Cryo-electron microscopy reconstruction of a poliovirus-receptor-membrane complex (original) (raw)
. Author manuscript; available in PMC: 2006 Jul 12.
Published in final edited form as: Nat Struct Mol Biol. 2005 Jun 19;12(7):615–618. doi: 10.1038/nsmb955
Abstract
To study non-enveloped virus cell entry, a versatile in vitro model system was developed in which liposomes containing nickel- chelating lipids were decorated with His-tagged poliovirus receptors and bound to virus. This system provides an exciting opportunity for structural characterization of the early steps in cell entry in the context of a membrane. Here we report the three-dimensional structure of a poliovirus-receptor-membrane complex solved by cryo-electron microscopy (cryo-EM) at a resolution of 32Å. Methods were developed to establish the symmetry of the complex objectively. This reconstruction demonstrates that receptor binding brings a viral five-fold axis close to the membrane. Density is clearly defined for the icosahedral virus, for receptors (including known glycosylation sites) and for the membrane bilayer. Apparent perturbations of the bilayer close to the viral five-fold axis may function in subsequent steps of cell entry.
Infection by a non-enveloped virus requires that either an inner nucleoprotein core or a viral genome crosses a cellular membrane, a process that remains poorly understood. Poliovirus provides an excellent model system to address this process. The virus has a simple organization, consisting of a protein shell of known structure1 enclosing an RNA genome. Its cellular receptor has been identified2, and the entry pathway has been characterized extensively. The poliovirus receptor (Pvr) has three extracellular immunoglobulin domains, a transmembrane region and a cytoplasmic tail2. The ectodomain can be transferred to a variety of anchors3, including a glycosylphosphatidylinositol anchor4, and remain fully functional, demonstrating that neither the transmembrane nor the cytoplasmic domain is essential for infection. At physiological temperature, receptor binding initiates uncoating by triggering an irreversible conformational change to form a 135S intermediate5-7. During this transition, VP4 and the N-terminal extension of VP1, both internal components of the viral capsid, become exposed and insert into membranes8,9.
Electrophysiology studies have shown that native virus bound to membrane-tethered Pvr alters the membrane conductance properties at room temperature4, and that further changes take place upon heating to physiological temperature. These changes have been attributed to the membrane insertion of VP4 and the VP1 N terminus after conversion to the 135S entry intermediate4,9,10. Genetic data for poliovirus have shown a direct correlation between channel formation and RNA release, suggesting that these channels function in cell entry9.
The structures of soluble forms of picornavirus cell entry intermediates have provided some insights into virus-receptor interactions and receptor-induced conformational rearrangements11-17 but lack the context of a membrane. Conversely, studies of virus entry by light or electron microscopy, or by electron cryo-tomography18 have not provided high-resolution information about the critical steps. Here we describe an in vitro liposome-based model system that offers an opportunity for detailed biochemical and structural characterization of a virus-receptor complex in the context of a membrane. In this model system, nitrilotriacetate (NTA)-containing lipids are used to produce liposomes decorated with His-tagged Pvr. At room temperature, receptor-decorated liposomes capture virus in its native form. Exposure of such complexes to physiological temperature is known to induce structural changes in the virus that are indistinguishable from those observed during infection (T. Tuthill (University of Leeds), D.B., D. Rowlands (University of Leeds) and J.M.H., unpublished data).
In this article we report the three-dimensional structure of the poliovirus-receptor-liposome complex. The complex was formed at room temperature and then flash frozen; its structure was determined by cryo-EM at a resolution of 32Å. The assembled complex represents a molecular machine poised to deliver a viral genome into its target cell.
RESULTS
Two-dimensional cryo-EM images
Examination of cryo-EM fields of virus-receptor-liposome complexes revealed a population of liposomes of fairly uniform size in which there are multiple instances of viral attachment (Fig. 1). Upon closer inspection of the point of attachment, where the virus particle and liposome are nearly side by side in the plane of the projection, density for the receptor is often visible between them (Fig. 1b). In these views receptor molecules appear to be splayed away from vector normal to the plane of the membrane, and the particle center is estimated to be 220Å from the membrane.
