Non-stimulatory peptides contribute to antigen induced CD8-TCR interaction at the immunological synapse (original) (raw)
. Author manuscript; available in PMC: 2006 Jan 27.
Published in final edited form as: Nat Immunol. 2005 Jun 26;6(8):785–792. doi: 10.1038/ni1220
Abstract
It is unclear if the interaction between CD8 and the TCR-CD3 complex is constitutive or antigen-induced. FRET microscopy between fluorescent chimeras of CD3ζ and CD8β showed that this interaction was induced by antigen recognition, within the immunological synapse. Non-stimulatory endogenous or exogenous peptides, presented simultaneously with antigenic peptides, increased the CD8-TCR interaction. This indicates that the interaction between the intracellular regions of a TCR-CD3 complex recognizing its cognate peptide-major histocompatibility complex (pMHC) antigen, and CD8 (plus Lck), is enhanced by a non-cognate CD8-MHC interaction. Thus the CD8 interaction with non-stimulatory pMHC helps mediate T cell recognition of antigen, improving the coreceptor function of CD8.
The αβ T cell receptor (TCR) is responsible for the affinity and specificity of antigen recognition1,2, whereas the coreceptors, CD8 or CD4, enhance the sensitivity of TCR recognition3. Disruption of coreceptor-pMHC interactions inhibit or change the quality of the T cell response3–5. Coreceptors act in two main ways. Firstly, they bind to non-polymorphic regions of MHC4. This can aid in adhesion, but the main role is generally believed to be increasing the sensitivity of T cell activation via the entropic facilitation of TCR-pMHC binding, rather than through energetic stabilization of the tri-molecular complex6–9). Secondly, CD4 and CD8 are associated with the kinase Lck. Coreceptor binding to pMHC recruits Lck close to the TCR, enabling it to phosphorylate components of the TCR’s signaling complex (CD3), thus enhancing signal transduction3.
There are conflicting data on the interaction between the TCR-CD3 complex and coreceptors, and in particular whether any interaction between these molecules is constitutive or induced by antigen recognition. Various co-immunoprecipitation and flow cytometry fluorescence resonance energy transfer (FRET) experiments suggest a constitutive interaction between some TCR-CD3 and coreceptors in unstimulated T cells7,10–13. Others show interaction only after T cell activation14–17. Co-capping and co-modulation experiments also support an interaction of coreceptor with TCR18–21. CD8αβ and CD4 reside on glycolipid-enriched microdomains or rafts, whereas TCR association with rafts is greatly increased upon stimulation22,23. It is therefore questionable whether all of these assays measure a direct interaction or simply co-localization of the molecules on the same rafts.
Whether the interaction between CD8 and TCR is constitutive or antigen-induced has important consequences for the role of CD8. Constitutive association implies that CD8 acts as a universal amplifier in antigen recognition. Inducible interaction suggests an extra level of fine-tuning, where CD8 could sharpen and amplify sensitivity and specificity of recognition. In order to address definitively whether CD8 and TCR interact, and to what extent the interaction is induced upon antigen recognition, we have used microscopy to measure FRET between CD8β-yellow fluorescent protein (YFP) and CD3ζ-cyan (C)FP during antigen recognition in live and fixed OT-I T-hybridoma cells. FRET can be used to address the question of proximity between molecules, because its effective range is less than 10nm.
T cells acquire a polarized morphology upon antigen recognition, in which certain surface and intracellular molecules become concentrated into the contact area between the T cell and antigen presenting cell (APC), known as the immunological synapse9,24. This is a dynamic structure whose exact role in T cell activation is contentious, although it is without doubt the site of antigen recognition and signaling. The coreceptor is brought into the synapse quickly after T cell-APC contact25–27. The CD8-MHC class I interaction is absolutely required for synapse formation, because a mutation in the CD8-binding site of MHC class I renders T cells unable to form T-APC conjugates28.
FRET experiments performed by flow cytometry cannot relate molecular interactions between TCR and coreceptors to the formation of the immunological synapse. Microscopic evaluation of FRET between fluorescent chimeric proteins therefore has a great advantage in that it allows spatio-temporal localization of molecular interactions with subcellular resolution in living cells. This makes it possible to ask whether the molecular interaction between TCR and CD8 drives CD8 recruitment to the synapse, or if the interaction is induced in the synapse. Recent FRET microscopy experiments on living cells showed that interaction between fluorescent chimeras of CD4 and CD3ζ was strongly induced by antigen recognition and occurred only in the synapse26.
Certain APCs support non-antigen-specific recruitment of coreceptor to the synapse by pMHC bearing non-stimulatory or even antagonist peptide9,26,29. Non-stimulatory, as well as antigenic pMHC class II, can contribute to T cell activation and synapse formation30,31. This effect was noted at low antigen concentration, and was not caused by antagonist pMHC ligands30. It involved interaction between TCR and the endogenous pMHC class II31.
Using FRET microscopy, we show that interaction between CD3ζ and CD8β is transiently induced at the synapse between a T cell and an APC loaded with agonist peptide, but not by a non-stimulatory peptide. The presence of non-stimulatory peptides enhances antigen recognition and increases CD8-CD3ζ interaction. Thus the majority endogenous pMHC enhance recognition of the rare antigenic pMHC via a non-cognate interaction between CD8 and MHC class I.
RESULTS
Biological activity of fluorescent chimeric proteins
In order to study the interaction between TCR and coreceptors during antigen recognition in living cells, we developed fluorescent chimeras of CD3ζ-CFP and CD8β-YFP, expressing them in a T cell hybridoma. FRET between CFP and YFP reports when these molecules are brought within 10nm of each other26,32. The OT-I hybridoma recognizes H-2Kb with an ovalbumin peptide (OVA)2 and its TCR binds to Kb-OVA with relatively high affinity compared to non-stimulatory peptides for this TCR, such as a peptide derived from vesicular stomatitis virus nucleoprotein (VSV)33. The C-termini of CD8β and CD3ζ were fused with YFP or CFP, respectively, with peptide linkers to allow proper folding and flexibility. The chimeric CD8β-YFP and CD3ζ-CFP genes were retrovirally transduced into OT-I hybridomas34 expressing wild-type (WT) CD8α, to obtain stable transfectants (OT-I.ZC.8βY) expressing surface CD8β at amounts similar to CD8+ splenocytes (anti-CD8β staining intensity: 738±200 vs. 622±136 above background, respectively). CD8β needs to pair with CD8α for expression on the cell surface35. CD8β-YFP did not reach the surface of cells lacking CD8α (data not shown), but was expressed on the surface of cells expressing CD8α WT (Fig.1a). Anti-CD8α (or anti-CD8β) staining showed complete co-localization and co-capping with CD8β-YFP (Fig.1b), confirming that CD8β-YFP formed a complex with CD8α. IL-2 release was undetectable in OT-I hybridomas lacking CD8. Transfection of CD8α WT partially restored antigen reactivity, whereas CD8β-YFP+CD8α WT gave 1000-fold stronger responsiveness than CD8α WT alone, similar to CD8αβ WT (Fig.1c), showing that CD8β-YFP retained the coreceptor properties of CD8β. Immunoprecipitation with anti-CD8β co-precipitated Lck (Fig.1d), showing that the YFP moiety did not interfere with Lck binding to the cytoplasmic domain of CD8α. This also confirms the CD8α-CD8β-YFP interaction, because Lck is associated with CD8α. Therefore the CD8β-YFP is biologically functional.
Figure 1.
