Hepatocyte growth factor promotes lymphatic vessel formation and function (original) (raw)
Abstract
The lymphatic vascular system plays a pivotal role in mediating tissue fluid homeostasis and cancer metastasis, but the molecular mechanisms that regulate its formation and function remain poorly characterized. A comparative analysis of the gene expression of purified lymphatic endothelial cells (LEC) versus blood vascular endothelial cells (BVEC) revealed that LEC express significantly higher levels of hepatocyte growth factor receptor (HGF-R). Whereas little or no HGF-R expression was detected by lymphatic vessels of normal tissues, HGF-R was strongly expressed by regenerating lymphatic endothelium during tissue repair and by activated lymphatic vessels in inflamed skin. Treatment of cultured LEC with HGF promoted LEC proliferation, migration and tube formation. HGF-induced proliferation of LEC did not require vascular endothelial growth factor receptor-3 activation, and HGF-induced cell migration was partially mediated via integrin alpha-9. Transgenic or subcutaneous delivery of HGF promoted lymphatic vessel formation in mice, whereas systemic blockade of HGF-R inhibited lymphatic function. These results identify HGF as a novel, potent lymphangiogenesis factor, and also indicate that HGF-R might serve as a new target for inhibiting pathological lymphangiogenesis.
Keywords: angiogenesis, HGF, integrins, lymphangiogenesis, tissue repair
Introduction
The lymphatic vascular system is composed of a dense network of thin-walled capillaries that drain protein-rich lymph from the extracellular spaces. Its major functions are the maintenance of tissue fluid homeostasis and mediation of the afferent immune response (Witte et al, 2001; Oliver and Detmar, 2002). Recent studies in mouse tumour models have revealed that lymphatic vessels also play an active role in the metastatic spread of malignant tumour cells to regional lymph nodes (Mandriota et al, 2001; Skobe et al, 2001; Stacker et al, 2001). Furthermore, recent studies have shown that the induction of lymphangiogenesis is a prognostic indicator of the metastatic risk of some human cancers, including malignant melanoma of the skin (Dadras et al, 2003).
Vascular endothelial growth factor (VEGF)-C and -D have been identified as specific lymphangiogenesis factors, acting via activation of the VEGF receptor-3 (VEGFR-3), which is expressed on lymphatic endothelial cells (LEC) (Jussila and Alitalo, 2002). VEGF-C-deficient mice fail to develop a functional lymphatic system (Karkkainen et al, 2004), and transgenic expression of a soluble VEGFR-3 results in pronounced lymphedema (Makinen et al, 2001). In animal tumour models, both VEGF-C and VEGF-D increase tumour lymphangiogenesis and lymphatic metastasis (Mandriota et al, 2001; Skobe et al, 2001; Stacker et al, 2001), and expression levels of VEGF-C and/or VEGF-D have been shown to correlate with metastasis in a large number of human tumour types (Stacker et al, 2002).
Owing to the excitement over the recent findings that lymphatic vessel formation is an important aspect of both tumour progression and inflammation (Oliver and Detmar, 2002), there has been a quest for additional lymphangiogenic growth factors. Fibroblast growth factor-2 (FGF-2) promotes lymphatic vessel growth in the mouse cornea, but these effects are believed to occur indirectly, via induction of VEGF-C expression and activation of VEGFR-3 signalling (Kubo et al, 2002; Chang et al, 2004). We and others have recently established that murine VEGF-A164 also induces lymphangiogenesis (Nagy et al, 2002; Hong et al, 2004b; Kunstfeld et al, 2004; Hirakawa et al, 2005b), probably through both direct and indirect mechanisms of action. Other non-VEGF family growth factors are likely to be involved in the developmental and pathological formation of lymphatic vessels, but identification of these factors has been hampered by the lack of suitable models for the discovery of vascular lineage-specific growth factor receptors, and also by the insufficient molecular characterization of LEC.
Hepatocyte growth factor (HGF; also known as scatter factor) is a heparin-binding glycoprotein produced by various cells of mesenchymal origin. HGF was first identified as a mediator of liver generation (Nakamura et al, 1989), but it also stimulates the proliferation and migration of several epithelial cell types, including mammary cells and kidney intestinal epithelial cells, as well as endothelial cells (Zarnegar and Michalopoulos, 1995). In vivo studies have shown that HGF plays a major role in tissue repair and promotes tumour invasiveness (Rosen et al, 1994; Silvagno et al, 1995; Jeffers et al, 1996; Matsumoto and Nakamura, 2001).
HGF's activities are mediated by a membrane-spanning receptor tyrosine kinase, the HGF receptor (HGF-R, also known as MET/c-met). HGF-R is a dimeric transmembrane polypeptide consisting of an α-chain that is exposed to the cell surface and a β-chain that spans the plasma membrane (Giordano et al, 1989). Increased expression of HGF-R has been found in papillary carcinomas of the thyroid gland and in carcinomas of the colon, pancreas and ovary. Point mutations of HGF-R have been identified in papillary renal carcinomas, hepatocellular and gastric carcinomas and in head and neck carcinomas (Danilkovitch-Miagkova and Zbar, 2002). A number of experimental and clinical studies have shown a correlation between expression levels of HGF-R, or the presence of HGF-R mutations, and metastasis. These findings indicate that HGF/HGF-R signalling promotes tumour invasiveness and progression, including metastasis to the lymph node (Birchmeier et al, 2003). These effects have primarily been attributed to the direct effects of HGF on tumour cells.
Using transcriptional profiling to compare gene expression patterns between cultured LEC and blood vascular endothelial cells (BVEC), we show that HGF-R is more highly expressed by LEC than by BVEC. In vivo, upregulation of HGF-R expression occurs in newly formed Prox1-positive lymphatic vessels during embryonic development and during tissue repair. Furthermore, HGF, the only known ligand for HGF-R, directly promotes proliferation, migration and tube formation of LEC, independently from activation of the VEGFR-3 pathway. In investigating the mechanisms of these activities, we found that the promigratory effects of HGF are mediated by integrin alpha-9, which is selectively expressed by LEC. Our study shows that HGF is a strong promoter of lymphatic vessel formation when overexpressed in transgenic mice and when delivered by HGF releasing implants in vivo, whereas HGF-R blockade impairs lymphatic vessel function.
Together, these results identify HGF as a novel and potent lymphangiogenesis factor. These findings have important implications for our understanding of the molecular basis of embryonic and pathological lymphatic vessel formation. The pivotal role of HGF in lymphatic vessel formation may also provide new insight into the molecular regulation of lymphangiogenesis and metastasis.
Results
Increased expression of HGF-receptor by cultured LEC as compared with BVEC
To identify genes that are specifically expressed or upregulated by LEC, compared to BVEC, we isolated and purified both LEC and BVEC from human neonatal foreskins of three independent donors, as previously described (Hirakawa et al, 2003). After five rounds of passage in culture, all three LEC lines expressed high levels of mRNAs encoding the major lymphatic lineage markers Prox1 (a homeobox transcription factor) and LYVE-1 (a hyaluronan receptor). These markers were either not expressed or expressed at very low levels by BVEC, which expressed instead the blood vascular lineage marker, Flt-1 (Figure 1A–C). The three LEC and BVEC cell lines were then subjected to transcriptional profiling by microarray analysis as described, using Affymetrix HU133 plus 2.0 arrays and the Affymetrix Suite5 software (Hong et al, 2004a). These studies revealed that HGF-R is expressed at higher levels by LEC, compared to BVEC (2.04±0.28-fold; _n_=3). This difference in gene expression was confirmed by quantitative TaqMan real-time RT–PCR, revealing a more than two-fold level of upregulation of HGF-R expression by LEC in all three independent pairs of cell lines (Figure 1D). Western blot analyses demonstrated that HGF-R protein expression was also strongly increased in LEC, compared to BVEC (Figure 1E). Moreover, treatment of LEC with 30 ng/ml of HGF resulted in HGF-R phosphorylation, indicating that the kinase activity of this receptor is active in these cells (Figure 1F).