Figure 1.

Poliovirus bound to receptor-decorated liposomes as visualized by cryo-EM. (a) Electron micrograph field showing a fairly uniform population of receptor-decorated liposomes and multiple instances of virus attachment. (b) Individually windowed particles in which density corresponding to receptor can be observed linking native virus to the membrane. (c) Model-based electron density was calculated from pseudo atomic models fitted by Belnap et al.14 into a cryo-EM reconstruction of receptor-decorated poliovirus. In this diagram, the distance between the virus center and the nearest membrane contact point (assuming a planar bilayer) is predicted to be 220Å for an approach along the viral five-fold axis.
Determining the symmetry of the complex
Symmetry constraints have made it possible to derive relatively high-resolution three-dimensional reconstructions from cryo-EM images of icosahedral viruses. However, in this study, the (inherently lower) symmetry of the complex was not known in advance, and the shape of the bilayer was expected to be variable away from the viral binding site. To address these problems, we temporarily introduced an unambiguous fiducial mark (a high-intensity ‘dot’) in a subset of 117 images (Fig. 2a). This dot identified the point that is one end of the shortest vector between the membrane surface and the center of the virus, and served as a proxy for the membrane contact point. In two-dimensional projection, a variety of vector lengths were observed, reflecting the diversity of particle orientations. The orientation of the virion in each of the undotted images was determined by standard single-particle methods, using a native virion as reference, and assuming icosahedral symmetry (see Supplementary Methods online). These orientations applied to dotted images produced a reconstruction that showed a prominent dot along each five-fold axis at 221Å from the virion center (Fig. 2b). This distance is similar to the estimate of 220Å for the distance derived from the fully decorated virus-receptor complex14, assuming that the virion binds five copies of the receptor about its five-fold axis (Fig. 1c) and is 20Å shorter than the distance predicted for a three-fold approach. The presence of a single well-defined dot in the three-dimensional reconstruction, its position and its statistical significance (Fig. 2) provide the first experimental validation of a widely accepted model in which the virus approaches the membrane along its five-fold axis12,17, and demonstrates that the complex forms a homogeneous population with a well-defined structure.
Figure 2.

Artificially dotted icosahedral reconstruction of a virus-receptor-liposome complex demonstrates that virus approaches membrane along its five-fold axis. (a) Two-dimensional images in various orientations were modified by adding dots to the liposome position closest to the virus. (b) Three-dimensional reconstruction computed from dotted images using icosahedral orientations from undotted images. (c) Density values sampled along various radial directions, measured from the virus center. The peak value at 221Å along the five-fold axis is significantly higher than in other directions. (d) Distribution of particle orientations in the final five-fold symmetric reconstruction. Each diamond represents one usable two-dimensional image in the space of unique orientations (triangle).
Five-fold symmetric reconstructions
A dot was added at the appropriate position to a native reconstruction, and this model was used as a reference in determining the orientations of complexes in the subset of 117 dotted images, assuming five-fold symmetry. When these orientations were applied to undotted images to create an unbiased (undotted) reconstruction, portions of the membrane and receptor began to appear. This additional density, together with density corresponding to a scaled and oriented native virus reconstruction, was included in search models with no further reference to dots in subsequent cycles. Additional images were added at several stages of this iterative reconstruction process. In the final cycles, the search model consisted entirely of the reconstruction from the previous cycle. Successive improvements in orientation parameters led to the appearance of better defined density for receptor molecules and membrane. The final map, calculated by standard methods19,20, was derived from 609 complexes and has a nominal resolution of 32Å, as assessed by Fourier shell correlation21 using a conservative 0.5 threshold value (see Methods and Supplementary Fig. 1 online).
The structure of the poliovirus-receptor-liposome complex
The reconstruction of the native poliovirus-receptor-liposome complex (Fig. 3, Supplementary Video 1 online) demonstrates that five receptor molecules bind to the virus and orient it with a five-fold axis directed toward the membrane. The density level for the receptor, together with the high local concentrations of both receptors and receptor binding sites, strongly suggest that all five receptor sites are fully occupied.
