Conformational changes in dynamin on GTP binding and oligomerization reported by intrinsic and extrinsic fluorescence (original) (raw)
Abstract
The effects of guanine nucleotides on the intrinsic and extrinsic fluorescence properties of dynamin were assessed. The intrinsic Trp (tryptophan) fluorescence spectra of purified recombinant dynamin-1 and -2 were very similar, with a maximum at 332 nm. Collisional quenching by KI was weak (∼30%), suggesting that the majority of Trp residues are buried. Binding of guanine nucleotides decreased intrinsic fluorescence by 15–20%. Titration of the effects showed that GTP and GDP bound to a single class of non-interacting sites in dynamin tetramers with apparent dissociation constants (_K_d) values of 5.4 and 7.4 μM (dynamin-1) and 13.2 and 7.1 μM (dynamin-2) respectively. Similar dissociation constant values for both nucleotides were obtained by titrating the quenching of IAEDANS [_N_-iodoacetyl-_N_′-(5-sulpho-1-naphthyl)ethylenediamine]-labelled dynamin-2. Despite the similar binding affinities, GTP and GDP result in different conformations of the protein, as revealed by sensitivity to proteinase K fragmentation. Dynamins contain five Trp residues, of which four are in the PH domain (pleckstrin homology domain) and one is in the C-terminal PRD (proline/arginine-rich domain). Guanine nucleotides quenched fluorescence emission from a truncated (ΔPRD) mutant dynamin-1 to the same extent as in the full-length protein, suggesting conformational coupling between the G (groove)-domain and the PH domain. Efficient resonance energy transfer from PH domain Trp residues to bound mant-GTP [where mant stands for 2′-(3′)-_O_-(_N_-methylanthraniloyl)] suggests that the G-domain and PH domain are in close proximity (5–6 nm). Promotion of dynamin-2 oligomerization, by reduction in ionic strength or increasing protein concentration, had little effect on intrinsic dynamin fluorescence. However, fluorescence emission from IAEDANS·dynamin-2 showed a significant spectral shift on oligomerization. In addition, energy transfer was observed when oligomerization was promoted in mixtures of IAEDANS·dynamin-2 and 4-(4-dimethylaminophenylazo)benzoic acid-coupled dynamin-2, an effect that was counteracted by GTP but not GDP.
Keywords: dynamin, fluorescence quenching, GTP binding, oligomerization, pleckstrin homology domain (PH domain), tryptophan fluorescence
Abbreviations: BS3, bis(succinimidyl)suberate; dabcyl, 4-(4-dimethylaminophenylazo)benzoic acid; DOL, degree of labelling; DTT, dithiothreitol; EF, elongation factor; GTP[β-S], guanosine 5′-[β-thio]triphosphate; GTP[γ-S], guanosine 5′-[γ-thio]triphosphate; IAEDANS, _N_-iodoacetyl-_N_′-(5-sulpho-1-naphthyl)ethylenediamine; mant, 2′-(3′)-_O_-(_N_-methylanthraniloyl); NATA, _N_-acetyltryptophanamide; PH domain, pleckstrin homology domain; PRD, proline/arginine-rich domain; RET, resonance energy transfer; Trp, tryptophan
INTRODUCTION
The dynamins comprise a family of large GTPases, many of which play critical roles in several types of vesicular fission events (for reviews, see [1–5]). Their participation in endocytosis at the plasma membrane is thought to be at the scission step where internalizing vesicles are finally severed from the plasma membrane. In most models, dynamin oligomers are thought to form a ring-like collar, or possibly a spiral, around the neck of the invaginating vesicle that either constricts to sever the connection or, in the case of the spiral model, increases pitch to ‘pop’ the vesicle from the membrane [4,5]. Alternatively, others believe that dynamin may be a regulatory ‘switch-like’ GTPase with no mechanochemical properties, comparable with small and heterotrimeric G-proteins [4]. The three major subspecies of dynamin itself (dynamin-1, -2 and -3) are closely related multidomain proteins of approx. 100 kDa, but a variety of less related, frequently smaller, proteins play disparate roles in other aspects of cell regulation [1–3].
Within the family of dynamins proper, significant progress has been made in elucidating the contribution of each domain to the overall function of the protein. The N-terminal nucleotide-binding domain (‘G-domain’) is responsible for GTP hydrolysis and resembles equivalent domains in other G-proteins such as Ras and EFs (elongation factors) [6–8]. However, the three-dimensional structure of the G-domain from Dictyostelium discoideum dynamin A reveals that this subfamily of G-proteins has a 55-amino- acid insertion that extends the core G-domain β-sheet from a six-stranded to an eight-stranded structure, with an additional interposed α-helix [9]. The C-terminal PRD (proline/arginine-rich domain) links dynamin to several other proteins that contain SH3 (Src homology-3) domains (e.g. Grb2, amphiphysin and endophilin; [10–12]), which may be important docking sites facilitating the precise localization of dynamin in the cell. The central PH domain (pleckstrin homology domain) probably controls dynamin binding to membrane phospholipids [13], and a coiled-coil domain (also called the GTPase effector domain or GED) that follows the PH domain may play an important role in dynamin self-assembly and regulation of GTPase activity [4,14]. Ligand interactions with the PH domain and PRD, as well as the GED, impact the GTPase activity of dynamin, suggesting that conformational transitions in these regions are transmitted to the G-domain to regulate catalysis.
Very little is known about the conformational behaviour of dynamin and structural information has been restricted so far to either isolated domains or low-resolution cryomicroscopy of truncated proteins [9,15]. Fluorescence measurements, from either intrinsic sources or extrinsic labels, are often useful for reporting conformational states of proteins [16]. For example, emission from native Trp (tryptophan) residues is extremely sensitive to perturbations in the local environment, generally due to different degrees of quenching. Extrinsic probes can give similar information but have increased flexibility, e.g. in the use of RET (resonance energy transfer). In the present study, we evaluate the effects of guanine nucleotide binding on intrinsic and extrinsic fluorescence properties of dynamin-1 and -2 and examine domain and subunit interactions using RET. Both GTP and GDP lead to substantial intrinsic and extrinsic fluorescence quenching, and together with RET analysis, this suggests that the nucleotide-binding domain is in close proximity to the PH domain. We also show that dynamin subunit interactions can be detected by RET techniques.
EXPERIMENTAL
Purification of recombinant dynamin-1 and -2
Baculovirus expressing N-terminal His6-tagged dynamin-1, prepared by insertion of the coding sequence of human dynamin-1 into pFastBac, was kindly provided by Dr K. Ferguson and Dr M. Lemmon (University of Pennsylvania, Philadelphia, PA, U.S.A.). An equivalent virus coding for rat dynamin-2 was a gift from Dr D. Binns and Dr J. Albanesi (University of Texas Southwestern Medical School, Dallas, TX, U.S.A.) and virus stock for a truncated version of dynamin-1 (dynamin-1·ΔPRD) was obtained from Dr S. Schmid (Scripps Research Institute, La Jolla, CA, U.S.A.). Viruses were amplified by infection of Sf9 insect cells maintained at 26 °C for 48–72 h on an orbital shaker. For protein expression, amplified virus was used to infect 1 litre cultures of Sf9 or Tn5 insect cells (ClonTech Laboratories, Palo Alto, CA, U.S.A.) grown in serum-free Excell medium (JRH Biosciences, Lenexa, KS, U.S.A.) at a multiplicity of infection of approx. 5×108. After 64 h of incubation, cells were harvested by centrifugation at 1000 g for 5 min. The pellet was washed once with 50 ml of homogenization buffer (0.2 M NaCl, 20 mM Hepes, pH 8.0, 1 mM PMSF, 100 μM leupeptin, 1 μg/ml pepstatin and 5%, v/v, glycerol), then resuspended in 20 ml of the same buffer and disrupted by brief (4×5 s) pulses of a probe sonifier (Branson Ultrasonic, Danbury, CT, U.S.A.). The homogenate was centrifuged at 48000 g for 40 min and the resultant supernatant clarified by re-centrifugation under the same conditions. For full-length dynamin-1 and -2, the final supernatant was diluted with homogenization buffer to a protein concentration of 2.5–3 mg/ml and was directly applied batchwise to a Talon metal chelate affinity resin (ClonTech Laboratories; resin/supernatant ratio of 1:10) equilibrated with the same buffer. The resin was washed twice with 10 vol. of a buffer containing 10 mM imidazole (pH 7.0) and 0.2 M NaCl. Recombinant dynamins were eluted with a buffer containing 100 mM imidazole and 0.2 M NaCl. The peak fractions were pooled, made up to 10% with respect to glycerol and 1 mM DTT (dithiothreitol) was added, after which the protein was divided into aliquots and frozen at −70 °C. The yield was approx. 12 mg of protein/109 cells for dynamin-1 and approx. 15 mg/109 cells for dynamin-2. For dynamin-1·ΔPRD, the cell supernatant was applied first to DEAE-Sepharose with elution using a 200 mM step. This pool was applied to a column of ceramic hydroxyapatite (Bio-Rad Laboratories, Hercules, CA, U.S.A.); after washing with 200 mM sodium phosphate buffer (pH 7.4), the purified protein was step eluted with 400 mM of the same buffer. The purity of these products was assessed as >95% by SDS/PAGE (7.5% polyacrylamide) (Figure 1, and results not shown); protein concentrations were determined by a dye-binding assay.