Figure 1.
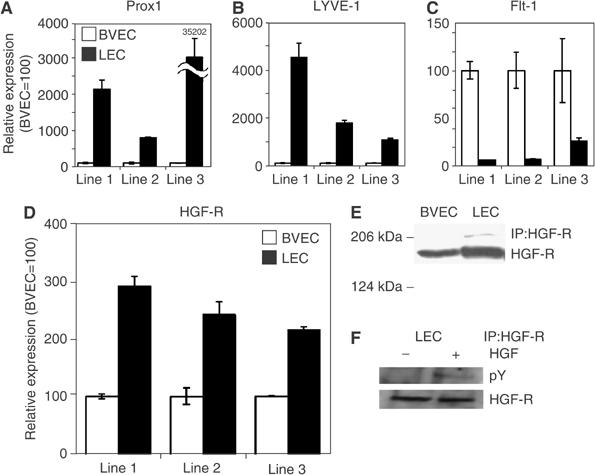
Increased expression of HGF-R by LEC. (A–C) Quantitative real-time RT–PCR confirmed that three independently established lines of primary LEC (black bars) expressed high levels of mRNA of the major lymphatic lineage markers Prox1 and LYVE-1, but expressed low levels of the blood vascular lineage marker Flt-1, as compared to BVEC (white bars). (D) LEC expressed over two-fold higher levels of HGF-R mRNA than BVEC, as determined by real-time RT–PCR. (E) Immunoprecipitation and Western blot analyses demonstrated that LEC (right lane) expressed much higher levels of HGF-R protein, compared with BVEC (left lane). (F) Treatment of LEC with 30 ng/ml HGF (right lane) resulted in increased phosphorylation of HGF-R, compared to untreated cells (left lane). IP: immunoprecipitate.
HGF-R is expressed by lymphatic vessels in vivo
We next performed differential immunofluorescence analyses of lymphatic vessels in mouse models of inflammation and tissue repair, and also during normal embryogenesis, using antibodies against HGF-R and against the lymphatic-specific hyaluronan receptor LYVE-1. Little or no expression of HGF-R was detected on quiescent lymphatic vessels in normal skin (Figure 2A–C). To investigate whether HGF-R might be upregulated by lymphatic endothelium during pathological processes, we examined its expression levels in chronically inflamed skin samples obtained from a mouse model of experimentally induced delayed-type hypersensitivity. These mice, which are created through transgenic expression of VEGF-A (VEGF-TG), are characterized by lymphatic vessel enlargement and increased LEC proliferation (Kunstfeld et al, 2004). At 7 days after induction of experimental skin inflammation in these mice, enlarged LYVE-1-positive lymphatic vessels were detected in VEGF-TG mice, but not in wild-type mice (Figure 2A and D). LYVE-1-positive lymphatic vessels expressed high levels of HGF-R, whereas little or no HGF-R expression was detected in the normal lymphatic vessels of wild-type mice (Figure 2A–F). Immunofluorescence stains for the lymphatic-specific transcription factor Prox1 (Wigle et al, 2002) and for HGF-R confirmed that Prox1-positive LECs also expressed HGF-R (Supplementary data 1, A and B).
Figure 2.
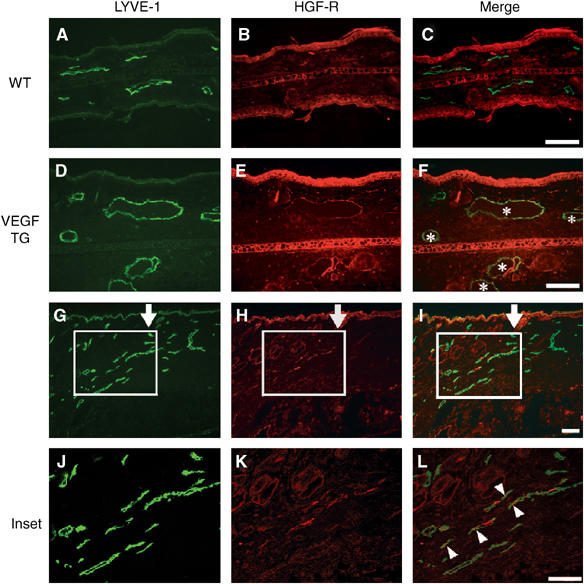
HGF-R is expressed by lymphatic vessels during inflammation and tissue repair. Double immunofluorescence stains for the lymphatic marker LYVE-1 (green) and for HGF-R (red) revealed little or no HGF-R expression by lymphatic vessels in normal mouse skin (A–C), whereas, in the inflamed skin of VEGF-A transgenic mice, indicated by LYVE-1-positive regions, enlarged lymphatic vessels (asterisks) also expressed HGF-R (D–F). HGF-R was also expressed by epidermal keratinocytes. At 21 days after induction of full-thickness wounds in mouse skin, several LYVE-1-positive, regenerating lymphatic vessels at the wound edge expressed HGF-R (G–I, inset H–L). Arrows (G–I) indicate the left border of the original wound area. Arrowheads indicate HGF-R-expressing lymphatic vessels. Scale bars: 100 μm.
We have previously reported the presence of pronounced lymphangiogenesis within the granulation tissue of experimentally induced full-thickness skin wounds in wild-type mice at 2 and 3 weeks after wounding (Hong et al, 2004b). Double immunofluorescence analyses of wound tissue at 21 days after wound formation revealed several LYVE-1-positive lymphatic vessels that also expressed HGF-R (Figure 2G–L). Overall, 39 out of 72 LYVE-1-positive lymphatic vessels (54.2%) examined also expressed HGF-R. We also examined the expression pattern of HGF-R during normal mouse embryogenesis. Mouse embryonic tissues were examined at embryonic days (E) 10.5–14.5, when active budding and proliferation of lymphatic vessel progenitors occurs (Oliver and Detmar, 2002). At E11.5, LYVE-1 expression was detected on endothelial cells of the anterior cardinal vein (CV), in agreement with previous results (Wigle et al, 2002). These cells expressed little or no HGF-R, whereas HGF-R expression was already detected within the pharyngeal region of the foregut and in mesenchymal cells (Figure 3A and B). However, at E12.5, HGF-R expression was clearly detectable on LYVE-1-positive endothelial cells of the anterior CV (Figure 3C and D), whereas HGF-R expression was only occasionally observed in the endothelial cells that line the primitive lymph sacs and also express LYVE-1 (Figure 3C and D). By E14.5, strong HGF-R expression was detected on the vast majority of LYVE-1-positive LECs (Figure 3E and F). At this stage, HGF-R was weakly expressed by endothelial cells that line the jugular vein and artery, which were LYVE-1-negative. Immunofluorescence stains of serial sections confirmed that—at E14.5—HGF-R was expressed by many Prox1-positive endothelial cells that had budded from the CV, as compared to little or no expression at E12.5 (Supplementary data 1, C–F).
Figure 3.
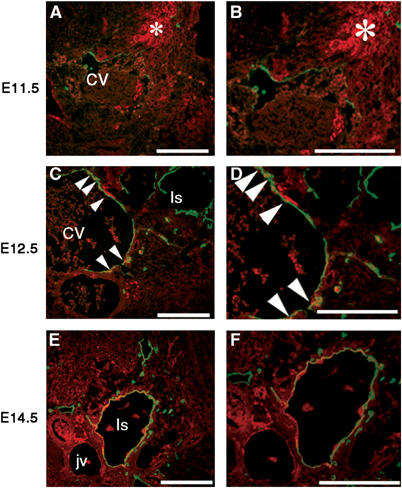
HGF-R is expressed by endothelial cells of lymph sacs during mouse embryonic development. (A, B) By E11.5, LYVE-1-expressing endothelial cells (green) of the anterior CV expressed little or no HGF-R (red), whereas HGF-R expression was detected within the pharyngeal region of the foregut and in mesenchymal cells (asterisk). (C, D) At E12.5, weak HGF-R expression was detected in some LYVE-1-positive endothelial cells of the anterior CV (arrowheads), whereas only occasional HGF-R expression was observed near the endothelium lining the primitive lymph sacs (ls). (E, F) At E14.5, HGF-R was expressed by the vast majority of LYVE-1-positive LEC. At this stage, HGF-R was weakly expressed by endothelial cells of the jugular vein (jv) and the artery, which were LYVE-1-negative. Scale bars: 100 μm.
