Molecular haplotyping at high throughput (original) (raw)
Abstract
Reconstruction of haplotypes, or the allelic phase, of single nucleotide polymorphisms (SNPs) is a key component of studies aimed at the identification and dissection of genetic factors involved in complex genetic traits. In humans, this often involves investigation of SNPs in case/control or other cohorts in which the haplotypes can only be partially inferred from genotypes by statistical approaches with resulting loss of power. Moreover, alternative statistical methodologies can lead to different evaluations of the most probable haplotypes present, and different haplotype frequency estimates when data are ambiguous. Given the cost and complexity of SNP studies, a robust and easy-to-use molecular technique that allows haplotypes to be determined directly from individual DNA samples would have wide applicability. Here, we present a reliable, automated and high-throughput method for molecular haplotyping in 2 kb, and potentially longer, sequence segments that is based on the physical determination of the phase of SNP alleles on either of the individual paternal haploids. We demonstrate that molecular haplotyping with this technique is not more complicated than SNP genotyping when implemented by matrix-assisted laser desorption/ionisation mass spectrometry, and we also show that the method can be applied using other DNA variation detection platforms. Molecular haplotyping is illustrated on the well-described β2-adrenergic receptor gene.
INTRODUCTION
Genotyping of single nucleotide polymorphisms (SNPs) has multiple applications that include human genetic studies, epidemiology, pharmacogenomics, marker assisted selection in animals and plants, and traceability. Intensive efforts to identify SNPs in humans (e.g. the SNP consortium) (1) and to provide basic information about linkage disequilibrium and haplotype patterns for these markers (e.g. the proposed Haplotype Map program) are underway (2). Parallel to these efforts, several high-throughput SNP genotyping methods have been demonstrated (3). These developments have rendered genome-wide association studies and systematic testing of variants of all human genes technically feasible. Applications of these approaches in genetics, pharmacogenomics and epidemiology are likely have a profound impact, for example in identifying the most effective medication for patients (4), even if many theoretical questions remain to be resolved regarding their power and limits (5–7). In addition, these applications require that genotyping become more affordable and/or that other strategies are adapted to reduce the costs, for example genotyping of small subsets of SNPs that identify major blocks of linkage disequilibrium (8).
A key approach in such studies is the ability to analyse haplotypes. Based on haplotypes, it should be possible to deduce what common DNA variants are likely to be present on chromosomes from two individuals from the identity of the alleles at a small number of pre-selected SNPs within a region of linkage disequilibrium. It has been suggested that haplotypes might be more appropriate for pharmacogenetic assessment (4,7,9), when SNPs contributing to some trait are not directly observed or the effects of observed SNPs are non-additive (10). In some instances, haplotypes can be inferred directly from genotype data with relatively high precision, for example when linkage disequilibrium is quite strong or families are available for study, but this is far from being possible in all circumstances of interest. Otherwise, haplotypes can be inferred from genotyping experiments with statistical algorithms, but due to their inherent uncertainty these procedures may not be ideal for extracting all the desired information. Thus, an alternative method to obtain haplotypes directly based on molecular studies (molecular haplotyping) would have wide application if easy and inexpensive to implement in comparison with standard methods for SNP genotype determination. Other applications of molecular haplotyping include the simplification of linkage and association calculations when studying many SNPs at a single locus in large pedigrees with missing data (J.O’Connell, University of Pittsburg, personal communication).
In the past, molecular haplotyping was only possible by the laborious cloning and sequencing of the two homologous chromosome regions (11). Limited information about one or two polymorphic positions can be obtained with techniques like allele-specific PCR (12–18) or heteroduplex analysis (19), the latter only giving information about insertions and deletions. Information about several positions can be obtained with the combination of allele-specific PCR and hybridization with sequence-specific oligonucleotides (20), or the combination of allele-specific PCR and restriction fragment length polymorphism analysis (21). Recently, a method for haplotyping within kilobase-size DNA fragments was presented (22). Atomic force microscopy in conjunction with carbon nanotubes was applied. Although this approach is of interest, it is not obvious how it could be applied in a high-throughput setting. Douglas et al. (23) recently presented a method for the isolation of entire chromosomes, however the procedure does not seem sufficiently easy to implement in high throughput to deal with cohorts of thousands of individuals as somatic cell hybrids have to be generated.