Figure 1. Purification of recombinant (His6) dynamins from baculovirus-infected Tn5 cells and analysis of holoenzyme structure by cross-linking.

After baculoviral infection and incubation for 48 h, Tn5 insect cells were lysed and the clarified supernatant was applied to and eluted from an immobilized metal-ion affinity chromatography column. (A) Samples (50 μg of lysate and 5 μg of purified product) were subjected to SDS/PAGE (7.5% polyacrylamide) and the gels were stained. Lane 1, lysate from dynamin-1 baculovirus-infected cells; lane 2, purified dynamin-1; lane 3, lysate from dynamin-2 baculovirus-infected cells; and lane 4, purified dynamin-2. (B) Native recombinant dynamin exists as a tetramer. Dynamin-1 (or -2) (10 μg) in 500 mM NaCl was treated with 5 mM BS3 for 60 min and the samples were processed for immunoblot analysis using anti-pan-dynamin antibodies. Molecular-mass standards (116 kDa: β-galactosidase; 408 kDa: cardiac plasmalemmal dystrophin) are indicated at right.
To establish that the recombinant full-length proteins existed in the tetrameric form in physiological salt, dynamins were cross-linked with 5 mM BS3 [bis(succinimidyl)suberate; Pierce, Rockford, IL, U.S.A.] and the products were separated by SDS/PAGE (4% polyacrylamide) and identified by immunoblotting using anti-dynamin IgGs, as described previously [17]. Additionally, we confirmed that reduction in ionic strength (from 100 to 25 mM NaCl) resulted in oligomerization of dynamin-2, as assessed by centrifugation at 100000 g for 30 min [18].
Dynamin GTPase assays and proteolysis
GTPase activity was determined by measuring the hydrolysis of [γ-32P]GTP (Amersham Biosciences). Reactions were performed at 37 °C in buffer A comprising 20 mM Hepes (pH 7.4), 1 mM MgCl2, 1 mM DTT, 0.2 mM PMSF, 40 μM leupeptin, 1 mM [γ-32P]GTP and 1–10 μM dynamin. NaCl concentrations were adjusted to maintain a final concentration of 25, 50, 75 or 100 mM. [32P]Pi produced by hydrolysis was separated from unchanged nucleotide using activated charcoal (Norit A) with the inclusion of appropriate buffer-only blanks. Taxol-stabilized microtubules were a gift from Dr E. W. Taylor (Northwestern University Medical School, Chicago, IL, U.S.A.) and were used at a concentration of 100 μg/ml.
To assess the proteolytic profile of dynamin-2 after limited digestion, we incubated the enzyme (preincubated with no nucleotide, 2 mM GDP[β-S] (guanosine 5′-[β-thio]triphosphate) or 2 mM GTP[γ-S] (guanosine 5′-[γ-thio]triphosphate) with proteinase K at a proteinase/dynamin-2 ratio of 1:1666. The reaction was carried out for 90 min on ice and the products were separated by SDS/PAGE (7.5% polyacrylamide) and identified by Coomassie Blue staining and immunoblotting.
Steady-state intrinsic fluorescence measurements
Intrinsic fluorescence measurements were performed at 22 °C on a Hitachi F2000 spectrofluorimeter set at an excitation wavelength (λex) of 295 nm (slit width=4 nm). At this wavelength, the contribution of tyrosine residues to dynamin fluorescence was negligible and no evidence of Raman emission lines was found. Emission spectra were collected from 310 to 460 nm and were corrected for the buffer blank. The proteins were diluted to a concentration of 0.5–2 μM in buffer A containing 20 mM Hepes, 1 mM MgCl2, 0.1 mM DTT, 0.2 mM PMSF, 40 μM leupeptin and 1 μg/ml pepstatin A. NaCl concentrations were adjusted to yield a final concentration of 25–100 mM. Five types of sample were analysed: 0.5 μM dynamin-1 in 100 mM NaCl (dynamin-1 tetramer); 2 μM dynamin-1 in 35 mM NaCl (dynamin-1 oligomer); 0.5 μM dynamin-2 in 100 mM NaCl (dynamin-2 tetramer) and either 0.5 μM dynamin-2 in 50 mM NaCl or 2 μM dynamin-2 in 100 mM NaCl (dynamin-2 oligomer).
Quenching experiments with KI or acrylamide were performed by addition of small aliquots of concentrated stock solutions to the cuvette; measurements were taken 30 s later and dilution was taken into account. Results were analysed using the Stern–Volmer equation or its reciprocal variant, the Lehrer equation: _F_o/Δ_F_=1/_f_a+(1/_f_a_K_SV)(1/[Q]), where F_o is the fluorescence without quencher, Δ_F is the reduction in fluorescence (Δ_F_=F_o–_F; where F is the intensity in the presence of quencher) after the addition of quencher, [Q] is the quencher concentration, _f_a is the fraction of fluorescence emission accessible to the quencher and _K_SV is the Stern–Volmer quenching constant (see [19] for details). Quenching experiments with various nucleotides (GTP, GTP[γ-S], GDP, GDP[β-S], ATP and ADP) were performed in a similar manner. To correct for the increased sample absorption due to guanine nucleotide addition (‘inner filter’ effect), we measured the influence of these compounds on the fluorescence emission of the model compound NATA (_N_-acetyltryptophanamide; Sigma; λex=295 nm; λem=332 nm) diluted in buffer A at each nucleotide concentration; the effect of nucleotides was linear with respect to concentration and the relevant values were then subtracted from those obtained with dynamin. The data were analysed using a modified form of the Michaelis–Menten equation
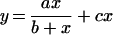
where y is the quenching at different guanine nucleotide concentrations, a is the maximal quenching, _x_=[GTP] or [GDP], _b_=‘_K_d’ and c is a constant representing a linear component. Non-linear least-squares best fits and associated parameters were generated using SigmaPlot software (Jandel Scientific, Madera, CA, U.S.A.).
Extrinsic fluorescence of IAEDANS·dynamin
Dynamin-1 or -2 was labelled with the fluorescent reagent IAEDANS [_N_-iodoacetyl-_N_′-(5-sulpho-1-naphthyl)ethylenediamine; Molecular Probes] under standard conditions. Briefly, dynamins at 10 μM in 20 mM Hepes (pH 7.0), 1 mM MgCl2, 500 mM NaCl and 10% glycerol were incubated for 3 h in the dark with a 50-fold molar excess of IAEDANS. The reaction was stopped by the addition of 200-fold molar excess of DTT and free dye was removed by extensive dialysis. This resulted in the incorporation of approx. 4 mol/mol of dynamin, as calculated at the λmax (excitation) of 336 nm using molar absorption coefficient ϵ=5700 M−1·cm−1. Labelling had no effect on the GTPase activity of dynamin (results not shown), suggesting that modification did not substantially perturb the overall protein function. Fluorescence emission of IAEDANS was evoked by illumination at 295 nm, resulting in energy transfer from Trp residues to the extrinsic label, or directly by illumination at 336 nm; emission was recorded at 310–550 nm.
