Dimerisation-dependent GTPase reaction of MnmE: how potassium acts as GTPase-activating element (original) (raw)
Abstract
MnmE, a Guanine nucleotide-binding protein conserved between bacteria and man, is involved in the modification of tRNAs. Here we provide biochemical and X-ray structural evidence for a new GTP-hydrolysis mechanism, where the G-domains of MnmE dimerise in a potassium-dependent manner and induce GTP hydrolysis. The structure in the presence of GDP-AlF_x_ and potassium shows how juxtaposition of the subunits induces a conformational change around the nucleotide which reorients the catalytic machinery. A critical glutamate is positioned such as to stabilise or activate the attacking water. Potassium provides a positive charge into the catalytic site in a position analogous to the arginine finger in the Ras-RasGAP system. Mutational studies show that potassium-dependent dimerisation and GTP hydrolysis can be uncoupled and that interaction between the G-domains is a prerequisite for subsequent phosphoryl transfer. We propose a model for the juxtaposition of G-domains in the full-length protein and how it induces conformational changes in the putative tRNA-modification centre.
Keywords: GAP, GTPase, GTP binding, MnmE, potassium
Introduction
MnmE (formerly TrmE) is a Guanine nucleotide-binding protein (GNBP) that is conserved in all three kingdoms of life. Unlike Ras-like proteins, which cycle between a GDP bound inactive and a GTP bound active state, in which they interact with effector proteins (Vetter and Wittinghofer, 2001), MnmE is apparently directly involved in an enzymatic reaction, the modification of the wobble uridine (U34) in tRNAs in bacteria, yeast and mammalia (i.e. tRNALys, tRNAGlu, tRNA4Leu, tRNA4Arg and probably tRNAGln). The modification to the hypermodified U34 involves several proteins performing different steps of the reaction. MnmE together with the protein MnmG, formerly GidA (Elseviers et al, 1984; Brégeon et al, 2001), catalyses the formation of the carboxymethylaminomethyl group at the 5 position (cmnm5U), although the precise role of either protein in the reaction is unknown. Final modification to 5-methylaminomethyl-uridine in bacteria, 5-carboxymethylaminomethyl-uridine in yeast and 5-taurinomethyl-uridine in human allows interaction with G and A, but restricts base-pairing with C and U (Yokoyama et al, 1979; Yokoyama et al, 1985; Yokoyama and Nishimura, 1995), thereby avoiding misincorporation of amino acids in mixed codon box families (Lys, Glu, Leu, Arg and Gln). Furthermore, the hypermodified U34 influences frame shifting during the translation process (Brierley et al, 1997; Hagervall et al, 1998; Bjork et al, 1999; Urbonavicius et al, 2001).
Mutant alleles of the MnmG and MnmE homologues Mto1 and MSS1 in Saccharomyces cerevisiae reveal a respiratory-deficient phenotype (Decoster et al, 1993; Colby et al, 1998). Recent studies on GTPBP3 and Mto1, the human homologues of MnmE and MnmG, lead to the suggestion that those proteins may also be involved in several human diseases like the nonsyndromic-deafness or different clinical forms of myofibrillar myopathy (MERRF: myoclonic epilepsy; ragged red fibres/MELAS: mitochondrial encephalomyopathy; lactic acidosis; stroke), which are based on mutations in mitochondrial tRNA genes (Suzuki et al, 2001; Li and Guan, 2002; Li et al, 2002).
Several mechanisms for the stimulation of the intrinsically slow GTPase reaction of GNBPs have so far been discovered. Ras proteins interact with a GAP specific for each subfamily. RasGAPs and RhoGAPs stimulate hydrolysis by supplying a catalytic residue, an ‘arginine finger', into the active site. This stabilises the endogenous glutamine residue which in turn stabilises the water molecule for the in-line attack while the arginine neutralises negative charge of β- and γ-phosphate oxygens in the transition state, and a similar mechanism is likely to hold for RabGAPs also. RanGAPs act by merely stabilising the catalytic machinery of Ran without using an arginine, similar to RGS proteins (regulator of G protein signalling) which stabilise the crucial endogenous Gln and Arg residues in the catalytically relevant conformation of Gα-proteins (Vetter and Wittinghofer, 2001; Seewald et al, 2002; Rehmann and Bos, 2004). Rap proteins do not have a glutamine residue corresponding to Gln61, and Rap1GAP provides an essential Asn residue, called ‘asparagine thumb' in analogy to the ‘arginine finger' to accelerate hydrolysis, presumably by stabilising the nucleophilic water (Daumke et al, 2004).
Stimulation of the GTPase reaction can also be achieved by dimer (or higher oligomer) formation. The large GTP-binding proteins dynamin and human guanylate-binding protein 1 (hGBP1) show a concentration-dependent hydrolysis reaction (Praefcke and McMahon, 2004). In hGBP1, a cis-Arg is relocated into the active site by dimerisation (Ghosh et al, 2006). The bacterial homologues of the signal recognition particle (SRP) and the signal recognition particle receptor (SR) form a heterodimeric complex by which they stimulate each others GTPase reaction. Structural analysis has shown that this is achieved by complementing each others active site, especially a mutual hydrogen bond between the ribose of one with the γ-phosphate of the other partner (Focia et al, 2004; Egea et al, 2004). GTPase stimulation by dimerisation is also known for the chloroplast translocon factor Toc34 (Sun et al, 2002).
Previously we showed that MnmE is a three-domain protein (Scrima et al, 2005). The N-terminal α/β-domain (1–118) induces dimerisation and binding of 5-formyl-tetrahydrofolate (5-formyl-THF). The central helical domain, which is only poorly conserved except for the C-terminal motif C(I/L/V)GK, forms a four-helix bundle. As shown by Yim et al (2003), Cys447 (Cys451 in MnmE from Escherichia coli) in this motif is essential for the tRNA modification activity of MnmE. The G-domain of MnmE (211–380) is inserted into the two halves of the central helical subdomain. It conserves the canonical Ras-like fold, with no insertion or deletion of secondary structural elements. In the structure, the G-domain is only loosely connected to the residual domains of MnmE, which may explain why the G-domain alone can be expressed and exhibits a GTPase activity similar to that of the full-length protein (Cabedo et al, 1999). The G-domain contains all canonical sequence elements, GxxxxGKS/T or P-loop (Saraste et al, 1990), T, DxxG, NKxD and SAK (Bourne et al, 1990, 1991) and exhibits a comparatively high hydrolysis rate, although it carries a hydrophobic residue at the position of the catalytic Q61 present in Ras. Previously, Yamanaka et al (2000) have shown that the hydrolysis reaction by MnmE from Thermotoga maritima is stimulated by potassium ions, although the mechanistic basis of this effect is unknown. We thus analysed the role of the G-domain and potassium in the function of MnmE.
Results
Potassium accelerates the GTPase by binding to the G-domain of MnmE
The GTPase reaction rate of full-length MnmE from E. coli (MnmE from now) is strongly accelerated in the presence of potassium ions, whereas sodium ions have no effect (Figure 1A), as shown previously for the T. maritima protein (Yamanaka et al, 2000). To analyse this selectivity for the cation in more detail and to confine the region in MnmE that is responsible for this selectivity, we used a deletion mutant of MnmE lacking the N-terminal dimerisation domain (ΔN-MnmE) and a construct encompassing the isolated G-domain of MnmE.
Figure 1.