Matrix-assisted laser desorption/ionisation mass spectrometry (MALDI) has revolutionised the instrumental analysis of biomolecules (24). Several protocols for SNP genotyping using MALDI as a detection method have been established (25–27). Some of these procedures rely on stringent purification procedures prior to MALDI analysis that do not lend themselves to easy automation and contribute a significant portion of the cost of an analysis. We recently introduced a sample preparation method for genotyping SNPs by MALDI mass spectrometry called the ‘GOOD assay’, which does not require any purification (28–31). Here, we show how the GOOD assay can be extended to high-throughput, low-cost and reliable molecular haplotyping across genomic regions of substantial size.
To demonstrate our molecular haplotyping method we chose the well-described β2-adrenergic receptor gene (9,32). The human β2-adrenergic receptor belongs to a family of seven _trans_-membrane receptors (33). SNPs in this receptor are associated with a genetic predisposition to hypertension and are found to be relevant for cardiovascular disease (32,34). Haplotypes of this gene have been suggested to play a role in the response to asthma treatment using β2-adrenergic receptor antagonists such as albuterol and to influence phenotypes such as bronchial hyper-responsiveness (9). The SNPs of the gene used for haplotyping here are at positions –654, and –47 in the promoter region, and 46 in the coding region. Because of strong linkage disequilibrium, or phase coupling, of the SNP at position –47 with other SNPs at positions –367, –20 and 79, an accurate determination of the principal haplotypes in Caucasian, African and Asian populations as presented by Drysdale et al. (9) can be obtained. Table 1 shows the groups of molecularly distinguishable haplotype groups along with the nomenclature for these as defined previously in the literature.
Table 1. Classification of the haplotypes by examination of the three positions in the β2-adrenergic receptor gene.

MATERIALS AND METHODS
Primers for PCR amplification were purchased from MWG (Ebersberg, Germany) and primers for primer extension reactions from Biotez (Berlin, Germany). Taq DNA polymerase was purchased from Roche Diagnostics (Basel, Switzerland), Stoffel Taq DNA polymerase from Applied Biosystems (Foster City, CA), AccuPrime™ Taq DNA polymerase system from Invitrogen Life Technologies (Carlsbad, CA), shrimp alkaline phosphatase (SAP) and Thermosequenase from Amersham (Little Calfont, UK), phosphodiesterase II (PDE; from calf spleen) from Worthington Biochemical Corp. (Lakewood, NJ), dNTPs from Hybaid (Ashford, UK) and α-S-ddNTPs from Biolog (Bremen, Germany). Standard reagents were either bought from Aldrich (Milwaukee, WI) or Merck (Darmstadt, Germany) in analysis grade, if not otherwise stated. Liquid handling was performed using a BasePlate Robot from The Automation Partnership (Royston, UK). Allele-specific PCRs were carried out in Eppendorf 96 or 384 Gradient Mastercycler (Hamburg, Germany), all other thermocycling procedures in Primus thermocyclers from MWG Biotech (Ebersberg, Germany). Mass spectrometric analyses were performed using an Autoflex MALDI mass spectrometer (Bruker Daltonik, Bremen, Germany).
‘GOOD assay’ protocol for haplotyping
PCR. For amplification of the DNA segment encompassing the three SNPs of the β2-adrenergic receptor 2.25 pmol each of forward and reverse primer and 1 µl of DNA (5 ng/µl) as template was used. The reaction conditions were 20 mM Trisbase (pH 8.4), 50 mM KCl, 1.5 mM MgCl2, 200 µM dNTPs, thermostable AccuPrime™ protein (all included in 10× PCR AccuPrime buffer II) and 0.03 µl AccuPrime™ Taq DNA polymerase [0.06 reactions (rxn)] in a 3 µl volume. The supplier of the AccuPrime™ Taq DNA polymerase makes no indication of units per microlitre, but rather rnx. Reactions were denatured for 3 min at 95°C, then thermocycled for 30 s at 95°C, 45 s at the respective annealing temperature and 60 s at 68°C, repeating the cycle 30 times. For the genotyping experiment the primers used were 5′-GTGCATGTCGGT GAGCTG (forward) and 5′-ACATTGCCAAACACGATGG (reverse) to yield a 856 bp product, with an annealing temperature of 64°C. For allele-specific PCR on the (A/G) SNP position –654 either the primer 5′-GCTGTGGTT CGGTATAAGTCTG (specific for the G genotype, annealing temperature 65°C) or 5′-GCTGTGGTTCGGTATAAGTCTA (specific for the A genotype, annealing temperature 62.5°C) was used with the same reverse primer as above to give a product of 808 bp. For allele-specific PCR on the (C/G) SNP position 79 either the primer 5′-CCACACCTCGTCCCTTTG (specific for the C genotype, annealing temperature 61°C) or 5′-CCACACCTCGTCCCTTTC (specific for the G genotype, annealing temperature 60°C) was used with the same forward primer as above to give a PCR product of 797 bp.