HGF directly promotes LEC proliferation and migration
HGF is the only known ligand of HGF-R and has been shown to induce proliferation and migration of HUVEC cells. We investigated whether the differential expression levels of HGF-R by LEC and BVEC might lead to their differential response toward HGF stimulation by examining the in vitro effects of this growth factor on these two cell types. HGF potently and dose-dependently induced LEC proliferation, compared to untreated control cells, at a minimal effective concentration of 1 ng/ml (P<0.01). Although this concentration of HGF also slightly increased the rate of BVEC proliferation, the extent of growth stimulation after treatment with HGF concentrations up to 30 ng/ml was consistently higher in LEC than in BVEC (Figure 4A).
Figure 4.
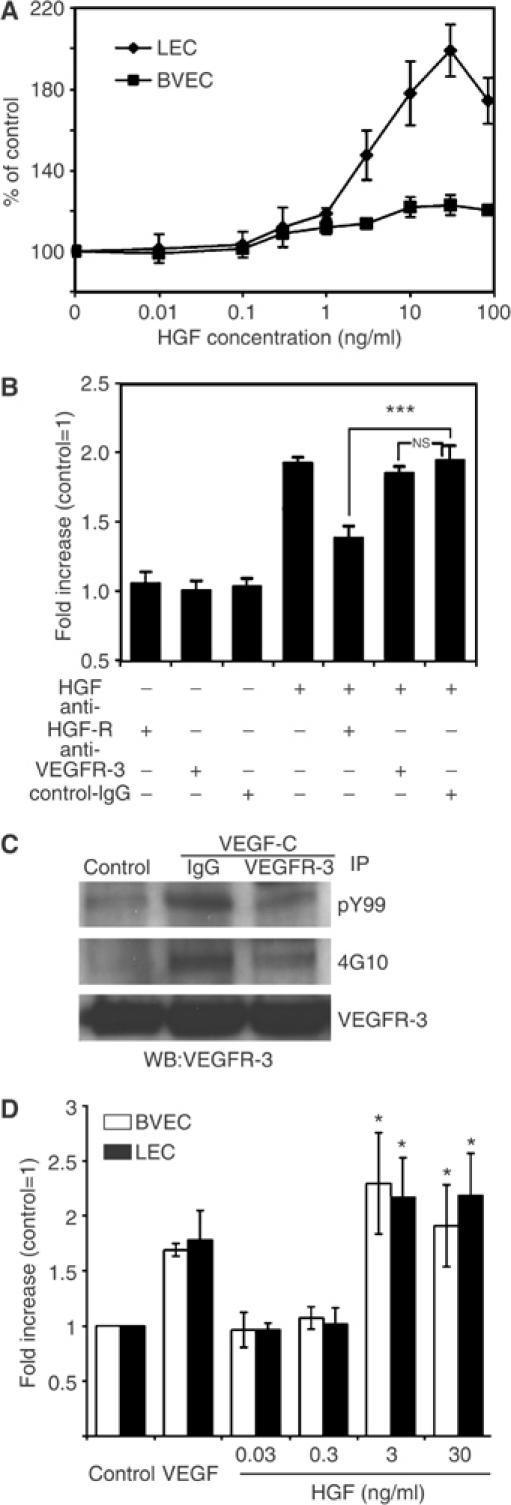
HGF promotes LEC proliferation and migration. (A) HGF induced proliferation of LEC (♦) in a dose-dependent manner, compared to untreated control cells. Proliferation was induced at a minimum effective concentration of 1 ng/ml HGF (P<0.01). HGF induced higher levels of proliferation in LEC than in BVEC (▪) at all concentrations. (B) An HGF-R-blocking antibody potently inhibited the HGF-induced stimulation of LEC proliferation (***P<0.001). Incubation with a VEGFR-3 blocking antibody or control IgG did not affect HGF-induced proliferation. (C) Treatment of LEC with VEGF-C (500 ng/ml) induced phosphorylation of VEGFR-3, which was inhibited by incubation with an anti-human VEGFR-3-neutralizing antibody. Lysates were immunoprecipitated with phosphotyrosine antibodies PY99 or 4G10 and immunoblotted with VEGFR-3. VEGFR-3 signals demonstrate the presence of equal amounts of protein. IP: Immunoprecipitate, WB: Western blot. (D) HGF induced migration of LEC and BVEC with a minimal effective concentration of 3 ng/ml. Data represent mean values ±s.e.m. of three independent experiments (*P<0.05).
Previously, in addition to PDGF-BB (Cao et al, 2004), VEGF-C and VEGF-D were the only growth factors shown to directly promote LEC proliferation, which occurs via activation of the VEGF receptor-3 (VEGFR-3) (Jussila and Alitalo, 2002). The reported ability of FGF-2 to induce lymphangiogenesis is believed to occur through its upregulation of VEGFR-3 ligands, because FGF-2 lymphangiogenic activity can be inhibited by blockade of the VEGFR-3 signalling pathway (Kubo et al, 2002; Chang et al, 2004). To investigate whether HGF directly or indirectly stimulates LEC proliferation, we treated LEC with HGF in the presence or absence of blocking antibodies against VEGFR-3 or HGF-R. Incubation of LEC with a HGF-R-blocking antibody blocked the stimulation of LEC proliferation by HGF (P<0.001). However, treatment of LEC with the VEGFR-3 antibody did not affect HGF-induced proliferation (Figure 4B), although VEGF-C-induced phosphorylation of VEGFR-3 was inhibited (Figure 4C). No transphosphorylation of VEGFR-3 by treatment with HGF or of HGF-R by treatment with VEGF-C was found in LEC (data not shown). Moreover, 1 ng/ml of HGF and 300 ng/ml of VEGF-C induced proliferation of LEC to a comparable extent (47.91±7.24%, P<0.001 and 47.02±6.61%, P<0.01, respectively), and no synergistic effect of HGF and VEGF-C on LEC proliferation was found (data not shown). These results indicate that HGF-induced LEC proliferation occurs independently from activation of the VEGFR-3 pathway, and is dependent upon the binding of HGF to its receptor.
HGF treatment also promoted migration of LEC and BVEC in a dose-dependent manner, starting at a minimal effective concentration of 3 ng/ml (Figure 4D). To investigate whether HGF stimulation might also promote the in vitro formation of lymphatic tubes, confluent LEC cultures were overlaid with type I collagen, as described previously (Hirakawa et al, 2003). HGF potently induced cord formation by LEC (Figure 5A and B) and BVEC (data not shown), with a minimal effective dose of 3 ng/ml (P<0.001). Untreated cells were used as controls.
Figure 5.