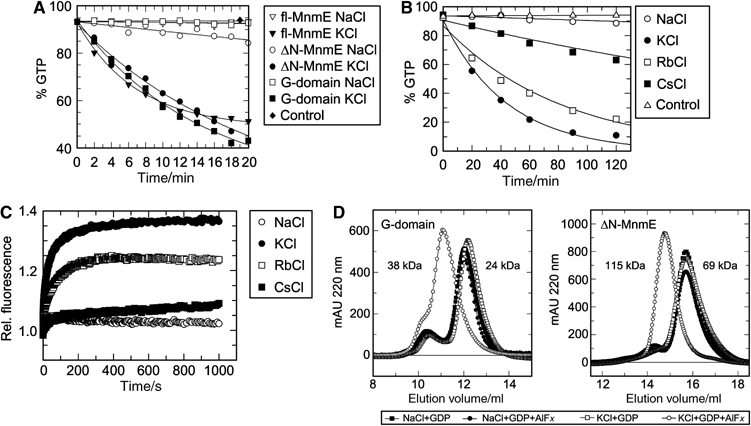
Biochemical characterisation of potassium effects. (A) GTPase activity of full-length (M1-K454), ΔN-MnmE (G102–K454) and the isolated G-domain (G216–G384) determined under multiturnover conditions in the presence of 100 mM NaCl or KCl, 1 μM protein, 200 μM GTP and analysing samples by HPLC. (B) Multiturnover analysis of the hydrolysis activity of the G-domain as in (A), in the presence of the indicated salts. (C) Stopped-flow analysis of mGDP-AlF_x_ binding to full-length MnmE in the presence of different salts. Preformed mGDP–protein complex (10 μM mGDP/60 μM MnmE) was mixed with 5 mM AlF_x_. The mant-group was excited at 360 nm and fluorescence change was monitored through a 408 nm cutoff filter. (D) Gel filtration analysis of the multimerisation behavior of ΔN-MnmE and the G-domain. The protein (270 μM G-domain or 120 μM ΔN-MnmE) was incubated with 200 μM GDP±1 mM AlF_x_ and with 100 mM Na+ or K+ as indicated. Apparent molecular masses corresponding to elution volumes are indicated.
The deletion mutant ΔN-MnmE (residues G102 to K454) elutes in gel filtration experiments as a monomer due to the lack of the N-terminal dimerisation domain, whereas full-length MnmE elutes as a dimer (Scrima et al, 2005). This N-terminally truncated protein retained the fast, potassium ion-dependent reaction (Figure 1A), indicating that the N-terminal dimerisation domain of MnmE is not involved in the selectivity for potassium. Supporting earlier observations, the G-domain alone (residues G216 to G384) can be stably expressed. It also shows a fast, KCl-dependent GTPase reaction (Figure 1A), indicating that the binding site for potassium is located in the G-domain of MnmE.
To narrow down the requirement for GTPase stimulation, we analysed the effect of the alkali metals rubidium and caesium on the hydrolysis activity of the isolated G-domain. While Rb+ has a similar activating effect as K+, Cs+ only showed a slight activation over control or the reaction in the presence of sodium (Figure 1B).
Potassium does not affect nucleotide binding
Considering the strong effect of potassium and as the GTPase reaction has an almost millimolar _K_m for GTP as compared to a micromolar affinity (_K_D), we asked whether potassium modulates the affinity towards guanine nucleotides. We analysed the effect of the cation on nucleotide binding by using fluorescent mant-nucleotides as described previously (Scrima et al, 2005). MnmE binds GDP with affinities of 570 nM in Na+- and 620 nM in K+-containing buffer. Binding affinity towards the GTP-analogue GppNHp is 5.82 and 5.83 μM in Na+ and K+ buffer, respectively. The N-terminally truncated construct and the G-domain bind the di- and triphosphate with an affinity in the range of 1–2 μM. The results of the equilibrium titration are summarised in Table I. Again, no significant difference can be detected in binding affinity in the presence or absence of potassium, independent of the length of the construct.
Table 1.
Affinities of MnmE constructs to mGDP/mGppNHp in presence of Na+ or K+
| | | _K_DNa+/μM | _K_DK+/μM | | | --------- | ---------- | --------- | --------- | | mGDP | | | | | fl-MnmE | M1–K454 | 0.57±0.05 | 0.62±0.05 | | ΔN-MnmE | G102–K454 | 0.83±0.12 | 0.92±0.11 | | G-domain | G216–G384 | 1.06±0.09 | 0.92±0.05 | | | | | | | | mGppNHp | | | | | fl-MnmE | M1–K454 | 5.82±0.79 | 5.83±0.67 | | ΔN-MnmE | G102–K454 | 1.67±0.20 | 1.71±0.21 | | G-domain | G216–G384 | 1.12±0.08 | 1.39±0.10 |
AlFx binding is dependent on potassium ions
Aluminum fluoride binds to the γ-phosphate-binding site of hydrolysis competent ATP- and GTP-hydrolysing proteins (Wittinghofer, 1997). For the small GTP-binding proteins of the Ras superfamily, AlF_x_ binding is only realised in the presence of their respective GAPs. Structural analysis of Ras, RhoA and heterotrimeric Gα-proteins has shown that AlF_x_ binding mimics the transition state of phosphoryl transfer, and that in these cases an ‘arginine finger', supplied in trans by GAP and in cis by Gα-proteins, stabilises the transition state by neutralising the negative charges of the β- and γ-phosphate oxygen groups (Vetter and Wittinghofer, 2001).
To show whether acceleration of MnmE-mediated GTPase is also due to stabilisation of the transition state by potassium, interaction of an MnmE·mGDP complex with aluminium fluoride was monitored by stopped-flow. We observe a 37% fluorescence increase (Figure 1C) in the presence of KCl, but not NaCl (3%). The fluorescence signal reaches a plateau after approximately 500 s leading to the conclusion that a stable complex of MnmE, K+ and mGDP-AlF_x_ is formed. An increase of fluorescence by 24% is detectable in the presence of Rb+ and by 9% with Cs+. These results fairly well match with the hydrolysis data and show that the catalytic activity correlates with the signal intensity detected in the AlF_x_-binding assay.
As the G-domain of MnmE is sufficient for potassium-dependent GTP hydrolysis, we tested both the ΔN-MnmE and the G-domain constructs for AlF_x_ binding in the presence of potassium. Both the truncated construct and the isolated G-domain showed an increase in fluorescence as well (see Figure 5A for ΔN-MnmE and Supplementary Figure 1 for G-domain) in line with the conclusion that the G-domain is required and sufficient for stabilisation of the transition state by potassium.
Figure 5.
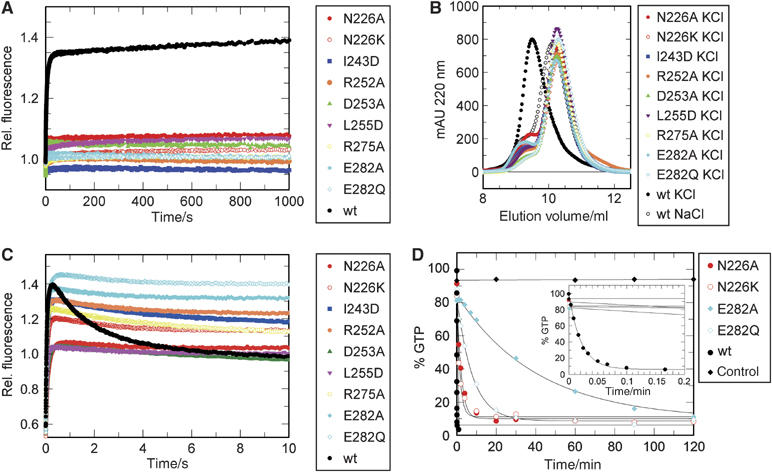
Mutational analysis. (A) mGDP-AlF_x_-induced dimerisation. Preformed complex of wild-type/mutant ΔN-MnmE (60 μM) and mGDP (10 μM) was mixed in a 1:1 ratio with 5 mM AlF_x_ in a stopped-flow apparatus. Mant fluorescence, excited at 360 nm and monitored through a 408 nm cutoff filter, was followed with time. (B) Analysis of dimerisation by gel filtration. ΔN-MnmE (120 μM) was incubated with 1 mM of GDP, 1 mM AlF_x_, and Na+ or K+ as indicated, and eluted from a gel filtration column. (C) Stopped-flow analysis of mGTP binding and hydrolysis. ΔN-MnmE (60 μM) was mixed with 10 μM of mGTP and the mant fluorescence emission was followed in real time. (D) GTPase activity of the wt and mutant ΔN-MnmE determined under single-turnover conditions in the presence of potassium (100 μM ΔN-MnmE+80 μM GTP), using quench-flow for analysis of the wild-type, and HPLC of mutant reactions, as described in Materials and methods.