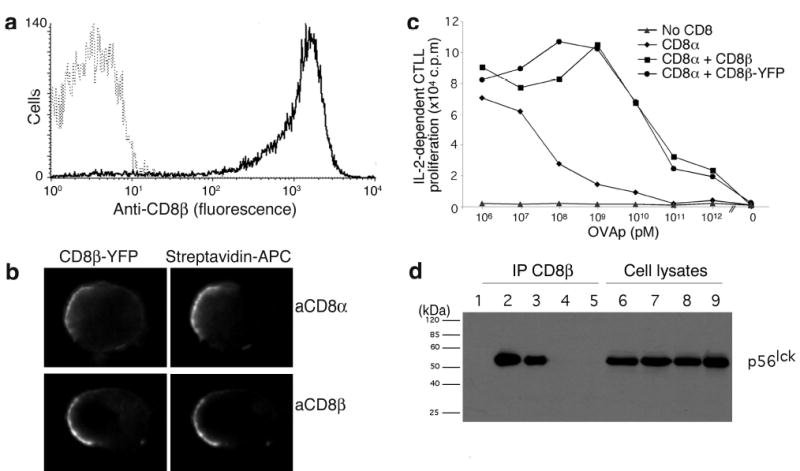
Function of fluorescent chimeric proteins. (a) OT-I hybridomas with CD8α WT (broken line) or with CD8α WT + CD8β-YFP (solid line) were stained with anti-CD8β and analyzed by flow cytometry. (b) OT-I.ZC.8βY cells were stained with biotinylated anti-CD8α or -CD8β antibodies and crosslinked with streptavidin-allophycocyanin. (c) OT-I hybridomas lacking CD8 (▴), expressing CD8α WT (♦), CD8α WT + CD8β-YFP (•), or CD8α WT + CD8β WT (▪) were cultured with irradiated B6 splenocytes loaded with titrated amounts of OVA. (d) OT-I hybridomas lacking CD8 (lanes 5,9), with CD8α WT (lanes 4, 8), with CD8α WT + CD8β WT (lanes 3, 7) or with CD8α WT + CD8β-YFP (lanes 2, 6) were immunoprecipitated with anti-CD8β (lanes 1-5, lane 1 is anti-CD8β Ab alone), and analyzed together with cell lysates (lanes 6-9) by SDS-PAGE, immunoblotted with anti-Lck and detected with anti-MIgG-HRP. These data are representative of ≥3 experiments, except (d), performed twice.
Antigen dependent and independent cell coupling
We quantitated the ability of antigenic or non-stimulatory peptides to induce stable conjugate formation, to assess the contribution of antigen recognition to the formation of stable conjugates26,29. RMA-S cells were labeled with Cy5 and loaded with OVA or VSV peptides at concentrations that caused equal expression of stabilized Kb on the cell surface36 (Fig.2a). The OT-I.ZC.8βY cells were incubated with the RMA-S cells at 37°C. At different time points, the cells were pipetted to separate any weakly conjugated cells, and fixed. The percentage of OT-I.ZC.8βY cells forming conjugates with RMA-S cells was assessed by flow cytometry (Fig.2b,c). In the absence of exogenously added peptides, less than 1% of the OT-I.ZC.8βY cells formed conjugates with RMA-S cells (Fig.2b). However, when these few putative conjugates were sorted and visualized with a microscope, 90% were found to be single cells (data not shown). Therefore, RMA-S cells without exogenously added peptides rarely formed any stable conjugates with OT-I.ZC.8βY cells.
Figure 2.
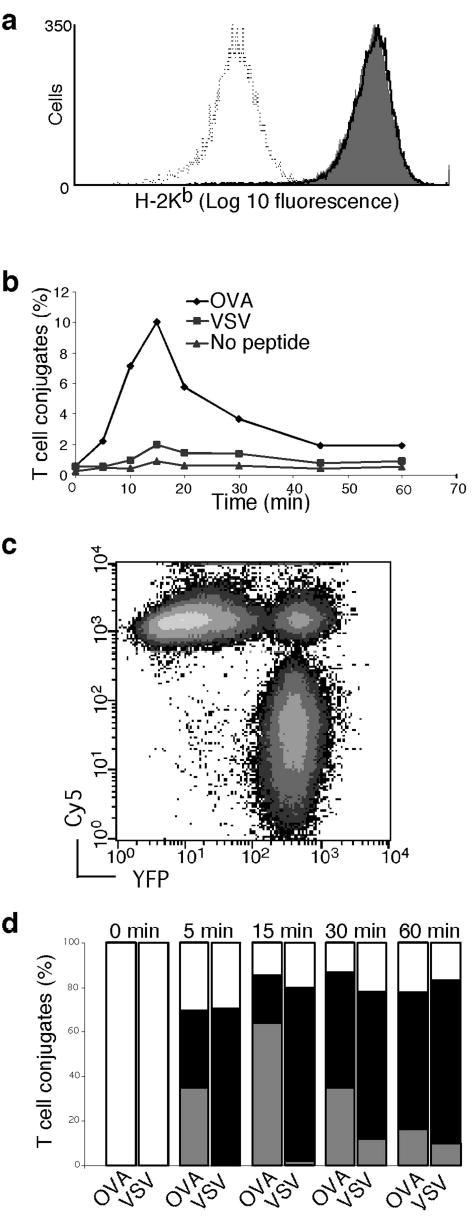
T-APC conjugate formation and CD3ζ and CD8β recruitment to the synapse. (a) RMA-S cells were loaded with OVA (36 μM, grey fill) or VSV (80 μM, solid line) peptides, or no peptide (dotted line). These concentrations had previously been determined by titration to give equal H-2Kb density, as shown by anti-Kb Ab staining. (b) Time course of conjugate formation with antigen and non-stimulatory pMHC complexes. OT-I.ZC.8βY cells were allowed to interact with Cy5 labeled RMA-S cells expressing the same MHC density of either OVA or VSV peptide, or no peptide. At different time-points, cells were fixed and conjugate formation was assessed by flow cytometry. An example is shown in (c). Results are representative of three independent experiments. (d) Proportion of conjugates that had recruited CD8β-YFP or CD3ζ–CFP at different time-points. Conjugates were sorted by flow cytometry, visualized by microscopy, and randomized samples were categorized into three groups: increased CD8β-YFP and CD3ζ-CFP in synapse compared to the rest of the cell surface (gray bars), increased CD8β-YFP only (filled bars), or no increase in either protein (open bars). The 0 min timepoint represents cells prior to conjugate formation. The results are averages of 34<n<108 cells from 2 independent experiments. See also Supplementary Fig.1 online.
T cell-APC conjugate formation with RMA-S cells loaded with the non-stimulatory peptide VSV was increased compared to RMA-S without peptide (Fig.2b). However conjugate formation was much stronger if the RMA-S expressed antigenic Kb-OVA (Fig.2b). Thus, conjugate formation was strongly induced by an antigen recognition-dependent mechanism. The weak but significant activity of Kb-VSV suggested that non-stimulatory (e.g. endogenous) pMHC complexes could be important for initial formation of T cell-APC conjugates. The conjugates were sorted, imaged, and grouped according to whether they had recruited CD8β-YFP with or without CD3ζ-CFP to the T-APC interface. About 70% of T cells in conjugates recruited CD8β-YFP, regardless of the peptide (Fig.2d). This contrasted to CD3ζ-CFP recruitment, which occurred mainly in cells forming conjugates with OVA-loaded APC, peaking around 15 min (Fig.2d). Representative images are shown (Supplementary Fig.1 online).
