0 as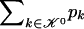 and _P_1 as
and _P_1 as 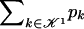 .
. - 3.
For each of the w+1 (or fewer) sets 𝒥+, compute the pair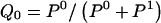 and
and 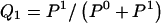 , and denote by
, and denote by 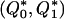 the pair of those quantities that maximizes
the pair of those quantities that maximizes 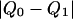 over the different sets 𝒥+.
over the different sets 𝒥+. - 4.
Fill in the hole Y i,j with a 0 for probability Q*0 and a 1 for probability Q*1. On filling in that hole, consider it, for the remainder of the time, as nonmissing data.
Holes in the data are filled in by the above routine in random order. Some of the missing data are due to unresolved heterozygosity at a SNP. In such a case, a procedure like the one above is used, but the pair of two chromosomes from the same individual are considered simultaneously, with one of them receiving a 0 and the other receiving a 1.
We do not use the data set with missing data imputed directly. Rather, to minimize the stochastic effects of imputing the missing data, we add another step in dealing with the missing data. This occurs at the stage when the number of different types in a block is being calculated and proceeds as follows: First, types in a block are defined as if the imputed data were real. Each of these types is defined as a complete sequence of 0s and 1s. There are 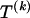 such sequences at the _k_th block. A chromosome in the sample is said to be “compatible” (borrowing the terminology from Zhang et al. 2002_b_) with such a sequence if it matches that sequence at all sites that are not missing data on the chromosome. Of the
such sequences at the _k_th block. A chromosome in the sample is said to be “compatible” (borrowing the terminology from Zhang et al. 2002_b_) with such a sequence if it matches that sequence at all sites that are not missing data on the chromosome. Of the 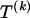 sequences, one of them will be compatible with more chromosomes than any other; all chromosomes compatible with that maximally compatible sequence are assigned the type of that sequence. Of the remaining _T_-1 sequences, one is compatible with the largest number of remaining chromosomes; all chromosomes compatible with it are assigned its sequence, and so forth, until there are no remaining chromosomes to be assigned. The number of sequences to which chromosomes were assigned is
sequences, one of them will be compatible with more chromosomes than any other; all chromosomes compatible with that maximally compatible sequence are assigned the type of that sequence. Of the remaining _T_-1 sequences, one is compatible with the largest number of remaining chromosomes; all chromosomes compatible with it are assigned its sequence, and so forth, until there are no remaining chromosomes to be assigned. The number of sequences to which chromosomes were assigned is 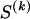 , which is typically less than
, which is typically less than 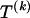 . Assignment of types in this way is a type of greedy algorithm for assignment of blocks with missing data to compatible types in a way that minimizes the entropy of the observed distribution of types in a block.
. Assignment of types in this way is a type of greedy algorithm for assignment of blocks with missing data to compatible types in a way that minimizes the entropy of the observed distribution of types in a block.
Appendix D: Notation for Variables, Parameters, and Functions
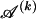 :
:
Set of unique sequences observed in the data at the _k_th block.
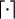 :
:
The ceiling function. 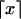 is the least integer greater than or equal to x.
is the least integer greater than or equal to x.
𝒟1,𝒟2,𝒟:
Description lengths of the first stage, the second stage, and both stages, respectively. 𝒟=𝒟1+𝒟2.
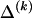 :
:
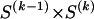 matrix of 0s and 1s. The value of each element
matrix of 0s and 1s. The value of each element 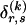 dictates how
dictates how 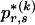 is determined by equation (2).
is determined by equation (2).
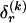 :
:
Vector 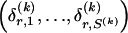 , which is the _r_th row of
, which is the _r_th row of 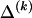 .
.
E:
Vector 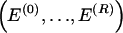 of right endpoints of the blocks. Note that
of right endpoints of the blocks. Note that 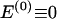 .
.
h:
Collection of vectors 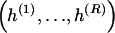 . Each vector is of the form
. Each vector is of the form 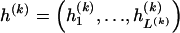 , where
, where 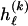 is the proportion of 1s among the sequences in
is the proportion of 1s among the sequences in 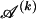 at the ℓth SNP in the _k_th block.
at the ℓth SNP in the _k_th block.