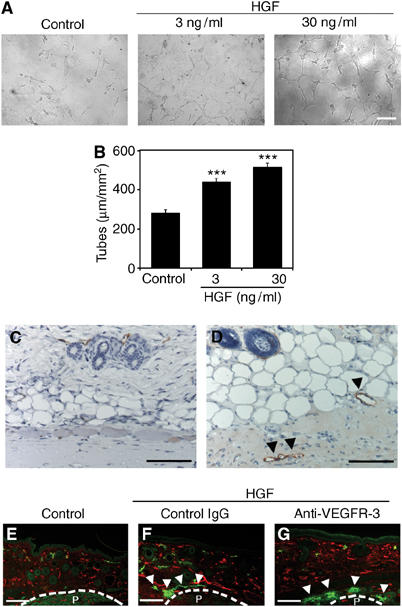
HGF promotes LEC tube formation in vitro and lymphatic vessel formation in vivo. (A, B) Treatment of LEC with 3 and 30 ng/ml HGF promoted cord formation in response to overlay with type I collagen, compared with untreated control cultures. Data are expressed as mean values ±s.d. (***P<0.001). Control Matrigels (C) and those that contain HGF (D) were subcutaneously implanted in FVB mice. Immunostaining for the lymphatic-specific glycoprotein podoplanin (red) revealed pronounced formation of new lymphatic vessels (D; arrowheads) within HGF-containing matrigels at day 7 after implantation, whereas no lymphatic vessel formation was observed within control matrigels. (E–G) Double immunofluorescence analyses of mouse ear sections for LYVE-1 (green) and CD31 (red) showed induction of LYVE-1-positive lymphatic vessel formation at 14 days after implantation of HGF-containing slow-release pellets (F and G; arrowheads), but not that of control pellets (E). Systemic treatment with a blocking anti-VEGFR-3 antibody (G) did not inhibit HGF-induced lymphatic vessel formation, as compared with control IgG treatment (F). Newly formed blood vessels were observed in all samples. P: Pellet. Scale bars: 100 μm (A, C–G).
HGF promotes lymphatic vessel formation in vivo
To investigate whether HGF might also induce lymphangiogenesis in vivo, we subcutaneously implanted HGF-containing matrigels into FVB mice, as described (Hirakawa et al, 2003). Immunostaining for the lymphatic-specific glycoprotein podoplanin (Schacht et al, 2003) revealed pronounced formation of new lymphatic vessels within HGF-containing matrigels by 7 days after implantation. No lymphatic vessels were observed within control matrigels, which did not contain HGF (Figure 5C and D). We next investigated the lymphatic vasculature in previously established metallothionin I promoter-driven HGF transgenic mice. We focused our analysis on the skin and the small intestine where lymphatic vessels are most abundant, and where abnormalities of the lymphatic system are most easily detected in genetic mouse models. We found vascular enlargement in the mucosa and submucosa of the ileum in HGF transgenic mice (Figure 6B), as compared with wild-type mice (Figure 6A). Immunofluorescence stains for the lymphatic-specific marker podoplanin revealed pronounced dilation of central lacteals and enlargement of lymphatic vessels in the submucosa of the ileum in HGF transgenic mice (Figure 6C and D). Podoplanin stains also revealed an increased number and an enlargement of lymphatic vessels in the skin of HGF transgenic mice (Figure 6G and H), whereas no major histological abnormalities were observed (Figure 6E and F). Enhanced lymphatic vessel formation and enlargement were also observed in the duodenum and liver of HGF transgenic mice (data not shown). To examine whether HGF promotes the formation of new lymphatic vessels directly or indirectly via the VEGFR-3 pathway, slow-release pellets—with or without HGF—were implanted subcutaneously into mouse ears, and mice were treated systemically with a blocking antibody against mouse VEGFR-3 or with control IgG. After 14 days, immunofluorescence stains for CD31 and LYVE-1 revealed pronounced lymphatic vessel formation surrounding HGF-containing pellets, but not surrounding control pellets (Figure 5E–G). However, treatment with an anti-VEGFR-3-blocking antibody did not prevent lymphatic vessel formation induced by HGF (Figure 5F and G). Mice implanted with HGF-containing pellets showed moderately enhanced formation of blood vessels.
Figure 6.
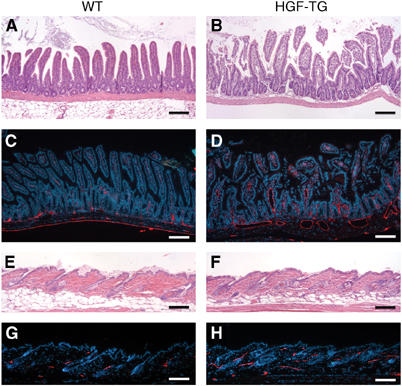
Enhanced formation of lymphatic vessels in HGF transgenic mice. Representative histologic images (H&E stain) of the ileum revealed vascular enlargement in HGF transgenic mice (B), as compared with wild-type mice (A). Immunofluorescence stains for the lymphatic vessel marker podoplanin (red) revealed enlarged central lacteals and enlarged submucosal lymphatic vessels in HGF transgenic mice (D), as compared with wild-type mice (C). Nuclei are labeled blue (Hoechst stain). (E–H) No major differences were observed in the histoarchitecture of the skin of HGF transgenic (F) and wild-type mice (E). However, podoplanin stains (red) revealed numerous enlarged lymphatic vessels in the skin of HGF transgenic mice (H) as compared with wild-type mice (G). Scale bars: 100 μm.
Systemic blockade of HGF-R inhibits lymphatic vessel enlargement during experimental skin inflammation
As HGF-R was strongly expressed by the enlarged lymphatic vessels that form in a mouse model of skin inflammation, and because expression of HGF, the ligand of HGF-R, was upregulated in the inflamed skin as compared to normal skin (Supplementary data 2, A and B), we investigated whether HGF might directly contribute to in vivo lymphatic vessel enlargement. Delayed-type hypersensitivity reactions were induced by topical application of oxazolone to mouse ears, as described (Kunstfeld et al, 2004). At 1 day prior to induction of experimental inflammation, 100 μg of a blocking antibody against HGF-R or an equal amount of control IgG was injected intraperitoneally. Immunofluorescence analyses of skin samples taken 24 h after the induction of inflammation revealed that the size of lymphatic vessels was greatly reduced in mice that had received treatment with the HGF-R-blocking antibody, as compared to mice that had received control IgG (Figure 7A and B). Computer-assisted morphometric analyses of sections stained for LYVE-1 and CD31 demonstrated that the average size of lymphatic vessels and the percentage of tissue area covered by lymphatic vessels were significantly decreased after injection of the HGF-R-blocking antibody (P<0.01), as compared with control IgG-treated mice (Figure 7C and D). The density of lymphatic vessels was not significantly different between the two treatment groups (Figure 7E). There were no significant differences of the area occupied by blood vessels (Figure 7F), the average vessel size (Figure 7G) and the number of the vessels (Figure 7H) between two groups. No significant differences of the number of infiltrating CD4-positive, CD8-positive, or CD11b-positive cells were found between mice treated with the blocking anti-HGF-R antibody (Supplementary data 2, C–H), as compared to control IgG-treated mice. Moreover, no major difference of epidermal thickness was observed between mice treated with blocking anti-HGF-R antibody (13.1±1.57 μm) and control IgG-treated mice (14.3±1.71 μm).
Figure 7.