Mant-GTP binding
The fluorescent GTP analogue mant-GTP [where mant stands for 2′-(3′)-_O_-(_N_-methylanthraniloyl)] was prepared by minor modification of the method of Hiratsuka [20]. All unchanged components were separated from mant-GTP using a Macro Prep High Q column; mant-GTP was eluted from the column with a triethylammonium bicarbonate gradient (0.1–1.5 M). The associated absorption _A_255/_A_356 ratio was 3.96 for the main component, and the purity of the material was confirmed by TLC. Dynamins (0.5 μM) were mixed with a 10-fold molar excess of mant-GTP, yielding a binding ratio of 0.73 for probe/protein. For energy transfer experiments, excitation was at 295 nm, while emission spectra were collected from 310 to 550 nm. For displacement experiments with GTP and GDP, excitation was at a λmax (excitation) of 356 nm to avoid ‘inner filter’ effects. The distance between Trp residues and mant-GTP was estimated spectroscopically by Förster energy transfer [16]. The efficiency (E) of RET was determined by comparing the intensity of the donor in the presence of acceptor (_F_DA) with the donor intensity in the absence of acceptor (_F_D). E is then given by:
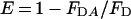
The distance between the fluorophores (R) is given by:
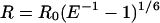
where R is calculated in Å and _R_0 is the Förster critical distance that gives 50% of the transfer efficiency. _R_0 is given by:
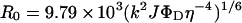
where _k_2 is an orientation factor for which the value of 2/3 is generally used, η is the refractive index of the buffer (in this case, 1.4), ΦD is the quantum yield of the donor and J is the overlap integral in cm3/M given by:
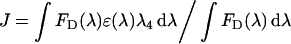
where λ is the wavelength in cm, _F_D(λ) is the corrected fluorescence of the unquenched donor and ϵ(λ) is the acceptor molar absorption coefficient in M−1·cm−1. J was obtained by numerical integration of normalized spectra at 1 nm intervals. The quantum efficiency of the donor Trp residues in dynamin was determined on the basis of the value of 0.13 for _N_-acetyl-L-tryptophanamide from the following relation:
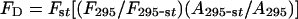
where _F_D is the quantum yield of Trp residues in dynamin, _F_st is the quantum yield of the standard, _F_295 and _F_295-st are the respective areas of the emission spectrum of the protein and standard when excited at 295 nm, and _A_295-st and _A_295 are the associated absorption values.
Energy transfer experiments to examine oligomerization
Separate samples of dynamin-1, dynamin-2 or dynamin-1·ΔPRD were labelled with IAEDANS (as above) or with the dye dabcyl [4-(4-dimethylaminophenylazo)benzoic acid; Molecular Probes]. The labelling procedure for dabcyl was similar to that for IAEDANS, and between 3 and 5 mol of dye/mol of dynamin was incorporated (as determined by emission at λmax=454 nm using molar absorption coefficient ϵ=32000 M−1·cm−1; this modification had no discernible effect on GTPase activity). Then equal amounts of IAEDANS·dynamin-1 (or -2) were mixed with dabcyl·dynamin-2 (or -1) or dabcyl·dynamin-1·ΔPRD and the [NaCl] was either left at 500 mM salt (no oligomerization control) or reduced to 50 mM salt to induce oligomerization. Illumination was at either 295 or 334 nm and quenching of donor IAEDANS emission by the acceptor dabcyl was recorded at 310–550 nm.
RESULTS
Intrinsic fluorescence properties of dynamin-1 and -2
Dynamin-1 and -2, His-tagged at their N-termini, were purified essentially to homogeneity from baculovirus-infected Sf9 or Tn5 cells (Figure 1A). The intrinsic basal GTPase activity of these preparations were 3.1 and 37.7 nmol·mg−1·min−1 respectively, and these values were increased to 350 and 300 nmol·mg−1·min−1 by addition of microtubules and to a lesser extent by oligomerization induced by ionic strength reduction (Table 1, and results not shown). Cross-linking experiments indicated that the predominant form of the expressed recombinant proteins was tetrameric (Figure 1B), as described previously for dynamin-1 ([21,22]; see the Discussion section). Dynamin-2 tetramers were found to oligomerize efficiently into higher-order structures when diluted into low-ionic-strength solutions or by increasing the protein concentration at 100 mM NaCl, as assessed by sedimentation analysis (see the Experimental section), whereas dynamin-1 had a much lower propensity to precipitate under similar conditions (results not shown; cf. [23]). These results indicate that the dynamins used in these experiments are functionally active.
Table 1. Parameters of GTP and GDP binding to dynamin-1, dynamin-1·ΔPRD and dynamin-2 derived from quenching of intrinsic Trp fluorescence.
Dissociation constants (_K_d) were determined from titration curves fitted to the Michaelis–Menten equation using SigmaPlot 5.0. Δ_F_max is the percentage of maximal fluorescence decrease in the presence of saturating concentrations of nucleotide. GTPase activity was measured as described in the Experimental section. Models: dynamin-1 tetramer (0.5 mM dynamin-1+100 mM NaCl); dynamin-1 oligomer (2.0 mM dynamin-1+35 mM NaCl); dynamin-2 tetramer (0.5 mM dynamin-2+100 mM NaCl); dynamin-2 oligomer (2.0 mM dynamin-2+100 mM NaCl); ΔPRD·dynamin-1 (0.5 mM ΔPRD·dynamin-1+100 mM NaCl).
| GTP | GDP | ||||
|---|---|---|---|---|---|
| Dynamin isoform | _K_d (μM) | Δ_F_max | _K_d (μM) | Δ_F_max | GTPase activity (nmol·mg−1·min−1) |
| Dynamin-1 | |||||
| Tetramer | 5.4 | 19.7 | 7.4 | 18.6 | 3.1 |
| Oligomer | 6.3 | 7.3 | 2.8 | 12.9 | 11.5 |
| Dynamin-2 | |||||
| Tetramer | 13.2 | 17.6 | 7.1 | 17.1 | 37.7 |
| Oligomer | 1.1 | 10.7 | 3.5 | 11.4 | 113.5 |
| ΔPRD·dynamin-1 | 6.0 | 15.9 | 3.5 | 22.3 | 15.9 |
The excitation–emission spectra of dynamin-2 are shown in Figure 2(A). The excitation maximum was at 276 nm; when excited at 295 nm (where contribution from tyrosine residues is minimal), the emission spectrum was broad, exhibiting a maximum at 332 nm. The spectral parameters of dynamin-1 were virtually identical (results not shown). To bring all Trp residues to the same polar environment, dynamin-2 was denatured with 8 M urea. This decreased the emission intensity by approx. 55% and the spectrum was markedly red-shifted to λmax (emission)∼344 nm (Figure 2B). These results indicate that most Trp residues in the native protein reside in an apolar environment. The disposition of the Trp residues in dynamin-1 is summarized in Figure 2(C).
Figure 2. Intrinsic spectral properties of dynamin-2.

(A) Excitation and emission spectra of 0.5 μM native dynamin-2, determined in buffer A plus 100 mM NaCl; λmax (excitation)=276 nm, λmax (emission)=332 nm (excitation at 295 nm). (B) Emission spectra of 1 μM native (–––) and denatured (▲) dynamin-2 (8 M urea, 1 h) were determined in buffer A using an excitation wavelength of 295 nm. The maximum fluorescence intensity was decreased by 54% and λmax (emission) was red-shifted to 344 nm following denaturation. (C) Location of Trp residues in dynamin. Left-hand panel shows linear domain structure of dynamin with positions of the five Trp residues indicated; M, middle domain. Right-hand panel shows disposition of four Trp residues (black) in the PH domain of dynamin-1 (model based on crystal structure).