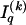 :
:
Indicator of which of the three distributions is used to calculate 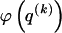 .
.
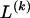 :
:
Number of SNPs included in the _k_th block. 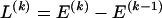 .
.
M:
Number of SNPs typed on each chromosome.
N:
Number of chromosomes typed in the sample.
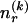 :
:
Number of 1s in the _r_th row of 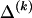 .
.
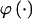 :
:
The code-length function.
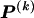 :
:
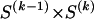 matrix of elements
matrix of elements 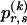 . Each
. Each 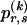 is the unknown population frequency of chromosomes having type r in block _k_-1 and type s in block k.
is the unknown population frequency of chromosomes having type r in block _k_-1 and type s in block k.
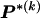 :
:
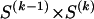 matrix of elements
matrix of elements 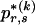 . The elements of this matrix are determined by
. The elements of this matrix are determined by 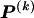 ,
, 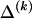 , and
, and 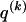 , as shown in equation (2), and are used in equation (4) to compute 𝒟2.
, as shown in equation (2), and are used in equation (4) to compute 𝒟2.
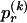 :
:
Vector 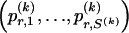 , which is the _r_th row of
, which is the _r_th row of 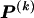 .
.
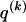 :
:
Vector 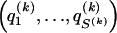 of unknown marginal frequencies in the sampled population of the
of unknown marginal frequencies in the sampled population of the 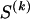 types at block k.
types at block k.
R:
Number of blocks.
S:
Vector 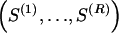 . Each
. Each  is the number of distinct haplotypes observed in the data at block k.
is the number of distinct haplotypes observed in the data at block k.
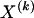 :
:
Vector 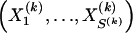 counting the number of times each of the
counting the number of times each of the 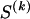 types is observed in the data at the _k_th block.
types is observed in the data at the _k_th block.
Y:
N_×_M matrix of observed SNP values. Y i,j is 0 or 1 according to the type of the _j_th SNP in the _i_th individual.
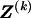 :
:
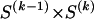 matrix of observed pairs of types at adjacent blocks.
matrix of observed pairs of types at adjacent blocks. 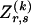 counts the number of chromosomes in the sample having type r at block _k_-1 and type s at block k.
counts the number of chromosomes in the sample having type r at block _k_-1 and type s at block k.
Electronic-Database Information
The URL for data presented herein is as follows:
- MDBlocks Home, http://ib.berkeley.edu/labs/slatkin/eriq/software/mdb_web/index.htm (for the MDBlocks program, as well as for information on the preparation of the 5q31 data of Daly et al. and the mtDNA data used in the analyses presented here)
References
- Cover TM, Thomas JA (1991) Elements of information theory. John Wiley & Sons, New York [Google Scholar]
- Cullen M, Perfetto SP, Klitz W, Nelson G, Carrington M (2002) High-resolution patterns of meiotic recombination across the human major histocompatibility complex. Am J Hum Genet 71:759–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES (2001) High-resolution haplotype structure in the human genome. Nat Genet 29:229–232 [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229 [DOI] [PubMed] [Google Scholar]
- Goldstein DB (2001) Islands of linkage disequilibrium. Nat Genet 29:109–111 [DOI] [PubMed] [Google Scholar]
- Greenspan G, Geiger D (2003) Model-based inference of haplotype block variation. Paper presented at the Seventh Annual International Conference on Research in Computational Molecular Biology—RECOMB, Berlin, April 10–13 [DOI] [PubMed] [Google Scholar]
- Hansen M, Yu B (2001) Model selection and the principle of minimum description length. J Am Stat Assoc 96:746–774 [Google Scholar]