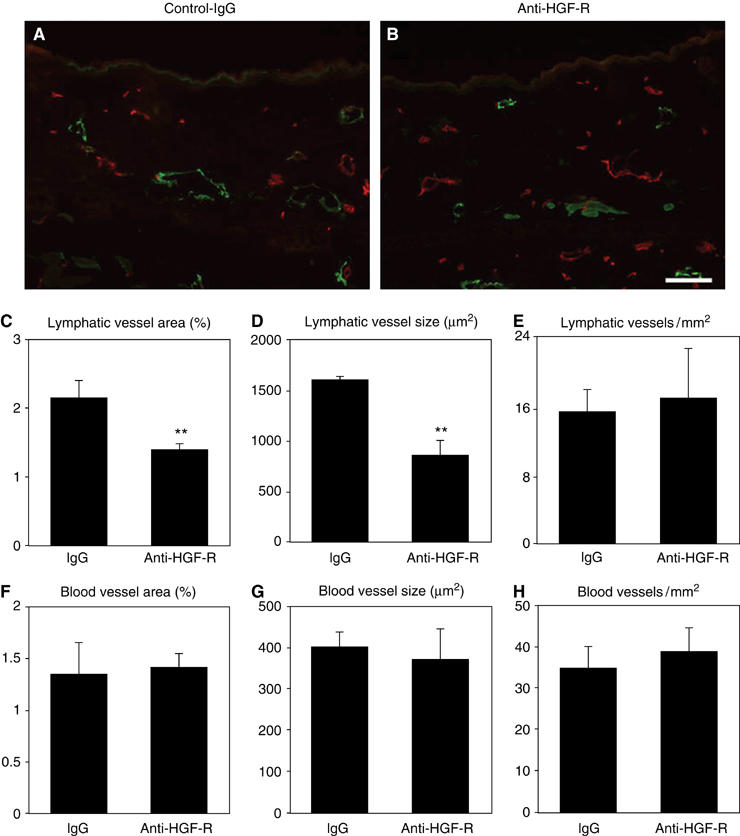
HGF-R blockade inhibits lymphatic but not blood vessel enlargement in an experimental model of skin inflammation. (A, B) Double immunofluorescence analysis for LYVE-1 (green) and CD31 (red) revealed that the characteristic enlargement of lymphatic vessels at 24 h after induction of inflammation was inhibited in mice treated with a blocking anti-HGF-R antibody (B), as compared with the control IgG-treated group (A). Scale bars: 100 μm. (C–H) Computer-assisted morphometric analyses demonstrated that the relative tissue area occupied by lymphatic vessels (C) and the average size of lymphatic vessels (D) were significantly reduced after blockade of HGF-R (**P<0.01), compared to control IgG-treated mice (C, D). No significant differences of the density of lymphatic vessels were found between both groups (E), and there were no significant differences of the area occupied by blood vessels (F), the average blood vessel size (G) and the number of blood vessels (H).
HGF promotes LEC migration via the integrin alpha-9
To identify mechanisms by which HGF promotes LEC proliferation, migration and lymphangiogenesis, we performed gene expression analysis—using the Affymetrix HU133 plus2.0. arrays—on two independent LEC lines that were incubated with 30 ng/ml HGF for 6 h, compared to untreated control cells (accession number E-MEXP-256; MIAMExpress: http://www.ebi.ac.uk/miamexpress/login.html). We found that stanniocalcin 1 was one of the most highly upregulated genes after HGF treatment, in agreement with recent results obtained in HUVEC cells (Zlot et al, 2003). Furthermore, the expression of the integrin alpha-9 was also significantly upregulated after HGF treatment. These results were confirmed by quantitative real-time RT–PCR analysis (Figure 8A).
Figure 8.
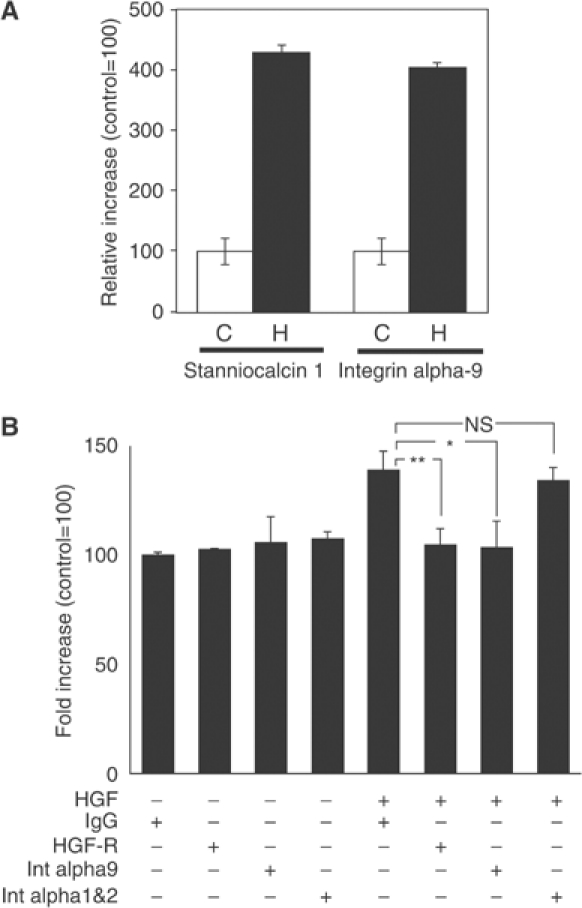
HGF promotes LEC migration via integrin alpha-9. (A) SYBR-Green-based quantitative real-time RT–PCR shows that the expression of stanniocalcin 1 and of integrin alpha-9 are significantly upregulated after treatment of LEC with HGF (H) as compared with control (C). The expression levels were normalized to those of β-actin. (B) Treatment of LEC with HGF induces cell migration, but coincubation of cells with an integrin alpha-9-specific blocking antibody prevents this migration (*P<0.05). Incubation with an HGF-R-blocking antibody also inhibited the induction of LEC migration by HGF (**P<0.01), whereas LEC migration in the absence of HGF was not affected by treatment with an integrin alpha-9-blocking antibody or an HGF-R-blocking antibody. Treatment with control IgG or combined treatment with blocking antibodies against the integrin alpha-1 and -2 had no effect on HGF-induced LEC migration. Data represent mean values ±s.e.m. (_n_=5 per group), and are representative of three independent experiments.
As integrin alpha-9 has been previously shown to be involved in lymphatic vessel formation (Huang et al, 2000), and because integrins are important mediators of endothelial cell migration (Senger et al, 1997), we looked to see whether integrin alpha-9 is required for HGF-induced LEC migration. Incubation of LEC with an integrin alpha-9-specific blocking antibody blocked HGF-induced migration (_P_=0.0143) to roughly the same extent as LEC treated with an HGF-R-specific blocking antibody (_P_=0.0074). In contrast, incubation with an HGF-R-blocking antibody or with an integrin alpha-9-blocking antibody did not affect the migration of LEC in the absence of HGF (Figure 8B). Treatment with a control IgG or with blocking antibodies against both the integrin alpha-1 and alpha-2—that have previously been shown to inhibit VEGF-A-induced LEC migration (Hong et al, 2004b)—did not inhibit LEC migration in response to HGF treatment. Together, these findings indicate that integrin alpha-9 is an important mediator of HGF-induced migration of LEC.
Discussion
In a search for regulators of lymphangiogenesis, we have used gene expression analysis, in vitro and in vivo studies to identify HGF as a potent lymphangiogenic factor. We found that the HGF-R is expressed by LEC more strongly than by BVEC in vitro, that HGF-R is expressed by activated lymphatic endothelium in vivo, and that HGF directly promotes proliferation, migration, and tube formation in cultured LEC. Furthermore, HGF promotes formation of new lymphatic vessels in vivo, and the promigratory effects of HGF on LEC are largely mediated by the integrin alpha-9. Blockade of the HGF–HGF-R signalling pathway might therefore serve as a new strategy to inhibit unwanted lymphatic vessel growth.
It has been a challenge to identify factors that regulate lymphangiogenesis because of a lack of reliable markers to distinguish between lymphatic and blood vascular differentiation. Moreover, the lack of suitable in vitro models for the selective cultivation of both vascular cell types prevented comparative functional studies. The recent identification of lymphatic-specific genes (for a review, see Oliver and Detmar, 2002) cleared the path for molecular investigations of lineage-specific vascular differentiation and function, and for the reliable isolation and expansion of LEC and BVEC.
FGF-2 has been shown to induce lymphangiogenesis indirectly, via stimulation of the release of the lymphangiogenic factor VEGF-C, and FGF-2's effects on lymphangiogenesis can be prevented by blockade of VEGFR-3 signalling (Kubo et al, 2002; Chang et al, 2004). HGF, in contrast, directly stimulates LEC proliferation, and does not require VEGFR-3 signalling, but can be completely prevented by blockade of the HGF-R. Moreover, HGF treatment did not increase LEC expression of the VEGFR-3 ligands VEGF-C or VEGF-D (data not shown). So, HGF–HGF-R signalling, in addition to the VEGF-C/-D–VEGFR-3 (Jussila and Alitalo, 2002) and the PDGF-BB–PDGFR pathways (Cao et al, 2004), acts as a third lymphangiogenesis growth factor system.