Figure 3.
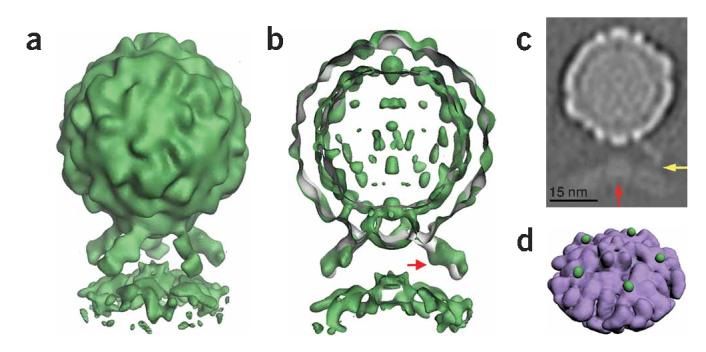
Five-fold symmetric three-dimensional reconstruction of native poliovirus bound to receptor-decorated liposomes. (a) In a surface rendering of the complex (green), the icosahedral features of the virus capsid are maintained, and the density for receptors and membrane are well defined. (b) A cut-away view shows that the three-dimensional reconstruction of the complex (green) superimposes well on electron density (gray) corresponding to the virus-receptor complex in solution. Extra density on the outer leaflet of the membrane near the viral five-fold axis could be due to a perturbation of the membrane. A red arrow indicates a glycosylation site on domain 2 of Pvr. (c) A gray-scale representation of a central slice of the reconstruction that is 27Å thick. This view shows the protrusion of the membrane toward the viral five-fold axis (red arrow) and a weak but continuous density connection between the C-terminal domain of the receptor and the membrane (yellow arrow). Note the clear separation between the inner and outer leaflets of the bilayer, in both the bulged and peripheral areas of the membrane. (d) An unobstructed view of the membrane (purple) shows the crownlike appearance of the apparent distortion. Before removing the receptor density, the small green spheres were located near the predicted C terminus of receptor domain 3. For clarity, d shows lower contour levels than a and b.
In the complex, receptor binding brings the prominent star-shaped feature on the virion surface close to the membrane. Densities for the virus, membrane and receptor, including prominent glycosylation sites in domain 2 of the receptor, are clearly defined. Although only five-fold symmetry was imposed in the reconstruction, the features of the virus closely resemble those seen in the icosahedrally symmetric native form1,14. In addition, virus and receptor densities in the current reconstruction overlay extremely well with electron density derived from a higher resolution reconstruction of receptor-decorated native poliovirus in solution14 (Fig. 3b). The quality of the density for the virus and receptor, the appearance of icosahedral features in the capsid and the consistency of the density for the receptor with the previously reported reconstruction of the soluble binary complex provide compelling objective confirmation of the validity of the methods used to solve this structure.
The morphology of the membrane component is striking. Near the periphery of the complex, the membrane appears to form a canonical bilayer of the expected thickness. However, where the bilayer approaches the virus, a strong density feature extends toward the virus (Fig. 3a-c), with projections that are substantially (B25Å) off-axis. The off-axis position of the density feature, together with the lack of unexplained density elsewhere along the five-fold axis, strongly suggest that this feature is not due to the accumulation of noise on a symmetry axis. If the reconstruction is contoured at a sufficiently low level (as in the Supplementary Video 1 online), an additional set of five spherical density features appears on the outer surface of the membrane (Fig. 3d). Although their significance is not completely certain, each feature is located immediately adjacent to the predicted position of the C terminus of domain 3 of the receptor14,16. These features may thus represent part of the connection between the ectodomain and its membrane anchor. The density connection becomes more apparent at lower contour levels, and in gray-level representations (Fig. 3c and Supplementary Video 1 online).