CD8 recruitment is not peptide-specific
The relationship between CD8 recruitment and pMHC density was assessed by analyzing the synapses formed between OT-I.ZC.8βY cells and RMA-S cells expressing Kb-OVA or Kb-VSV. The intensity of CD8β-YFP in the contact area was compared to CD8β-YFP expression in the rest of the membrane (Fig.3a). pMHC molecule expression on RMA-S cells was quantitated by comparing anti-H-2Kb-PE staining with reference beads loaded with known quantities of PE. When RMA-S cells expressing equally high amounts of Kb-OVA or Kb-VSV (26,000 molecules/cell) were added to the OT-I.ZC.8βY cells, a much smaller percentage of T cells formed conjugates with VSV (Fig.2b). However, in T cell-APC conjugates, the same 2.5 fold increase of CD8 in the synapse relative to the rest of the cell membrane was observed with both OVA and VSV. Therefore conjugate formation depends on an early signal, that is infrequently generated in the absence of antigen. The equivalent ability of non-stimulatory and agonist peptides to induce CD8β-YFP clustering showed that CD8 recruitment itself was not purely an antigen-dependent process, but rather was driven by the CD8-MHC class I interaction. When Kb-OVA (Fig.3a) or Kb-VSV (Supplementary Fig.2 online) were titrated, CD8β-YFP recruitment decreased. Thus, the amount of CD8 recruitment depends on MHC class I density, whereas the cell-to-cell frequency of T-APC conjugate formation is antigen-specific.
Figure 3.
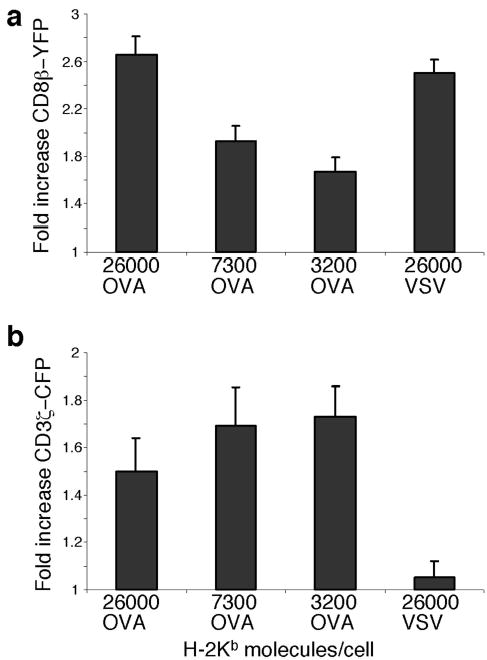
CD8 clustering to the synapse is MHC-driven whereas CD3ζ clustering is antigen driven. OT-I.ZC.8βY cells were allowed to interact with RMA-S cells expressing equivalent amounts of Kb-OVA or Kb-VSV complexes or different amounts of Kb-OVA. The number of pMHC molecules expressed by the RMA-S cells is indicated. OT-I.ZC.8βY cells were assessed for their fold increase in CD8β-YFP in the synapse (a) or fold increase in CD3ζ-CFP in the synapse (b), by comparing the fluorescence intensity in the contact area to that in the rest of the cell membrane. The results are shown as mean + SE, for n>10. In (a) p<0.05 between all the samples except for 26,000 OVA vs. 26,000 VSV and 7300 OVA vs. 3200 OVA. In (b) p< 0.05 between VSV vs. all of the OVA samples, but not between the different OVA samples. Results are representative of three independent experiments.
In contrast, TCR-CD3ζ recruitment to the synapse is antigen-dependent, as shown previously24,26,37 (Figs.2d,3b). In the presence of antigenic pMHC, CD3ζ expression in the synapse was increased 1.5–1.7-fold compared to the rest of the cell membrane. This increased amount of CD3ζ-CFP was not reduced as the level of Kb-OVA was reduced (below ~3000 Kb molecules per cell, too few T:APC conjugates were formed to analyze). The lower CD3ζ-CFP level at the highest concentration of OVA was probably due to faster, stronger TCR downregulation at such high antigen density (see also Figs.4f,6). Thus, above a certain threshold, the degree of CD3ζ recruitment was not dependent on MHC density per se.
Figure 4.
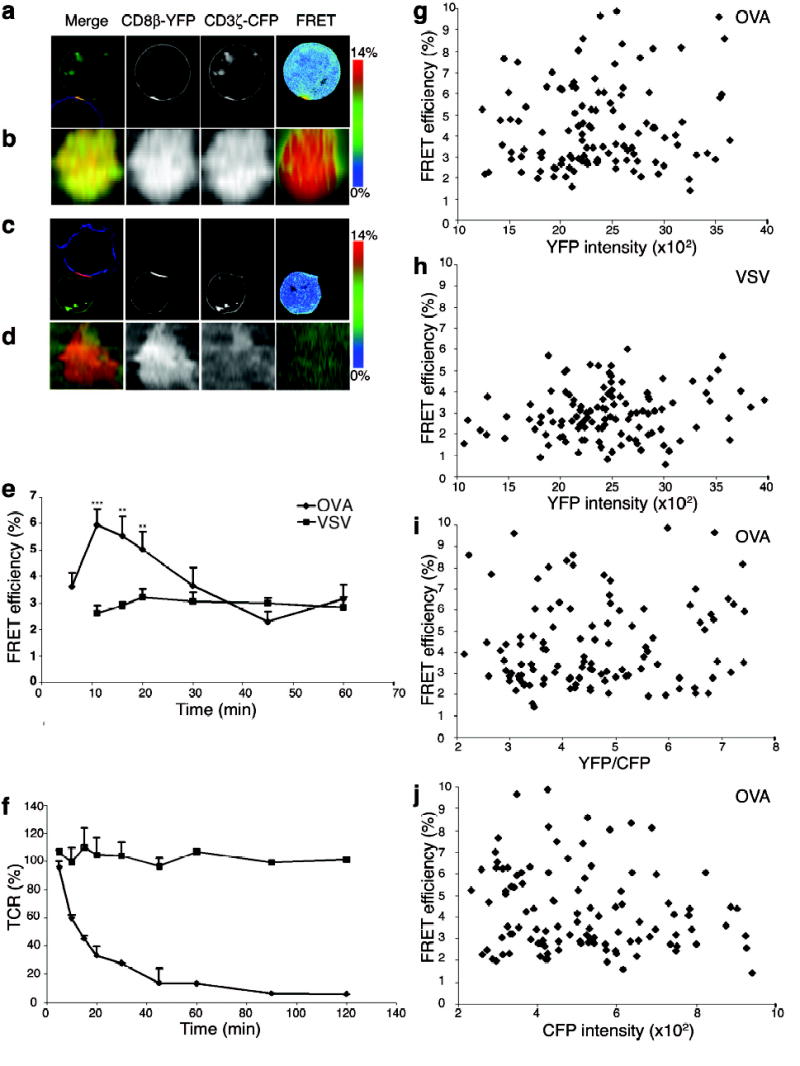
Agonist peptide induces co-localization and interaction between CD8β-YFP and CD3ζ-CFP. Live OT-I hybridomas expressing CD3ζ-CFP, CD8α WT plus CD8β-YFP (OT-I.ZC.8βY) were incubated with Cy5 stained APC loaded with OVA (a, b) or VSV (c, d). (a,c) mid-cell sections, (b,d) en face projections. Merged images (left) show CD8β-YFP (red), CD3ζ-CFP (green) and Cy5 (blue). FRET efficiency images are shown in the right column (see scale bar). Contact areas were defined in 3D by the T cell-APC fluorescence overlap. (e) OT-I.ZC.8βY cells were incubated with OVA or VSV loaded RMA-S cells for indicated times, fixed and average FRET efficiency + SE (18<n<38, except OVA 30 min n=13, VSV 20 min n=11, VSV 30 min n=15) was assessed from the synapse. *** p<0.00002; ** p<0.01 between OVA and VSV. For VSV there were too few 5 min T-APC conjugates to measure. (f) As (e): cell surface TCR was assessed with anti-Vβ5. (g-j) OT-I.ZC.8βY cells were incubated with OVA (g,i,j) or VSV (h) loaded RMA-S cells for 12 min and FRET efficiency was assessed from the synapses. (g,h) FRET efficiency versus YFP intensity, (i) FRET efficiency versus YFP/CFP ratio (The molar ratio= intensity ratio/2.2), (j) FRET efficiency versus CFP intensity.