How does HGF mediate its effects on LEC migration? Based on transcriptional profiling studies, we identified the integrin alpha-9 as a factor that is highly upregulated in LEC after HGF treatment. Previous studies have shown that integrin alpha-9 is required for the normal development of the lymphatic system (Huang et al, 2000), and that mice that lack this transmembrane signalling protein develop chylothorax—a sign of impaired lymphatic transport. Our finding that HGF-induced LEC migration, but not migration of LEC in the absence of HGF, was inhibited by a blocking antibody against integrin alpha-9, along with recent findings that integrin alpha-9 is specifically upregulated in cultured LEC (Petrova et al, 2002), indicate that this integrin might be required for the LEC motility that occurs during lymphangiogenesis. In fact, interaction of the integrin alpha-9 with the lymphangiogenic factor VEGF-C has been recently reported (Vlahakis et al, 2005). Other integrins, such as integrins alpha-1 and alpha-2, have been shown to be expressed by cultured LEC and to mediate VEGF-A-induced LEC migration (Hong et al, 2004b); however, blockade of these integrins did not inhibit HGF-induced LEC migration. The distinct functional roles and in vivo expression patterns of these individual integrins in different lymphatic vessel types remain to be established.
What is the role of HGF-R signalling during normal embryonic development? During embryogenesis, Prox1-positive lymphatic progenitor cells bud from the CV and migrate out beginning at E10.5 to form the embryonic lymph sacs and, consequently, the lymphatic vascular network (Wigle et al, 2002). We found that HGF-R was expressed on LYVE-1-positive endothelial cells of the anterior CV by E12.5, but also occasionally by the LYVE-1-positive LEC that line the primitive lymph sacs. By E14.5, HGF-R was strongly expressed by the majority of LYVE-1- and Prox1-positive endothelial cells of the lymph sacs, revealing that HGF-R is expressed at a later stage of lymphatic development than Prox1 or LYVE-1. This expression pattern is similar to that of neuropilin-2 receptor and podoplanin (Yuan et al, 2002; Schacht et al, 2003), indicating a possible role of HGF-R during the later stages of lymphatic network formation and maturation.
HGF also appears to function in the lymphangiogenesis that occurs during cutaneous tissue repair. In a model of mouse wound healing (Hong et al, 2004b), we found that lymphangiogenic LYVE-1-positive vessels expressed high levels of HGF-R, whereas lymphatic vessels in normal skin expressed little or no HGF-R. Similarly, we found that HGF-R was strongly expressed by enlarged lymphatic vessels in a mouse model of chronic skin inflammation (Kunstfeld et al, 2004). Together, these findings indicate that HGF-R is preferentially expressed by activated, proliferating lymphatic endothelium, but not by quiescent lymphatic vessels in normal skin, a finding with important implications for the potential use of targeted HGF-R-blocking therapeutic strategies.
Our findings that transgenic overexpression of HGF, as well as implantation of HGF-containing Matrigels or slow-release pellets, induced pronounced formation of new lymphatic vessels are the first demonstration that HGF acts as a potent lymphangiogenesis factor in vivo. HGF also has important effects on lymphatic vessel function, since blockade of HGF-R signalling prevents lymphatic enlargement during cutaneous inflammation and also impairs lymphatic transport. Alternatively, direct intradermal injection of HGF into ear skin promotes lymphatic flow, as demonstrated by in vivo confocal microcopy (K Kajiya, unpublished results). Importantly, HGF-R blockade has only minor effects on blood vessel activation in the same experimental models, in accordance with our findings that activation of HGF-R by HGF stimulated LEC proliferation more potently than BVEC proliferation. Similarly, it has been found that VEGF-C, the first identified lymphangiogenesis factor, specifically promotes LEC proliferation, with only minor effects on BVEC (Alitalo and Carmeliet, 2002).
The expression of the lymphangiogenic factors VEGF-C and -D has been correlated with metastasis of many human tumour types (Stacker et al, 2002). Our preliminary data that tumour-associated LYVE-1-positive lymphatic vessels strongly express HGF-R (K Kajiya, unpublished results), together with the correlation between tumour expression of HGF and metastasis of human tumours to the lymph nodes (Birchmeier et al, 2003), raise the possibility that HGF, in addition to its direct effects on some tumour cells, also contributes to tumour progression by promoting lymphangiogenesis. Therefore, HGF and HGF-R might represent promising new targets for the therapeutic blockade of lymphatic cancer spread.
Materials and methods
Cells
Human dermal BVEC and LEC were isolated from neonatal human foreskins, as previously described (Hirakawa et al, 2003), with slight modifications. Briefly, in order to remove fibrocytes, CD45-negative selection was performed using an immunomagnetic beads-conjugated anti-human CD45 antibody (Dynal, Lake Success, NY) before isolating CD34-positive BVEC. Thereafter, the remaining CD34-negative cells were incubated with an immunomagnetic beads-conjugated anti-human CD31 antibody (Dynal) to isolate LEC.
Quantitative real-time RT–PCR
The methods used are described in Supplementary data 3.
Immunoblotting
For Western blot analyses of HGF-R and phosphotyrosine, confluent BVEC and LEC were homogenized in lysis buffer, and protein concentrations were determined using the BCA-Kit (Pierce Biotechnology, Rockford, IL). The lysates (300 μg total protein each) were immunoprecipitated with a goat polyclonal antibody against HGF-R (R&D Systems, Minneapolis, MN) and then immunoblotted with a rabbit polyclonal antibody against HGF-R (Santa Cruz Biotechnology, Santa Cruz, CA). To assess tyrosine phosphorylation levels, LEC were cultured with HGF (30 ng/ml) for 15 min, followed by homogenization in lysis buffer. Untreated cells were prepared as controls in the same manner. Cell lysates (300 μg total protein each) were immunoprecipitated with a goat polyclonal antibody against HGF-R (R&D Systems) and then immunoblotted with an anti-phosphotyrosine antibody (PY99; Santa Cruz Biotechnology). In additional studies, LEC were incubated with 1 μg/ml of anti-human VEGFR-3 antibody (clone hF4-3C5, kind gift of Dr B Pytowski, Imclone Systems Inc., New York, NY) or with 1 μg/ml of control IgG for 2 h, and were then treated or not with 500 ng/ml VEGF-C (R&D Systems) for 10 min, followed by homogenization in lysis buffer and immunoprecipitation (300 μg total protein) with antiphosphotyrosine antibodies PY99 or 4G10 (Upstate, Lake Placid, NY) or with an anti-VEGFR-3 antibody (Santa Cruz Biotechnology), and then immunoblotted with anti-VEGFR-3 antibody. Specific binding was detected by the enhanced chemiluminescence system (Amersham Biosciences, Piscataway, NJ).
Immunofluorescence analyses
Immunofluorescence analyses were performed on 6-μm cryostat sections of mouse tissues or on 10-μm sections of mouse embryos as described (Hong et al, 2004b; Kunstfeld et al, 2004), using polyclonal antibodies against murine LYVE-1 (kindly provided by Dr D Jackson, Oxford, UK), murine HGF-R (R&D Systems), murine CD4, CD8, CD11b and CD31 (all from BD Biosciences, Bedford, MA), HGF (Santa Cruz Biotechnology, Santa Cruz, CA) and Prox1 (kindly provided by Dr K Alitalo, Helsinki, Finland). Immunohistochemical analyses for podoplanin were performed on 6-μm 4% paraformaldehyde-fixed skin sections as described previously (Schacht et al, 2003), using the hamster antibody 8.1.1 (Developmental Studies Hybridoma Bank, University of Iowa). Paraffin sections were also obtained from the formaldehyde-fixed skin, duodenum, liver and ileum of 8-weeks-old transgenic FVB mice, with overexpression of HGF under control of the mouse metallothionin I promoter (_n_=4; kindly provided by Dr Glenn Merlino, National Institutes of Health, Bethesda, USA) (Takayama et al, 1996) and from age-matched wild-type FVB mice, and were stained for podoplanin. Corresponding secondary antibodies were labeled with AlexaFluor488 or AlexaFluor594 (Molecular Probes, Eugene, OR). Sections were examined by a Nikon E-600 microscope (Nikon, Melville, NY) and images were captured with a SPOT digital camera (Diagnostic Instruments, Sterling Heights, MI). Computer-assisted morphometric vessel analyses of representative LYVE-1 and CD31 double-stained sections were performed as described (Hirakawa et al, 2005b).