Influence of collisional quenchers on Trp fluorescence
To probe further the influence of environment on the dynamin-1 emission spectrum, we assessed the effects of the ionic and neutral quenchers, I− and acrylamide. KI was more efficacious over the entire concentration range (0.1–1 M), reducing emission by approx. 30%, whereas acrylamide had a much smaller (<10%) effect. Both KI (Figure 3) and acrylamide (results not shown) yielded downward-curving Stern–Volmer plots, probably due to differential quenching of distinct Trp residues. KI also caused a narrowing of the emission spectrum, from 75 to 60 nm at half-peak (results not shown). Reciprocal (Lehrer) plots (Figure 3, inset) yielded a _K_SV of 130 mM and a fractional ratio of emission accessible to quencher (_f_a) of 0.34, suggesting that only one or two of the five Trp residues was efficiently quenched by KI (but see [19] for discussion of the complexities in these plots). We suspected that the lone Trp in the C-terminal PRD of dynamin might be exposed and highly susceptible to KI quenching, hence we titrated the effects of [KI] on fluorescence emission from dynamin-1·ΔPRD, a truncated form that is enzymatically active and thus it is probably folded in a manner similar to the parent protein (see, e.g., [15]). The _f_a value decreased to 0.27 (Figure 3 and inset), suggesting that the PRD Trp is probably exposed to the environment in the full-length protein. These data confirm that the majority of Trp residues are buried away from the solvent in the intact dynamin molecule.
Figure 3. Quenching of dynamin-1 and dynamin-1·ΔPRD fluorescence by KI.

Stern–Volmer plots of fluorescence quenching by KI of dynamin-1 (●) and dynamin-1·ΔPRD (○). Straight lines represent the best fit (generated by SigmaPlot) to the Stern–Volmer equation, yielding _K_SV (dynamin-1)=130 mM, _K_SV (dynamin-1·ΔPRD)=100 mM. The inset (Lehrer plot) yields the fraction of fluorophores (_f_a) accessible to KI: _f_a (dynamin-1)=0.34, _f_a (dynamin-1·ΔPRD)=0.27. Conditions: buffer A (pH 7.0), 100 mM NaCl and 1 μM dynamin-1 or dynamin-1·ΔPRD.
Quenching of dynamin fluorescence by guanine nucleotides
Several studies have shown that guanine nucleotide binding affects the conformation of diverse G-proteins, and this is reflected in intrinsic fluorescence measurements (e.g. [24–26]). We investigated this possibility with dynamin with the goal of determining the apparent affinities of various guanine nucleotides. Addition of either GTP or GDP led to significant quenching of native dynamin-1 and -2 fluorescence (Figures 4A and 4C). Because quenching was manifest at micromolar concentrations of nucleotides, we took into account the ‘inner filter’ effect (loss of fluorescence due to absorption of incident light by nucleotide) by linearly subtracting obtained values from titration of similar nucleotide concentrations on the fluorescence emission of NATA (results not shown). With this correction, titration of nucleotide quenching yielded hyperbolic relationships (Figures 4B and 4D) with apparent _K_d values for all species in the low μM range (Table 1). A complicating aspect was the presence of a significant, apparently linear, component of quenching in all titrations even after the subtraction of ‘inner filter’ effects. Such a component was also found in quenching experiments with the extrinsic fluorophore IAEDANS (see below), so it is unlikely to be artifactual and may be indicative of non-specific binding or further low-affinity nucleotide-binding sites; this component was not investigated further. Transformation of the quenching data into logarithmic plots yielded approximately linear relationships with slopes (Hill coefficients) of <1 (Figures 4B and 4D, insets). These results suggest a single class of non-interacting binding sites for GTP and GDP. Very similar curves were obtained when the corresponding non-hydrolysable analogues of GTP (GTP[S]) and GDP (GDP[β-S]) were used (for _K_d values, see Table 1), but no quenching was observed after the addition of 0.5 mM ATP, ADP or p[NH]ppA (adenosine 5′-[β,γ-imido]triphosphate), indicating that dynamin does not bind adenine nucleotides even at high concentrations.
Figure 4. Guanine nucleotide quenching of intrinsic dynamin fluorescence.

(A, C) Corrected spectra of dynamin-1 (A) and dynamin-2 (C) in the presence or absence (control) of 100 μM GTP or GDP. Conditions: buffer A, 100 mM NaCl, 0.5 μM tetrameric dynamin-1 (dynamin-2), λmax (excitation)=295 nm, λmax (emission)=332 nm in the absence of nucleotide and λmax (emission)=330 nm in the presence of nucleotides. Spectra with nucleotides were corrected for the ‘inner filter’ effect as described in the Experimental section. (B, D) Titration of quenching effects with increasing concentrations of GTP or GDP. The percentage fluorescence decrease %Δ_F_=100−(%_F_dynamin+GTP(GDP)−%_F_NATA+GTP(GDP)) was determined by integration of the emission spectrum (from 310 to 500 nm, background subtracted to account for the ‘inner filter’ effect) and plotted as a function of nucleotide concentration. All fits contained linear components, and are interpreted as ‘non-specific binding’ here (see text). Insets: logarithmic plots of the data in (A, C) had slopes of <1, suggesting a single class of non-interacting nucleotide-binding sites.
Slight differences in nucleotide effects emerged when quenching analysis was conducted at higher dynamin concentrations that favour the oligomeric state. Oligomerization caused only a marginal decrease in native fluorescence emission of both dynamin-1 and -2 (results not shown), but reduced the quenching efficiency of both GTP and GDP (Table 1). These results suggest that oligomerization may reduce the exposure of Trp residues that are responsive to guanine nucleotides. Strikingly, GTP was a significantly more potent quencher at 2 μM dynamin-2, where a large fraction of the protein becomes oligomeric, but oligomerization had little effect on the apparent affinity of GTP for dynamin-1. The apparent affinity for GDP also slightly increased under these conditions for both dynamin isoforms.
Quenching of fluorescence by guanine nucleotides was also apparent in extrinsically labelled dynamin. Tetrameric dynamin-1 and -2 were tagged with the thiol-specific fluorophore IAEDANS to 4–5 mol/mol (there are seven cysteine residues in dynamin); this had no effect on GTPase activity and did not alter oligomerization induced by dilution into low-ionic- strength buffers (see below), suggesting that little structural perturbation occurs. Guanine nucleotides quenched emission from IAEDANS·dynamin by approx. 25% at 100 μM. Titrations of these effects yielded hyperbolic relationships, but with distinct linear components as found with intrinsic fluorescence (results not shown). In the present study, excitation was conducted at λmax (excitation) of IAEDANS, where ‘inner filter’ effects due to high nucleotide concentrations are negligible. _K_d values from these experiments were similar to those found above using intrinsic fluorescence (Table 2) and are consistent with dynamin having equivalent affinity for GTP and GDP.
Table 2. Nucleotide binding to dynamin-2: comparison of three methods.
Binding constants were determined from the titration curves fitted to the Michaelis–Menten equation using SigmaPlot 5.0. _K_d represents the nucleotide concentration yielding half-maximal quenching. n.d., not determined. Models: dynamin-2 tetramer (0.5 mM dynamin-2+100 mM NaCl); dynamin-2 oligomer (2.0 mM dynamin-2+100 mM NaCl).
| _K_d (μM) | ||||
|---|---|---|---|---|
| Dynamin-2 | Nucleotide | Trp fluorescence | IAEDANS | Mant-GTP |
| Tetramer | GTP | 13.2 | 8.3 | 10.6 |
| GTP[γ-S] | 7.5 | 4.5 | n.d. | |
| GDP | 7.1 | 27.4 | 59.0 | |
| GDP[β-S] | 5.6 | n.d. | n.d. | |
| Oligomer | GTP | 1.1 | 3.5 | 1.6 |
| GTP[γ-S] | 2.4 | n.d. | n.d. | |
| GDP | 3.5 | 5.3 | 8.2 | |
| GDP[β-S] | 2.9 | n.d | n.d. |
The GTP- and GDP-bound states of dynamin are conformationally distinct
Despite the similarity in apparent affinity for GDP and GTP from quenching studies, by analogy to all other G-proteins, we suspected that dynamin adopted different conformations in the presence of these two nucleotides. To probe this further, we conducted limited proteolysis experiments on proteins loaded with either GDP or GTP and compared the pattern with unliganded protein. As shown in Figure 5, incubation of dynamin-2 in low concentrations of proteinase K resulted in fragmentation of the nucleotide-free protein into several distinct products. Occupation of the G-domain by GDP[β-S] did not alter cleavage significantly, but GTP[γ-S] resulted in subtle changes in the product pattern. In particular, certain peptides below 49 kDa were reduced, with some disappearing altogether, along with a concomitant increase in the amount of a 68 kDa fragment. Differences were also noted when proteolytic profiles were analysed using immunoblotting with a dynamin-2 antibody (results not shown). These results suggest that the conformation of dynamin induced by GTP differs from that adopted in the presence of GDP. Similar results with a comparable digestion method were previously reported for the dynamin-related protein MxA [27].