SAP digest. Two microlitres containing 0.25 U SAP in 44 mM Tris buffer (pH ∼8.0) were added to the PCR reaction and incubated for 1 h at 37°C, followed by denaturation at 90°C for 10 min.
Primer extension reaction. 8.75 pmol of extension primer (5′-CACCCTGGCAGACATGPTCCTPTT) for position –654, 5 pmol of extension primer (5′-CCGCCGTGGGTCCGPT CCTPTC) for position –47 and 2.75 pmol of extension primer (5′-GTCCGGCGCATGGCTPTTCTPTC) for position 46, and 0.8 U Thermosequenase were added. MgCl2 was adjusted to a final concentration of 4.3 mM and 50 µM, respectively, α-S-ddCTP and α-S-ddTTP were used in a reaction volume of 7 µl. An initial denaturing step for 2 min at 95°C was used followed by 44 cycles of 10 s at 95°C, 15 s at 58°C and 10 s at 72°C.
Extension primers carry N-modified bases at the second base from the 3′-end. This base is bracketed by two phosphorothioate bridges (PT). The amino function is used to attach charge tags (CT). Charge tagging of the N-modified oligonucleotides was done according to the procedure described by Sauer et al. (28) with 6-trimethylammoniumhexyryl-_N_-hydroxy-succinimidylester for primers query ing positions –47 and 46 and 6-diethyl-methylammoniumhexyryl-_N_-hydroxy-succinimidylester for the primer querying position –654. Using different CTs gives the allele pairs of each of the SNPs their discrete mass window. The quality of oligonucleotides was checked before and after charge tagging by MALDI mass spectrometry.
PDE digestion. A sample of 0.5 µl of 0.5 M acetic acid and 1.5 µl of PDE were added to the reaction and incubated for 60 min at 37°C.
Alkylation reaction. A mixture of 11 µl acetonitrile, 2.75 µl 2 M triethylammoniumhydrogencarbonate buffer (pH ∼7.5) and 5.25 µl iodomethane was added and incubated at 40°C for 25 min. Upon cooling a biphasic system was obtained. Ten microlitres of water were added and let stand for 5 min at room temperature. Five microlitres of the upper layer were taken off and diluted with 10 µl of 40% acetonitrile.
Sample preparation. A 1.5% solution of α-cyano-4-hydroxy-cinnamic acid methyl ester in acetone was used to prepare the matrix and 0.5 µl of sample was spotted on top with the BasePlate robot (The Automation Partnership, Royston, UK).
Mass spectrometric analysis. Spectra were recorded automatically on a Bruker Autoflex MALDI time-of-flight mass spectrometer using the AutoXecute software. Spectra were recorded in positive ion linear time-of-flight mode. Typical acceleration potentials were 18 kV. The ion extraction was delayed by 200 ns. The Genotools SNP manager was used to automatically analyse the spectra and generate allele calls (35).
Sequencing. Sequencing was performed using standard procedures on an ABI 3700 96 capillary sequencer (Foster City, CA). Ten microlitre PCRs were performed using 5 ng of genomic DNA, 0.1 µl AccuPrime™ Taq DNA polymerase (0.2 rxn) and the same concentrations of the other reagents as described above. PCR products were purified by G50 (Sephadex) gelfiltration and sequencing reactions were performed using standard procedures with nested primers 5′-GAGTGTGCAGGACGAGTC (forward) and 5′-CAAT GGCACGCCGGCAG (reverse). Analysis was performed using in-house developed software called Genalys (downloadable from www.cng.fr).