AlFx induces dimerisation of the G-domain
In the X-ray structure of the MnmE dimer model (Scrima et al, 2005), the nucleotide-binding sites of the G-domains face each other, but are not in close proximity. The magnitude of the fluorescence change of mGDP after AlF_x_ addition suggested a major conformational change such as a close juxtaposition of the G-domains. We thus measured the nucleotide-dependent oligomerisation state of the latter in presence of various ions. As MnmE is stably dimerised due to the N-terminal domain, it shows no significant shift in the elution profile when using GDP or GDP+AlF_x_ in NaCl and KCl-containing buffers (not shown). Furthermore, when using NaCl, ΔN-MnmE elutes as a monomer with an apparent molecular mass of 69 kDa in the presence of GDP or GDP-AlF_x_ (Figure 1D). With KCl, however, ΔN-MnmE forms a dimer (115 kDa apparent molecular mass) in the presence of GDP-AlF_x_ (Figure 1D), but not with GDP alone. This property is retained for the isolated G-domain, demonstrating that the G-domains form a stable, potassium-dependent dimer (38 kDa) in the presence of GDP-AlF_x_, but not GDP (24 kDa). We additionally analysed the dimerisation behaviour for the isolated G-domain in the presence of potassium using the triphosphate analogue GTPγS (see Supplementary Figure 2). These studies suggest that GTP- and potassium-dependent dimerisation is required for efficient GTP hydrolysis. As GTP and GTPγS (rate constant: 0.42/min) are hydrolysed during gel filtration, we used a mutant E282A that shows a strongly reduced hydrolysis activity (see below). As expected, GTP but not GDP induces dimerisation of ΔN-MnmE in the presence of potassium (Supplementary Figure 3). Summarising, GTP as well as the transition state mimic GDP-AlF_x_ induce a potassium-dependent dimerisation via the G-domain of MnmE.
X-ray structure of the G-domain dimer
To get structural insights into the mechanism by which oligomerisation and catalysis of MnmE are mediated by potassium, we crystallised the G-domain of MnmE of E. coli in the presence of potassium, GDP and AlF_x_. As, surprisingly, molecular replacement using the G-domain of the apo-structure of full-length MnmE did not allow phasing, phase determination was performed by single-wavelength anomalous dispersion (SAD) after incorporation of seleno-methionine. A resolution of 1.7 Å obtained with those crystals in combination with the phase information allowed model building (Table II).
Table 2.
Data collection, phasing and refinement statistics
| Data collection | G-domain SeMet 1 | G-domain SeMet 2 | G-domain native rubidium chloride | G-domain native ammonium sulphate |
|---|---|---|---|---|
| X-ray source | ESRF ID14-3 | SLS X10SA | SLS X10SA | SLS X10SA |
| Space group | P2(1) | P2(1) | P2(1) | C222(1) |
| Cell parameters | _a_=57.44 Å | _a_=57.54 Å | _a_=57.24 Å | _a_=71.28 Å |
| _b_=69.88 Å | _b_=70.23 Å | _b_=70.10 Å | _b_=90.65 Å | |
| _c_=91.10 Å | _c_=91.30 Å | _c_=90.21 Å | _c_=138.47 Å | |
| β=95.40° | β=95.50° | β=95.76° | ||
| Resolution (Å) | 20.0–2.1 (2.2–2.1) | 20.0–1.7 (1.8–1.7) | 20.0–2.0 (2.1–2.0) | 20.0–1.85 (1.9–1.85) |
| Wavelength (Å) | 0.931 | 0.934 | 0.815 | 0.950 |
| Completeness (%) | 98.6 (98.2) | 99.6 (99.8) | 98.3 (94.3) | 98.7 (98.0) |
| Unique reflections | 42 090 (5433) | 155 913 (24701) | 92 686 (12120) | 38 157 (2875) |
| Redundancy | 4.0 (3.2) | 2.3 (2.3) | 2.1 (1.7) | 3.7 (3,8) |
| _R_sym (%) | 5.9 (21.7) | 8.2 (33.3) | 7.0 (23.5) | 6.9 (33.3) |
| I/σ_I_ | 20.23 (6.92) | 8.25 (3.47) | 8.43 (3.15) | 14.18 (5.19) |
| Phasing | ||||
| Resolution (Å) | 20.0–3.0 | |||
| No. of sites | 24 | |||
| Anomalous phasing power | 2.4 | |||
| _R_cullis (acen) | 0.63 (0.79) | |||
| Lack of Closure | 0.16 | |||
| Overall FOM | 0.48 | |||
| Overall FOM after SOLOMON | 0.77 | |||
| Refinement | ||||
| PDB code | 2GJ8 | 2GJ9 | 2GJA | |
| _R_worka (%) | 20.6 | 24.9 | 19.6 | |
| _R_freeb (%) | 25.6 | 29.9 | 23.0 | |
| Reflections (work/free) | 72 145/3797 | 45 423/2391 | 36 249/1908 | |
| R.m.s.d./av. (sigma) | ||||
| Bond length (Å) | 0.015/0.022 | 0.019/0.021 | 0.014/0.021 | |
| Bond angle (deg) | 1.579/1.994 | 1.854/1.996 | 1.501/1.989 | |
| Ramachandran plot | ||||
| Core (%) | 91.9 | 91.2 | 94.4 | |
| Additionally (%) | 7.9 | 8.4 | 5.6 | |
| Generously (%) | 0.2 | 0.4 | 0.0 | |
| Values in parentheses are for last resolution shell. | ||||
| a_R_work=∑_h_∣_F_o−_F_c∣/∑_hF_o, where _F_o and _F_c are the observed and calculated structure factor amplitudes of reflection h. | ||||
| b_R_free is the same as _R_work, but calculated on the reflections set aside from refinement. |
The asymmetric unit of the crystals contains four molecules organised as two identical homodimers (Figure 2A) with GDP, aluminium fluoride and potassium in the nucleotide-binding site. G-proteins contain two regions called switch I and II that sense the nucleotide state of the protein by forming two invariant main chain hydrogen bonds to the γ-phosphate. As expected, the switch regions of MnmE, which were highly flexible in the apo-form, are now fixed and restructured (Figure 2B). The protein shows a seven-stranded β-sheet with four α-helices, as compared to the canonical six β-strands and five α-helices in the apo-form. This is due to a remarkable conformational change whereby helix Gα4 is converted into a β-strand (Gβcon). Owing to the rearrangement of switch II, there is a large change in Gβ2/3 and an even larger one in Gα2, which is elongated by two additional turns and tilted by approximately 33 degree compared to the apo-form (Figure 2B). The large conformational changes thus provide an explanation for the inability to solve the structure by molecular replacement.
Figure 2.
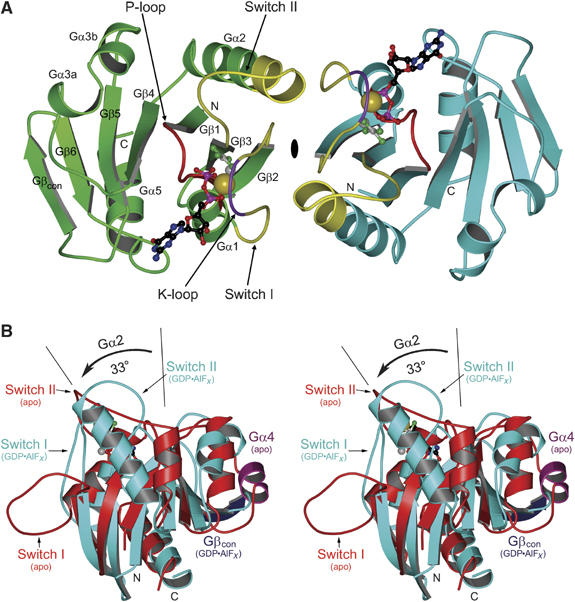
MnmE G-domain dimer structure. (A) Ribbon presentation of the MnmE G-domain dimer (G216–G384), with molecules A and B in green and cyan, respectively, with switch I and II (yellow), P-loop (red) and K-loop (purple) highlighted, and GDP, AlF4−, Mg2+ and K+ as ball-and-stick models. (B) Stereo view of the superimposition of the nucleotide-free and GDP-AlF_x_-bound structures of the G-domain, highlighting potassium-induced conformational changes in helix Gα2, the switches and Gα4 (purple), which is converted into a β-strand Gβcon (blue).