Figure 6.
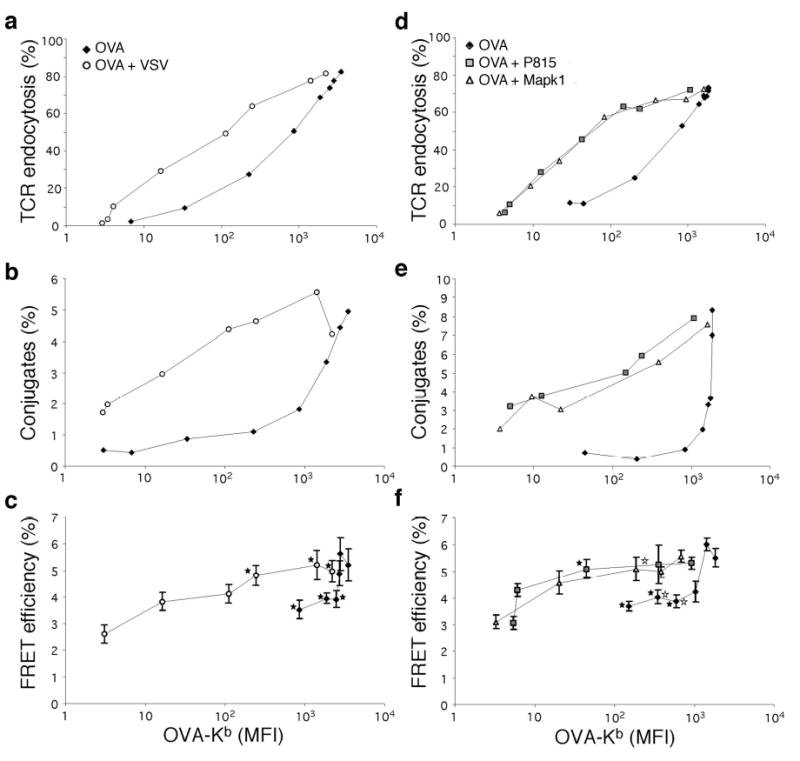
Non-stimulatory peptides increase antigen induced interaction between CD8 and TCR. RMA-S cells were loaded with different amounts of OVA (♦), OVA+VSV (○), OVA+P815 (□), or OVA+Mapk1 (Δ). The amount of Kb-OVA was determined using anti-Kb-OVA Ab 25-D1.1640 (x-axis). The total number of Kb molecules for the OVA-only group was estimated from staining with PE labeled anti-Kb (AF6-88.5), as in Fig.3. 26,000 Kb molecules corresponds to 3515 MFI on the Kb-OVA scale, 7300 Kb to 987 MFI and 3200 Kb to 433 MFI. OVA+VSV, +P815 or +Mapk1-loaded RMA-S cells all expressed ~26,000 Kb molecules. (a,d) OT-I.ZC.8βY cells were incubated with OVA or OVA+VSV, +P815 or +Mapk1 loaded RMA-S cells for 1h and stained with anti-Vβ5. (b,e) OT-I.ZC.8βY cells were incubated with Cy5 labeled RMA-S cells for 12 min, fixed and conjugate formation assessed by flow cytometry. (c,f) FRET efficiency was assessed from synapses. Results are shown as average FRET efficiency +SE (17<n<96 (c), 12<n<48 (f), naverage=30). ★ FRET efficiency values significantly different between OVA and OVA+VSV groups (c), or between OVA and OVA+P815 groups (f). ⋆ significantly different between OVA and OVA+Mapk1 groups (f). (For clarity, not all significant differences are marked).
Antigen induced CD8β-CD3ζ interaction
The interaction between two molecules (strictly, their molecular proximity) can be assessed by FRET imaging32. Upon CFP excitation, FRET leads to decreased fluorescence of CFP and increased fluorescence of YFP, as energy is transferred from CFP to YFP. OT-I.ZC.8βY cells were allowed to interact with OVA- or VSV-loaded RMA-S cells at 37°C, and the interaction of CD3ζ-CFP and CD8β-YFP was assessed by FRET efficiency imaging26,38. Upon stimulation with OVA-loaded cells, both CD3ζ-CFP and CD8β-YFP were recruited to the synapse, where the FRET signal between CD8β-YFP and CD3ζ-CFP increased (Fig.4a,b). FRET intensity did not correlate with donor:acceptor ratio (Fig.4i), YFP intensity (Fig.4g,h), nor CFP intensity (Fig.4j). Thus, FRET between CD8β-YFP and CD3ζ-CFP was not due to non-specific, diffusion-driven interactions caused by increased crowding of the molecules.
The FRET signal was quantitated over time (Fig.4e). To get accurate time-points, and equivalent experimental conditions for FRET analysis, we fixed the conjugates, resulting in lower FRET efficiency compared to live cells. FRET peaked at 10–12 min after initiation of stimulation, showing that interaction between CD8 and TCR was transiently induced upon antigen recognition. The non-stimulatory peptide VSV did not induce an increase in the FRET signal (Fig.4c–e). It should be noted that basal CD3ζ-CFP expression is high enough to give a FRET signal (Fig.4j), showing that the lack of extra CFP does not explain the failure to increase the FRET signal26. A low basal FRET signal (Eapp ~2.5%) was detectable on the cell surface in the absence of any APC. Whether this was due to low constitutive interaction between CD8β and CD3ζ is unclear. TCR downregulation was assessed after stimulation of OT-I.ZC.8βY cells with RMA-S loaded with OVA or VSV. OVA induced rapid TCR downregulation whereas this was absent when VSV was used (Fig.4f). TCR downregulation and FRET signal downmodulation showed similar kinetics.
Immobilized pMHC induces CD8-CD3ζ interaction
To study whether the interaction between CD8β and CD3ζ can be induced by pMHC complexes alone, we refolded soluble Kb with peptide in vitro, biotinylated the Kb molecule, and bound the complex to avidin-coated non-fluorescent beads. Both Kb-VSV and Kb-OVA-coated beads readily interacted with the T cells at 37°C, causing accumulation of CD8β-YFP at the T cell-bead interface (Fig.5a,b). CD3ζ-CFP became concentrated at the interface with Kb-OVA but not Kb-VSV-coated beads. Recognition of Kb-OVA caused rapid engulfment of the beads, as noted previously39. The FRET signal was increased on the contact interfaces of Kb-OVA but not Kb-VSV beads (Fig.5a–c). Thus, CD8-CD3ζ interaction was induced by pMHC complexes alone in the absence of costimulatory molecules.
Figure 5.