DISCUSSION
The reconstruction supports previous proposals that the viral five-fold axis is involved in interactions of externalized polypeptide sequences with membranes and is the probable site of RNA translocation12. Although more than five receptors may be recruited to the virus as it is enveloped by membrane during cell entry, the current reconstruction of the minimal complex provides a model for the initial interaction of virus with receptor on the cell surface, and it establishes the preferred orientation of the virus for attachment. The structure demonstrates how receptor binding positions, orients and primes the viral machinery for cell entry.
Binding of the virus to membrane-anchored receptor seems to perturb the canonical bilayer in a way that may be relevant to early events in membrane penetration and cell entry. The appearance of a strong density feature protruding toward the virus is consistent with a local perturbation of the bilayer, which could explain the changes in membrane conductance during pore formation induced by receptor binding at room temperature4. There are several possible explanations for the apparent perturbation: (i) Formation of the virus-receptor complex could cause a structural distortion of the outer leaflet of the bilayer; (ii) complex formation could induce selective partitioning of lipids within the footprint of the complex, here, the electron-dense Ni2+-chelating NTA lipids, which were used to anchor the receptor. Either of these could prime the membrane for the insertion of viral peptides during subsequent steps in cell entry. (iii) It is also possible that receptor binding at room temperature facilitates breathing similar to that observed for free virus at 37 1C (ref. 22), resulting in transient global or local exposure of viral peptides, which then associate with membrane.
The reconstruction of native virus bound to receptor-decorated NTA-liposomes represents a proof of principle demonstrating that the three-dimensional structures of membrane-bound complexes of mixed or unknown symmetry can be determined. The versatile nature of the NTA-liposome interaction with His-tagged receptor, combined with the new structural methods demonstrated here, will allow this system to be applied broadly to study other virus-receptor complexes, internalized virus-receptor complexes and late cell entry intermediates in the context of membrane.
METHODS
Sample preparation. The type 1 Mahoney strain of poliovirus was propagated in HeLa cell suspension culture and purified by CsCl density gradient as described23.
Soluble poliovirus receptor (sPvr), consisting of the ectodomain (amino acids 1-337) with a His6 tag at the C terminus, but lacking the transmembrane and cytoplasmic domains, was expressed and purified as described24. Purified sPvr was a gift from Vincent Racaniello.
A lipid mixture containing phosphatidylethanolamine, phosphatidylcholine, sphingomyelin, cholesterol and phosphatidic acid in molar ratios of 1:1:1:1.5:0.3 with 10% (w/w) DOGS-NTA and 1% (w/w) rhodamine-PE was dried under argon gas and desiccated overnight under vacuum. The lipid film was rehydrated in 50 mM HEPES, pH 7.3, 50 mM NaCl and extruded through a membrane with 0.1-μm pores. Then, 50 μl of 1 mg ml-1 liposomes were incubated with 0.03 μg of sPvr for 10 min before the addition of 16.5 μg of native virus. An aliquot of 2.5 μl of the virus-receptor-liposome mixture was incubated for 1 min at room temperature and loaded onto a glow discharged Quantifoil holey carbon grid (SPI) and flash frozen in liquid ethane as described25. Because the complex is maintained well below the temperature required to produce 135S particles, the virus is expected to remain in its native conformation.
Image acquisition. 122 micrographs were recorded at a magnification of 50,000 using a FEI Tecnai F20 microscope at a defocus range of 2-4 μm underfocus. Micrographs were digitized using a SCAI scanner (Z/I Imaging) at a step size of 7 μm corresponding to 2.69Å per pixel.
Image processing. A total of 1,049 complexes were windowed individually in X3D (ref. 26) and corrected for the phase reversal of the contrast transfer function with Bsoft27. Origins and orientation parameters for each complex were determined using a modification of PFT19 altered to use both phase and amplitude information in orientation selection. Three-dimensional reconstructions were computed using a Fourier-Bessel algorithm in EM3DR (refs. 19, 20).