- Hudson RR (2002) Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18:337–338 [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kaplan N (1985) Statistical properties of the number of recombination events in the history of a sample of sequences. Genetics 111:147–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingman M, Kaessmann H, Pääbo S, Gyllensten U (2000) Mitochondrial genome variation and the origin of modern humans. Nature 408:708–713 [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Kauppi L, Neumann R (2001) Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet 29:217–222 [DOI] [PubMed] [Google Scholar]
- Johnson GCL, Esposito L, Barratt BJ, Smith AN, Heward J, Di Genova G, Ueda H, Cordell HJ, Eaves IA, Dudbridge F, Twells RCJ, Payne F, Hughes W, Nutland S, Stevens H, Carr P, Tuomilehto-Wolf E, Tuomilehto J, Gough SCL, Clayton DG, Todd JA (2001) Haplotype tagging for the identification of common disease genes. Nat Genet 29:233–237 [DOI] [PubMed] [Google Scholar]
- Johnson NL, Kotz Z, Balakrishnan N (1997) Discrete multivariate distributions. John Wiley & Sons, New York [Google Scholar]
- Kauppi L, Sajantila A, Jeffreys AJ (2003) Recombination hotspots rather than population history dominate linkage disequilibrium in the MHC class II region. Hum Mol Genet 12:33–40 [DOI] [PubMed] [Google Scholar]
- Koivisto M, Perola M, Varilo R, Hennah W, Ekelund J, Lukk M, Peltonen L, Ukkonen E, Mannila H (2003) An MDL method for finding haplotype blocks and for estimating the strength of haplotype block boundaries. Pac Symp Biocomput 8:502–513 [DOI] [PubMed] [Google Scholar]
- Lanterman AD (2001) Schwarz, Wallace, and Rissanen: intertwining themes in theories of model selection. Int Stat Rev 69:185–212 [Google Scholar]
- Lee TCM (2001) An introduction to coding theory and the two-part minimum description length principle. Int Stat Rev 69:169–183 [Google Scholar]
- Maca-Meyer N, Gonzalez AM, Larruga JM, Flores C, Cabrera VM (2001) Major genomic mitochondrial lineages delineate early human expansions. BMC Genetics 2:13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu TH, Qin ZHS, Xu XP, Liu JS (2002) Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms. Am J Hum Genet 70:157–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M, Tavare S (2002) Linkage disequilibrium: what history has to tell us. Trends Genet 18:83–90 [DOI] [PubMed] [Google Scholar]
- Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, Kautzer CR, Lee DH, Marjoribanks C, McDonough DP, Nguyen BTN, Norris MC, Sheehan JB, Shen N, Stern D, Stokowski RP, Thomas DJ, Trulson MO, Vyas KR, Frazer KA, Fodor SPA, Cox DR (2001) Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294:1719–1723 [DOI] [PubMed] [Google Scholar]
- Phillips MS, Lawrence R, Sachidanandam R, Morris AP, Balding DJ, Donaldson MA, Studebaker JF, et al (2003) Chromosome-wide distribution of haplotype blocks and the role of recombination hot spots. Nat Genet 33:382–387 [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA, Holmes EC (2002) Recombination in evolutionary genetics. Ann Rev Genet 36:75–97 [DOI] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Ritcher DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES (2001) Linkage disequilibrium in the human genome. Nature 411:199–204 [DOI] [PubMed] [Google Scholar]
- Rissanen J (1978) Modeling by shortest data description. Automatica 14:465–471 [Google Scholar]
- ——— (1989) Series in computer science. Vol 15: Stochastic complexity in statistical inquiry. World Scientific, London [Google Scholar]
- Stephens M, Donnelly P (2000) Inference in molecular population genetics. J R Stat Soc B 62:605–635 [Google Scholar]
- Wang N, Akey JM, Zhang K, Chakraborty R, Jin L (2002) Distribution of recombination crossovers and the origin of haplotype blocks: the interplay of population history, recombination, and mutation. Am J Hum Genet 71:1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Calbrese P, Nordborg M, Sun F (2002_a_) Haplotype block structure and its application to association studies: power and study designs. Am J Hum Genet 71:1386–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Deng M, Chen T, Waterman M, Sun F (2002_b_) A dynamic programming algorithm for haplotype block partitioning. Proc Natl Acad Sci USA 99:7335–7339 [DOI] [PMC free article] [PubMed] [Google Scholar]