Figure 5. GTP induces a different conformation of dynamin: proteolytic analysis.

Limited proteolytic cleavage products from dynamin-2 in the presence of different nucleotides. Dynamin-2 (10 μg) was incubated in proteinase K as indicated in the Experimental section, and the pattern of breakdown products in the presence of different nucleotides was analysed by SDS/PAGE (7.5% polyacrylamide) and staining. M, molecular-mass standards (kDa); C, native protein before digestion; −, digested in the absence of nucleotide; GDP, digested in the presence of 2 mM GDP[β-S]; GTP, digested in the presence of 2 mM GTP[γ-S]. Note the absence of bands at 39 and 35 kDa and the diminution of bands at 20 and 17 kDa in the GTP[γ-S]-containing sample. A slight increase in the band at 68 kDa is apparent in this lane.
Energy transfer between the G-domain and PH domain
Of the five Trp residues in dynamin, four reside in the PH domain (Figure 2C). To ascertain if ligation of the G-domain influences the conformation of the PH domain, we studied the effects of GTP and GDP on fluorescence emission from dynamin-1·ΔPRD, which lacks the fifth Trp residue located in the C-terminus. Both nucleotides effectively quenched emission from the truncated protein, to a similar degree and with similar affinities to those seen with the full-length protein (Table 1). These results suggest that the conformational effects of guanine nucleotide bound to the G-domain are transmitted to the PH domain in the protein. Such conformational coupling may imply proximity between the G-domain and PH domain in the tertiary structure of dynamin. To evaluate this possibility, we determined if energy transfer from PH-domain Trp residues to bound GTP could be observed. Previous studies using anthranoyl (mant) derivatives of GTP with full-length dynamin-2 have shown significant energy transfer [22,28]. We confirmed that energy transfer of a similar magnitude occurred in both full-length and truncated versions of dynamin-1, suggesting that radiative emission from PH-domain Trp residues was the major contributor to mant-GTP excitation (Figure 6A). Theory predicts that for energy transfer to occur, donor and acceptor moieties must be within 10 nm of each other [16]. Accordingly, calculation of the average (Förster) distance between the Trp residues in the PH domain and bound mant-GTP gave values of 5.48 nm for dynamin-1 and 5.23 nm for dynamin-1·ΔPRD (see the Experimental section). These results suggest that the G-domain and PH domain lie in close proximity in the intact tetramer. We do not know if our value reflects the proximity of the PH domain and G-domain in dynamin monomers or whether in the tetrameric state the PH domain from one subunit may lie adjacent to the G-domain of another. Recent analytical ultracentrifugation experiments lend support to a monomer–tetramer equilibrium model of dynamin self-association [28]. Our cross-linking results, while not equilibrium data, indicate that all the dynamin in the present experiments could be cross-linked to tetramers at 100 mM NaCl, even at protein concentrations as low as 0.5 μM (E. Solomaha and H. C. Palfrey, unpublished work).
Figure 6. Energy transfer from endogenous Trp residues to bound mant-GTP in dynamin-1 and dynamin-1·ΔPRD and effect of guanine nucleotides on mant-GTP binding.

(A) Mant-GTP emission spectra were recorded in the absence or presence of dynamin-1 or dynamin-1·ΔPRD (both 1 μM in buffer A+100 mM NaCl); λmax (excitation) was 295 nm for all spectra. Solid line represents fluorescence of mant-GTP (10 μM) alone in the absence of protein, and other curves in the presence of the respective proteins. Note that the quenching of donor Trp emission (recorded at 332 nm) and enhancement of acceptor mant-GTP fluorescence (recorded at 438 nm) are virtually identical for the full-length and truncated dynamin-1 species, indicating that PH-domain Trp residues are largely responsible for energy transfer in the full-length protein. (B) Mant-GTP was bound to dynamin-2 (ratio: 0.73 mol/mol of dynamin-2) then emission spectra were collected following direct excitation [λmax (excitation)=356 nm, λmax (emission)=438 nm] in the presence or absence of either GTP or GDP (both 50 μM). Lower solid curve is emission from free mant-GTP at the same concentration. Conditions: buffer A+100 mM NaCl, 2 μM dynamin-2 and 10 μM mant-GTP. (C) Titration of GTP and GDP competition for mant-GTP-binding sites. Experiment as in (B) except that increasing concentrations of nucleotides were used (Δ_F_ is percentage reduction in fluorescence emission of directly excited mant-GTP compared with zero nucleotide control). The continuous lines represent best fits to rectangular hyperbolas derived using SigmaPlot. Note the absence of a linear component (compare Figures 4B and 4D). For _K_d values, see Table 2.
Binding of mant-GTP to dynamins also offered an alternative way to probe guanine nucleotide affinity in the form of displacement assays. GTP competed effectively for mant-GTP binding (Figure 6B) and the apparent affinity compared favourably with those determined by intrinsic fluorescence (Figures 6C; Table 2). However, GDP was a weaker competitor for mant-GTP binding to dynamin-1 (Figures 6B and 6C) with an apparent _K_d of ∼59 μM (Table 2).
Effects of oligomerization on extrinsic fluorescence of dynamins: fluorescence RET assays of dynamin association
On dilution into a low-ionic strength solution, tetrameric dynamin holoenzymes undergo a self-association event, resulting in a stimulation of GTPase activity [18,23]; this may possibly mimic some aspects of the in vivo behaviour of the protein. We confirmed that dilution also stimulated the GTPase activity of both dynamin-1 and -2 GTPase (Table 1), with dynamin-2 being more active in this regard, as described previously [23]. As we did not find significant changes in the intrinsic spectral properties of dynamin under conditions of increased oligomerization, we turned to the use of extrinsic probes. Oligomerization caused a marked blue shift in the fluorescence emission spectrum of IAEDANS·dynamin (Figure 7A), suggesting that at least some of the labelled cysteine residues become buried during oligomerization. The spectral shift was reversed by the addition of GTP, strongly suggesting that the effect was due to oligomerization.
Figure 7. Oligomerization of dynamin-2 detected by spectral shift and RET: inhibitory effect of GTP but not GDP.

(A) Dynamin-2 was labelled with IAEDANS [DOL (degree of labelling)=3.4] and illuminated at 334 nm (excitation maximum for IAEDANS). Fluorescence [recorded at λmax (emission)=478 nm] was determined at a dynamin-2 concentration of 0.5 μM in buffer A+100 mM NaCl, where dynamin-2 is largely tetrameric (–––), or at 2 μM, where dynamin is largely oligomeric (········); note the shift in emission maximum (to 470 nm) and quenching. Addition of GTP (100 μM) to the oligomeric protein led to oligomer disassembly as reflected in a shift back to the original λmax (emission) as well as substantial quenching (----). Inset: precipitation assays of dynamin-2 assembly state: lanes 1 and 2, 0.5 μM and 100 mM NaCl (tetramer); lanes 3 and 4, 0.5 μM and 25 mM NaCl (oligomer); lanes 5 and 6, 2 μM and 100 mM NaCl (oligomer); s, supernatant; and p, pellet of 100000 g spin (30 min). (B) Quenching of IAEDANS·dynamin-2 fluorescence on oligomerization with dabcyl·dynamin-2. Top spectrum: 0.5 μM IAEDANS·dynamin-2 in 50 mM NaCl; bottom spectrum (–––): 0.5 μM IAEDANS·dynamin-2+0.5 μM dabcyl·dynamin-2, and 100 mM NaCl, showing IAEDANS fluorescence decrease upon dynamin-2 oligomerization. Addition of GTP (100 μM) partially reverses energy transfer, indicating reduced dynamin-2 oligomerization (----) but GDP (100 μM) has no effect (········) (DOLIAEDANS=3.43 and DOLdabcyl=1.44). (C) Detection of oligomerization using RET from native Trp residues to IAEDANS. Emission spectra of IAEDANS·dynamin-2 either alone or mixed with an equimolar amount of dabcyl·dynamin-2 are shown. Illumination was at λmax (excitation)=295 nm; note efficient energy transfer from Trp residues to IAEDANS·dynamin-2 and quenching of emission with addition of dabcyl·dynamin-2 (increase in Trp fluorescence is due to doubling of dynamin-2 concentration).