Five-fold symmetric reconstruction. While determining that the complex had five-fold symmetry (see Supplementary Methods online), an icosahedrally symmetric reconstruction of the virus was produced (Fig. 2b) having 12 intense dots, one above each of the five-fold peaks of the virus. One of these dots was masked out and added to a cryo-EM reconstruction of native virus15, scaled and oriented appropriately. The resulting construct was temporarily used as a reference object for a five-fold symmetric (C5) orientation search in PFT to determine preliminary orientation parameters for the dotted images. The resulting orientations were then applied to the original unmodified images using the reconstruction program EM3DR to obtain a five-fold symmetric three-dimensional reconstruction of the virus-receptor-liposome complex with no artificial modifications. Although this reconstruction could serve as a reference model for subsequent orientation searches, better statistics were obtained when the virus component was replaced with the more feature-filled 23-Å 160S reconstruction15. That model was used to determine the orientations of complexes in the larger data set, and the resulting orientations, in a five-fold symmetric reference frame, were used to calculate an improved three-dimensional reconstruction. This procedure was iterated until receptor density and icosahedral features of the virus were resolved. After that, orientation searches implementing a model composed entirely from the previous reconstruction resulted in the calculation of a new reconstruction with no loss in resolution, as judged by a Fourier shell correlation <0.5 (see Supplementary Fig. 1 online). Several weighting schemes were examined, and it was determined empirically that better results were obtained by multiplying the intensity of the virus component of the reference by three. In the final round of orientation determination a mask was applied to the reference object.
Windowed particles included in the final three-dimensional reconstruction were selected on the basis of their correlation coefficients (compared with reference projections) and confirmed by the correct positioning of the virus-membrane contact point (controlled by two of the Eulerian angles), which was assessed visually. The final reconstruction, based on commonly used and well-established reconstruction methods19,20, was calculated from 609 selected images. These 609 projections produce a fairly uniform sampling of the set of possible orientations in a five-fold symmetric reference frame (Fig. 2d). The reconstruction has a nominal resolution of 32Å (29Å for virus alone, 30Å for virus and receptor).
Supplementary Material
2
3
4
ACKNOWLEDGMENTS
We thank S. Lanzavecchia for discussions, D. Gohara for rendering Figure 3d, T. Walz for access to instruments in the Harvard Medical School Electron Microscopy laboratory and Y. Cheng for instruction and assistance in their use. This work is supported by National Institutes of Health grant AI20566 (to J.M.H.) and National Science Foundation predoctoral fellowship (to D.B.).
Footnotes
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
References
- 1.Hogle JM, Chow M, Filman DJ. Three-dimensional structure of poliovirus at 2.9Å resolution. Science. 1985;229:1358–1365. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- 2.Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56:855–865. doi: 10.1016/0092-8674(89)90690-9. [DOI] [PubMed] [Google Scholar]
- 3.Selinka HC, Zibert A, Wimmer E. Poliovirus can enter and infect mammalian cells by way of an intercellular adhesion molecule 1 pathway. Proc. Natl. Acad. Sci. USA. 1991;88:3598–3602. doi: 10.1073/pnas.88.9.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tosteson M, Wang H, Naumov A, Chow M. Poliovirus binding to its receptor in lipid bilayers results in particle-specific, temperature-sensitive channels. J. Gen. Virol. 2004;86:1581–1589. doi: 10.1099/vir.0.19745-0. [DOI] [PubMed] [Google Scholar]
- 5.Lonberg-Holm K, Gosser LB, Kauer JC. Early alteration of poliovirus in infected cells and its specific inhibition. J. Gen. Virol. 1975;27:329–345. doi: 10.1099/0022-1317-27-3-329. [DOI] [PubMed] [Google Scholar]
- 6.De Sena J, Mandel B. Studies on the in vitro uncoating of poliovirus. II. Characteristics of the membrane-modified particle. Virology. 1977;78:554–566. doi: 10.1016/0042-6822(77)90130-1. [DOI] [PubMed] [Google Scholar]