RESULTS
In principal, allele-specific PCR combined with multiplex high-throughput detection is the key to this large-scale haplotyping method. Two strategies can be adopted to carry out the procedure. The more elaborate strategy involves two steps. First, a stretch of genomic DNA containing several SNPs is amplified by a single PCR, and the SNPs are genotyped. If two or more SNP sites on the PCR fragment are heterozygous, the sample is of interest for haplotyping. In the second step, one of the heterozygous SNPs on the boundary is used to prepare two allele-specific PCR products for such a sample, and the SNPs are re-genotyped using these products as template. The genotypes for each of these products correspond to one of two possible alleles at each site, which conveys the phase of the SNPs, while the accumulation of the two allele-specific results should be equivalent to the original genotypes. This provides an internal control for the experiment. Selecting DNA samples (cherry picking) for haplotyping is time consuming and tedious. The more direct strategy involves only the second step. A highly polymorphic SNP on the boundary of the DNA segment to be haplotyped is used for two allele-specific PCRs on all samples. For the sake of demonstration we combined both strategies, all samples were first genotyped and then all samples haplotyped.
To implement this as an effective approach, we found it necessary to undertake extensive investigation of several strategies and DNA polymerases for allele-specific PCR. For example, primers containing GC-tails can be used at elevated annealing temperatures to increase the allele-specificity and the introduction of additional mismatches in the 3′ region of the primers (16,36–38). However, optimisation of PCR products >200 bp proved difficult by these strategies, thus not providing the ability to haplotype across substantial genomic regions. At low reaction volumes (typically in SNP genotyping production in our group PCR is carried out in 2 µl volume), we found that the false positive rate of these systems increases substantially, and becomes a prohibitive factor. A large number of different DNA polymerases were tested, and generally they were not compatible with the aim to provide an accessible and high-throughput method for molecular haplotyping. However, one of these, AccuPrime™ Taq DNA polymerase, developed for multiplex PCRs, distinguishes itself from other polymerase systems by an additional DNA binding protein, permitting amplification only if the primer matches completely with the target sequence in the template. With this polymerase, allele-specific PCR using primers with the 3′ base matching one of the alleles was found to be very effective. It does not tolerate additional mismatches in positions –2 to –4 from the 3′ end of the allele-specific PCR primer. PCR products up to a length of 2 kb were obtained without much optimisation. Critically, no false positive results were observed at the chosen annealing temperatures even in small volume PCRs. As only a very small amount of enzyme is used for the PCRs, this procedure is also very cost effective.
For the analysis of the β2-adrenergic receptor gene, we chose to amplify the sequence encompassing the SNPs from position –654 to position 79, as these allow an accurate division into the principal haplotypes described previously in three major ethnic populations. The quality and performance of our haplotyping procedure is demonstrated below in a series of experiments: (i) allele-specificity of the PCR; (ii) sequencing of the allele-specific PCR products; (iii) MALDI analysis of primer extension products of the SNPs on the allele-specific products; (vi) test for transmission of haplotypes using 384 CEPH family DNAs in a automated format.
Agarose gel analysis of allele-specific PCR products is shown in Figure 1. Previously sequenced DNA samples homozygous for the G allele (lane 1–4), homozygous for the C allele (6–9) or heterozygous (lanes 11–14) for position 79 of the β2-adrenergic receptor gene were PCR amplified using a primer matching the G allele (Fig. 1A) or the C allele (Fig. 1B) with its 3′ terminal base.
Figure 1.
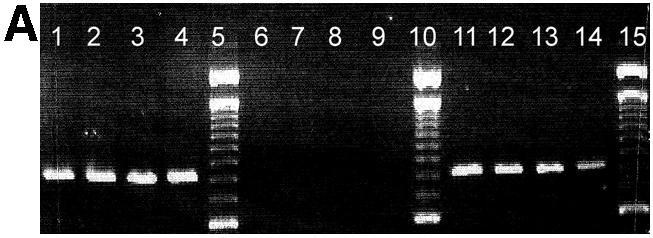
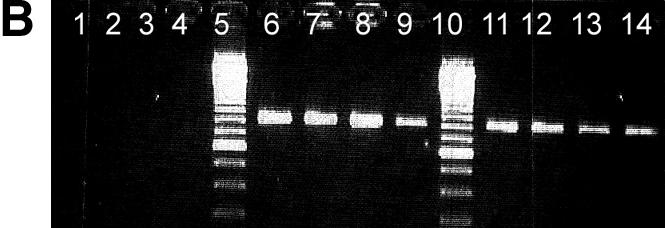
Agarose gel separation of a 797 bp PCR product generated by allele-specific PCR using a primer matching the G allele of the polymorphic position 79 (A) and the C allele of the same position, respectively (B). Previously sequenced DNA from CEPH families from individuals homozygous for the G allele (lanes 1–4), homozygous for the C allele (lanes 6–9) or heterocygous (C/G) (lane 11–14) was used. Lanes 5, 10 and 15 are reference ladders. Products are missing where the 3′ terminal base is not complementary to the template strand.