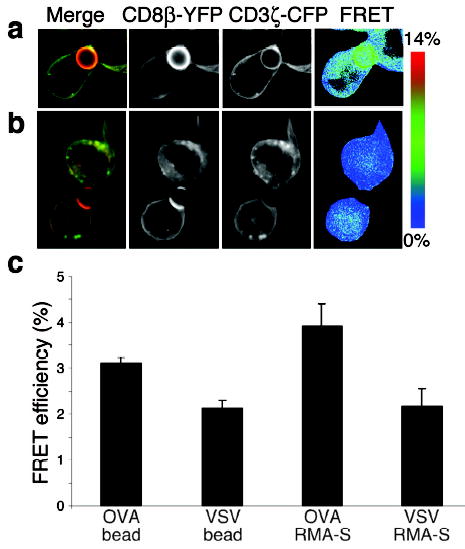
Close molecular proximity of CD8β and CD3ζ can be induced by pMHC molecules alone. OT-I.ZC.8βY cells were incubated with Kb-OVA coated beads (a) or Kb-VSV coated beads (b). The beads are not themselves fluorescent. Kb-OVA induced the recruitment of both CD8β-YFP and, to a lesser extent, CD3ζ-CFP (a), whereas Kb-VSV allowed only the recruitment of CD8β-YFP to the contact interface with the bead (b). The cells almost engulf the beads, as seen previously39. The FRET signal was specifically induced at the interface with Kb-OVA beads but not with Kb-VSV beads. (c) statistics of fixed cells are shown. The results are shown as average FRET efficiency + SE (_n_=36 for OVA coated beads, _n_=36 for VSV coated beads, _n_=19 for OVA RMA-S and _n_=10 for VSV RMA-S). _p_=2.7 x 10−6 between OVA and VSV beads, _p_=0.032 between OVA beads and VSV RMA-S, and _p_=0.123 between OVA beads and OVA RMA-S.
Endogenous non-stimulatory peptides enhance antigen recognition
Endogenous pMHC complexes accumulate in the synapse between a CD4 T cell and APC, in the presence of agonist pMHC. This enhances T cell activation, indicating that simultaneous recognition of self and foreign peptide on the same APC can affect the response to a foreign ligand30. To study how a non-stimulatory peptide influences antigen recognition and CD8-CD3ζ interaction, we used the mAb 25-D1.16 that specifically recognizes Kb-OVA40. This enabled comparison of T cell responses to RMA-S cells expressing various levels of Kb-OVA in the presence or absence of additional non-stimulatory ligand (Kb-VSV) (Fig.6a–c). The amount of OVA peptide was titrated and the amount of VSV peptide was varied so that the total MHC density on cells loaded with OVA+VSV was kept constant. Conjugate formation, TCR down-regulation and FRET efficiency were plotted as a function of anti-Kb-OVA fluorescence. Therefore, any difference in the dose response curves between the two groups was caused by the presence of the non-stimulatory pMHC. TCR down-regulation in response to OVA was enhanced by the presence of VSV (Fig.6a). Similarly, the formation of cell conjugates was greatly enhanced if the VSV peptide was also present (Fig.6b). Similar results were obtained at time points between 6–30 min (data not shown). Therefore, very small amounts of agonist peptide were needed to induce conjugate formation in the presence of excess non-stimulatory peptides. In the absence of non-stimulatory peptides, quite high amounts of the cognate antigen were required for conjugate formation. Most strikingly, the non-stimulatory peptide was able to increase interaction between CD8 and CD3ζ, as measured by FRET (Fig.6c). We performed similar experiments with six endogenous Kb-binding peptides that do not activate OT-I T cells41; P815, Mapk1p (Fig.6d–f), Stat3, Ndufa4, Slc2a3 and Hcph (Supplementary Fig.3d–f online). Similar to VSV (Figs.2,3), these peptides alone induced more T-APC conjugates compared to RMA-S cells without peptide, and were as active as OVA in recruiting CD8 to the synapse (Supplementary Fig.3b,c online). When used in combination with titrated amounts of OVA, these peptides, like VSV, enhanced conjugate formation, TCR downmodulation, and FRET. Thus, the ability of a non-stimulatory peptide to increase antigen recognition when presented simultaneously with OVA, was not limited to the viral-derived VSV peptide but also occurred with endogenous peptides. Thus, non-cognate binding of CD8 to MHC class I molecules bearing a non-stimulatory ligand, enhanced recognition of antigenic ligands.
DISCUSSION
There is considerable disagreement in the literature about whether the interaction between CD8 and the TCR-CD3 complex is constitutive or induced during antigen recognition. Interaction between TCR-CD3 and CD8 was detectable by co-immunoprecipitation from unstimulated cells, and therefore judged constitutive7,10–13. Other data indicated a constitutive TCR-CD3-CD8 interaction that was increased by T cell activation15,16. Immunoprecipitation analyses detect interactions between small proportions of the molecular species involved. These may not be representative of the majority of the molecules. Our FRET analysis showed that there was little interaction, as measured by FRET, between the CD3ζ-CFP and CD8β-YFP on unstimulated T cells. On antigen recognition by the TCR, interaction between CD3ζ-CFP and CD8β-YFP was triggered within the synapse, and an increased FRET signal was observed. Thus most of the TCR-CD3 and CD8 molecules do not interact in the absence of TCR stimulation, although we cannot say that there is absolutely no interaction. The low background FRET signal may represent such an interaction, but even so, the strong TCR-CD3-CD8 interaction induced in the immunological synapse by antigen recognition is striking.
Two recent experiments used FRET in flow cytometry to investigate TCR-CD8 interaction. In one case monomeric or tetrameric pMHC induced FRET between anti-CD3ɛ and anti-CD8 bound to T cells17. In the absence of pMHC, no FRET was observed, indicating that the TCR-CD3 interaction with CD8 was induced by the interaction of pMHC with both TCR and CD8. In the other case, FRET between anti-CD3ɛ and anti-CD8β ± saturating amounts of soluble monomeric pMHC was interpreted as showing constitutive TCR-CD3-CD8 interaction13. Potential reasons for the discrepancies between these and our results include the use of Abs (large, flexible molecules) as the fluorescent species in FRET experiments. These carry the inherent risk that the distance between the target molecules is much greater than the 10nm suggested by a FRET signal. Also, Abs could potentially induce cellular signaling. Previous experiments measured FRET between Abs to the extracellular portions of CD8 and TCR, whereas we measured FRET between intracellular portions of the molecules. The previous studies reflect on the ability of pMHC to couple TCR and CD8, whereas our results reflect more on the ability of cytoplasmic portions important in signal transduction to be brought together. Induction of FRET between directly fluorescent CD3 and CD8 by purified pMHC on beads confirms that pMHC is sufficient to induce the TCR-CD3-CD8 interaction17, and shows that this applies to the intracellular portions of the molecules.
The interaction between CD8 and CD3ζ peaked within the first ten minutes and then gradually faded with time. The transient nature of the TCR-CD3-CD8 interaction may be important in regulating signal transduction. The similar kinetics of the FRET response, TCR downregulation and number of conjugates, suggests that the loss of the TCR-CD3-CD8 interaction is due to loss of TCR from the cell surface, and that as stimulation gets lower, the T cells dissociate from the APC.
We found non-antigen specific recruitment of CD8 into the synapse, as reported earlier for CD426. Antigen-independent synapses have been observed between naïve T cells and dendritic cells26,29. We found that RMA-S cells presenting non-stimulatory peptide formed more T-APC conjugates than RMA-S cells without peptide (and therefore with little or no MHC class I). The frequency of conjugate formation was strongly increased in the presence of antigenic compared to non-stimulatory peptide. However, the non-stimulatory peptides induced CD8 clustering to the synapse as efficiently as the antigenic peptide in the conjugates. This indicated that CD8 clustering is driven by pMHC density and suggests that the weak CD8:MHC class I interaction6 is nonetheless sufficient to pull this ligand-receptor pair into the interface between two cells, as discussed previously42. This would have the effect of concentrating CD8 and pMHC molecules in the synapse where they can affect responses to antigen-stimulation. Clustering of CD8 to the synapse may explain earlier findings showing increased adhesion between CD8 and MHC class I molecules on CTLs, upon stimulation with soluble anti-TCR or antigen43.