As the spectral shift was quite small, we searched for a more versatile optical tool to investigate dynamin oligomerization. RET has proven to be useful in the assessment of polymerization kinetics in the case of actin and offers the possibility that both homo- and hetero-oligomerization can be investigated. We used IAEDANS·dynamin for such an assay, as IAEDANS fluorescence can be quenched by non-fluorescent acceptors such as the aliphatic amine-reactive compound dabcyl [16]. Initially, we labelled separate pools of dynamin-2 with the two probes, mixed equimolar amounts and recorded the change in fluorescence emission on reduction in ionic strength. We found that IAEDANS fluorescence was efficiently quenched under these conditions (Figure 7B), indicating proximity between emitter- and quencher-containing tetramers in the oligomer. GTP, but not GDP, partially reversed this quenching, indicating that GTP can inhibit oligomerization under these conditions, while GDP is inactive (Figure 7B). Similar results were obtained when IAEDANS fluorescence was activated by energy transfer from intrinsic Trp residues (Figure 7C). The RET method further allowed us to determine if co-polymerization between different species of dynamin could take place in vitro. Separate pools of IAEDANS·dynamin-1 and dabcyl·dynamin-2 were mixed and the salt concentration was reduced to induce assembly. We found efficient quenching of IAEDANS fluorescence, indicating co-assembly of dynamin-1 and -2 (Figure 8A). Confirmation of co-polymerization was obtained by precipitation assays followed by immunoblotting with dynamin-1-specific antibodies (Figure 8A, inset). Co-polymerization was blocked in 500 mM NaCl, resulting in no quenching (Figure 8B). Dynamin-1 could be induced to assemble in 100 mM NaCl with dynamin-1·ΔPRD (Figure 8C), and this was similarly inhibited at high salt concentration (Figure 8D).
Figure 8. Oligomerization of dynamin-1 with dynamin-2 or dynamin-1·ΔPRD detected by RET.

(A) IAEDANS·dynamin-2 was mixed with equimolar amounts of dabcyl·dynamin-1 and emission spectra determined with λmax (excitation)=295 nm; note efficient energy transfer from Trp residues to IAEDANS·dynamin-2 and quenching of emission with addition of dabcyl·dynamin-1 (increase in Trp fluorescence is due to doubling of dynamin concentration). Conditions: buffer A and 100 mM NaCl. DOLIAEDANS=3.43 (dynamin-2), DOLdabcyl=3.1 (dynamin-1). Inset: immunoblots showing increased precipitation of dynamin-1 when co-oligomerized with dynamin-2, as detected by immunoblotting with an anti-dynamin-1-specific IgG; s, supernatant; p, pellet from a 100000 g centrifugation. (B) Same as (A) except that excitation of IAEDANS was direct [λmax (excitation)=334 nm].········, no quenching was observed if IAEDANS·dynamin-2 was mixed with equimolar dabcyl·dynamin-1 in the presence of 500 mM NaCl, under which oligomerization is prohibited. (C, D) Emission spectra of IAEDANS·dynamin-1 either alone or in the presence of dabcyl·dynamin-1 or dabcyl·dynamin-1·ΔPRD. Illumination was at λmax (excitation, Trp)=295 nm (C) or at 334 nm (D). Note efficient energy transfer from Trp residues to IAEDANS·dynamin-1 in (C) and quenching of emission with addition of either full-length or truncated dabcyl dynamin-1 [in C, fluorescence decrease was 47% with full-length and 39% with truncated]. Increase in Trp fluorescence is due to doubling of dynamin-2 concentration. In (D), dotted line indicates emission from IAEDANS·dynamin-1+dabcyl·dynamin-1·ΔPRD in the presence of 500 mM NaCl, where oligomerization does not occur. Conditions: 0.5 μM IAEDANS·dynamin-1±0.5 μM dabcyl·dynamin-1 or dabcyl·dynamin-1·ΔPRD, in buffer A+100 mM NaCl. DOLIAEDANS=5.56 (dynamin-1), DOLdabcyl=7.48 (dynamin-1) and DOLdabcyl=9.82 (dynamin-1·ΔPRD).
DISCUSSION
Dynamin has been a well-studied protein from both biochemical and cell biological standpoints [1–5], but less is known about its structure and conformational transitions. In the present study, we have used both intrinsic and extrinsic fluorescence analyses of two dynamin isoforms, as well as a truncated dynamin-1, to investigate the effects of guanine nucleotide binding and oligomerization on conformation. Dynamin's five Trp residues are located in different environments as revealed by the effects of collisional quenchers. KI, for example, maximally quenched only approx. 30% of the fluorescence emission, suggesting that a majority of the Trp residues are buried. The single Trp in the C-terminal PRD is probably the most exposed, as dynamin-1·ΔPRD showed less quenching at similar concentrations of KI. The remaining four Trp residues are all in the PH domain, providing a useful internal reporter for conformational effects on this domain. In the present study, we show that the intrinsic fluorescence of the PH domain is quenched by guanine nucleotide binding to the G-domain, and that energy transfer between the PH-domain Trp residues and bound GTP occurs. We presume that guanine nucleotide quenching is probably due to the exposure of PH- domain Trp residues to a more polar environment, as evidenced by the reduced quenching efficacy of KI after nucleotide addition (results not shown). Quantification of energy transfer between PH-domain Trp residues and bound mant-GTP suggests that the G-domain and PH domain are within 5.23 nm in the dynamin-1 holoenzyme. This is closer than the approx. 10 nm between the G-domain and PH domain estimated from electron cryomicroscopy of a truncated dynamin [15], but this structure was elucidated in the presence of lipid that probably displaces the PH domain relative to other domains in the protein. Our results clearly support the notion that, in the intact protein, the PH domain and G-domain are in close proximity and are conformationally coupled.
A close interaction between the G-domain and PH domain is probably important for dynamin function. Indeed, as with the G-domain, mutation of critical residues within the PH domain affects the participation of dynamin in endocytosis [29], and the isolated PH domain was found to be a dominant-negative inhibitor of rapid endocytosis [30]. Alongside many other proteins, the dynamin PH domain is thought to link the protein to the membrane, primarily (although perhaps not exclusively) via phospholipid head groups [31]. Direct association of the dynamin PH domain with various phosphoinositides has been shown in vitro and exhibits specificity for di- or tri-phosphorylated species [13]. Binding may be strongly dependent on the oligomerization state of the parent protein, as dimeric PH domains have a much increased avidity for PtdIns(4,5)_P_2-containing surfaces compared with monomers. PtdIns(4,5)_P_2 binding to the PH domain markedly increases dynamin GTPase activity [32], suggesting that lipid association induces a conformational change that is transmitted through the molecule to the G-domain. Removal of the PH domain in dynamin-1 results in an increase in basal GTPase activity, suggesting that in the absence of phospholipid binding the PH domain acts as a tonic inhibitor of GTPase activity [21]. Whether this reflects a direct or indirect interaction between the two domains is not known. In some small GTPases (e.g. cdc42), the PH domain of the corresponding exchange factor does contact the GTPase directly and controls the exchange reaction [33].
Dynamin is a GTPase with relatively weak affinity for GTP, but with a high basal turnover rate compared with heterotrimeric and small G-proteins. In this respect, it resembles other large GTPases, such as EF-2, of protein synthesis. Because of the micromolar dissociation constants, it has been difficult to assess directly the binding of unmodified guanine nucleotides. In particular, the affinity for GDP has not been determined, although it was generally assumed that the _K_d is much higher than that for GTP (e.g. 0.5 mM GDP does not interfere with GTP hydrolysis by dynamin-1 [34]). However, using quenching of intrinsic fluorescence as a probe, we show that GTP and GDP bind with similar micromolar affinities for dynamin-1, dynamin -2 and dynamin-1·ΔPRD. Apparent _K_d values obtained with this assay were notably different for dynamin-1 and -2; we also found that oligomerization of dynamin-2, but not of dynamin-1, resulted in an increased affinity for GTP. These results reinforce previous observations of significant biochemical as well as functional differences between the two isoforms [11,23,35,36]. The _K_d value for GTP in dynamin-1 (5.4 μM for the tetrameric form) is in line with equilibrium constants derived from GTPase activity measurements on this enzyme (e.g. 12 μM, [29]; 7.8 μM, [37]), but diverges slightly from the data of Warnock et al. [23], who found _K_m values of 36 μM for dynamin-1 and 12 μM for dynamin-2 for the ΔPRD versions of these proteins.