To demonstrate the reliability of allele-specific PCR the four allele-specific PCR products (two allele-specific PCR products for position –654 and two for position 79) of DNA samples from two CEPH families were sequenced (96 samples). Results were checked for allele transmission through generations and by generating the same haplotypes with forward and reverse allele-specific primers for individuals heterozygous on both SNPs used for the allele-specific PCR. No false positive results were observed. This constitutes a new, easy way to determine haplotype information by sequencing without cloning.
Haplotyping using the GOOD assay was then performed on nine unrelated DNA samples, three Caucasian, three African and three Asian. The results of the genotyping experiment (Table 2) were confirmed by sequencing. The success of allele-specific PCR is confirmed by querying the SNP used for allele-specific PCR (internal control). Position –654 is typed with an extension primer in the opposite direction to the amplification primer, position 79 is controlled by querying position –47, which is in complete linkage disequilibrium with position 79 in all known haplotypes. Figure 2 shows examples of spectra obtained by MALDI analysis with the different allele-specific PCRs. They allow unambiguous assignment of the different haplotypes with the queried positions. In all three populations haplotypes of groups C and E were found, the frequent haplotypes in these populations. Group A was not found in African samples, as it is uncommon in this ethnic group.
Table 2. Results of haplotyping in different populations.

Figure 2.


(Previous page and above) Schematic representation of procedure for haplotyping and resulting mass spectra for Caucasian DNA A008GG5 (A), which is heterozygous for all three positions queried, and Asian DNA A006K5U (B), which is heterozygous for positions –654 and 46 and homozygous for position –47 (79, respectively). The allele-specific PCR in forward and reverse direction gives the same results for DNA A008GG5, while in the case of DNA A006K5U the reverse PCR is not able to split the two haplotypes. The MALDI spectra show the results of genotyping and haplotyping the SNPs –654, –47 and 46 in the human β2-adrenergic receptor gene. Product masses of the alleles are GptCCTptCptC with 1418 Da (I) and GptCCTptCptT Da with 1433 Da (II) for the SNP –47, GptCCTptTptC with 1461 Da (III) and GptCCTptTptT with 1476 Da (IV) for the SNP –654 and TptTCTptCptC with 1509 Da (V) and TptTCTptCptT with 1524 Da (VI) for the SNP 46. The peak marked with an asterisk corresponds to incomplete 5′ phosphodiesterase digestion of the primer querying position 46.
Finally, we performed the haplotyping with the GOOD assay on 384 individuals from the CEPH DNA collection in a high-throughput format. All liquid handling was done using a BasePlate liquid handling robot. The accumulation and analysis of spectra were done unattended using the software provided with the Autoflex mass spectrometer. After an initial genotyping experiment, haplotypes were generated using the four allele-specific PCRs (two on the two alleles of position –654 in sense and two on the two alleles of position 79 in antisense). We found the three different haplotypes groups, A, C and E, in the CEPH families. These are the main haplotypes in Caucasians. Six different genotypes were found with frequencies of AA = 18.9%, AC = 29.4%, AE = 28.5%, CC = 4.4%, CE = 15.7% and EE = 3.2%. The haplotype frequencies, obtained from all individuals without taking account of family relationships, were A = 47.8%, C = 26.9% and E = 25.3, and the genotype frequencies were in agreement with Hardy–Weinberg expectations (Fig. 3). The alleles found in the different families are shown in Table 3. The first pass return was in excess of 94%. Failures were probably due to DNA template of low quality. Genotypes and haplotypes of 96 of the individuals were confirmed by sequencing. Sequencing of allele-specific PCR products gave similar quality to regular sequencing. This leads us to believe that any DNA sequence interrogation (SNP genotyping) method that follows PCR amplification could be used for haplotyping by including the described allele-specific PCR. We tested inferring haplotypes based on preliminary estimation with the EM algorithm, followed by assignment of the most probable haplotypes to individuals. Results of the statistical and the experimental analysis matched completely here, as was expected because the system was chosen for its simplicity so that the molecular results could be easily confirmed.
Figure 3.

Distribution of alleles and genotypes in the 384 CEPH samples analysed.