The dimer interface
The buried interface (probe radius 1.5 Å) of 1790 Å2 is composed of three highly conserved patches of polar and hydrophobic contacts (Figure 3A) formed by switch I and II. Residues from the first (E240, A241, I243, T245) and second patch (T250 to R256) from switch I are involved in several main-chain/main-chain and main-chain/side-chain interactions with patch II, and mostly hydrophobic interactions with patch III residues (V281, I284, R288) from switch II in the second molecule. Most of the interactions are mediated by patch II including the 249GTTRD253 motif conserved in all MnmE homologues (Figure 3B). Of these, T251, R252 and D253 have a large number of polar interactions with the symmetry-related patch II. D253 contacts four counter-residues (A241 and I243 directly, R252 and D253 via a bridging water) via its side chain and D253 via the main chain. L255 make symmetric hydrophobic contacts with each other and additionally position the important D253. G249 is not involved in contacting the second molecule, but appears essential for the conformation of the loop as its torsion angles are unfavoured for non-glycine residues. Residues of the third patch (V281, I284) contribute to the interface via hydrophobic interaction and R288 by a main-chain interaction (Figure 3A).
Figure 3.
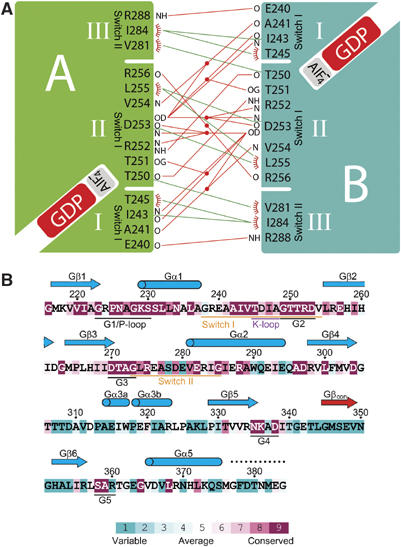
The dimer interface. (A) Schematic representation of the three interaction patches in the dimer interface. Polar interactions are indicated by a red line, hydrophobic interactions by a green line. Water molecules involved in the polar interactions are shown as red dots. The labels N and O indicate main-chain interactions. (B) Secondary structure assignment as deduced from the GDP-AlF_x_ structure and sequence conservation of the MnmE G-domain, with degree of conservation as indicated by the coloured bar. Sequence conservation has been calculated with ConSeq (Berezin et al, 2004) based on 50 sequences. The flexible region at the C-terminus with weak electron density is depicted as a dashed line.
Nucleotide binding and active site
The active site contains GDP and aluminium fluoride, which from the electron density we identify as AlF4−. The nucleotide is bound in a way typical for G-proteins (summarised in Figure 4A). The base is contacted by the NKxD motif of which N335 contacts the N7 position of the base, the specificity residue D338 forms a double hydrogen bond with the N1 hydrogen and 2-amino-group of the guanine base. The base is further stabilised by K336 on one and R360 of the SAR motif on the other side. S358 contacts the 6-oxygen of the base and additionally positions the D338 side chain. The hydrogen bond between the main chain NH of A359 and the O6-oxygen is the second major specificity determinant that excludes adenine binding.
Figure 4.
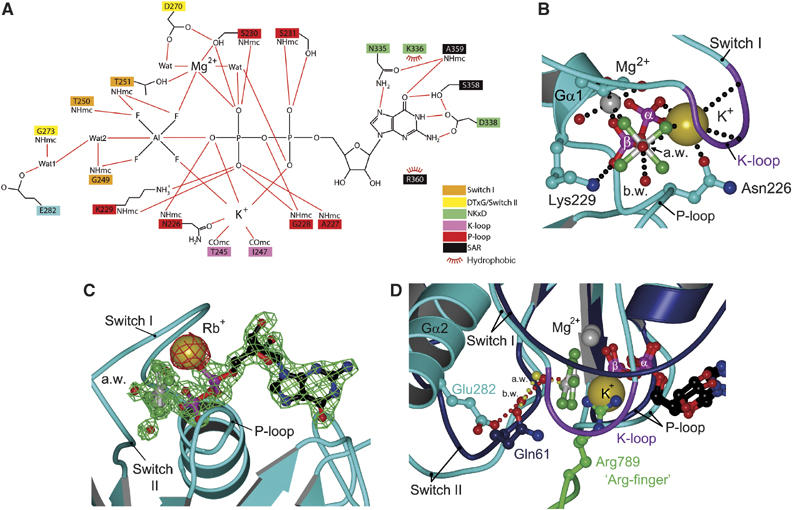
Nucleotide binding and catalysis. (A) Schematic representation of the binding site of GDP-AlF4− and potassium. (B) The catalytic centre as viewed from the in-line attacking water towards AlF4−. The K-loop together with the phosphates, the AlF4− and Asn226 coordinates the potassium ion and shields it from the surrounding solvent. The three positive charges of the P-loop Lys229, Mg2+ and K+ form a triangle around what represents the β–γ-bridging oxygen of GTP. (C) Structural analysis with RbCl. The anomalous density (contoured at 2σ) of the Rb+ position is shown in red, the 2_F_O−F_C map surrounding the GDP-AlF_x and Mg2+ (contoured at 2σ) in green. (D) Superimposition of the Ras-RasGAP (Ras: blue; RasGAP: green) structure and the GDP-AlF4−-bound G-domain (cyan). Red and yellow water molecules belong to MnmE and Ras-RasGAP, respectively. Note the close juxtaposition of the Gln61 side-chain oxygen and the bridging water (b.w.) from MnmE, and the superimposition of the guanidinium group from the Arg-finger (Arg789) of RasGAP and the potassium ion from MnmE.
Mg2+ is hexa-coordinated by the β-phosphate oxygen and a fluoride, the side-chain OH of S230 (P-loop) and T251 (switch I) and two water molecules, one of which is coordinated by D270 from the DTxG motif. Residues from the P-loop contact the phosphate oxygens by main- and side-chain interactions in the conventional way. Fluorides from AlF4− are bound by main chain hydrogen bonds from G249, T250 and T251 from the 249GTTRD253 motif.
As in most other triphosphate structures or AlF_x_ mimics thereof, there is a water molecule in a position suitable for in-line attack on the γ-phosphate. It is stabilised by a main-chain interaction of G249 from the P-loop. Unlike in Ras proteins (except Rap), where the attacking water is stabilised by a Gln, in MnmE this water is contacted by another water molecule, which in turn is hydrogen bonded by G273 from the DTxG motif and E282. The latter residue is totally conserved and could potentially activate the nucleophilic water in a way similar to the mechanism suggested for F1-ATPase (see discussion).
Potassium as a GTPase-activating element
There is an additional strong electron density peak located between the β-phosphate and the AlF4−, which we assign to a potassium ion (see below). It is coordinated to the main-chain CO of T245 and I247 that are located in switch I and precede the 249GTTRD253 motif. We named this region (245–248) of switch I the K-loop. It contacts potassium and forms a cap that shields the ion from solvent. The hexagonal coordination of potassium is complemented by the side-chain CO of Asn226 (P-loop), the α- and β-phosphate oxygens of GDP, where the latter represents the former β−γ-bridging oxygen of GTP and a fluoride ion (Figure 4A and B). The coordination bond lengths of the potassium ion to MnmE and the phosphates of GDP are between 2.67 and 2.99 Å, typical for the coordination of potassium (Supplementary Figure 4). The coordination to AlF4− is weaker with a distance of 3.34 Å. To further confirm the position of the potassium ion, we crystallised the G-domain by replacing KCl with RbCl, which has similar biochemical effects. The incorporation of Rb+ allowed collection of an anomalous data set owing to the anomalous scattering behaviour of Rb+. The anomalous density of the cation is shown in Figure 4C, contoured at 2σ, with a 2_F_O−F_C map surrounding the GDP-AlF_x and Mg2+. As expected, the anomalous signal overlaps the position assigned to the potassium-binding site.
The active site structure of MnmE in the transition state provides an explanation for the potassium-induced increase of activity. By adding another positive charge to the active site, the β-phosphate is surrounded by a total of three positively charged moieties: Lys229 from the P-loop, the bivalent magnesium and the monovalent potassium ion (Figure 4B). When viewed along the axis from the attacking water to the AlF4−, the positive charges form a planar triangle around what represents the former β−γ-bridging oxygen of GTP. Mg2+ and Lys neutralise the negative charges of phosphates in the ground state, whereas potassium, which is not required for binding of GTP, additionally reduces the developing negative charge in a presumed associative transition state. The GTPase-activating function of potassium is further supported by the superimposition with the Ras-RasGAP-structure (1WQ1; Scheffzek et al, 1997; Figure 4D), where the potassium ion is located at the position of the ‘arginine finger' (Arg789) guanidinium group in RasGAP, shown to be responsible for a 2000-fold acceleration of the Ras GTPase.
