The in vivo expansion rate of properly stimulated transferred CD8+ T cells exceeds that of an aggressively growing mouse tumor (original) (raw)
. Author manuscript; available in PMC: 2006 Aug 21.
Abstract
It has been hypothesized that rapidly dividing tumor cells can outpace adoptively transferred anti-tumor lymphocytes when tumors are large. However, this hypothesis is at odds with clinical observations indicating that bulky tumors can be destroyed by small numbers of adoptively transferred anti-tumor T cells. We sought to measure the relative growth rates of T cells and tumor cells in a model using transgenic CD8+ T cells specific for the gp10025–33 H-2Db epitope (called pmel-1) to treat large, well-established subcutaneous B16 melanoma. We tested the impact of the immunization using an altered-peptide ligand vaccine alone or in combination with IL-2 by analyzing the kinetics of T cell expansion using direct enumeration. We found that pmel-1 T cells proliferated explosively during a 5-day period following transfer. Calculations from net changes in population suggest that at the peak of cell division, pmel-1 T cells divide at a rate of 5.3 hours/cell division, which was much faster than B16 tumor cells during optimal growth (24.9 hours/cell division). These results clearly indicate that the notion of a kinetic “race” between the tumor and lymphocyte is no contest when adoptively transferred cells are stimulated with immunization and IL-2. When appropriately stimulated, tumor-reactive T cell expansion can far exceed the growth of even an aggressively growing mouse tumor.
Keywords: Cell proliferation, Tumor immunity, Cytotoxic T cells, Cytokines, Vaccination
Introduction
Multiple approaches have been applied to cancer immunotherapy (1–6). Adoptive transfer of autologous lymphocytes with antitumor reactivity is one of the potential immunotherapeutic means currently under investigation for the treatment of cancer (7). The scientific foundation for these studies is comprised of data from murine tumor systems that model the treatment effects of clinical adoptive therapy. Many of these models include systems utilizing transgenic lymphocytes targeting highly immunogenic model antigens expressed on transduced tumor cell lines (8–11). Other groups have described similar adoptive transfer models, which utilize transgenic (8) or tumor-challenge derived lymphocytes (12, 13) that target naturally-occurring tumor antigens. Together, these scientific reports have demonstrated that adoptive immunotherapy can be successful in treating tumors in murine systems. However, the treatment of large, vascularized tumors is generally successful only when targeting highly immunogenic “model” antigens. Systems utilizing naturally expressed tumor-associated antigens generally are only effective in the treatment of smaller tumors. One potential explanation for the lower efficacy of these systems may lie in the fact that the bulk of these studies have focused on the adoptive transfer of tumor-reactive cells alone, without further activation by immunization or cytokine support.
Based on these models, some have suggested that the adoptive transfer of tumor-specific T cells fails to successfully treat larger, well-established tumor masses because the tumors contain too large a number of rapidly dividing cells (8). This, in turn, was taken to indicate that the biological reason for the failure of adoptive cell transfer in some settings was due to a kinetic disparity between the growth of the malignancy and the accumulation of anti-tumor effector cells – the interpretation being that tumor cells simply outgrew any anti-tumor response (8). However, this reasoning is at odds with results of recent clinical trials, which indicate that relatively small numbers of adoptively transferred T cells (1010 cells) can treat tumor volumes that can be measured in kilograms (>1012 tumor cells) (7, 14, 15). These studies have shown that a number of the responder patients exhibited profound lymphocytosis within 8 days after receiving the cells (15). In addition, a number of animal studies have also shown a rapid proliferation of adoptively transferred T cell response to viral infection (16–18). However, these studies do not address the kinetics of the adoptively transferred T cell response in the context of its potential for immunotherapeutic treatment of cancer.
In the current clinical trials, polyclonal tumor-infiltrating lymphocytes (TIL) that are widely heterogenous for avidity, activation and differentiation status are the primary source of cells for adoptive transfer. However, measurement of TIL expansions is greatly complicated by their heterogenicity. Each clonotype can become activated, undergo expansion, and become inactivated or apoptotic at rates that vary widely from one clonotype to another. Thus, the use of a TCR transgenic mouse derived from a single clonotype, normal anti-tumor T lymphocyte minimizes intra-clonal diversity and serves as an optimal model to study the kinetics of a T cell response responsible during an active, anti-tumor response.