The TCR must recognize a few antigenic ligands in a vast excess of self-derived peptides. Such endogenous peptides mediate important survival signals and can enhance antigen recognition in CD4 T cells when presented simultaneously with an antigenic peptide31. So far, endogenous peptides have not been shown to aid antigen recognition by CD8+ T cells. Indeed, a prior attempt to demonstrate functional enhancement by endogenous peptides in CD8+ T cells failed44. In this study, we found that the simultaneous presence of non-stimulatory (exogenous or endogenous) and agonist pMHC on the same APC enhanced CD8+ T cell responses. Non-stimulatory peptides presented simultaneously with an antigenic peptide increased interaction between CD8 and TCR-CD3, specifically between their cytoplasmic domains. Thus our FRET data demonstrate that the non-cognate CD8-MHC interaction enhances the interaction between CD8 and TCR during recognition of cognate pMHC.
The simplest model for coreceptor function has TCR and CD8 binding to the same antigenic pMHC, with the cytoplasmic region of coreceptor providing Lck to phosphorylate CD3. However, this classical model cannot explain how endogenous or other non-stimulatory peptides enhance antigen recognition. The classical model was initially questioned by structural studies suggesting that it might be impossible for coreceptor to interact with the same MHC molecule as is contacted by the TCR45 (although too little is known of the CD8 stalk to say this with any certainty). This suggested that coreceptor might bind pMHC with its extracellular domain, yet interact intracellularly with a TCR recognizing a different pMHC, thus bridging two TCRs. This “pseudodimer” model could explain how T cell activation requires TCR cross-linking to at least a dimer, yet an individual antigenic pMHC is sufficient to induce activation46. The coreceptor must interact with the MHC molecule presenting antigen, suggesting that coreceptor interacts intracellularly with TCR binding to endogenous pMHC31. Experiments in MHC class II-restricted cells showed a role for TCR-recognition of the endogenous pMHC, because recruitment of endogenous pMHC was CD4-independent30, and because only some endogenous pMHC worked to enhance T cell activation31. One model suggested that endogenous pMHC can amplify the T cell response to a few antigenic pMHC by virtue of Lck phosphorylation of TCRs binding transiently to the endogenous pMHC complexes47. The few cognate TCR-pMHC interactions would recruit coreceptor plus Lck. Thus the Lck would be in the vicinity of the frequent (but very short-lived) TCR interactions with endogenous pMHC, and ready to phosphorylate these TCR complexes47. An artificial heterodimer of agonist and certain endogenous pMHC complexes was sufficient to activate MHC class II-restricted T cells, suggesting that a TCR recognizing antigenic pMHC and one recognizing endogenous pMHC, are bridged by the coreceptor CD431.
These models for amplification of recognition by endogenous pMHC could explain our data, except that we do not see a requirement for TCR interaction with endogenous pMHC. In our study on CD8+ cells, all seven non-stimulatory peptides enhanced antigen recognition. Only two of six peptides enhanced CD4+ antigen recognition strongly (plus two weakly)31. CD8 and CD4 are structurally very different45, so the higher order structures formed by these molecules may be different. It is therefore possible that the MHC class I restricted response is not dependent on TCR interacting with endogenous pMHC in the same way as the MHC class II response. The non-cognate interaction between CD8 and MHC class I could enhance the cognate TCR-CD3-CD8 interaction (and therefore antigen recognition) by concentrating pMHC and CD8 to the synapse. Thus CD8-Lck is concentrated in the vicinity of any cognate interaction of TCR with antigenic-pMHC, making it easier for CD8 to bind antigenic-pMHC and thus to phosphorylate CD3 and start the T cell activation cascade. Also, pMHC is concentrated in the synapse so that the TCR has a more concentrated set of pMHC species to sort through to find the few antigenic-pMHC. These mechanisms would result in enhancement of coreceptor function, whether CD8 binds to the same pMHC as the TCR, or whether the CD8 associated Lck interacts in trans with ITAMs of CD3ζ.
METHODS
Peptides and Antibodies
Peptides OVA (SIINFEKL), VSV (RGYVYQGL), P815 (HIYEFPQL), Mapk119–26 (VGPRYTNL), Stat353–60 (ATLVFHNL), Ndufa461–68 (VNVDYSKL), Slc2a3314–321 (VNTIFTVV) and Hcph503–510 (AQYKFIYV) were synthesized and purified as described33,41. Anti-CD8β (H35-17.2), anti-H-2Kb (AF6-88.5), anti-Vβ5 (MR9-4), and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG were from BD Pharmingen (San Diego, CA).
Constructs and Cells
Hybridomas expressing the OT-I TCR ±CD8α and β were made by retroviral transfection of TCR-deficient 58α−β− cells34. Chimeric genes for CD8β-YFP (Supplementary methods online) and CD3ζ-CFP26 were constructed, inserted into retroviral vector pBMN-Z (S. Kinoshita and G. Nolan, www.stanford.edu/group/nolan), and expressed in Phoenix packaging cells. The supernatant was used to transduce OT-I hybridomas (Supplementary methods online). For optimal FRET sensitivity and to avoid false negative results, the acceptor (YFP) should be in excess over donor (CFP). OT-I cell clones were selected based on optimal YFP/CFP molar ratio for FRET i.e. acceptor:donor molar ratio of 1:1 to 3:1.
Microscopy
Imaging was carried out with a dual camera system specifically designed for FRET imaging, allowing simultaneous acquisition of donor emission and acceptor emission during donor excitation, and fast changes between donor and acceptor excitation. This consisted of two CoolSnapHQ cameras (Roper, Tuscon, AZ), attached to a Zeiss 200M microscope through a beam-splitter (custom 510LPXR, Chroma) and stationary emission filters. Rapid wavelength switching was performed with a DG4 galvo illuminator customized with a 300W xenon lamp (Sutter, Novato, CA). YFP excitation was attenuated to 20% by appropriate positioning of the exit mirror. The system was run by Slidebook 4.0.3.9tz software (3I Corp., Denver, CO). The optical filters were: YFP excitation 510/20 (center/bandpass), YFP emission 550/50nm, CFP excitation 430/25, CFP emission 470/30, as well as Cy5 excitation 622/36, emission 700/75. Dichroic mirror was JP4. Exposure times were 0.2–0.5 s with 2x2 binning, 60x1.4 oil objective, and software flat field correction was used. 3D images were reconstructed from 15 z-sections 0.468 microns apart, collected with 60x1.4 oil objective with 1x1 binning. Live cells were imaged in HEPES–buffered 199 (low riboflavin autofluorescence) medium (Gibco, Grand Island, NY) with 5% FBS, no antibiotics, maintained at 37°C by the FCS2 live imaging chamber and objective heater (Bioptechs, Butler PA). T cells and APC were mixed and added into a prewarmed imaging chamber coated with poly-D-lysine (Sigma). For fixed cell imaging, cells were mounted in Slowfade Light antifade mounting media (Molecular Probes, Eugene, Oregon).
FRET analysis
A 3-filter set algorithm for the crosstalk compensation and extraction of donor-normalized FRET was used as described earlier26,38. Details are provided (Supplementary Methods online). The FRET image was masked to accept only regions where CFP intensity was >4x noise level. Cells with CD8β-YFP/CD3ζ-CFP ratios outside the 1:1 to 3:1 stoichiometric range, as well as movement artifacts, were excluded from analysis. The average FRET was calculated from the synapse. Statistical differences were calculated using the mean difference hypothesis of Student’s two tailed t-test assuming different variances and confidence level of 95%.