Potassium binding also stabilises the dimer of the G-domains by inducing a number of conformational changes. The most relevant is the rearrangement of helix Gα2, the first turn of which contains the conserved residue Glu282 implicated in catalysis by a proton relay system. In the superimposition (Figure 4D), the side-chain oxygen of Gln61 in Ras is very close to the bridging water in MnmE (1.3 Å). Thus, the function of Gln61 to orient the nucleophilic water is taken over by Glu282 and the bridging water in MnmE. As GAPs like RasGAP and RhoGAP also act by repositioning and/or stabilising the crucial catalytic glutamine in Ras/Rho, this argues for potassium being the major GTPase-activating element for MnmE, although the details of mechanism are profoundly different.
Dissecting the dimerisation and GTPase reactions
In order to get more insight into the mechanism and to see whether it is possible to uncouple dimer formation and GTPase activation, we used the X-ray structure to design two types of mutants: those aiming to disrupt the dimerisation, and those that should not interfere with the dimerisation but should affect the GTPase activity. The first class of mutants concerns mutations of residues forming the dimer interface such as I243, D253, L255, the second E282, which is assumed to be involved in catalysis and seems to have no function in dimer formation. As N226 in the P-loop contacts potassium, its mutation should disrupt ion binding and thus affect both dimerisation and catalytic activity. We obtained crystals of a different space group in (NH4)2SO4 that contained an ammonium ion instead of a potassium ion in the active site, but showed the same dimeric structure as the potassium ion bound form. As ammonium shows comparable GTP hydrolysis and mGDP-AlF_x_-binding activity (results not shown), we reasoned that an N226K mutant might position its N-ɛ ammonium group into the active site and show a potassium-independent fast GTPase activity.
To rule out any appreciable effect of the mutations on nucleotide binding, we determined the affinities to mGDP and mGppNHp for the mutants, in the background of ΔN-MnmE (Table III). The affinity to mGDP was only reduced maximally approximately five-fold in the case of the N226A mutant. In general, the affinity was in the range of 0.38–2.59 μM compared to 0.92 μM for wt. For mGppNHp, the maximum effect due to the mutations was not larger than six-fold, except for L255D, which showed a 15-fold decrease in affinity.
Table 3.
Affinities of wt and mutant ΔN-MnmE to mGDP/mGppNHp
| | mGDP | mGppNHp | | | ---------------------------------------------- | --------- | ---------- | | | _K_D/μM | KD/μM | | | wta | 0.92±0.11 | 1.71±0.21 | | N226A | 4.28±0.57 | 3.91±0.42 | | N226K | 2.08±0.21 | 0.91±0.07 | | I243D | 0.81±0.08 | 10.23±0.97 | | D253A | 0.44±0.04 | 2.78±0.21 | | R252A | 0.79±0.06 | 7.31±0.54 | | L255D | 1.38±0.33 | 26.00±3.74 | | R275A | 0.38±0.05 | 5.35±0.39 | | E282A | 1.17±0.23 | 2.77±0.29 | | E282Q | 2.59±0.33 | 9.85±1.61 | | aValues for wt were taken from Table I. | | |
We then analysed the effect of mutations on AlF_x_-induced dimerisation of the G-domains by the mGDP-AlF_x_ assay, under saturating conditions. As shown in Figure 5A, none of the mutants was able to induce dimerisation in the presence of mGDP, AlFx and potassium, as evident from the absence of fluorescence increase. These results are confirmed by analytical gel filtration (Figure 5B), where none of the mutants elutes as a dimer in the presence of potassium and GDP-AlF_x_. The conclusion would therefore be that the mutants either cannot dimerise or that they dimerise but can no longer stabilise the transition state.
To find out, GTP hydrolysis was monitored by fluorescence using mGTP and stopped-flow kinetics (Figure 5C). Rapid mixing with mGTP leads to an initial increase in fluorescence due to binding of the labelled nucleotide to the protein, with a signal increase from approximately 0.6 to 1.0 relative fluorescence. For wt ΔN-MnmE (black), besides the signal due to nucleotide binding we monitor an additional fluorescence increase correlated with dimerisation and an amplitude change from 1.0 to 1.4 (compare Figure 5A and C), and a slower decay after hydrolysis due to dissociation of the G-domains. The N226A mutant, where potassium binding should be compromised, shows no additional fluorescence increase beyond that for nucleotide binding, whereas the E282A and E282Q mutants (cyan) show both fast fluorescence changes, confirming that dimerisation is not influenced, but only a very slow decay. The interface mutants (and N226K) show behaviours between that of wild-type and the E282 mutants. The studies show that dimerisation precedes catalysis and that dimerisation and hydrolysis can be uncoupled by appropriate mutations.
To verify that the fluorescence decay really monitors the chemical step, we analysed the single turnover GTPase reaction directly by monitoring the decay of GTP and/or the increase of GDP. For wild-type protein, the reaction is too fast to be measured by HPLC and was analysed by quench-flow with radioactive GTP. The reaction is complete after 6–8 s (rate constant 47.4/min), well in line with the fluorescence data (compare Figure 5C and D). The E282A and Q mutants were analysed with HPLC and show a 1925- and 371-fold reduction in _k_cat, respectively, whereas the other mutants show an intermediate phenotype (Table IV). As neither potassium binding nor dimerisation in the presence of GTP is influenced by these mutations, whereas AlF_x_-mediated transition state formation is (Figure 5A and B), we conclude that E282 has an important purely catalytic function, such as that of a general base that acts to activate or stabilise the nucleophilic water via an intermediate water molecule. According to this, mutation of E282 to Q should be less drastic than the alanine mutation as it should still be able to stabilise the hydrogen-bonding network. In the GAP catalysed reaction of Ras and Rho, a catalytic glutamine stabilises the position of the attacking water directly, and its mutation drastically reduces the reaction rate.
Table 4.
GTPase activities of wt and mutant ΔN-MnmE
| | _k_catKCl/min | Fold reduction compared to wt | | | ---------------- | ----------------------------- | ----- | | wt | 47.379±1.757 | | | E282Q | 0.128±0.011 | x371 | | E282A | 0.025±0.001 | x1925 | | N226A | 0.484±0.038 | x98 | | N226K | 0.658±0.081 | x72 |
Mutation of N226 is expected to interfere with potassium binding. Correspondingly, N226A shows no fluorescence increase and a reduced hydrolysis rate of 0.484/min, corresponding to a 98-fold reduction compared to wt. Contrary to expectation, the N226K mutant shows a strong decrease in activity comparable to the N226A mutation (72-fold reduction). Mutations in the dimer interface (I243D, D253A, L255D) do inhibit the dimerisation in presence of GDP-AlF_x_ and potassium (Figure 5A and B) and also show a reduced dimerisation in presence of GTP, as deduced from the lower amplitude of fluorescence increase as compared to wt (Figure 5C). As described by Martinez-Vicente et al (2005), the two arginine residues R252 and R275 are not directly involved in catalysis, which is supported by our data. In the structure, R252 is located in the dimer interface, responsible for part of the binding specificity, whereas R275 stabilises the position of the catalytic E282 by polar interactions. These mutants are unable to stabilise the GDP-AlF_x_ transition state and show a lower amplitude and slower decay of the fluorescence signal in the stopped-flow GTPase reaction.
Discussion
Phosphoryl transfer reactions are most frequently used in biology. This is due to the fact that phosphate monoesters or phospho-anhydrides are kinetically stable due to high-energy barriers, which require a delicate enzymatic machinery to achieve fast reaction rates. GTP-binding proteins have been shown to adopt a variety of mechanisms to accelerate cleavage of GTP, which is intrinsically very slow and is stimulated in a variety of ways. Here, we show that a potassium ion is a central element required for a stable dimerisation of the G-domains of MnmE and its stimulated hydrolysis. Dimerisation stabilises the switch region and reorients the catalytic Glu282, which in turn positions and/or stabilises the attacking water. As mutation of Glu282 to alanine reduces the hydrolysis rate by 2000-fold as compared to the 370-fold reduction of E282Q, we conclude that Glu282 may act as general base or to just stabilise the attacking water. A similar mechanism of water-mediated proton transfer to an acidic side chain has been proposed for the F1-ATPase using computational simulation (Dittrich et al, 2004). In that scenario, the proton is eventually transferred to the γ-phosphate similar to what has also been discussed as substrate-assisted catalysis for Ras and Rab (Wittinghofer, 2006). General base catalysed proton transfer is also proposed for other phosphoryl transfer reactions such as those of protein kinases, where the mutation of an invariant Asp completely inactivates the enzyme (Gibbs and Zoller, 1991; Johnson et al, 1996).