Proliferation, migration and tube formation assays
BVEC or LEC (2 × 103) were seeded onto fibronectin-coated 96-well plates. Quinduplicate wells were treated or not with several concentrations of recombinant human HGF (R&D Systems) in EBM containing 2% fetal bovine serum. LEC were also incubated with HGF (30 ng/ml) together with anti-human HGF-R (R&D Systems), anti-human Flt4 (kind gift of Dr B Pytowski, Imclone Systems Inc., New York, NY), or control IgG (1 μg/ml, respectively). After 72 h, cells were incubated with 5-methylumbelliferylheptanoate as described (Detmar et al, 1992). The fluorescence intensity, proportional to the number of viable cells, was measured using a Victor2 Fluorometer (Perkin-Elmer, Boston, MA). Haptotatic cell migration was performed as described (Hong et al, 2004b), using 24-well FluoroBlok inserts of 8 μm pore size (Falcon, Franklin Lakes, NJ). The bottom sides of the inserts were coated with 10 μg/ml fibronectin (BD Biosciences, Bedford, MA) for 1 h, followed by incubation with 100 μg/ml bovine serum albumin (BSA). Cells (105 cells in 100 μl) were seeded in serum-free EBM containing 0.2% delipidized BSA into the upper chambers, and were incubated for 3 h at 37°C in the presence or absence of human recombinant VEGF (20 ng/ml) or HGF (0.03–30 ng/ml). In additional studies, cells were incubated with 10 μg/ml of a blocking anti-integrin alpha-9 antibody (clone Y9A2, Chemicon, Temecula, CA), an anti-HGF-R antibody (R&D Systems), blocking antibodies against the integrins alpha-1 (Upstate) and alpha-2 (Chemicon, Temecula, CA), or control IgG for 60 min. Cells were then seeded into the upper chambers and were incubated for 3 h in the presence or absence of human recombinant HGF (30 ng/ml). Cells on the underside of inserts were stained with Hoechst 33342 (Molecular Probes). Three different digital images per well were taken, and the number of migrated cells was counted. Tube formation assays were performed as described (Schacht et al, 2003). LEC or BVEC were grown on fibronectin-coated 24 well plates until confluence. In all, 0.5 ml of neutralized isotonic bovine dermal collagen type I (Vitrogen, Palo Alto, CA) in the absence or presence of HGF (3 or 30 ng/ml) was added to the cells. After incubation at 37°C for 6 h, cells were fixed with 4% paraformaldehyde for 30 min at 4°C. Representative images were captured and the total length of tube-like structures per area was measured using the IP-LAB software. All studies were performed in triplicate. Statistical analyses were performed using the unpaired Student's _t_-test.
In vivo lymphangiogenesis and HGF-R blocking assays
FVB wild-type mice (male, 10 weeks old) were subcutaneously injected with 250 μl of Matrigel (BD Bioscience; 9 mg/ml) containing or not 1 mg/ml HGF. After 7 days, mice were killed and tissues were fixed for 24 h in 4% paraformaldehyde and then embedded in paraffin. Immunohistochemistry for mouse podoplanin was performed as described above. In addition, delayed-type hypersensitivity reactions were induced in FVB mice as described (Kunstfeld et al, 2004). Mice (female, 8 weeks old) were sensitized by topical application of oxazolone to the paws and the shaved abdomen 5 days before challenge with topical application of oxazolone to the ears (Kunstfeld et al, 2004). At 1 day before oxazolone challenge, 100 μg of a goat anti-mouse HGF-R antibody (R&D Systems) or of control goat IgG was intraperitoneally injected (_n_=5 per group). At 1 day after challenge, mice were killed and ears were embedded and frozen in OCT compound (Sakura Finetek, Torrance, CA). To confirm the blocking activity of the anti-HGF-R antibody, murine B16 melanoma cells (0.5 × 103) were seeded onto 96-well plates and quinduplidate wells were treated or not with 3 ng/ml of HGF (R&D Systems) together with the anti-mouse HGF-R antibody or with control IgG (10 μg/ml) in DMEM containing 5% FBS. After 72 h, cell proliferation was assessed as described above for LEC. HGF treatment promoted B16 cell proliferation (33.6±5.08%), whereas incubation with the anti-HGF antibody prevented HGF-induced stimulation of cell proliferation (6.26±7.72%; P<0.01). In additional studies, slow-release pellets containing or not recombinant HGF (500 ng/pellet) were implanted subcutaneously into the ears of 8-weeks-old female FVB mice as described previously (Hirakawa et al, 2005a). Beginning at 4 days after implantation, mice bearing HGF pellets were injected i.p. with 600 μg of control IgG (Sigma) or of rat anti-mouse VEGFR-3 neutralizing antibody (clone mF4-31C1, Imclone Inc.) (Pytowski et al, 2005) every 2 days. Mice with control pellets were injected with control IgG (_n_=5 per group). Mouse ears were harvested at 14 days after implantation and were snap-frozen for histological analysis. All animal studies were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Supplementary Material
Supplemental Data 1
Supplementary data 2
Supplementary Material 3:
Acknowledgments
We thank M Constant and L Janes for expert technical assistance. This work was supported by NIH grants CA69184, CA86410, CA92644 (MD), American Cancer Society Research Project Grant 99-23901 (MD), Swiss National Fund project grant 3100A0-108207 (MD), FWF grant S9408-B11, and by the Cutaneous Biology Research Center through the Massachusetts General Hospital/Shiseido Co. Ltd Agreement (MD).