Table 3. Haplotype groups found in the DNA of the CEPH families, the alleles found in each generation for each family are shown, allowing the verification of the assay by allele transmission.

DISCUSSION
In this study we successfully demonstrated the first high-throughput method for molecular haplotyping. In this method, the utilization of GC rich tails and/or internal mismatches in the amplification primers for allele-specific PCRs have been discarded as strategies, and replaced by a novel DNA polymerase system (16,36,37). As a result of the application of the highly specific AccuPrime™ Taq DNA polymerase system, the laborious procedures of optimizing allele-specific PCRs to isolate one of the alleles was not necessary. The use of this or similar specific polymerase systems will have great influence on the analysis of haplotypes by a broad variety of detection platforms. With its ease of optimization it makes automation and therewith the high-throughput format feasible in a way that was not possible before.
Combining the described procedure with SNP genotyping and MALDI mass spectrometric detection is an ideal match and opens the door for molecular haplotyping at high throughput in absence of family information. The major strength of MALDI mass spectrometry is the speed and accuracy of analysis. An entire 384 well plate of samples takes significantly <1 h to analyse. Mass spectrometric methods for SNP genotyping and haplotyping in contrast to gel-based techniques such as conventional DNA sequencing have the advantage that each allele found gives an absolute, measured mass—a physical property. This allows a suitable degree of multiplexing and avoids artifacts in allele determination that have been observed in gel-based detection systems (39). As haplotyping requires multiplex analysis to determine the phase of SNPs, our assay makes optimal use the multi-channel detection capability of mass spectrometers. We demonstrated our method as a triplex assay, as this is suitable for the discrimination of the major haplotypes of the β2-adrenergic receptor gene within most populations, but greater multiplex factors are feasible with MALDI detection and have already been demonstrated for SNP genotyping (27). Based on manufacturers specification, we believe that it will be possible to use AccuPrime DNA polymerase for the amplification of 4 kb fragments, and improved enzyme systems may make it possible to extend beyond 4 kb fragments. Assays of overlapping fragments will also allow larger segments to be targeted for haplotyping as desired.
We chose to demonstrate our molecular haplotyping method on the β2-adrenergic receptor gene due to the known pharmacogenetic effect associated with haplotypes in this system and due to the fact that it is very well described allowing us to control for many parameters. However, we believe that molecular haplotying by a method like ours will be particularly valuable for large-scale association studies, where it could be applied to the boundaries of haplotype blocks, thus allowing determination of the alleles on either side and the phase (2). With the current efforts underway to generate a complete haplotype map of the human genome the method presented here provides a very powerful tool (40).
Acknowledgments
ACKNOWLEDGEMENTS
O.B. was supported by a fellowship from the European Community through LEONARDO (Technische Universität, Berlin, Germany). This work was supported by the Ministère de l’Education, Recherche et Technologie of the French Government (MENRT).
REFERENCES
- 1.Sachidanandam R., Weissman,D., Schmidt,S.C., Kakol,J.M., Stein,L.D., Marth,G., Sherry,S., Mullikin,J.C., Mortimore,B.J., Willey,D.L. et al. (2001) A map of human sequence variation containing 1.42 million single nucleotide polymorphisms. Nature, 409, 928–933. [DOI] [PubMed] [Google Scholar]
- 2.Gabriel S.B., Schaffner,S.F., Nguyen,H., Moore,J.M., Roy,J., Blumenstiel,B., Higgins,J., DeFelice,M., Lochner,A., Faggart,M. et al. (2002) The structure of haplotype blocks in the human genome. Science, 296, 2225–2229. [DOI] [PubMed] [Google Scholar]
- 3.Gut I.G. (2001) Automation in genotyping of single nucleotide polymorphisms. Hum. Mutat., 17, 475–492. [DOI] [PubMed] [Google Scholar]
- 4.McCarthy J.J. and Hilfiker,R. (2000) The use of single-nucleotide polymorphism maps in pharmacogenomics. Nat. Biotechnol., 18, 505–508. [DOI] [PubMed] [Google Scholar]
- 5.Kruglyak L. (1999) Prospect for whole-genome linkage disequilibrium mapping of common disease genes. Nature Genet., 22, 139–144. [DOI] [PubMed] [Google Scholar]
- 6.Weiss K.M. and Terwilliger,J.D. (2000) How many diseases does it take to map a gene with SNPs? Nature Genet., 26, 151–157. [DOI] [PubMed] [Google Scholar]
- 7.Davidson S. (2000) Research suggests importance of haplotypes over SNPs. Nat. Biotechnol., 18, 1134–1135. [DOI] [PubMed] [Google Scholar]
- 8.Schafer A.J. and Hawkins,J.R. (1998) DNA variation and the future of human genetics. Nat. Biotechnol., 16, 33–39. [DOI] [PubMed] [Google Scholar]
- 9.Drysdale C.M., McGraw,D.W., Stack,C.B., Stephens,J.C., Judson,R.S., Nandabalan,K., Arnold,K., Ruano,G. and Liggett,S.B. (2000) Complex promoter and coding region β2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc. Natl Acad. Sci. USA, 97, 10483–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Judson R. and Stephens,J.C. (2001) Notes from the SNP vs. haplotype front. Pharmacogenomics, 2, 7–10. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Arias R., Bertranpetit,J. and Comas,D. (2002) Determination of haploid DNA sequences in humans: application to the glucocerebrosidase pseudogene. DNA Seq., 13, 9–13. [DOI] [PubMed] [Google Scholar]
- 12.Newton C.R., Graham,A., Heptinstall,L.E., Powell,S.J., Summers,C., Kalsheker,N., Smith,J.C. and Markham,A.F. (1989) Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res., 17, 2503–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo Y.M., Patel,P., Newton,C.R., Markham,A.F., Fleming,K.A. and Wainscoat,J.S. (1991) Direct haplotype determination by double ARMS: specificity, sensitivity and genetic applications. Nucleic Acids Res., 19, 3561–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruano G. and Kidd,K.K. (1989) Direct haplotyping of chromosomal segments from multiple heterozygotes via allele-specific PCR amplification. Nucleic Acids Res., 17, 8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarkar G. and Sommer,S.S. (1991) Haplotyping by double PCR amplification of specific alleles. Biotechniques, 10, 436–440. [PubMed] [Google Scholar]
- 16.Okimoto R. and Dodgson,J.B. (1996) Improved PCR amplification of multiple specific alleles (PAMSA) using internally mismatched primers. Biotechniques, 21, 20–22. [DOI] [PubMed] [Google Scholar]
- 17.Koss K., Fanning,G.C., Welsh,K.I. and Jewell,D.P. (2000) Interleukin-10 gene promoter polymorphism in English and Polish healthy controls. Polymerase chain reaction haplotyping using 3′ mismatches in forward and reverse primers. Genes Immun., 1, 321–324. [DOI] [PubMed] [Google Scholar]
- 18.Eitan Y. and Kashi,Y. (2002) Direct micro-haplotyping by multiple double PCR amplifications of specific alleles (MD-PASA). Nucleic Acids Res., 30, e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang F.M. and Kidd,K.K. (1997) Rapid molecular haplotyping of the first exon of the human dopamine D4 receptor gene by heteroduplex analysis. Am. J. Med. Genet., 74, 91–94. [DOI] [PubMed] [Google Scholar]
- 20.Boldt A.B. and Petzl-Erler,M.L. (2002) A new strategy for mannose-binding lectin gene haplotyping. Hum. Mutat., 19, 296–306. [DOI] [PubMed] [Google Scholar]
- 21.McDonald O.G., Krynetski,E.Y. and Evans,W.E. (2002) Molecular haplotyping of genomic DNA for multiple single-nucleotide polymorphisms located kilobases apart using long-range polymerase chain reaction and intramolecular ligation. Pharmacogenetics, 12, 93–99. [DOI] [PubMed] [Google Scholar]
- 22.Woolley A.T., Guillemette,C., Cheung,C.L., Housman,D.E. and Lieber,C.M. (2000) Direct haplotyping of kilobase-size DNA using carbon nanotube probes. Nat. Biotechnol., 18, 760–763. [DOI] [PubMed] [Google Scholar]
- 23.Douglas J.A., Boehnke,M., Gillanders,E., Trent,J.M. and Gruber,S.B. (2001) Experimentally-derived haplotypes substantially increase the efficiency of linkage disequilibrium studies. Nature Genet., 28, 361–364. [DOI] [PubMed] [Google Scholar]
- 24.Karas M. and Hillenkamp,F. (1988) Laser desorption ionization of proteins with molecular masses exceeding 10.000 Daltons. Anal. Chem., 60, 2299–2301. [DOI] [PubMed] [Google Scholar]
- 25.Tang K., Fu,D.J., Julien,D., Braun,A., Cantor,C.R. and Köster,H. (1999) Chip-based genotyping by mass spectrometry. Proc. Natl Acad. Sci. USA, 96, 10016–10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griffin T.J., Hall,J.G., Prudent,J.R. and Smith,L.M. (1999) Direct genetic analysis by matrix-assisted laser desorption/ionization mass spectrometry. Proc. Natl Acad. Sci. USA, 96, 6301–6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ross P., Hall,L., Smirnov,I. and Haff,L. (1998) High level multiplex genotyping by MALDI-TOF mass spectrometry. Nat. Biotechnol., 16, 1347–1351. [DOI] [PubMed] [Google Scholar]
- 28.Sauer S., Lechner,D., Berlin,K., Lehrach,H., Escary,J.L., Fox,N. and Gut,I.G. (2000) A novel procedure for efficient genotyping of single nucleotide polymorphisms. Nucleic Acids Res., 28, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sauer S., Lechner,D., Berlin,K., Plancon,C., Heuermann,A., Lehrach,H. and Gut,I.G. (2000) Full flexibility genotyping of single nucleotide polymorphisms by the GOOD assay. Nucleic Acids Res., 28, e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sauer S., Lechner,D. and Gut,I.G. (2001) The GOOD assay. In Housby,J.N. (ed.), Mass Spectrometry and Genomic Analysis. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp. 50–65.
- 31.Sauer S., Gelfand,D.H., Boussicault,F., Bauer,K., Reichert,F. and Gut,I.G. (2002) Facile method for automated genotyping of single nucleotide polymorphisms by mass spectrometry. Nucleic Acids Res., 30, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor D.R. and Kennedy,M.A. (2001) Genetic variation of the β2-adrenoreceptor. Its functional and clinical importance in bronchial asthma. Am. J. Pharmacogenom., 1, 165–174. [DOI] [PubMed] [Google Scholar]
- 33.Johnson M. (1998) The beta adrenoreceptor. Am. J. Respir. Crit. Care Med., 158, 146–153. [DOI] [PubMed] [Google Scholar]
- 34.Timmermann B., Mo,R., Luft,F.C., Gerdts,E., Busjahn,A., Omvik,P., Li,G.H., Schuster,H., Wienker,T.F., Hoehe,M.R. and Lund-Johansen,P. (1998) Beta-2 adrenoceptor genetic variation is associated with genetic predisposition to essential hypertension: the Bergen Blood Pressure Study. Kidney Int., 53, 14514–14560. [DOI] [PubMed] [Google Scholar]
- 35.Pusch W., Kraeuter,K.-O., Froehlich,T., Stalgies,Y. and Kostrzewa,M. (2001) Genotools SNP MANAGER: a new software for automated high-throughput MALDI-TOF mass spectrometry SNP genotyping. Biotechniques, 30, 210–215. [DOI] [PubMed] [Google Scholar]
- 36.Liu Q., Thorland,E.C., Heit,J.A. and Sommer,S.S. (1997) Overlapping PCR for bidirectional PCR amplification of specific alleles: a rapid one-tube method for simultaneously differentiating homozygotes and heterozygotes. Genome Res., 7, 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sommer S.S., Groszbach,A.R. and Bottema,C.D. (1992) PCR amplification of specific alleles (PASA) is a general method for rapidly detecting known single-base changes. Biotechniques, 12, 82–87. [PubMed] [Google Scholar]
- 38.Kwok S., Kellogg,D.E., McKinney,N., Spasic,D., Goda,L., Levenson,C. and Sninsky,J.J. (1990) Effects of primer-template mismatches on the polymerase chain reaction: human immunodeficiency virus type 1 model studies. Nucleic Acids Res., 18, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ronaghi M., Nygren,M., Lundeberg,J. and Nyren,P. (1999) Analyses of secondary structures in DNA by pyrosequencing. Anal. Biochem., 267, 65–71. [DOI] [PubMed] [Google Scholar]
- 40.Reich D.E., Cargill,M., Bolk,S., Ireland,J., Sebeti,P.C., Richter,D.J., Lavery,T., Kouyoumjian,R., Farhadian,S.F., Ward,R. and Lander,E.S. (2001) Linkage disequilibrium in the human genome. Nature, 411, 199–204. [DOI] [PubMed] [Google Scholar]