In previously analysed GTPase reactions, there are two positive charges that are absolutely required for the reaction, the Mg2+ ion forming a bi-dentate coordination with the β-γ-phosphate oxygens and the totally conserved lysine residue from the P-loop. The GTPase-activating factor for the MnmE-mediated GTP hydrolysis is a suitably positioned potassium ion, which is hexa-coordinated by the nucleotide and residues from the P-loop and K-loop. Our experiments show that the binding site is suitable for monovalent cations with an ionic radius in the range of 138 to 152 pm (K+: 138 pm; NH4+: 144 pm; Rb+: 152 pm). Cations smaller or larger in size (Na+: 99 pm; Cs+: 169 pm) do either not bind or do not have the effects described for potassium.
The potassium as a GTPase-activating element has so far never been described for any GTP-binding protein. It has however been observed in the structures of the ATPase domain of the Hsc70 and GroEL chaperones. For many small molecule kinases, an influence of monovalent cations on the enzymatic activity was shown biochemically (reviewed in Suelter, 1970). The ATPase of Hsc70 is stimulated 5–10-fold by potassium and the structure of the N-terminal ATPase-fragment of Hsc70 (Wilbanks and McKay, 1995; Sousa and McKay, 1998) shows two potassium-binding sites close to the phosphates. One of these is coordinated by the β-phosphate, whereas the second is located close to the free-phosphate/γ-phosphate. Interestingly, the site 1 potassium in Hsc70 replaces a conserved lysine residue in actin that contacts the α-β-phosphates comparable to the P-loop lysine in GNBPs that coordinates the β-γ-phosphates. The closest structural match to the potassium of MnmE is found in the ADP-AlF_x_ structure of GroE (1PCQ, Chaudhry et al, 2003; 1KP8, Wang and Boisvert, 2003). In GroE, the monovalent cation induces a 104-fold increase in ATP hydrolysis.
The GTP-binding proteins Era and EngA that belong to the same protein family as MnmE (Caldon et al, 2001) show a high degree of similarity in the G1 and G2 motif. To Era, a function as cell-cycle regulator and translation factor has been assigned and the protein EngA, which contains two G-domains in a tandem organisation, is essential for growth of Neisseria gonorrhoeae. Both proteins contain the conserved G1 motif GRPNxGKS and the GTTRD motif (Era: QTTRH, EngA1: GLTRD, EngA2: GTTRD; Caldon et al, 2001), suggesting that they also show potassium-dependent GTPase activation.
Recently Martinez-Vicente et al (2005) have extensively analysed the MnmE-specific motif 249GTTRD253 in switch I. Alanine mutation of the 249GTTRD253 affected the GTPase, AlF_x_-induced complex formation and tRNA modification activity of MnmE, although dimerisation of the G-domains was not described. Considering the central role of the 249GTTRD253 motif for the dimerisation interface as deduced from the structure (Figure 3A), it becomes obvious that mutations of these and neighbouring residues have an indirect effect on the GTPase activity (Figure 5). Structural analysis also suggests why the G249A (20-fold) and T251A (92-fold) mutations have the most drastic effect on GTPase activity. Gly249 assumes the role of the canonical Gly residue from the DxxG motif. It is bound to the γ-phosphate (here AlF4−) via a main-chain H-bond and is additionally involved in orienting the attacking water (Wat2). Gly273 from the switch II DxxG motif in turn orients the bridging water molecule (Wat1, Figure 4A). As proposed by Martinez-Vicente et al, Thr251 (and not Thr250) does indeed correspond to the canonical threonine from switch I and contacts Mg2+ and AlF4− (corresponding to the γ-phosphate).
We have shown here that potassium induces GTP hydrolysis not only in the G-domain but also in the full-length protein. This would require the G-domains of native MnmE to come closer to each other than observed in the structural model of nucleotide-free full-length MnmE (Scrima et al, 2005; 1XZP/1XZQ), with a distance of 54 Å between glycines (G223) in the P-loop, which is reduced to 32 Å in the GDP-AlF_x_ structure (Figure 6B). In order for the G-domains to move into place, one would have to propose a major conformational change. This could be mediated by rigid body movements of the domains relative to each other using the linker between the domains as hinges (Figure 6A, black arrows). Structural independence of the domains is supported by the fact that the ΔN-MnmE and the G-domain can be isolated and retain the enzymatic properties of the full-length protein. Proteolytic digestion of MnmE (Yim et al, 2003) results in fragments of approximately 39 and 36 kDa that could correspond to MnmE molecules lacking most (residues 1–102) or all (1–118) of the N-terminal fragment, and has also been found in proteolysed crystals of MnmE (Scrima et al, 2005). Proteolysis in the presence of GTP leads to additional proteolytic fragments, supporting a nucleotide-dependent conformational reorientation between the domains.
Figure 6.
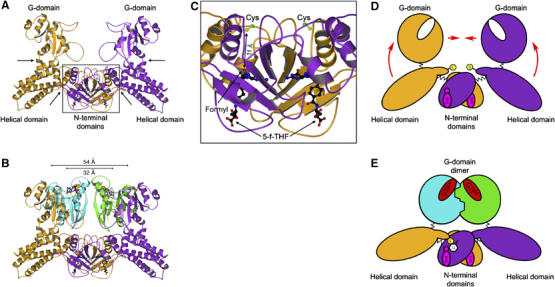
Conformational changes in MnmE upon juxtaposition of G-domains. (A) The homodimer model of full-length MnmE (orange/purple) in the apo-form. The catalytic Cys and 5-formyl-THF are shown as ball-and-stick model. Putative hinge regions for the conformational change induced by the G-domain dimerisation are marked by arrows. The box depicts the region shown in (C). (B) Model of the G-domain dimer (green/cyan) in the context of full-length MnmE, showing that the G-domains have to move by 22 Å, from 54 to 32 Å, as measured for the Cα-positions of G223. (C) Detailed view onto the putative tRNA modification centre harbouring the catalytic Cys and the C1-group donor 5-formyl-THF, which are 11 Å apart in the full-length MnmE structure (1XZQ). (D, E) Proposed model for the activation of MnmE. The potassium-dependent dimerisation of the G-domains during GTP hydrolysis (represented by GTP* in E) could lead to an upward movement of the helical domains (D, red arrows), thereby bringing the C-terminal catalytic cysteine (represented as a thiolate-anion S− in yellow) and the C1-group donor 5-formyl-THF (magenta) in juxtaposition allowing the activation of the uridine base (U) and the formyl transfer.
We have previously shown that the putative active site cysteine of MnmE and the C1 donor group of methylene tetrahydrofolate (5-formyl-THF) are 11 Å apart in the structure of THF-bound nucleotide-free MnmE (Figure 6C) and could therefore not be simultaneously bound to the same uridine base as required for the proposed tRNA modification scheme (Scrima et al, 2005). The conformational change induced by the dimerisation could close the active site, bringing Cys and the formyl group in juxtaposition to allow the modification reaction to take place (Figure 6C–E). Further structural and biochemical studies on the complete modification reaction with full-length protein, tRNA and MnmG/GidA are required to support such a model.
Materials and methods
Plasmids and proteins
MnmE E.coli was expressed from plasmid pIC933, a kind gift of ME Armengod. The expression constructs for ΔN-MnmE (G102–K454) and the G-domain (G216–G384) were made by PCR using pIC933 as template and cloned into a pET14b plasmid. Mutagenesis was performed using the Quik-Change method by Stratagene or by the method of overlapping PCR. Overexpression and purification of MnmE full-length, His6-tagged ΔN-MnmE and the mutants have been performed as described in Scrima et al (2005), the G-domain was prepared as described for His6-tagged ΔN-MnmE.