References
- Alitalo K, Carmeliet P (2002) Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell 1: 219–227 [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF (2003) Met, metastasis, motility and more. Nat Rev Mol Cell Biol 4: 915–925 [DOI] [PubMed] [Google Scholar]
- Cao R, Bjorndahl MA, Religa P, Clasper S, Garvin S, Galter D, Meister B, Ikomi F, Tritsaris K, Dissing S, Ohhashi T, Jackson DG, Cao Y (2004) PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell 6: 333–345 [DOI] [PubMed] [Google Scholar]
- Chang LK, Garcia-Cardena G, Farnebo F, Fannon M, Chen EJ, Butterfield C, Moses MA, Mulligan RC, Folkman J, Kaipainen A (2004) Dose-dependent response of FGF-2 for lymphangiogenesis. Proc Natl Acad Sci USA 101: 11658–11663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadras SS, Paul T, Bertoncini J, Brown LF, Muzikansky A, Jackson DG, Ellwanger U, Garbe C, Mihm MC, Detmar M (2003) Tumor lymphangiogenesis: a novel prognostic indicator for cutaneous melanoma metastasis and survival. Am J Pathol 162: 1951–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilkovitch-Miagkova A, Zbar B (2002) Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest 109: 863–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmar M, Tenorio S, Hettmannsperger U, Ruszczak Z, Orfanos CE (1992) Cytokine regulation of proliferation and ICAM-1 expression of human dermal microvascular endothelial cells in vitro. J Invest Dermatol 98: 147–153 [DOI] [PubMed] [Google Scholar]
- Giordano S, Di Renzo MF, Narsimhan RP, Cooper CS, Rosa C, Comoglio PM (1989) Biosynthesis of the protein encoded by the c-met proto-oncogene. Oncogene 4: 1383–1388 [PubMed] [Google Scholar]
- Hirakawa S, Fujii S, Kajiya K, Yano K, Detmar M (2005a) Vascular endothelial growth factor promotes sensitivity to ultraviolet B-induced cutaneous photodamage. Blood 105: 2392–2399 [DOI] [PubMed] [Google Scholar]
- Hirakawa S, Hong YK, Harvey N, Schacht V, Matsuda K, Libermann T, Detmar M (2003) Identification of vascular lineage-specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am J Pathol 162: 575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF, Detmar M (2005b) VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med 201: 1089–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YK, Foreman K, Shin JW, Hirakawa S, Curry CL, Sage DR, Libermann T, Dezube BJ, Fingeroth JD, Detmar M (2004a) Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat Genet 36: 683–685 [DOI] [PubMed] [Google Scholar]
- Hong YK, Lange-Asschenfeldt B, Velasco P, Hirakawa S, Kunstfeld R, Brown LF, Bohlen P, Senger DR, Detmar M (2004b) VEGF-A promotes tissue repair-associated lymphatic vessel formation via VEGFR-2 and the alpha1beta1 and alpha2beta1 integrins. FASEB J 18: 1111–1113 [DOI] [PubMed] [Google Scholar]
- Huang XZ, Wu JF, Ferrando R, Lee JH, Wang YL, Farese RV Jr, Sheppard D (2000) Fatal bilateral chylothorax in mice lacking the integrin alpha9beta1. Mol Cell Biol 20: 5208–5215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers M, Rong S, Vande Woude GF (1996) Enhanced tumorigenicity and invasion-metastasis by hepatocyte growth factor/scatter factor-met signalling in human cells concomitant with induction of the urokinase proteolysis network. Mol Cell Biol 16: 1115–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jussila L, Alitalo K (2002) Vascular growth factors and lymphangiogenesis. Physiol Rev 82: 673–700 [DOI] [PubMed] [Google Scholar]
- Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K (2004) Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol 5: 74–80 [DOI] [PubMed] [Google Scholar]
- Kubo H, Cao R, Brakenhielm E, Makinen T, Cao Y, Alitalo K (2002) Blockade of vascular endothelial growth factor receptor-3 signaling inhibits fibroblast growth factor-2-induced lymphangiogenesis in mouse cornea. Proc Natl Acad Sci USA 99: 8868–8873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunstfeld R, Hirakawa S, Hong YK, Schacht V, Lange-Asschenfeldt B, Velasco P, Lin C, Fiebiger E, Wei X, Wu Y, Hicklin D, Bohlen P, Detmar M (2004) Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood 104: 1048–1057 [DOI] [PubMed] [Google Scholar]
- Makinen T, Jussila L, Veikkola T, Karpanen T, Kettunen MI, Pulkkanen KJ, Kauppinen R, Jackson DG, Kubo H, Nishikawa S, Yla-Herttuala S, Alitalo K (2001) Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med 7: 199–205 [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, Banerji S, Huarte J, Montesano R, Jackson DG, Orci L, Alitalo K, Christofori G, Pepper MS (2001) Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J 20: 672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Nakamura T (2001) Hepatocyte growth factor: renotropic role and potential therapeutics for renal diseases. Kidney Int 59: 2023–2038 [DOI] [PubMed] [Google Scholar]
- Nagy JA, Vasile E, Feng D, Sundberg C, Brown LF, Detmar MJ, Lawitts JA, Benjamin L, Tan X, Manseau EJ, Dvorak AM, Dvorak HF (2002) Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J Exp Med 196: 1497–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, Sugimura A, Tashiro K, Shimizu S (1989) Molecular cloning and expression of human hepatocyte growth factor. Nature 342: 440–443 [DOI] [PubMed] [Google Scholar]
- Oliver G, Detmar M (2002) The rediscovery of the lymphatic system: old and new insights into the development and biological function of the lymphatic vasculature. Genes Dev 16: 773–783 [DOI] [PubMed] [Google Scholar]
- Petrova TV, Makinen T, Makela TP, Saarela J, Virtanen I, Ferrell RE, Finegold DN, Kerjaschki D, Yla-Herttuala S, Alitalo K (2002) Lymphatic endothelial reprogramming of vascular endothelial cells by the Prox-1 homeobox transcription factor. EMBO J 21: 4593–4599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pytowski B, Goldman J, Persaud K, Wu Y, Witte L, Hicklin DJ, Skobe M, Boardman KC, Swartz MA (2005) Complete and specific inhibition of adult lymphatic regeneration by a novel VEGFR-3 neutralizing antibody. J Natl Cancer Inst 97: 14–21 [DOI] [PubMed] [Google Scholar]
- Rosen EM, Knesel J, Goldberg ID, Jin L, Bhargava M, Joseph A, Zitnik R, Wines J, Kelley M, Rockwell S (1994) Scatter factor modulates the metastatic phenotype of the EMT6 mouse mammary tumor. Int J Cancer 57: 706–714 [DOI] [PubMed] [Google Scholar]
- Schacht V, Ramirez MI, Hong YK, Hirakawa S, Feng D, Harvey N, Williams M, Dvorak AM, Dvorak HF, Oliver G, Detmar M (2003) T1alpha/podoplanin deficiency disrupts normal lymphatic vasculature formation and causes lymphedema. EMBO J 22: 3546–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger DR, Claffey KP, Benes JE, Perruzzi CA, Sergiou AP, Detmar M (1997) Angiogenesis promoted by vascular endothelial growth factor: regulation through alpha1beta1 and alpha2beta1 integrins. Proc Natl Acad Sci USA 94: 13612–13617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvagno F, Follenzi A, Arese M, Prat M, Giraudo E, Gaudino G, Camussi G, Comoglio PM, Bussolino F (1995) In vivo activation of met tyrosine kinase by heterodimeric hepatocyte growth factor molecule promotes angiogenesis. Arterioscler Thromb Vasc Biol 15: 1857–1865 [DOI] [PubMed] [Google Scholar]
- Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K, Detmar M (2001) Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med 7: 192–198 [DOI] [PubMed] [Google Scholar]
- Stacker SA, Achen MG, Jussila L, Baldwin ME, Alitalo K (2002) Lymphangiogenesis and cancer metastasis. Nat Rev Cancer 2: 573–583 [DOI] [PubMed] [Google Scholar]
- Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S, Kubo H, Achen MG (2001) VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med 7: 186–191 [DOI] [PubMed] [Google Scholar]
- Takayama H, La Rochelle WJ, Anver M, Bockman DE, Merlino G (1996) Scatter factor/hepatocyte growth factor as a regulator of skeletal muscle and neural crest development. Proc Natl Acad Sci USA 93: 5866–5871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahakis NE, Young BA, Atakilit A, Sheppard D (2005) The lymphangiogenic vascular endothelial growth factors VEGF-C and -D are ligands for the integrin alpha9beta1. J Biol Chem 280: 4544–4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigle JT, Harvey N, Detmar M, Lagutina I, Grosveld G, Gunn MD, Jackson DG, Oliver G (2002) An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J 21: 1505–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte MH, Bernas MJ, Martin CP, Witte CL (2001) Lymphangiogenesis and lymphangiodysplasia: from molecular to clinical lymphology. Microsc Res Tech 55: 122–145 [DOI] [PubMed] [Google Scholar]
- Yuan L, Moyon D, Pardanaud L, Breant C, Karkkainen MJ, Alitalo K, Eichmann A (2002) Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development 129: 4797–4806 [DOI] [PubMed] [Google Scholar]
- Zarnegar R, Michalopoulos GK (1995) The many faces of hepatocyte growth factor: from hepatopoiesis to hematopoiesis. J Cell Biol 129: 1177–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlot C, Ingle G, Hongo J, Yang S, Sheng Z, Schwall R, Paoni N, Wang F, Peale FV Jr, Gerritsen ME (2003) Stanniocalcin 1 is an autocrine modulator of endothelial angiogenic responses to hepatocyte growth factor. J Biol Chem 278: 47654–47659 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data 1
Supplementary data 2
Supplementary Material 3: