Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation (original) (raw)
Abstract
Cardiac failure is a frequent cause of death in the aging human population. Telomere attrition occurs with age, and is proposed to be causal for the aging process. To determine whether telomere shortening leads to a cardiac phenotype, we studied heart function in the telomerase knockout mouse, Terc–/–. We studied Terc–/– mice at the second, G2, and fifth, G5, generation. Telomere shortening in G2 and G5 Terc–/– mice was coupled with attenuation in cardiac myocyte proliferation, increased apoptosis and cardiac myocyte hypertrophy. On a single-cell basis, telomere shortening was coincidental with increased expression of p53, indicating the presence of dysfunctional telomeres in cardiac myocytes from G5 Terc–/– mice. The impairment in cell division, the enhanced cardiac myocyte death and cellular hypertrophy, are concomitant with ventricular dilation, thinning of the wall and cardiac dysfunction. Thus, inhibition of cardiac myocyte replication provoked by telomere shortening, results in de-compensated eccentric hypertrophy and heart failure in mice. Telomere shortening with age could also contribute to cardiac failure in humans, opening the possibility for new therapies.
Keywords: aging/heart failure/p53/telomerase/telomeres
Introduction
It has been a general belief that myocyte growth in the adult heart is restricted to cellular hypertrophy. Recently, this notion has been challenged and evidence for cardiac myocyte replication has been obtained in animals and humans (Kajstura et al., 1998; Beltrami et al., 2001; Anversa and Nadal-Ginard, 2002; Quaini et al., 2002). If this were the case, cardiac myocyte hypertrophy and regeneration become the determinants of the growth reserve of the heart with aging and pathological states. The relative contribution of these two cellular adaptations to the changes in size and function of the aging heart can only be determined when one of these two growth processes is severely inhibited. Additionally, cell death typically occurs in the adult heart and increases with age (Kajstura et al., 1996; Guerra et al., 1999). In the absence of cell proliferation, chronic myocyte loss may lead to a critical decrease in the total number of ventricular myocytes, resulting in the deterioration of cardiac performance. Whether the imbalance between cell growth and cell death promotes an age-dependent increase in cardiac myocyte hypertrophy, precocious myocardial aging, and heart failure and death currently is unknown. This is a relevant issue because impaired cardiac function could be the limiting factor to maximum life span in mammals.
Telomeres are chromatin structures composed of tandem G-rich repeats bound to an array of proteins that cap the ends of eukaryotic chromosomes and which are essential for chromosomal stability and cell viability (reviewed in Chan and Blackburn, 2002; de Lange, 2002). Proliferation of human cells is dependent on having a functional telomere (reviewed in Collins and Mitchell, 2002). Telomere shortening coupled to cell division in the absence of telomerase activity is one of the major causes of telomere dysfunction in human cells (Harley et al., 1990; Bodnar et al., 1998). Telomere shortening to a critically short length results in chromosome end-to-end fusions, and triggers cell arrest and/or apoptosis (Espejel et al., 2002; Goytisolo and Blasco, 2002). Telomerase re-introduction prevents complete telomere erosion and chromosomal instability, as well as the loss of viability associated with critically short telomeres (Bodnar et al., 1998; Hemann et al., 2001; Samper et al., 2001). A telomerase RNA knockout (Terc–/–) mouse has been generated that lacks telomerase activity and exhibits shortening of telomeres at a rate of 3–5 kbp per generation (Blasco et al., 1997; Lee et al., 1998; Herrera et al., 1999). This progressive loss of telomeric sequences eventually leads to telomere dysfunction and to loss of organismal viability after 3–6 generations depending on the genetic background (Lee et al., 1998; Herrera et al., 1999). Phenotypic changes consisting of infertility, abnormal hematological profile, atrophy of the spleen and small intestine, hair graying, reduced angiogenic potential, attenuated bone marrow stem cell proliferation and a decreased life span are apparent in Terc–/– mice of the fifth generation (G5). This occurs in the case of the original mixed genetic background (Lee et al., 1998; Rudolph et al., 1999; Herrera et al., 2000; Franco et al., 2002; Samper et al., 2002). Conversely, a normal phenotype is detected in early generation Terc–/– mice, which show sufficiently long telomeres (Lee et al., 1998). From these studies, it was concluded that telomere dysfunction could contribute to some of the pathologies associated with human aging, in particular those affecting highly proliferative or regenerative tissues. The impact of telomere shortening in Terc–/– mice on cardiovascular and neurodegenerative diseases, which are central to organismal aging, however, has not been addressed to date.
Here, the effects of ablation of telomerase on cardiac myocyte size, number, proliferative potential and death have been studied in combination with cardiac function in G2 and G5 Terc–/– mice of the original mixed background and compared with wild-type (WT) mice in the same background (Blasco et al., 1997). These parameters were determined with the expectation that telomeric shortening and attenuation in cardiac myocyte division as a function of generation and age characteristically led to enhanced myocyte apoptosis, reactive hypertrophy, restructuring of the wall and depression of ventricular hemodynamics. This question is of great relevance since heart failure is the major cause of death in the elderly (www.americanheart.org/statistics).
Results and discussion
Telomere shortening in isolated late generation Terc–/– cardiac myocytes
Progressive telomere shortening in mouse cells lacking telomerase has been documented in different cell types (reviewed in Goytisolo and Blasco, 2002). To measure telomere length in cardiac myocytes from successive generations of Terc–/– mice, cardiac myocytes were dissociated enzymatically from the hearts of WT, G2 and G5 Terc–/– mice in the mixed genetic background at either a young (2–4 months old) or older (6–8 months old) age (Materials and methods). It is important to note that, in the case of the older G5 Terc–/– mice, they often showed sudden losses in body weight and physical activity. These manifestations appeared over a period of 24 h and culminated in death of the animals in 7–12 h as also noticed previously (see Herrera et al., 1999). These moribund G5 Terc–/– mice were studied immediately after the detection of these signs and age-matched WT and G2 Terc–/– mice were analyzed in parallel as controls. In ∼40% of the moribund mice, no obvious histopathological alterations were observed and the cause of death remained undetermined (Herrera et al., 1999). It is a rather common event that the development of heart failure mediated by chronic cardiac myocyte loss is rapid and may evolve quickly into death of the organism, suggesting a cardiac dysfunction phenotype in G5 Terc–/– mice. Rhythm dis turbances coupled with severe ventricular decompensation could also be the cause of sudden death.
To estimate the length of TTAGGG repeats at telomeres in Terc–/– cardiomyocytes, we performed quantitative fluorescence in situ hybridization (Q-FISH) using a peptide nucleic acid (PNA) telomere-specific probe on isolated WT, G2 and G5 Terc–/– cardiomyocytes from the two age groups (Materials and methods) (Leri et al., 1999). This approach consisted of the assessment of the aggregate fluorescence of telomeres in each isolated cardiomyocyte nucleus (see representative image in Figure 1A). Telo mere fluorescence was determined by digital analysis of the captured images and expressed as arbitrary units of fluorescence (a.u.f.) as described previously (Herrera et al., 1999) (Materials and methods). More than 6000 telomere fluorescence values (obtained from >150 myocyte nuclei) were obtained for each genotype and age group (Figure 1B and C). Average telomere length values for individual nuclei were plotted in histograms to visualize the population of cardiac myocytes with decreased telomere fluorescence (Figure 1B and C). As shown in Figure 1B and C, the frequency of nuclei with low telomere fluorescence was higher in G2 Terc–/– mice than in WT mice, and it was increased further in G5 Terc–/– mice compared with the G2 Terc–/– mice. This was seen in mice from both age groups (2- to 4-month-old and 6- to 8-month-old animals), and the differences between genotypes were highly significant as indicated by Student’s _t_-test values P < 0.001 (Figure 1B and C). In summary, Q-FISH analysis of telomere fluorescence indicates the presence of cardiac myocytes in G5 Terc–/– mice that have significantly shorter telomeres than those from G2 Terc–/– animals in the case of both young (2–4 months old) and older (6–8 months old) animals (Figure 1B and C). It is also relevant to address whether cardiac myocytes suffer further telomere shortening as G5 Terc–/– mice age, since these mice show decreased viability with age (Herrera et al., 1999). Figure 1D shows a comparison of relative telomere fluorescence values of cardiac myocytes from young (2–4 months old) and older (6–8 months old) G5 Terc–/– mice (ratio of G5 Terc–/– interphase fluorescence/WT average fluorescence). A higher number of cardiac myocytes with low telomere fluorescence ratios was detected in the 6- to 8-month-old G5 Terc–/– mice compared with the younger animals (2–4 months old). The difference is statistically significant (P < 0.0001). These results suggest that cardiac myocytes from older G5 Terc–/– mice have significantly shorter telomeres than those from younger counterparts.
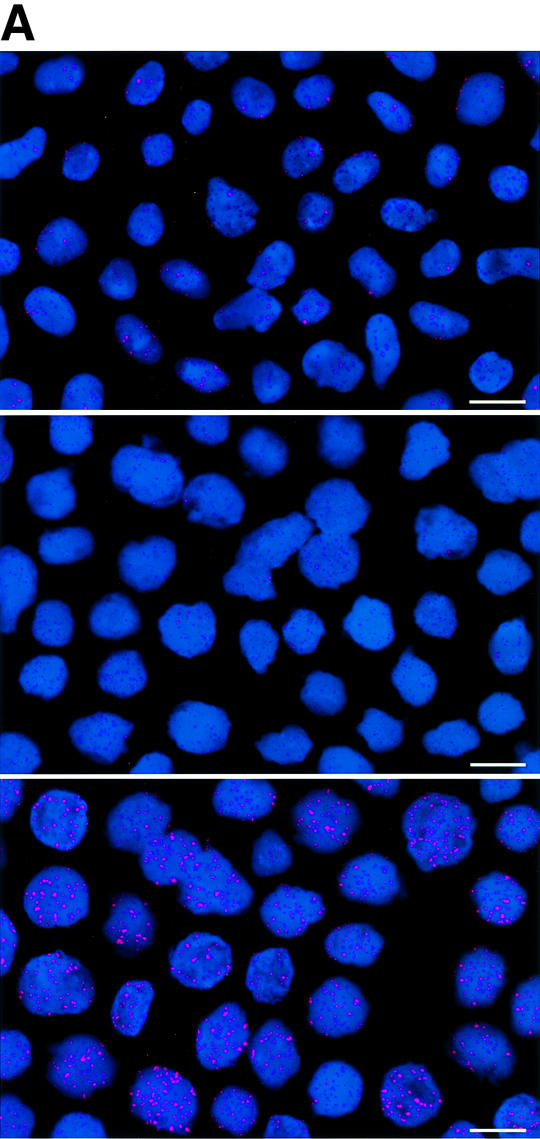



Fig. 1. Telomere length analysis of WT and Terc–/– cardiac myocytes. (A) Representative confocal microscopy image showing FITC–PNA telomeric fluorescence on isolated nuclei (see Materials and methods). Cardiomyocyte nuclei were obtained from a G5 Terc–/– mouse (top panel). Lymphoma cells with short telomeres (L5178Y-S cells, 7 kbp; middle panel) and lymphoma cells with long telomeres (L5178Y cells, 48 kbp; bottom panel) are shown for comparison (McIlrath et al., 2001). Nuclei are illustrated by the blue fluorescence of propidium iodide (PI), and the red fluorescent dots correspond to telomeres. Bar = 10 µm. (B and C) Telomere fluorescence frequency histograms of WT, G2 and G5 Terc–/– cardiomyocytes derived from either young (2–4 months old, B) or older (6–8 months old, C) mice after Q-FISH with a Cy3-labeled telomere-specific probe (Materials and methods). Telomere length is shown as a.u.f. Three mice of each genotype were used for the analysis. The total numbers of nuclei and telomere dots used for the analysis are also indicated. Average telomere fluorescence values for each genotype expressed as a.u.f. are also shown together with the corresponding standard deviation. Note a higher frequency of nuclei with shorter telomeres in the G2 Terc–/– cardiac myocytes compared with the corresponding wild-types. This frequency is increased further in G5 Terc–/– cardiac myocytes. Statistical significance calculations are also shown. (D) Comparison of relative telomere fluorescence in young (2–4 months old) and aged (6–8 months old) G5 Terc–/– cardiac myocytes (ratio G5 Terc–/– interphase fluorescence/wild-type average fluorescence). A higher number of cardiac myocytes show low telomere fluorescence ratio values in the aged G5 Terc–/– mice compared with the younger animals. The difference is statistically significant (P < 0.0001).
p53 up-regulation in cardiac myocytes with short telomeres in late generation Terc–/– mice
The tumor suppressor protein p53 has been proposed to be an important mediator of telomere dysfunction in mouse, and has been shown to be up-regulated in some cell types from Terc–/– mice as telomeres reach a critically short length (Chin et al., 1999; González-Suárez et al., 2000). To evaluate telomere function in G5 Terc–/– cardiac myocytes further, the co-localization of telomeric length and p53 protein was determined by confocal microscopy in preparations of isolated myocyte nuclei from WT, G2 and G5 Terc–/– mice. By this approach, it was possible to establish that p53 was present in nuclei with shorter telomeres (Figure 2A–D). As expected, this interaction was much more prominent in G5 mice (Figure 2E), which had the shortest telomeres. Additionally, the expression of p53 in cardiac myocyte nuclei was evaluated in left ventricular tissue sections from the three groups of mice. Again, p53 labeling involved a much larger fraction of myocyte nuclei from G5 Terc–/– mice (Figure 2F–H). These results are consistent with the notion that telomere shortening in mice activates a p53 response, which modu lates both apoptosis and growth arrest (Chin et al., 1999; González-Suárez et al., 2000; Smogorzewska and De Lange, 2002).


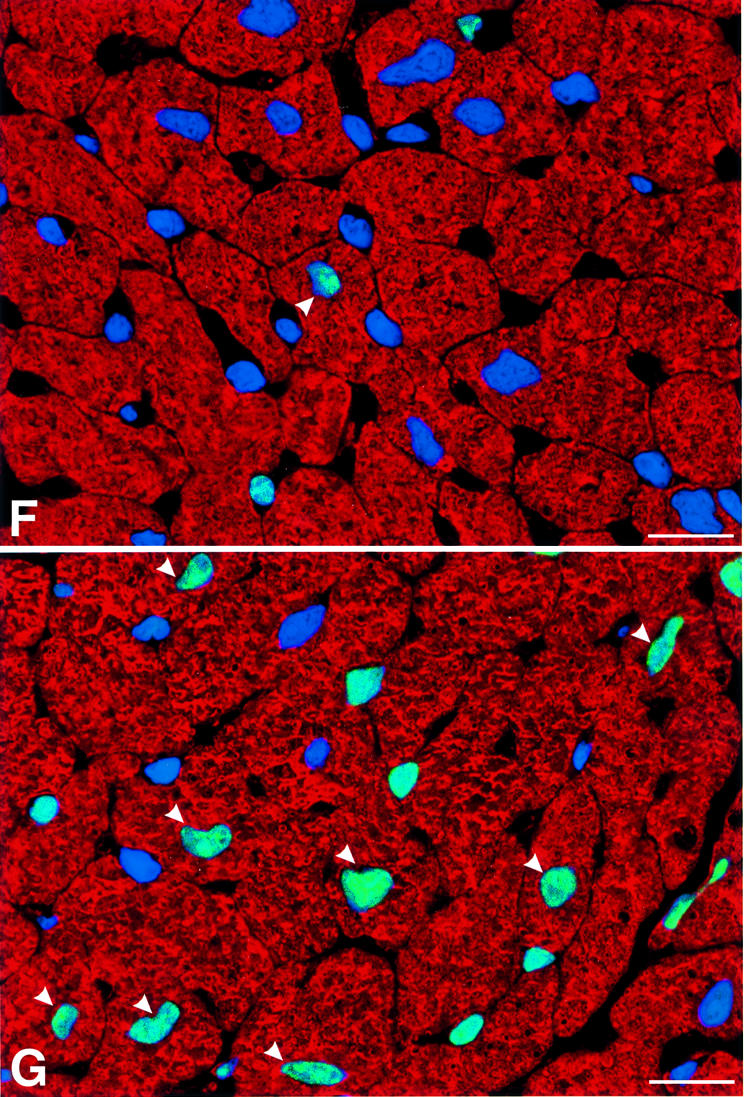

Fig. 2. Telomere length and p53 expression in myocyte nuclei. Myocyte nuclei are stained by a PNA probe (red fluorescence) and p53 (green fluorescence). Blue fluorescence corresponds to PI labeling. (A and B) Myocyte nuclei from the left ventricle of wild-type mice. (C and D) Myocyte nuclei from the left ventricle of G5 Terc–/– mice. Arrowheads point to nuclei with short telomeres and which are positive for p53. Bar = 10 µm. (E) The frequency distribution of telomere length in wild-type, G2 Terc–/– and G5 Terc–/– mice (open bars). The corresponding values for each subclass of nuclei expressing p53 are shown by solid bars. (F and G) p53 labeling of nuclei (bright fluorescence) in sections of the left ventricle of a wild-type (F) and G5 Terc–/– (G) mouse. Blue fluorescence corresponds to PI labeling and red fluorescence depicts α-sarcomeric actin antibody staining of the myocyte cytoplasm. Arrowheads point to p53 labeling of myocyte nuclei. Bar = 10 µm. (H) Percentages of myocyte nuclei labeled by p53 in wild-type (WT), G2 Terc–/– (G2) and G5 Terc–/– (G5) mice. Results are means ± SD. * and † indicate a difference, P < 0.005, versus WT and G2, respectively.
Abnormal cardiac hemodynamics in late generation Terc–/– mice
We next examined cardiac hemodynamics in WT, G2 and G5 Terc–/– mice at 6–8 months of age (see Materials and methods). G5 Terc–/– mice suffered from a severe left ventricular (LV) failure (Figure 3A–D; see Materials and methods). Ventricular decompensation in these mice was characterized by a marked elevation in LV end-diastolic pressure and a significant decrease in LV developed pressure, and + and – dP/dt (Materials and methods). Add itionally, diastolic wall stress markedly increased in G5 Terc–/– hearts (see Supplementary figures available at The EMBO Journal Online). These functional abnormalities were not detected in G2 Terc–/– mice, suggesting that they were associated with critical telomere shortening in G5 Terc–/– cardiac myocytes rather than with telomerase deficiency per se. It cannot be excluded that systemic alterations due to telomere attrition in other organs may have contributed to the impairment in cardiac performance.


Fig. 3. Ventricular function and anatomy. (A–D) LV hemodynamics in WT (n = 10), G2 (n = 18) and G5 (n = 8) mice at sacrifice. (E–H) LV anatomy in WT (n = 8), G2 (n = 12) and G5 (n = 8) mice. Results are means ± SD. * and †, P < 0.05 versus WT and G2, respectively.
The functional data, in Figure 3A–D, and the anatomical parameters discussed in the next section, in Figure 3E–H, show the results as means ± SD. The lack of overlap between the values of SDs in G5 Terc–/– and WT mice indicates that all animals in G5 had similar indices of cardiac failure and anatomy. All together, these results strongly suggest that critical telomere loss in G5 Terc–/– cardiac myocytes is the most likely cause underlying the cardiac phenotype of these mice.
Abnormal heart anatomy in late generation Terc–/– mice
In comparison with WT and G2 Terc–/– mice, heart and LV weights were significantly decreased in G5 Terc–/– mice (Figure 3E–H). The reduction in LV mass was accompanied by a decrease in LV mass:chamber volume ratio dictated by an abnormal increase in cavitary volume (Figure 3E–H). These anatomical alterations were indicative of decompensated eccentric LV hypertrophy in the absence of an absolute increase in ventricular weight in G5 Terc–/– mice (Anversa and Olivetti, 2001). To identify the cellular basis of this apparent inconsistency, the size and number of LV myocytes were measured in each animal group as described previously (Orlic et al., 2001a,b; see Materials and methods and Supplementary figures). These determinations were obtained in mononucleated, binucleated and multinucleated myocytes (see Supplementary figures), which comprise different proportions of the mouse heart (Orlic et al., 2001a,b).
With respect to WT, the volume of binucleated cardio myocytes, which constituted nearly 90% of the entire cardiac myocyte population of the LV, increased by 24% in G2 and 52% in G5 Terc–/– mice (Figure 4A). Corresponding increases in the volume of mononucleated cells, which represented ∼6% of LV myocytes, were 39 and 43% (Figure 4B). However, multinucleated cells, which comprised 4% of LV myocytes, were similar in volume in WT and Terc knockout mice (Figure 4C). Although LV weight was comparable in WT and G2 Terc–/– mice and was decreased in G5 Terc–/– mice, hypertrophy at the cellular level was demonstrated in both generations of Terc–/– mice. In addition, cellular hypertrophy was accompanied by a reduction in cardiomyocyte number in Terc–/– mice. The aggregate numbers of mononucleated, binucleated and multinucleated cardiomyocytes (see Supplementary figures) were combined to yield the total number of cells in the LV of each group of mice (Figure 4D). In comparison with WT, cardiomyocyte number decreased 16% (NS) in G2 Terc–/– mice and 49% in G5 Terc–/– mice (Figure 4D). This latter change was statistically significant (see asterisk in Figure 4D). It is noteworthy that a dilated cardiac myopathy developed in G5 Terc–/– mice as a result of absence of telomerase activity and critical telomere shortening in cardiac myocytes. This observation has no precedent in animals since a similar anatomical condition has only been obtained by genetic manipulation consisting of deletion or overexpression of myocyte proteins. This was not the case here where the diseased heart in late generation Terc–/– mice mimicked the end-stage dilated cardiac myopathy in humans (Kajstura et al., 1998).


Fig. 4. Cardiomyocyte volume and number. (A–C) Average volume of each LV myocyte class. (D) Total number of LV myocytes. WT (n = 10), G2 (n = 10) and G5 (n = 8) mice. Results are means ± SD. * and †, P < 0.05 versus WT and G2, respectively.
Decreased proliferation and increased apoptosis in Terc–/– cardiac myocytes
On the basis of the quantitative measurements of cardio myocyte volume and number, it was evident that ablation of telomerase and critical telomere shortening resulted in a change in the phenotypic characteristics of the heart in G2 Terc–/– mice and in a more pronounced form in G5 Terc–/– mice. In G2 Terc–/– mice, myocyte hypertrophy was modest and the loss of ventricular myocytes did not reach statistical significance. However, in G5 Terc–/–mice, the increase in volume and the reduction in number of binucleated cardiomyocytes were 2.2- and 3.1-fold greater than in G2 Terc–/– mice, respectively (see above). The question then concerned whether enhanced cell death and/or impaired cell proliferation were the underlying mechanisms responsible for the alterations in cardiomyocytes from G2 and G5 Terc–/– mice. In this regard, other pathologies present in late generation Terc–/– mice, such as severe testicular atrophy, have been found to be associated with p53-mediated increased apoptosis and decreased proliferation of male germ cells (Lee et al., 1998; Chin et al., 1999).
To determine cardiac myocyte proliferation, bromodeoxyuridine (BrdU) incorporation and Ki67 labeling (Materials and methods) of cardiomyocyte nuclei were measured (Figure 5A–H). BrdU was injected once a day for 6 days before sacrifice in age-matched mice in each group. The two markers of cell division clearly showed that cardiomyocyte replication was reduced in G2 and G5 Terc–/– hearts (Figure 5G and H). The impairment in cell growth was nearly 2-fold higher in G5 than in G2 Terc–/– mice. Moreover, cardiomyocyte death by apoptosis (Materials and methods) was 63 and 39% greater in G5 Terc–/– mice than in WT and G2 Terc–/– hearts, respectively (Figure 5I–L). Cardiomyocyte necrosis (Materials and methods) was only detected occasionally in the LV of each group of mice. Importantly, the degree of apoptosis was not different in WT and G2 Terc–/– hearts. Therefore, deletion of the gene for the RNA template of telomerase induced phenotypic changes of the heart, which differed in magnitude in the second and fifth generation, in agreement with shorter telomeres in cardiac myocytes from G5 Terc–/– mice (see above). Cardiomyocyte renewal was moderately affected in G2 Terc–/– mice. In contrast, the severe inability of cells to replicate and apoptotic myocyte death were both involved in the significant decrease in ventricular myocytes of G5 Terc–/– mice. The significant decrease in cell proliferation in the adult G5 Terc–/– mouse is consistent with the inhibition of cell growth postnatally. This attenuation in cardiomyocyte division during maturation contributed to the initiation of ventricular dysfunction. Embryonic and fetal events could have participated in the creation of a cellular background that facilitated the impact of altered cell growth on cardiac decompensation in the adult mouse.
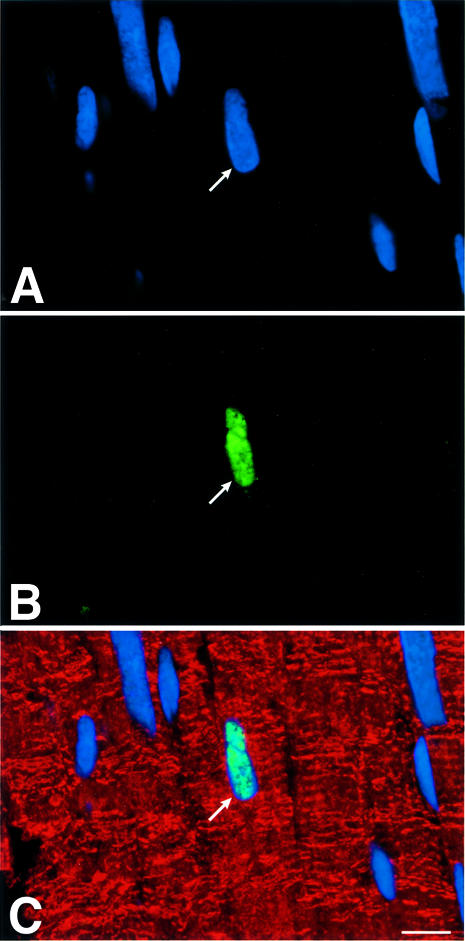
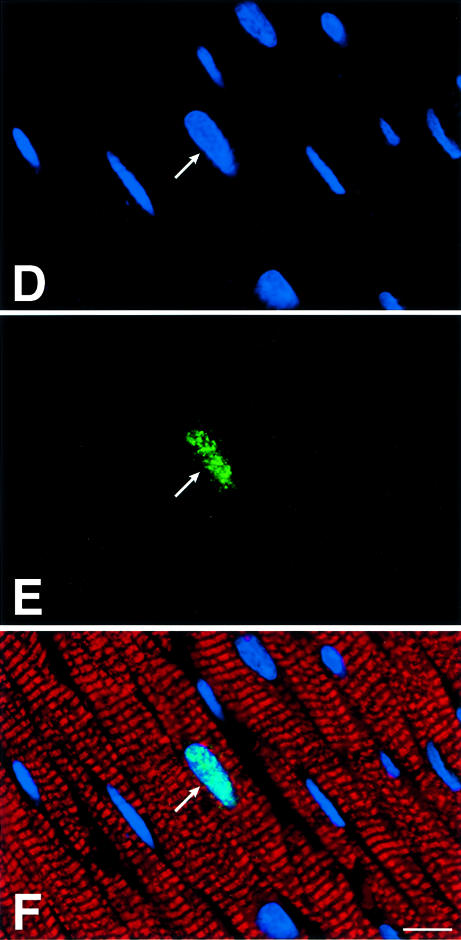



Fig. 5. Cardiomyocyte proliferation and death. BrdU (A–C) and Ki67 (D–F) labeling of LV myocytes from G2 mice. Nuclei are stained by PI (blue; A and D), and by BrdU (green; B) and Ki67 (green; E). Cardiomyocyte cytoplasm is stained by α-sarcomeric actin antibody (red; C and F). Bright fluorescence reflects the combination of PI and BrdU (C) or PI and Ki67 (F) labeling of nuclei. Bar = 10 µm. (G and H) Effects of the lack of telomerase on cardiomyocyte proliferation. WT (n = 8), G2 (n = 10 for BrdU and n = 9 for Ki67) and G5 (n = 9) mice. Results are means ± SD. * and †, P < 0.05 versus WT and G2, respectively. (I–K) Cardiomyocyte apoptosis in a G5 mouse. Nuclei are stained by PI (blue; I), and by hairpin 1 (green; J). Cardiac myocyte cytoplasm is stained by α-sarcomeric actin antibody (red; K). Bright fluorescence reflects the combination of PI and hairpin 1 (K) labeling of a myocyte nucleus. Bar = 10 µm. (L) Effects of the lack of telomerase on myocyte apoptosis. WT (n = 6), G2 (n = 9) and G5 (n = 9) mice. Results are means ± SD. *, P < 0.05 versus WT.
In conclusion, the current results demonstrate that impaired cardiomyocyte regeneration coupled with cardio myocyte death, as a consequence of telomerase deficiency and telomere shortening, leads to pathological cardiac remodeling and severe ventricular dysfunction in late generation Terc–/– mice. Hence, telomere shortening with increasing age in the human heart could be a critical biological determinant of heart failure in the elderly. Importantly, the molecular basis of this process has been recognized as an up-regulation of p53 in cells with the shortest telomeres. This novel information points to the critical role that telomere shortening and p53 activation in ventricular myocytes play in the development of premature cellular aging and heart failure in mammals.
It is of relevance to note that the critically short telomeres present in late generation Terc–/– mice can be rescued by re-introducing telomerase (Samper et al., 2001). In particular, we have shown that telomerase re-introduction into the mouse germ line is sufficient specifically to elongate short telomeres and rescue the phenotypes associated with telomere dysfunction in these mice (Samper et al., 2001). This opens up the possibility of using telomerase-based therapies for treatment of pathological states of the heart and, equally important, may provide a novel approach for the extension of maximum life span in mammals. The recent description of a mouse model that overexpresses transgenic telomerase may also help to evaluate this issue (reviewed in Blasco, 2002). Most importantly, the present study establishes the telomerase knockout mouse as a model for testing the efficacy of both telomerase-based and stem cell-based therapies for heart dysfunction.
Materials and methods
Mice
Female WT, G2 and G5 Terc–/– mice at either 2–4 or 6–8 months of age were studied. Wild-type and Terc–/– mice used in the study were generated as described previously (Blasco et al., 1997). In the case of the 2- to 4-month-old females, body weight was 20.7 ± 1.5 g in WT (n = 3), 21.3 ± 4.3 g in G2 (n = 3; NS), and decreased to 17.5 ± 3.6 g in G5 (n = 3) mice. In the case of the 6- to 8-month-old females, body weight was 26.8 ± 2.7 g in WT (n = 8) and decreased to 24.8 ± 3.5 g in G2 (n = 12; NS) and 21.1 ± 3.5 g in G5 (n = 8; P < 0.01 versus WT) mice.
Isolation of cardiac myocytes
LV myocytes were isolated by collagenase from 10 WT, 10 G2 and eight G5 Terc–/– mice as described previously (Leri et al., 1999). Intact cells obtained from the LV were enriched by repeated centrifugation to remove non-myocytes and cellular debris. Consistent with previous results, non-myocytes constituted 1–2% of the isolated cell population. The average yield of LV myocytes was 1–2 × 106.
Telomere length measurement in isolated cardiac myocytes
Freshly isolated cardiomyocyte and bone marrow cell suspensions were incubated in hypotonic buffer (0.03 M sodium citrate), fixed in methanol/acetic acid (3:1) and dropped onto wet slides. After drying overnight, FISH was performed as described previously (Herrera et al., 1999). For telomere length quantification, Cy3 and DAPI images were captured with a Leica Leitz DMRB microscope equipped with a COHU CCD camera at 100× magnification, and the telomere fluorescence was integrated using spot IOD analysis in the TFL-TELO program. For each mouse, 50 interphase nuclei were analyzed by Q-FISH and the data were plotted in histograms of telomere length frequencies. Confocal microscopy was also used to visualize telomeres (Figure 1A), but not for quantification of telomere fluorescence which was performed as described above (Figure 1B–D). For confocal images, a fluorescein isothiocyanate (FITC)–PNA telomere probe was used (Leri et al., 2001).
Telomeric length and p53 labeling
Myocyte nuclei were stained first by Q-FISH and, subsequently, were exposed to p53 antibody (FL-353, Santa Crux, Santa Cruz, CA). Cy5-conjugated secondary antibodies were employed. A total of 200–250 nuclei were measured in each group of animals: WT, G2 Terc–/– and G5 Terc–/– mice.
Determination of cardiac hemodynamics
For hemodynamics, mice were anesthetized with chloral hydrate (400 mg/kg body weight), and a Millar microtip pressure transducer connected to a chart recorder was advanced into the LV for the evaluation of pressures and + and – dP/dt in the closed chest preparation. Developed pressure corresponds to peak systolic pressure minus end-diastolic pressure.
Anatomical determinations
For anatomical measurements, eight WT, 12 G2 and eight G5 Terc–/– mice were utilized. The heart was arrested in diastole with CdCl2, and the myocardium was perfused with 10% formalin. The LV chamber was filled with fixative at a pressure equal to the in vivo measured end-diastolic pressure. The LV intracavitary longitudinal axis was determined, and the mid-section was used to obtain LV thickness and chamber diameter. LV cavitary volume was also computed. Diastolic wall stress was derived from the anatomical measurements of wall thickness and chamber diameter and the in vivo determination of LV end-diastolic pressure. These procedures have been employed repeatedly in our laboratory (Orlic et al., 2001a,b). LV myocytes isolated from 10 WT, 10 G2 and eight G5 Terc–/– mice were fixed in 10% buffered formalin for the determination of myocyte volume. Cardiomyocyte nuclei were stained by propidium iodide (PI), and the fraction of mononucleated, binucleated and multinucleated cells was determined by examining 500 cells from each LV. Cardiomyocyte length and width were measured with a computerized image analysis system: 200 binucleated and 50 mononucleated and multinucleated cardiomyocytes from each LV were assessed. Since isolated cells assume a cross-sectional area that resembles a flattened ellipse, the ratio of the minor to the major axis of the ellipse was obtained by confocal microscopy. Cell volume was calculated assuming an elliptical cross-section with the major axis equivalent to cell width while the minor axis was computed from the measured ratios. Additionally, cardiomyocyte volume in each cell class was confirmed by three-dimensional optical section reconstruction by confocal microscopy. In each animal, five mononucleated, 20 binucleated and five multinucleated cardiomyocytes were measured in this manner. Since comparable data were obtained, all values were combined (Orlic et al., 2001a,b).
Cross-sections of the LV were stained with hematoxylin and eosin and examined at 1000× with a reticle containing 42 sampling points to determine the volume fraction of cardiomyocytes. The total volume of myocytes in the LV was calculated from the product of LV volume and the volume percent of myocytes. The volume fraction of cardiac myocytes and the proportion of mononucleated, binucleated and multinucleated myocytes were utilized to compute the volume percent of each myocyte class in the myocardium. The number of mononucleated, binucleated and multinucleated cells in the LV was calculated from the quotient of their aggregate volume and corresponding myocyte cell volume (Orlic et al., 2001a,b).
Determination of cardiac myocyte proliferation
The proliferation studies included animals at 6–8 months of age in all groups. Heart sections were incubated with BrdU or Ki67 antibodies. Cardiomyocyte cytoplasm was labeled with a mouse monoclonal anti-α sarcomeric actin. Nuclei were stained by PI. The fractions of cardiomyocytes labeled by BrdU and Ki67 were obtained by confocal microscopy (Leri et al., 2001). Nuclei sampled for BrdU were: WT = 210 501, G2 = 204 016, G5 = 192 537. Corresponding values for Ki67 were: WT = 234 536, G2 = 282 237, G5 = 378 455.
Tissue sections were also stained with p53 antibody (CM5, Novocastra, New Castle upon Tyne, UK) and the localization of p53 in myocyte nuclei was determined quantitatively. The magnitude of sampling included 2459, 2537 and 2183 myocyte nuclei in WT, G2 and G5 Terc–/– mice.
Determination of cardiac myocyte apoptosis
Hairpin oligonucleotide probes, with a single base 3′ overhang (hairpin 1), specifically and sensitively detect double-strand DNA breaks in apop totic cells. Conversely, hairpin oligonucleotide probes with blunt ends (hairpin 2) selectively identify a form of DNA damage typically present in nuclei of cells undergoing necrosis. These probes were employed for in situ ligation as described previously (Guerra et al., 1999). Sections were treated with protease K and then incubated overnight with the biotinylated hairpin probes (Synthetic Genetics). Hairpin oligonucleotides were ligated to DNA using T4 DNA ligase. Ligated probes were detected with FITC–avidin. Cardiomyocyte cytoplasm was recognized by α-sarcomeric actin antibody staining. Nuclei sampled to assess apoptosis by hairpin 1 were: WT = 526 762, G2 = 527 880, G5 = 543 369. More than 300 000 nuclei were examined to assess necrosis by hairpin 2 in each group of mice.
Supplementay data
Supplementary data available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
This work was supported by National Institutes of Health Grants HL-38132, HL-39902, AG-15756, AG-17042, HL-66923 (to P.A.), HL-65577 (to A.L.) and HL-65573 (to J.K.). S.F. is a fellow of the Fondo de Investigaciones Sanitarias (FIS), Spain. The laboratory of M.A.B. is funded by The Cancer Swiss-Bridge Award 2000, by the Ministry of Science and Technology (PM97-0133), Spain, by EU grants EURATOM/991/0201, FIGH-CT-1999-00002 and FIS5-1999-00055, and by the Department of Immunology and Oncology (DIO). The DIO is funded by the Spanish Council for Scientific Research and by Pharmacia Corporation.
References
- Anversa A. and Nadal-Ginard,B. (2002) Myocyte renewal and ventricular remodelling. Nature, 415, 240–243. [DOI] [PubMed] [Google Scholar]
- Anversa P. and Olivetti,G. (2001) Cellular basis of physiologic myocardial growth. In Page,E., Fozzard,H. and Solaro,J. (eds), Handbook of Physiology, Section 2: The Cardiovascular System, Volume I: The Heart. Oxford University Press, New York, NY, 75–144.
- Beltrami A.P. et al. (2001) Evidence that human cardiac myocytes divide after myocardial infarction. N. Engl. J. Med., 344, 1750–1757. [DOI] [PubMed] [Google Scholar]
- Blasco M.A. (2002) Telomerase beyond telomeres. Nat. Rev. Cancer, 2, 627–632. [DOI] [PubMed] [Google Scholar]
- Blasco M.A., Lee,H.W., Hande,M.P., Samper,E., Lansdorp,P.M., DePinho,R.A. and Greider,C.W. (1997) Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell, 91, 25–34. [DOI] [PubMed] [Google Scholar]
- Bodnar A.G. et al. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- Chan S.W.-L. and Blackburn,E.H. (2002) New ways not to make ends meet: telomerase, DNA damage proteins and heterochromatin. Oncogene, 21, 553–563. [DOI] [PubMed] [Google Scholar]
- Chin L., Artandi,S.E., Shen,Q., Tam,A., Lee,S.L., Gottlieb,G.J., Greider,C.W. and DePinho,R.A. (1999) p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell, 97, 527–538. [DOI] [PubMed] [Google Scholar]
- Collins K. and Mitchell,J.R. (2002) Telomerase in the human organism. Oncogene, 21, 564–579. [DOI] [PubMed] [Google Scholar]
- De Lange T. (2002) Protection of mammalian telomeres. Oncogene, 21, 532–540. [DOI] [PubMed] [Google Scholar]
- Espejel S., Franco,S., Rodriguez-Perales,S., Bouffle,S.D., Cigudosa,J.C. and Blasco,M.A. (2002) Mammalian Ku86 mediates chromosomal fusions and apoptosis caused by critically short telomeres. EMBO J., 21, 2207–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco S., Segura,I., Riese,H. and Blasco,M.A. (2002) Decreased B16F10 melanoma growth and impaired vascularization in telomerase-deficient mice with critically short telomeres. Cancer Res., 62, 552–559. [PubMed] [Google Scholar]
- González-Suárez E., Samper,E., Flores,J.M. and Blasco,M.A. (2000) Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat. Genet., 26, 114–117. [DOI] [PubMed] [Google Scholar]
- Goytisolo F.A. and Blasco,M.A. (2002) Many ways to telomere dysfunction: in vivo studies using mouse models. Oncogene, 21, 584–591. [DOI] [PubMed] [Google Scholar]
- Guerra S., Leri,A., Wang,X., Finato,N., Di Loreto,C., Beltrami,C.A., Kajstura,J. and Anversa,P. (1999) Myocyte death in the failing human heart is gender dependent. Circ. Res., 85, 856–866. [DOI] [PubMed] [Google Scholar]
- Harley C.B., Futcher,A.B. and Greider,C.W. (1990) Telomeres shorten during ageing of human fibroblasts. Nature, 345, 458–460. [DOI] [PubMed] [Google Scholar]
- Hemann M.T., Strong,M.A., Hao,L.Y. and Greider,C.W. (2001) The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell, 107, 67–77. [DOI] [PubMed] [Google Scholar]
- Herrera E., Samper,E., Martin-Caballero,J., Flores,J.M., Lee,H.W. and Blasco,M.A. (1999) Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J., 18, 2950–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera E., Martinez,A.C. and Blasco,M.A. (2000) Impaired germinal center reaction in mice with short telomeres. EMBO J., 19, 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajstura J. et al. (1996) Necrotic and apoptotic myocyte cell death in the aging heart of Fischer 344 rats. Am. J. Physiol., 271, H1215–H1228. [DOI] [PubMed] [Google Scholar]
- Kajstura J., Leri,A., Finato,N., Di Loreto,C., Beltrami,C.A. and Anversa,P. (1998) Myocyte proliferation in end-stage cardiac failure in humans. Proc. Natl Acad. Sci. USA, 95, 8801–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.W., Blasco,M.A., Gottlieb,G.J., Horner,J.W., Greider,C.W. and DePinho,R.A. (1998) Essential role of mouse telomerase in highly proliferative organs. Nature, 392, 569–574. [DOI] [PubMed] [Google Scholar]
- Leri A., Liu,Y., Wang,X., Kajstura,J., Malhotra,A., Meggs,L.G. and Anversa,P. (1999) Overexpression of insulin-like growth factor-1 attenuates the myocyte renin–angiotensin system in transgenic mice. Circ. Res. 84, 752–762. [DOI] [PubMed] [Google Scholar]
- Leri A., Barlucchi,L., Limana,F., Deptala,A., Darzynkiewicz,Z., Hintze,T.H., Kajstura,J., Nadal-Ginard,B. and Anversa,P. (2001) Telomerase expression and activity are coupled with myocyte proliferation and preservation of telomeric length in the failing heart. Proc. Natl Acad. Sci. USA, 98, 8626–8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlrath J. et al. (2001) Telomere length anormalities in mammalian radiosensitive cells. Cancer Res., 61, 912–915. [PubMed] [Google Scholar]
- Orlic D. et al. (2001a) Bone marrow cells regenerate infarcted myocardium. Nature, 410, 701–705. [DOI] [PubMed] [Google Scholar]
- Orlic D. et al. (2001b) Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc. Natl Acad. Sci. USA, 98, 10344–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaini F., Urbanek,K., Beltrami,A.P., Finato,N., Beltrami,C.A., Nadal-Ginard,B., Kajstura,J., Leri,A. and Anversa,P. (2002) Chimerism of the transplanted heart. N. Engl. J. Med., 346, 5–15. [DOI] [PubMed] [Google Scholar]
- Rudolph K.L., Chang,S., Lee,H.W., Blasco,M., Gottlieb,G.J., Greider,C. and DePinho,R.A. (1999) Longevity, stress response and cancer in aging telomerase-deficient mice. Cell, 96, 701–712. [DOI] [PubMed] [Google Scholar]
- Samper E., Flores,J.M. and Blasco,M.A. (2001) Restoration of telomerase activity rescues chromosomal instability and premature aging in Terc–/– mice with short telomeres. EMBO Rep., 2, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samper E., Fernández,P., Martín-Rivera,L., Bernad,A., Blasco,M.A. and Aracil,M. (2002) Long-term repopulating ability of telomerase-deficient murine hematopoietic stem cells. Blood, 99, 2767–2775. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A. and De Lange,T. (2002) Different telomere damage signaling pathways in human and mouse cells. EMBO J., 21, 4338–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]