Crystallography
Crystals of G-domain dimer were obtained using the hanging drop/vapour diffusion method. The protein was incubated in 50 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2, 5 mM DTE and additionally 1 mM of GDP, 1 mM AlCl3 and 10 mM of NaF. Drops (1 μl) of 10 mg/ml MnmE solution were mixed with 1 μl of reservoir solution (16–19% PEG 3350, 100 mM MES pH 6.0, 200 mM MgCl2 at 20°C). Crystals with ammonium sulphate at 20°C had 1.5 M ammonium sulphate and 100 mM HEPES pH 7.0 in the reservoir. Crystals obtained from the PEG condition were cryoprotected in reservoir solution with 30% PEG 3350. Cryoprotection for ammonium sulphate crystals was obtained with 25% glycerol. For the Rb+-bound structure, buffers contained RbCl instead of KCl.
Collected data were processed with XDS (Kabsch, 1993). Initial heavy atom sites for SAD phasing were identified with SHELXD (Usón and Sheldrick, 1999). The heavy atom sites were completed using CNS (Brunger et al, 1998). Refinement of the initial sites, phase determination and density modification with SHARP (de La Fortelle and Bricogne, 1997) led to an interpretable density map. The atomic model was built using XtalView/Xfit (McRee, 1999); refinement was carried out with REFMAC5 (Murshudov et al, 1997). Figures were generated using MolScript (Kraulis, 1991) and POV-Ray (www.povray.org).
Preparation of nucleotide-free protein
For the removal of nucleotide from His6-tagged MnmE, 20 mg of protein were incubated at 4°C in 50 mM Tris/HCl pH 7.5, 100 mM NaCl and 5 mM DTE with 40 U of calf intestine alkaline phosphatase (Roche Diagnostics). Nucleotide degradation to GMP or Guanosine was monitored by HPLC, after which the protein was applied to an Ni-NTA column and washed with 50 mM Tris pH 7.5, 100 mM NaCl, 20 mM Imidazol and 3 mM β-ME to remove the GMP/Guanosine and alkaline phosphatase. The protein was eluted with 20 ml 50 mM Tris pH 7.5, 100 mM NaCl, 250 mM Imidazol and 3 mM β-ME, concentrated and buffer was exchanged to 50 mM Tris pH 7.5, 5 mM MgCl2, 100 mM NaCl and 5 mM DTE. Aliquots of the protein were flash-frozen in liquid nitrogen and stored at −80°C.
Fluorimetry/equilibrium titration
Determination of the equilibrium dissociation constant _K_d was performed as described in Scrima et al (2005). Experiments were carried out at 20°C in 50 mM Tris pH 7.5, 5 mM MgCl2, 5 mM DTE and 100 mM NaCl or KCl.
Stopped-flow kinetics
In stopped-flow experiments to analyse AlF_x_-binding, 60 μM protein was preincubated with 10 μM of mGDP. This preformed protein–mant–nucleotide complex was mixed in the stopped-flow apparatus in a 1:1 ratio with 5 mM AlF_x_ (5 mM AlCl3 plus 50 mM NaF). Hydrolysis of mGTP was analysed by mixing 10 μM of mGTP with 60 μM of protein. The mant-fluorophor was excited at 360 nm and change in fluorescence was monitored through a 408 nm cutoff filter (SM-17; Applied Photophysics). Experiments were carried out at 20°C in 50 mM Tris pH 7.5, 5 mM MgCl2, 5 mM DTE and 100 mM of the respective alkali salt.
Analytical gel filtration
All analytical gel filtration experiments were performed in 50 mM Tris pH 7.5, 5 mM MgCl2, 5 mM DTE and 100 mM NaCl or KCl, and, as indicated, the respective nucleotide and 1 mM of AlF_x_ (1 mM AlCl3 plus 10 mM NaF). In all, 1 mg of protein (corresponding to 270 μM of the G-domain or 120 μM of ΔN-MnmE) was incubated for 15 min on ice with nucleotide as stated in the figure legend (and 1 mM AlF_x_) before application onto the Superdex 75 10/30 or 200 10/30 columns.
Quench-flow experiment with _γ_32P-labelled GTP
For measuring the single-turnover GTPase of wild-type ΔN-MnmE, 200 μM of protein was mixed in a quench-flow apparatus with 160 μM of GTP containing 20 nCu of γ32P-labelled GTP (Amersham Biosciences) per μl of nucleotide solution. The experiment was carried out at 20°C (50 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2, 5 mM DTE), stopped at various time points with 1 M perchloric acid and neutralised with 8 M potassium-acetate. Precipitated protein and salt was removed by centrifugation and a volume of 0.6 μl of the supernatant was applied to a silica thin-layer-chromatography plate. The mobile phase was 0.65 M KH2PO4 pH 6.5. Free 32P-phosphate and uncleaved γ32P-labelled GTP were monitored using a FLA-3000 PhosphoImager-System (Fuji, Kanasawa, Japan). Determination of spot intensity was performed with the Software Aida (Raytest GmbH). After background correction, percentage of uncleaved GTP was calculated using the intensity of γ32P-GTP divided by the intensity of γ32P-GTP plus the intensity of free 32P-phosphate (corresponding to GDP).
HPLC analysis of GTPase and nucleotide content
GTP hydrolysis of mutants was analysed by HPLC (Beckman-Coulter, System Gold). Nucleotides were separated on a hydrophobic C18-column (Beckman-Coulter) with 100 mM potassium-phosphate pH 6.5, 10 mM tert-butyl-ammonium-bromide and 7.5% acetonitril as polar, mobile phase. Reaction conditions for the single-turnover conditions were analogous to the quench-flow experiment. In total, 100 μM of protein was incubated with 80 μM of nucleotide in 50 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2, 5 mM DTE at 20°C. At various time points, 30 μl aliquots were flash-frozen in liquid nitrogen to stop the reaction. Aliquots were incubated at 95°C for 2 min, denatured protein was removed by centrifugation (1 min, 13000 r.p.m.) and the supernatant applied to the HPLC. For multi-turnover analysis, 1 μM of protein was incubated with 200 μM GTP in 50 mM Tris pH 7.5, 5 mM MgCl2, 5 mM DTE at 20°C containing 100 mM of the respective salt. Further treatment of the samples was analogous to the single-turnover experiment.
Coordinates
Atomic coordinates and structure factors have been deposited at the Protein Data Bank.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
This work was performed at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland. We also gratefully acknowledge the use of beamline ID14-3 at the ESRF Grenoble. We thank the beamline staff for professional support. We thank Ilme Schlichting, Eckhard Hofmann, Michael Weyand, Olena Pylypenko and Karin Kühnel for data collection; Michael Weyand and Ingrid R Vetter for crystallographic assistance and Simone Kunzelmann for assistance with quench-flow. We thank Dorothee Kühlmann for technical assistance.