The apparent equivalence of GDP and GTP binding with the dynamin family is not inherently surprising when viewed in the context of other G-proteins. For example, the GDP affinity of small G-proteins is generally as high or higher than that for GTP (e.g. [38]), and some other large GTPases also have lower _K_d values for GDP than that for GTP (e.g. EF-2: 1.5 versus 2.8 μM, [25]). Such data have been interpreted to suggest that guanosine ring interactions contribute substantially to the overall binding energy between nucleotides and the G-domain in various GTPases. In contrast, terminal phosphate interactions seem to predominate in the mechanochemical ATPases, where ADP affinities are usually much lower than that of ATP. Nevertheless, as GDP exerts minimal inhibition of the GTPase activity of dynamin [29], we further tested the ability of GDP to displace mant-GTP from the nucleotide-binding site. Here, GDP proved to be considerably weaker than GTP with dynamin-1, suggesting that although both nucleotides can occupy the active site, GDP is a poorer competitor of GTP binding. Although we do not fully understand the differences in apparent affinity of dynamin-1 for GDP based on the two assays, it is interesting to note that a similar discrepancy previously cropped up in the case of the dynamin-like Mx proteins. Filter-binding studies suggested that MxA exhibited a much higher affinity for GDP than GTP [39], but subsequent mant-GTP competition analyses suggested that the opposite was true [40]. Binns et al. [28] estimated the affinities of the analogues mant-dGTP and mant-dGDP for dynamin-2 by stopped-flow fluorimetric measurement of association and dissociation rates and obtained values of approx. 0.5 and 19 μM respectively. Displacement experiments suggested that GTP might have an affinity greater than mant-dGTP, while GDP might have a lower affinity than mant-dGDP, although some ambiguity is present in these data, as the association and dissociation of mant-dGTP were not monophasic and low concentrations of the fluorescent nucleotides were used. These authors [28] also concluded that GDP release from the protein after hydrolysis was very rapid, suggesting that hydrolysis rather than nucleotide exchange is the rate-limiting step in the enzyme cycle. In any event, with the concentrations of GTP and GDP prevalent in most cells (GTP∼470 μM; GDP∼160 μM [41]), the present results taken together with previous data imply that GDP occupancy would be very short-lived in vivo and probably not rate-limiting for dynamin function. This makes it unlikely that dynamin would require a guanine nucleotide-exchange factor to promote nucleotide cycling, as is the case with the small G-proteins that have picomolar affinities for GDP.
In other G-proteins, it is quite clear that GTP and GDP binding results in different conformations of the protein. This has been most successfully worked out at the atomic level with respect to small G-proteins such as Ras [42], where specific ‘switch’ regions of the protein are controlled by ligation of the terminal phosphate residue. While such a structural analysis has not yet been carried out with dynamin, the presence of homologous switch features strongly suggests that similar interactions occur in this family of proteins. We show here that the GTP and GDP forms of dynamin-2 differ, while the GDP form is indistinguishable from the empty form, using limited proteolysis as a criterion. The recently solved structures of D. discoideum DynA G-domain also showed no differences between the GDP and empty forms, but unfortunately the GTP-bound structure is not yet available for comparison [9]. Other works supporting GTP-dependent conformational changes in mammalian dynamins arose by examining the structures of lipid-bound oligomers before and after GTP hydrolysis. Hydrolysis led to either a reduction in diameter or an increase in pitch of dynamin rings; observations that gave rise to the competing ‘pinchase’ and ‘poppase’ models of dynamin function respectively (see [3–5] for reviews). Further work on the various molecular states of dynamin will be needed to understand fully the nature of dynamin function in endocytosis.
Another feature of dynamin that presumably involves conformational change is its ability to homo-oligomerize. Oligomerization of dynamin into ring and spiral structures can be induced in vitro by simple reduction in tonicity [26] and similar structures are found to be associated with lipid membranes and may well occur in vivo. We found that oligomerization exerted little effect on the intrinsic fluorescence of dynamin. This is not inherently surprising, as work on mutants in which the PH domain has been deleted [21] suggests that this domain does not contribute to dynamin–dynamin interactions and that its presence may even exert an inhibitory influence on oligomerization. Thus we resorted to extrinsic fluorescence to devise models that could be employed to investigate the characteristics of subunit behaviour during assembly. IAEDANS-labelled dynamin was fully active in GTPase and guanine nucleotide-binding assays, as well as functionally operational in oligomerization assays following dilution. Moreover, oligomerization caused a blue shift in IAEDANS·dynamin emission and significant quenching. However the effects were small. To increase the sensitivity of such a method to detect oligomerization, we adopted a heterogeneous labelling strategy and RET in which proximal protomers would exhibit quenching. Such a strategy may be useful in evaluating the kinetics of oligomerization and the effects of guanine nucleotides and other associated proteins on the process.
Acknowledgments
We thank J. Hahm for technical assistance and Dr S. Meredith for use of his fluorimeter. We appreciate the provision of reagents by Dr K. Ferguson, Dr M. Lemmon, Dr D. Binns, Dr J. Albanesi and Dr S. L. Schmid. This work was partially supported by a grant from the National Science Foundation.
References
- 1.van der Bliek A. M. Functional diversity in the dynamin family. Trends Cell Biol. 1999;9:96–102. doi: 10.1016/s0962-8924(98)01490-1. [DOI] [PubMed] [Google Scholar]
- 2.McNiven M. A., Cao H., Pitts K. R., Yoon Y. The dynamin family of mechanoenzymes: pinching in new places. Trends Biochem. Sci. 2000;25:115–120. doi: 10.1016/s0968-0004(99)01538-8. [DOI] [PubMed] [Google Scholar]
- 3.Hinshaw J. E. Dynamin and its role in membrane fission. Annu. Rev. Cell Dev. Biol. 2000;16:483–519. doi: 10.1146/annurev.cellbio.16.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sever S., Damke H., Schmid S. L. Garrotes, springs, ratchets, and whips: putting dynamin models to the test. Traffic. 2000;1:385–392. doi: 10.1034/j.1600-0854.2000.010503.x. [DOI] [PubMed] [Google Scholar]
- 5.Praefcke G. J., McMahon H. T. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- 6.Warnock D. E., Schmid S. L. Dynamin GTPase, a force-generating molecular switch. BioEssays. 1996;18:885–893. doi: 10.1002/bies.950181107. [DOI] [PubMed] [Google Scholar]
- 7.Bourne H. R., Sanders D. A., McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature (London) 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 8.Kjeldgaard M., Nyborg J., Clark B. F. C. The GTP binding motif: variations on a theme. FASEB J. 1996;10:1347–1368. [PubMed] [Google Scholar]
- 9.Niemann H. H., Knetsch M. L., Scherer A., Manstein D. J., Kull F. J. Crystal structure of a dynamin GTPase domain in both nucleotide-free and GDP-bound forms. EMBO J. 2001;20:5813–5821. doi: 10.1093/emboj/20.21.5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gout I., Dhand R., Hiles I. D., Fry M. J., Panayatou G., Das P., Truong O., Totty N. F., Hsuan J., Booker G. W., et al. The GTPase dynamin binds to and is activated by a subset of SH3 domains. Cell (Cambridge, Mass.) 1993;75:25–36. [PubMed] [Google Scholar]