We have successfully modeled human adoptive immunotherapy in mice using a transgenic T cell expressing the T cell receptor (TCR) specific for the mouse melanoma antigen, gp10025–33 H-2Db epitope, called pmel-1. Upon the adoptive transfer of in vitro cultured pmel-1 cells followed by recombinant fowlpox virus encoding hgp100 (FP-hgp100) immunization and high dose of IL-2 administration into sub-lethally irradiated recipient mice, regression of large cutaneous murine B16 melanoma were achieved (19). This model targets a naturally expressed tumor antigen and therefore provides us a unique opportunity to study the population kinetics of an adoptively transferred self/tumor reactive CD8+ T cell responses necessary to mediate regression of large tumors.
In this study, we found a clear association of the initial kinetic response of adoptively transferred, self/tumor-reactive T cells and the regression of established B16 melanoma. We also investigated the influence of immunization and IL-2 administration on the kinetics of the transgenic cell population. Finally, we mathematically determined net rates of division at different stages of the T cell response and compare these growth rates with that of established B16 tumor to investigate the issue of kinetic disparity between tumor growth and T cell expansion.
Materials and Methods
Animals and cell culture
We obtained C57BL/6 recipient mice from National Cancer Institute-Frederick, MD. Pmel-1 TCR transgenic mice have been described previously (13). Pmel-1 mice are on a C57BL/6 background and express a gp10025–33 H-2Db epitope-specific TCR. We crossed pmel-1 mice with syngenic mice expressing the secondary thy1.1 (CD90.1) congenic marker to aid in the detection of transgenic cell populations. To culture pmel-1 T cells for adoptive transfer, splenocytes from pmel-1 transgenic mice were cultured in RPMI media with 10% FCS and 60 IU/ml recombinant human IL-2 (Chiron Corp., Emeryville, CA) and stimulated with 1 μg/ml hgp10025–33 peptide for 6 – 7 days. Cells cultured in this fashion result in a >95% pure CD8+ cell population that expresses both the transgenic TCR and thy1.1 markers as measured by fluorescent cell sorting analysis for CD8+, TCR Vβ13+, and thy1.1+ surface staining of the cells.
Adoptive transfer of pmel-1 T cells and in vivo immunization
Prior to adoptive transfer of cells, we sub-lethally irradiated C57BL/6 recipient mice at 500 rads the day of adoptive transfer to deplete endogenous T cell populations that might interfere with subsequent cell analysis of donor T cells. We then delivered cultured pmel-1 T cells or Bg-1 transgenic T cells (specific for Kb restricted β-galacosidase protein 96–103, used as a control) by tail vein injection at a concentration of 1×107 cells in 0.5 ml PBS. Following adoptive transfer of pmel-1 T cells, mice are immunized i.v. with 2×107 plaque forming units (PFU) of recombinant fowlpox-hgp100 (FP-hgp100) or -βgal virus (Therion Biologics, Cambridge, MA) in 0.5 ml PBS. In addition, recombinant human IL-2 was administered i.p. (600,000 IU in 0.5 mls PBS, twice daily for a total of 6 injections). For tumor treatment experiments, 1×105 B16.F10 melanoma cells were injected subcutaneously (s.c.) into C57BL/6 mice 14 days prior to adoptive transfer.
Harvesting and quantifying transgenic T cells
We harvested spleens at selected time points from 2–5 mice given identical treatments as described in figure legends. The total numbers of splenocytes were derived from a manual cell count of single-cell suspensions and average splenocyte populations for single spleens were calculated. Samples of each cell population were then stained for surface Vβ13 TCR chain and the congenic marker thy1.1 using FITC-labeled anti-mouse Vβ13 TCR and Phycoerythrin (PE) anti-mouse CD90.1 antibodies (BD Biosciences, San Diego, CA), respectively, and analyzed by flow cytometry. We determined the total splenocyte count by multiplying the total splenic pmel-1 T cell population by the percentage of the splenocytes positively stained for Vβ13 and thy1.1.
Depletion of transferred pmel-1 T cells
B16-bearing C57BL/6 recipient mice were irradiated and given standard adoptive transfer of pmel-1 T cells, immunization and IL-2 treatment described in previous sections. We depleted CD8+ T cells by injection of 150μg of purified anti-mouse CD8α antibody (clone 53-6.7, BD Biosciences, San Diego, CA) i.p. at indicated time points. Control antibody, purified Rat IgG2a, κ(BD Biosciences), was injected into recipient mice at the identical amount and concentration as CD8-depleting antibody. Two days after antibody injection, cell staining analysis of harvested splenocytes from depleted mice exhibited >95% depletion of the transgenic cell population (CD8+, thy1.1+).