References
- Berezin C, Glaser F, Rosenberg Y, Paz I, Pupko T, Fariselli P, Casadio R, Ben-Tal N (2004) ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics 20: 1322–1324 [DOI] [PubMed] [Google Scholar]
- Bjork GR, Durand JM, Hagervall TG, Leipuviene R, Lundgren HK, Nilsson K, Chen P, Qian Q, Urbonavicius J (1999) Transfer RNA modification: influence on translational frameshifting and metabolism. FEBS Lett 452: 47–51 [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F (1990) The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348: 125–132 [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F (1991) The GTPase superfamily: conserved structure and molecular mechanism. Nature 349: 117–127 [DOI] [PubMed] [Google Scholar]
- Brégeon D, Colot V, Radman M, Taddei F (2001) Translational misreading: a tRNA modification counteracts a +2 ribosomal frameshift. Genes Dev 15: 2295–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I, Meredith MR, Bloys AJ, Hagervall TG (1997) Expression of a coronavirus ribosomal frameshift signal in Escherichia coli: influence of tRNA anticodon modification on frameshifting. J Mol Biol 270: 360–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Cabedo H, Macian F, Villarroya M, Escudero JC, Martinez-Vicente M, Knecht E, Armengod ME (1999) The Escherichia coli trmE (mnmE) gene, involved in tRNA modification, codes for an evolutionarily conserved GTPase with unusual biochemical properties. EMBO J 18: 7063–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldon CE, Yoong P, March PE (2001) Evolution of a molecular switch: universal bacterial GTPases regulate ribosome function. Mol Microbiol 41: 289–297 [DOI] [PubMed] [Google Scholar]
- Chaudhry C, Farr GW, Todd MJ, Rye HS, Brunger AT, Adams PD, Horwich AL, Sigler PB (2003) Role of the gamma-phosphate of ATP in triggering protein folding by GroEL-GroES: function, structure and energetics. EMBO J 22: 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby G, Wu M, Tzagoloff A (1998) MTO1 codes for a mitochondrial protein required for respiration in paromomycin-resistant mutants of Saccharomyces cerevisiae. J Biol Chem 273: 27945–27952 [DOI] [PubMed] [Google Scholar]
- Daumke O, Weyand M, Chakrabarti PP, Vetter IR, Wittinghofer A (2004) The GTPase-activating protein Rap1GAP uses a catalytic asparagine. Nature 429: 197–201 [DOI] [PubMed] [Google Scholar]
- Decoster E, Vassal A, Faye G (1993) MSS1, a nuclear-encoded mitochondrial GTPase involved in the expression of COX1 subunit of cytochrome c oxidase. J Mol Biol 232: 79–88 [DOI] [PubMed] [Google Scholar]
- de La Fortelle E, Bricogne G (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol 276: 472–494 [DOI] [PubMed] [Google Scholar]
- Dittrich M, Hayashi S, Schulten K (2004) ATP hydrolysis in the betaTP and betaDP catalytic sites of F1-ATPase. Biophys J 87: 2954–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea PF, Shan SO, Napetschnig J, Savage DF, Walter P, Stroud RM (2004) Substrate twinning activates the signal recognition particle and its receptor. Nature 427: 215–221 [DOI] [PubMed] [Google Scholar]
- Elseviers D, Petrullo LA, Gallagher P (1984) Novel E. coli mutants deficient in biosynthesis of 5-methylaminomethyl-2-thiouridine. Nucleic Acids Res 12: 3521–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focia PJ, Shepotinovskaya IV, Seidler JA, Freymann DM (2004) Heterodimeric GTPase core of the SRP targeting complex. Science 303: 373–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Praefcke GJ, Renault L, Wittinghofer A, Herrmann C (2006) How guanylate-binding proteins achieve assembly-stimulated processive cleavage of GTP to GMP. Nature 440: 101–104 [DOI] [PubMed] [Google Scholar]
- Gibbs CS, Zoller MJ (1991) Rational scanning mutagenesis of a protein kinase identifies functional regions involved in catalysis and substrate interactions. J Biol Chem 266: 8923–8931 [PubMed] [Google Scholar]
- Hagervall TG, Pomerantz SC, McCloskey JA (1998) Reduced misreading of asparagine codons by Escherichia coli tRNALys with hypomodified derivatives of 5-methylaminomethyl-2-thiouridine in the wobble position. J Mol Biol 284: 33–42 [DOI] [PubMed] [Google Scholar]
- Johnson LN, Noble ME, Owen DJ (1996) Active and inactive protein kinases: structural basis for regulation. Cell 85: 149–158 [DOI] [PubMed] [Google Scholar]
- Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26: 795–800 [Google Scholar]
- Kraulis PJ (1991) MolScript—a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Li X, Guan MX (2002) A human mitochondrial GTP binding protein related to tRNA modification may modulate phenotypic expression of the deafness-associated mitochondrial 12S rRNA mutation. Mol Cell Biol 22: 7701–7711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Li R, Lin X, Guan MX (2002) Isolation and characterization of the putative nuclear modifier gene MTO1 involved in the pathogenesis of deafness-associated mitochondrial 12 S rRNA A1555G mutation. J Biol Chem 277: 27256–27264 [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Yim L, Villarroya M, Mellado M, Perez-Paya E, Bjork GR, Armengod ME (2005) Effects of mutagenesis in the switch I region and conserved arginines of Escherichia coli MnmE protein, A GTPase involved in tRNA modification. J Biol Chem 280: 30660–30670 [DOI] [PubMed] [Google Scholar]
- McRee DE (1999) XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J Struct Biol 125: 156–165 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Praefcke GJ, McMahon HT (2004) The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol 5: 133–147 [DOI] [PubMed] [Google Scholar]
- Rehmann H, Bos JL (2004) Signal transduction: thumbs up for inactivation. Nature 429: 138–139 [DOI] [PubMed] [Google Scholar]
- Saraste M, Sibbald PR, Wittinghofer A (1990) The P-Loop—a common motif in ATP- and GTP-binding proteins. TIBS 15: 430–434 [DOI] [PubMed] [Google Scholar]
- Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F, Wittinghofer A (1997) The Ras–RasGAP complex: structural basis for GTPase activation and its loss in oncogenic ras mutants. Science 277: 333–338 [DOI] [PubMed] [Google Scholar]
- Scrima A, Vetter IR, Armengod ME, Wittinghofer A (2005) The structure of the TrmE GTP-binding protein and its implications for tRNA modification. EMBO J 24: 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seewald MJ, Körner C, Wittinghofer A, Vetter IR (2002) RanGAP mediates GTP hydrolysis without an arginine finger. Nature 415: 662–666 [DOI] [PubMed] [Google Scholar]
- Sousa MC, McKay DB (1998) The hydroxyl of threonine 13 of the bovine 70-kDa heat shock cognate protein is essential for transducing the ATP-induced conformational change. Biochemistry 37: 15392–15399 [DOI] [PubMed] [Google Scholar]
- Suelter CH (1970) Enzymes activated by monovalent cations. Science 168: 789–795 [DOI] [PubMed] [Google Scholar]
- Sun Y-J, Forouhar F, Li H-M, Tu S-L, Kao S, Shr H-L, Chou C-C, Hsiao C-D (2002) Crystal structure of pea Toc34—a novel GTPase of the chloroplast protein translocon. Nat Struct Biol 9: 95–100 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Suzuki T, Wada T, Saigo K, Watanabe K (2001) Novel taurine-containing uridine derivatives and mitochondrial human diseases. Nucleic Acids Res Suppl 1: 257–258 [DOI] [PubMed] [Google Scholar]
- Urbonavicius J, Qian Q, Durand JMB, Hagervall TG, Björk GR (2001) Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J 20: 4863–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usón I, Sheldrick GM (1999) Advances in direct methods for protein crystallography. Curr Opin Struct Biol 9: 643–648 [DOI] [PubMed] [Google Scholar]
- Vetter IR, Wittinghofer A (2001) The guanine nucleotide-binding switch in three dimensions. Science 294: 1299–1304 [DOI] [PubMed] [Google Scholar]
- Wang J, Boisvert DC (2003) Structural basis for GroEL-assisted protein folding from the crystal structure of (GroEL-KMgATP)14 at 2.0 A resolution. J Mol Biol 327: 843–855 [DOI] [PubMed] [Google Scholar]
- Wilbanks SM, McKay DB (1995) How potassium affects the activity of the molecular chaperone Hsc70. II. Potassium binds specifically in the ATPase active site. J Biol Chem 270: 2251–2257 [DOI] [PubMed] [Google Scholar]
- Wittinghofer A (1997) Signaling mechanistics: aluminum fluoride for molecule of the year. Curr Biol 7: R682–R685 [DOI] [PubMed] [Google Scholar]
- Wittinghofer A (2006) Phosphoryl transfer in Ras proteins, conclusive or elusive? TIBS 31: 20–23 [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Hwang J, Inouye M (2000) Characterization of GTPase activity of TrmE, a member of a novel GTPase superfamily, from Thermotoga maritima. J Bacteriol 182: 7078–7082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim L, Martinez-Vicente M, Villarroya M, Aguado C, Knecht E, Armengod ME (2003) The GTPase activity and C-terminal cysteine of the Escherichia coli MnmE protein are essential for its tRNA modifying function. J Biol Chem 278: 28378–28387 [DOI] [PubMed] [Google Scholar]
- Yokoyama S, Nishimura S (1995) Modified nucleosides and codon recognition. In tRNA: Structure, Biosynthesis and Function, Söll D, RajBhandary UL (eds) pp 207–223. Washington, DC: American Society for Microbiology [Google Scholar]
- Yokoyama S, Watanabe T, Murao K, Ishikura H, Yamaizumi Z, Nishimura S, Miyazawa T (1985) Molecular mechanism of codon recognition by tRNA species with modified uridine in the first position of the anticodon. Proc Natl Acad Sci USA 82: 4905–4909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Yamaizumi Z, Nishimura S, Miyazawa T (1979) 1H NMR studies on the conformational characteristics of 2-thiopyrimidine nucleotides found in transfer RNAs. Nucleic Acids Res 6: 2611–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4