- 11.Okamoto P. M., Herskovits J. S., Vallee R. B. Role of the basic, proline-rich region of dynamin in Src homology 3 domain binding and endocytosis. J. Biol. Chem. 1997;272:11629–11635. doi: 10.1074/jbc.272.17.11629. [DOI] [PubMed] [Google Scholar]
- 12.Solomaha E., Szeto F., Yousef M., Palfrey H. C. Kinetics of SH3 domain association with the proline rich domain of dynamins: specificity, occlusion and the effects of phosphorylation. J. Biol. Chem. 2005;280:23147–23156. doi: 10.1074/jbc.M501745200. [DOI] [PubMed] [Google Scholar]
- 13.Klein D. E., Lee A., Frank D. W., Marks M. S., Lemmon M. A. The pleckstrin homology domains of dynamin isoforms require oligomerization for high affinity phosphoinositide binding. J. Biol. Chem. 1998;273:27725–27733. doi: 10.1074/jbc.273.42.27725. [DOI] [PubMed] [Google Scholar]
- 14.Sever S., Muhlberg A. B., Schmid S. L. Impairment of dynamin's GAP domain stimulates receptor-mediated endocytosis. Nature (London) 1999;398:481–486. doi: 10.1038/19024. [DOI] [PubMed] [Google Scholar]
- 15.Zhang P., Hinshaw J. E. Three-dimensional reconstruction of dynamin in the constricted state. Nat. Cell Biol. 2001;3:922–926. doi: 10.1038/ncb1001-922. [DOI] [PubMed] [Google Scholar]
- 16.Lakowicz J. 2nd edn. New York: Plenum Press; 1999. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 17.Artalejo C. R., Henley J. R., McNiven M. A., Palfrey H. C. Rapid endocytosis coupled to exocytosis in adrenal chromaffin cells involves Ca2+, GTP and dynamin but not clathrin. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8328–8332. doi: 10.1073/pnas.92.18.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warnock D. E., Hinshaw J. E., Schmid S. L. Dynamin self-assembly stimulates its GTPase activity. J. Biol. Chem. 1996;271:22310–22314. doi: 10.1074/jbc.271.37.22310. [DOI] [PubMed] [Google Scholar]
- 19.Eftink M. R., Ghiron C. A. Fluorescence quenching studies with proteins. Anal. Biochem. 1981;114:199–227. doi: 10.1016/0003-2697(81)90474-7. [DOI] [PubMed] [Google Scholar]
- 20.Hiratsuka T. New ribose-modified fluorescent analogs of adenine and guanine nucleotides available as substrates for various enzymes. Biochim. Biophys. Acta. 1983;742:496–508. doi: 10.1016/0167-4838(83)90267-4. [DOI] [PubMed] [Google Scholar]
- 21.Muhlberg A. B., Warnock D. E., Schmid S. L. Domain structure and intramolecular regulation of dynamin. EMBO J. 1997;16:6676–6683. doi: 10.1093/emboj/16.22.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Binns D. D., Barylko B., Grichine N., Atkinson M. A., Helms M. K., Jameson D. M., Eccleston J. F., Albanesi J. P. Correlation between self association modes and GTPase of dynamin. J. Protein Chem. 1999;18:277–290. doi: 10.1023/a:1021083211267. [DOI] [PubMed] [Google Scholar]
- 23.Warnock D. E., Baba T., Schmid S. L. Ubiquitously expressed dynamin-II has a higher intrinsic GTPase activity and a greater propensity for self-assembly than neuronal dynamin-I. Mol. Biol. Cell. 1997;8:2553–2562. doi: 10.1091/mbc.8.12.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higashijima T. H., Ferguson K. M., Sternweis P. C., Ross E. M., Smigel M. D., Gilman A. G. The effect of activating ligands on the intrinsic fluorescence of guanine nucleotide-binding regulatory proteins. J. Biol. Chem. 1987;262:752–756. [PubMed] [Google Scholar]
- 25.Sontag B., Reboud A. M., Divita G., Di Pietro A., Guillot D., Reboud J. P. Intrinsic tryptophan fluorescence of rat liver elongation factor eEF-2 to monitor the interaction with guanylic and adenylic nucleotides and related conformational changes. Biochemistry. 1993;32:1976–1980. doi: 10.1021/bi00059a014. [DOI] [PubMed] [Google Scholar]
- 26.Pan J. Y., Sanford J. C., Wessling-Resnick M. Effect of guanine nucleotide binding on the intrinsic tryptophan fluorescence properties of Rab5. J. Biol. Chem. 1995;270:24204–24208. doi: 10.1074/jbc.270.41.24204. [DOI] [PubMed] [Google Scholar]
- 27.Varne A., Muthukumaraswamy K., Jatiani S. S., Mittal R. Conformational analysis of the GTP-binding protein MxA using limited proteolysis. FEBS Lett. 2002;516:129–132. doi: 10.1016/s0014-5793(02)02519-x. [DOI] [PubMed] [Google Scholar]
- 28.Binns D. D., Helms M. K., Barylko B., Davis C. T., Jameson D. M., Albanesi J. P., Eccleston J. F. The mechanism of GTP hydrolysis by dynamin II: a transient kinetic study. Biochemistry. 2000;39:7188–7196. doi: 10.1021/bi000033r. [DOI] [PubMed] [Google Scholar]
- 29.Achiriloaie M., Barylko B., Albanesi J. P. Essential role of the dynamin pleckstrin homology domain in receptor-mediated endocytosis. Mol. Cell. Biol. 1999;19:1410–1415. doi: 10.1128/mcb.19.2.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Artalejo C. R., Lemmon M. A., Schlessinger J., Palfrey H. C. Functional role of the PH domain of dynamin-1 in the regulation of rapid endocytosis in adrenal chromaffin cells. EMBO J. 1997;16:1565–1574. doi: 10.1093/emboj/16.7.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemmon M. A., Ferguson K. M. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem. J. 2000;350:1–18. [PMC free article] [PubMed] [Google Scholar]
- 32.Barylko B., Binns D. D., Lin K. M., Atkinson M. A., Jameson D. M., Yin H. L., Albanesi J. P. Synergistic activation of dynamin GTPase by Grb2 and phosphoinositides. J. Biol. Chem. 1998;273:3791–3797. doi: 10.1074/jbc.273.6.3791. [DOI] [PubMed] [Google Scholar]
- 33.Rossman K. L., Worthylake D. K., Snyder J. T., Siderovski D. P., Campbell S. L., Sondek J. A crystallographic view of interactions between Dbs and Cdc42: PH domain-assisted guanine nucleotide exchange. EMBO J. 2002;21:1315–1326. doi: 10.1093/emboj/21.6.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shpetner H. S., Vallee R. B. Dynamin is a GTPase stimulated to high levels of activity by microtubules. Nature (London) 1992;355:733–735. doi: 10.1038/355733a0. [DOI] [PubMed] [Google Scholar]
- 35.Altschuler Y., Barbas S. M., Terlecky L. J., Tang K., Hardy S., Mostov K. E., Schmid S. L. Redundant and distinct functions for dynamin-1 and dynamin-2 isoforms. J. Cell Biol. 1998;143:1871–1881. doi: 10.1083/jcb.143.7.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Artalejo C. R., Elhamdani A., Palfrey H. C. Sustained stimulation shifts the mechanism of endocytosis from dynamin-1-dependent rapid endocytosis to clathrin- and dynamin-2-mediated slow endocytosis in chromaffin cells. Proc. Natl. Acad. Sci. U.S.A. 2002;99:6358–6363. doi: 10.1073/pnas.082658499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marks B., Stowell M. H., Vallis Y., Mills I. G., Gibson A., Hopkins C. R., McMahon H. T. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature (London) 2001;410:231–235. doi: 10.1038/35065645. [DOI] [PubMed] [Google Scholar]
- 38.John J., Sohmen R., Feuerstein J., Linke R., Wittinghofer A., Goody R. S. Kinetics of interaction of nucleotides with nucleotide-free H-ras p21. Biochemistry. 1990;29:6058–6065. doi: 10.1021/bi00477a025. [DOI] [PubMed] [Google Scholar]
- 39.Melen K., Ronni T., Lotta T., Julkunen I. Enzymatic characterization of interferon-induced antiviral GTPases murine Mx1 and human MxA proteins. J. Biol. Chem. 1994;269:2009–2015. [PubMed] [Google Scholar]
- 40.Richter M. F., Schwemmle M., Herrmann C., Wittinghofer A., Staeheli P. Interferon-induced MxA protein. GTP binding and GTP hydrolysis properties. J. Biol. Chem. 1995;270:13512–13517. [PubMed] [Google Scholar]
- 41.Traut T. W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 42.Vetter I. R., Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]