Antigen presenting cell preparation
RMA-S cells have a defect in the TAP2 gene, and cannot bind endogenous peptides to newly synthesized MHC class I molecules. Addition of synthetic peptides able to bind to Kb or Db at low temperature leads to assembly and stable expression of pMHC on the cell surface. The peptide-loaded molecules remain stable at physiological temperature48. Different peptides have different abilities to stabilize the cell surface pMHC. In order to have equal numbers of pMHC complexes on the cell surface for both OVA and VSV, we titrated the amount of peptide required to give equal pMHC loading by both peptides, as described36. RMA-S cells were stained with Cy5 20h prior to experiments by incubating cells with 0.1 mg/ml of Cy5 monomeric succinimidyl ester (Amersham Biosciences, UK) in RPMI at RT for 5 min, washing with RPMI, and quenching with 10% FBS in RPMI. The RMA-S cells were incubated at 29°C overnight, pulsed with peptides for 30 min at 29°C, incubated at 37°C for 3h and washed once. For pMHC quantitation, RMA-S cells were stained with anti-H-2Kb-PE. QuantiBRITE PE fluorescence quantitation kit (Becton Dickinson) was used to calculate the number of molecules.
TCR down-regulation and conjugate formation assays
OT-I hybridomas and peptide pulsed RMA-S cells (1x105 each in 50 μl) were incubated in flat bottom wells at 37°C. After incubation, the cells were stained for Vβ5 and examined by flow cytometry. The data are shown as a percentage of Vβ5 expression on the surface of cells compared to cells incubated with RMA-S without peptide, or as percentage of TCR endocytosed. For the conjugate formation assay, the cells were pipetted up and down three times after incubation, fixed in 2% paraformaldehyde, washed in PBS, and the paraformaldehyde inactivated by 10 mM Tris pH 7.4 in PBS. The cell conjugates were examined by flow cytometry based on simultaneous expression of YFP (OT-I hybridoma) and Cy5 (Cy5 labeled RMA-S cell).
Immunoprecipitation and Western blotting
Cells (107) were lysed in 1% Brij96, 50 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM Na3VO4, and protease inhibitor cocktail, precleared with sepharose G beads and immunoprecipitated overnight with anti-CD8β-conjugated beads. After washing, samples were run on 8% reducing SDS-PAGE gel, blotted and developed with anti-Lck (3A5; Upstate Biotechnology, Lake Placid, NY) followed by anti-MIgG-HRP.
Preparation of beads coated with pMHC
H-2Kb with a BirA biotinylation sequence and human β2-microglobulin were individually produced in E. coli, refolded and biotinylated, as described33,36,49. Biotinylated pMHC was added to streptavidin-coated 6 μm carboxylated microspheres (Polysciences, Warrington, PA) at twice the saturating concentration, and incubated for 16h at 4°C with constant agitation. The beads were washed three times before use.
Supplementary Material
sup1
sup2
sup3
sup4
sup5
Acknowledgments
We thank S. Jameson (University of Minnesota, MN) for his collection of endogenous Kb-binding peptides. We also thank the following for their generous gifts of reagents: E. Palmer, R. Germain, G. Nolan, and J. Altman. We are grateful to W. Havran, L. Sherman and C. Lotz for their critical reading of the manuscript. Supported by NIH grants GM065230, DK61329 and GM039476 to NRJG and by SNAPS core grant P30MH62261. T.Z. was supported by NIH training grant T32 AI07290. This is manuscript 16479-IMM from The Scripps Research Institute.
Footnotes
Competing interests statement
The authors declare that they have no competing financial interests.
References
- 1.Davis MM, et al. Ligand recognition by αβ T cell receptors. Annu Rev Immunol. 1998;16:523–534. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 2.Gascoigne NRJ, Zal T, Alam SM. T-cell receptor binding kinetics in T-cell development and activation. Exp Rev Mol Med. 2001 Feb 12;2001:1–17. doi: 10.1017/S1462399401002502. ( http://www.expertreviews.org/01002502h.htm) [DOI] [PubMed] [Google Scholar]
- 3.Zamoyska R. CD4 and CD8: Modulators of T-cell receptor recognition of antigen and of immune responses? Curr Opin Immunol. 1998;10:82–87. doi: 10.1016/s0952-7915(98)80036-8. [DOI] [PubMed] [Google Scholar]
- 4.Potter TA, Rajan TV, Dick RF, 2nd, Bluestone JA. Substitution at residue 227 of H-2 class I molecules abrogates recognition by CD8-dependent, but not CD8-independent, cytotoxic T lymphocytes. Nature. 1989;337:73–75. doi: 10.1038/337073a0. [DOI] [PubMed] [Google Scholar]
- 5.Madrenas J, Chau LA, Smith J, Bluestone JA, Germain RN. The efficiency of CD4 recruitment to ligand-engaged TCR controls the agonist/partial agonist properties of peptide-MHC molecule ligands. J Exp Med. 1997;185:219–229. doi: 10.1084/jem.185.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wyer JR, et al. T cell receptor and coreceptor CD8αα bind peptide-MHC independently and with distinct kinetics. Immunity. 1999;10:219–225. doi: 10.1016/s1074-7613(00)80022-9. [DOI] [PubMed] [Google Scholar]
- 7.Arcaro A, et al. CD8β endows CD8 with efficient coreceptor function by coupling T cell receptor/CD3 to raft-associated CD8/p56(lck) complexes. J Exp Med. 2001;194:1485–1495. doi: 10.1084/jem.194.10.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiong Y, Kern P, Chang HC, Reinherz EL. T cell receptor binding to a pMHCII ligand is kinetically distinct from and independent of CD4. J Biol Chem. 2001;276:5659–5667. doi: 10.1074/jbc.M009580200. [DOI] [PubMed] [Google Scholar]
- 9.Gascoigne NRJ, Zal T. Molecular interactions at the T cell-antigen-presenting cell interface. Curr Opin Immunol. 2004;16:114–119. doi: 10.1016/j.coi.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 10.Gallagher PF, Fazekas de St Groth B, Miller JFAP. CD4 and CD8 molecules can physically associate with the same T-cell receptor. Proc Natl Acad Sci USA. 1989;86:10044–10048. doi: 10.1073/pnas.86.24.10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beyers AD, Spruyt LL, Williams AF. Molecular associations between the T-lymphocyte antigen receptor complex and the surface antigens CD2, CD4, or CD8 and CD5. Proc Natl Acad Sci USA. 1992;89:2945–2949. doi: 10.1073/pnas.89.7.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki S, Kupsch J, Eichmann K, Saizawa MK. Biochemical evidence of the physical association of the majority of CD3 δ chains with the accessory/co-receptor molecules CD4 and CD8 on nonactivated T lymphocytes. Eur J Immunol. 1992;22:2475–2479. doi: 10.1002/eji.1830221002. [DOI] [PubMed] [Google Scholar]
- 13.Doucey MA, et al. CD3δ establishes a functional link between the T cell receptor and CD8. J Biol Chem. 2003;278:3257–3264. doi: 10.1074/jbc.M208119200. [DOI] [PubMed] [Google Scholar]
- 14.Mittler RS, Goldman SJ, Spitalny GL, Burakoff SJ. T-cell receptor-CD4 physical association in a murine T-cell hybridoma: Induction by physical antigen receptor ligation. Proc Natl Acad Sci USA. 1989;86:8531–8535. doi: 10.1073/pnas.86.21.8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anel A, Martinez-Lorenzo MJ, Schmitt-Verhulst AM, Boyer C. Influence on CD8 of TCR/CD3-generated signals in CTL clones and CTL precursor cells. J Immunol. 1997;158:19–28. [PubMed] [Google Scholar]