- 7.De Sena J, Mandel B. Studies on the in vitro uncoating of poliovirus. I. Characterization of the modifying factor and the modifying reaction. Virology. 1976;70:470–483. doi: 10.1016/0042-6822(76)90288-9. [DOI] [PubMed] [Google Scholar]
- 8.Fricks CE, Hogle JM. Cell-induced conformational change of poliovirus: Externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 1990;64:1934–1945. doi: 10.1128/jvi.64.5.1934-1945.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danthi P, Tosteson M, Li QH, Chow M. Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. J. Virol. 2003;77:5266–5274. doi: 10.1128/JVI.77.9.5266-5274.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tosteson MT, Chow M. Characterization of the ion channels formed by poliovirus in planar lipid membranes. J. Virol. 1997;71:507–511. doi: 10.1128/jvi.71.1.507-511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hewat EA, Neumann E, Blass D. The concerted conformational changes during human rhinovirus 2 uncoating. Mol. Cell. 2002;10:317–326. doi: 10.1016/s1097-2765(02)00603-2. [DOI] [PubMed] [Google Scholar]
- 12.Hogle JM. Poliovirus cell entry: common structural themes in viral cell entry path-ways. Annu. Rev. Microbiol. 2002;56:677–702. doi: 10.1146/annurev.micro.56.012302.160757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hewat EA, Blaas D. Cryoelectron microscopy analysis of the structural changes associated with human rhinovirus type 14 uncoating. J. Virol. 2004;78:2935–2942. doi: 10.1128/JVI.78.6.2935-2942.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belnap DM, et al. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc. Natl. Acad. Sci. USA. 2000;97:73–78. doi: 10.1073/pnas.97.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belnap DM, et al. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J. Virol. 2000;74:1342–1354. doi: 10.1128/jvi.74.3.1342-1354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Y, et al. Interaction of the poliovirus receptor with poliovirus. Proc. Natl. Acad. Sci. USA. 2000;97:79–84. doi: 10.1073/pnas.97.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolatkar PR, et al. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 1999;18:6249–6259. doi: 10.1093/emboj/18.22.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bohm J, et al. FhuA-mediated phage genome transfer into liposomes: a cryo-electron tomography study. Curr. Biol. 2001;11:1168–1175. doi: 10.1016/s0960-9822(01)00349-9. [DOI] [PubMed] [Google Scholar]
- 19.Baker TS, Cheng RH. A model-based approach for determining orientations of biological macromolecules imaged by cryo-electron microscopy. J. Struct. Biol. 1996;116:120–130. doi: 10.1006/jsbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- 20.Crowther RA, Amos LA, Finch JT, DeRosier DJ, Klug A. Three dimensional reconstructions of spherical viruses by Fourier synthesis from electron micrographs. Nature. 1970;226:421–425. doi: 10.1038/226421a0. [DOI] [PubMed] [Google Scholar]
- 21.Saxton WO, Baumeister W. The correlation averaging of regularly arranged bacterial cell envelope protein. J. Microsc. 1982;127:127–138. doi: 10.1111/j.1365-2818.1982.tb00405.x. [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Yafal AG, Lee YM-H, Hogle J, Chow M. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J. Virol. 1994;68:3965–3970. doi: 10.1128/jvi.68.6.3965-3970.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeates TO, et al. Three-dimensional structure of a mouse-adapted type 2/type 1 poliovirus chimera. EMBO J. 1991;10:2331–2341. doi: 10.1002/j.1460-2075.1991.tb07772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDermott BM, Rux AH, Eisenberg RJ, Cohen GH, Racaniello VR. Two distinct binding affinities of poliovirus for its cellular receptor. J. Biol. Chem. 2000;275:23089–23096. doi: 10.1074/jbc.M002146200. [DOI] [PubMed] [Google Scholar]
- 25.Dubochet J, et al. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- 26.Conway JF, Steven AC. Methods for reconstructing density maps of "single" particles from cryoelectron micrographs to subnanometer resolution. J. Struct. Biol. 1999;128:106–118. doi: 10.1006/jsbi.1999.4168. [DOI] [PubMed] [Google Scholar]
- 27.Heymann JB. Bsoft: image and molecular processing in electron microscopy. J. Struct. Biol. 2001;133:156–169. doi: 10.1006/jsbi.2001.4339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
2
3
4