- 16.Osono E, Sato N, Yokomuro K, Saizawa MK. Changes in arrangement and in conformation of molecular components of peripheral T-cell antigen receptor complex after ligand binding: analyses by co-precipitation profiles. Scand J Immunol. 1997;45:487–493. doi: 10.1046/j.1365-3083.1997.d01-427.x. [DOI] [PubMed] [Google Scholar]
- 17.Block MS, Johnson AJ, Mendez-Fernandez Y, Pease LR. Monomeric class I molecules mediate TCR/CD3ɛ/CD8 interaction on the surface of T cells. J Immunol. 2001;167:821–826. doi: 10.4049/jimmunol.167.2.821. [DOI] [PubMed] [Google Scholar]
- 18.Takada S, Engleman EG. Evidence for an association between CD8 molecules and the T cell receptor complex on cytotoxic T cells. J Immunol. 1987;139:3231–3235. [PubMed] [Google Scholar]
- 19.Anderson P, Blue ML, Schlossman SF. Comodulation of CD3 and CD4. Evidence for a specific association between CD4 and approximately 5% of the CD3:T cell receptor complexes on helper T lymphocytes. J Immunol. 1988;140:1732–1737. [PubMed] [Google Scholar]
- 20.Rojo JM, Saizawa K, Janeway CA., Jr Physical association of CD4 and the T-cell receptor can be induced by anti-T-cell receptor antibodies . Proc Natl Acad Sci USA. 1989;86:3311–3315. doi: 10.1073/pnas.86.9.3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwan Lim GE, McNeill L, Whitley K, Becker DL, Zamoyska R. Co-capping studies reveal CD8/TCR interactions after capping CD8β polypeptides and intracellular associations of CD8 with p56lck. Eur J Immunol. 1998;28:745–754. doi: 10.1002/(SICI)1521-4141(199802)28:02<745::AID-IMMU745>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 22.Montixi C, et al. Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J. 1998;17:5334–5348. doi: 10.1093/emboj/17.18.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xavier R, Brennan T, Li QQ, McCormack C, Seed B. Membrane compartmentation is required for efficient T cell activation. Immunity. 1998;8:723–732. doi: 10.1016/s1074-7613(00)80577-4. [DOI] [PubMed] [Google Scholar]
- 24.Bromley SK, et al. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 25.Krummel MF, Sjaastad MD, Wülfing C, Davis MM. Differential clustering of CD4 and CD3ζ during T cell recognition. Science. 2000;289:1349–1352. doi: 10.1126/science.289.5483.1349. [DOI] [PubMed] [Google Scholar]
- 26.Zal T, Zal MA, Gascoigne NRJ. Inhibition of T-cell receptor-coreceptor interactions by antagonist ligands visualized by live FRET imaging of the T-hybridoma immunological synapse. Immunity. 2002;16:521–534. doi: 10.1016/s1074-7613(02)00301-1. [DOI] [PubMed] [Google Scholar]
- 27.Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 2004;5:524–530. doi: 10.1038/ni1058. [DOI] [PubMed] [Google Scholar]
- 28.Potter TA, Grebe K, Freiberg B, Kupfer A. Formation of supramolecular activation clusters on fresh ex vivo CD8+ T cells after engagement of the T cell antigen receptor and CD8 by antigen-presenting cells. Proc Natl Acad Sci USA. 2001;98:12624–12629. doi: 10.1073/pnas.221458898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Revy P, Sospedra M, Barbour B, Trautmann A. Functional antigen-independent synapses formed between T cells and dendritic cells. Nat Immunol. 2001;2:925–931. doi: 10.1038/ni713. [DOI] [PubMed] [Google Scholar]
- 30.Wulfing C, et al. Costimulation and endogenous MHC ligands contribute to T cell recognition. Nat Immunol. 2002;3:42–47. doi: 10.1038/ni741. [DOI] [PubMed] [Google Scholar]
- 31.Krogsgaard M, et al. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 2005;434:238–243. doi: 10.1038/nature03391. [DOI] [PubMed] [Google Scholar]
- 32.Zal T, Gascoigne NRJ. Using live FRET imaging to reveal early protein-protein interactions during T cell activation. Curr Opin Immunol. 2004;16:418–427. doi: 10.1016/j.coi.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 33.Alam SM, et al. T cell receptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 34.Stotz SH, Bolliger L, Carbone FR, Palmer E. T cell receptor (TCR) antagonism without a negative signal: Evidence from T cell hybridomas expressing two independent TCRs. J Exp Med. 1999;189:253–263. doi: 10.1084/jem.189.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorman SD, Sun YH, Zamoyska R, Parnes JR. Molecular linkage of the Ly-3 and Ly-2 genes. Requirement of Ly-2 for Ly-3 surface expression. J Immunol. 1988;140:3646–3653. [PubMed] [Google Scholar]
- 36.Holmberg K, Mariathasan S, Ohteki T, Ohashi PS, Gascoigne NRJ. TCR binding kinetics measured with MHC class I tetramers reveal a positive selecting peptide with relatively high affinity for TCR. J Immunol. 2003;171:2427–2434. doi: 10.4049/jimmunol.171.5.2427. [DOI] [PubMed] [Google Scholar]
- 37.Monks CRF, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 38.Zal T, Gascoigne NRJ. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys J. 2004;86:3923–3939. doi: 10.1529/biophysj.103.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freiberg BA, et al. Staging and resetting T cell activation in SMACs. Nat Immunol. 2002;3:911–917. doi: 10.1038/ni836. [DOI] [PubMed] [Google Scholar]
- 40.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide–MHC class I complexes using a monoclonal antibody. Immunity. 1997;6:715–726. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 41.Santori FR, et al. Rare, structurally homologous self-peptides promote thymocyte positive selection. Immunity. 2002;17:131–142. doi: 10.1016/s1074-7613(02)00361-8. [DOI] [PubMed] [Google Scholar]
- 42.Kupfer A, Singer SJ, Janeway CA, Jr, Swain SL. Coclustering of CD4 (L3T4) molecule with the T cell receptor is induced by specific direct interaction of helper T cells and antigen-presenting cells. Proc Natl Acad Sci USA. 1987;84:5888–5892. doi: 10.1073/pnas.84.16.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Rourke AM, Rogers J, Mescher MF. Activated CD8 binding to class I protein mediated by the T cell receptor results in signalling. Nature. 1990;346:187–189. doi: 10.1038/346187a0. [DOI] [PubMed] [Google Scholar]
- 44.Sporri R, Reis e Sousa C. Self peptide/MHC class I complexes have a negligible effect on the response of some CD8+ T cells to foreign antigen. Eur J Immunol. 2002;32:3161–3170. doi: 10.1002/1521-4141(200211)32:11<3161::AID-IMMU3161>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 45.van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 46.Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- 47.Li QJ, et al. CD4 enhances T cell sensitivity to antigen by coordinating Lck accumulation at the immunological synapse. Nat Immunol. 2004;5:791–799. doi: 10.1038/ni1095. [DOI] [PubMed] [Google Scholar]
- 48.Ljunggren HG, et al. Empty MHC class I molecules come out in the cold. Nature. 1990;346:476–480. doi: 10.1038/346476a0. [DOI] [PubMed] [Google Scholar]
- 49.Altman JD, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
sup1
sup2
sup3
sup4
sup5