Bromodeoxy-uracil (BrdU) labeling and proliferation assay
Analysis of proliferation were done utilizing a Fluorescein Isothiocyanate (FITC) conjugated BrdU flow kit (BD Biosciences, San Diego, CA). We injected mice intra-peritoneally with 1 mg of BrdU in 0.1 ml of PBS 16 hours prior to harvesting, fixing and permeablizing splenocytes with Cytofix/Cytoperm (BD Pharmingen). We then treated the cells with DNase I to cleave genomic DNA from fixed cells and intracellular staining with FITC-labeled anti-BrdU antibodies was done at 4°C for 30 min to detect presence of incorporated BrdU. We then stained cell samples for the congenic marker thy1.1 as described above to identify the transgenic cell population using flow cytometry. Histograms were gated on thy1.1+ lymphocyte populations.
Results
Rapid expansion of transferred pmel-1 T cells occurs early during tumor treatment
We employed a model of the treatment of the highly aggressive, poorly immunogenic B16 melanoma, which grew and vascularized for 14 days after implantation. We then adoptively transferred pmel-1 T cells alone or together with a recombinant fowlpox virus expressing an altered peptide ligand of the gp10025–33 epitope and/or IL-2 cytokine (19). Although there was evidence of minor tumor regression in mice receiving pmel-1 cells and FP-hgp100 immunization without additional IL-2, maintenance of B16 regression past 2 weeks following treatment was only observed with the full complement of adoptive transfer of the pmel-1 tumor-reactive cells, immunization with FP-hgp100, and IL-2 cytokine administration (Fig. 1A).
Figure 1.
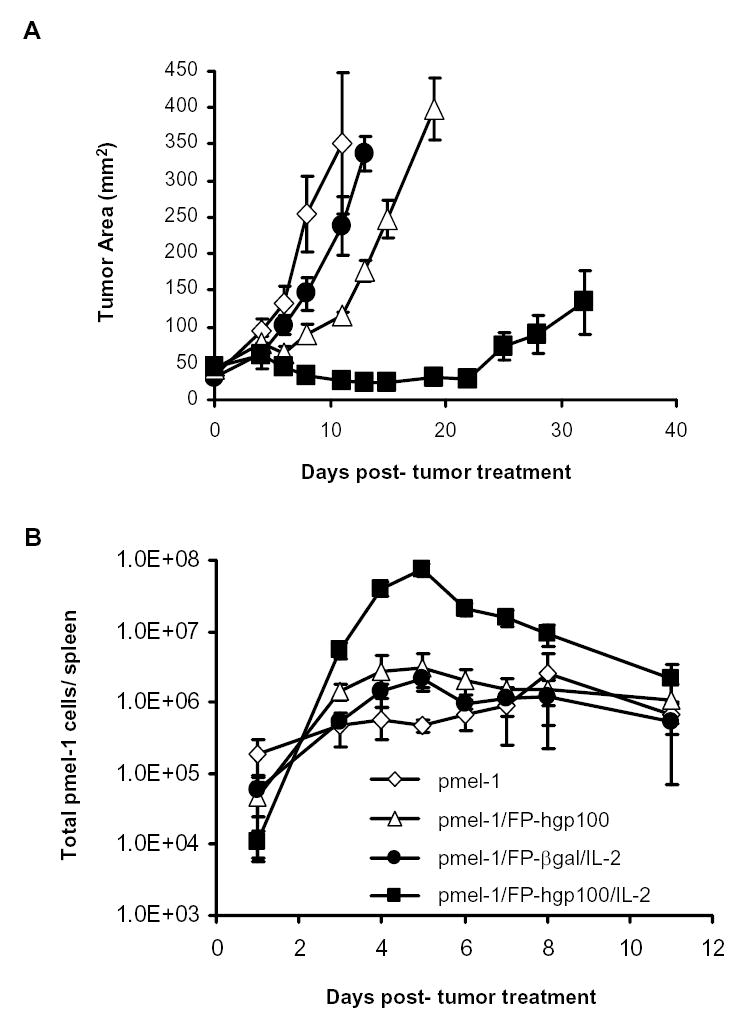
Kinetics of the expansion of pmel-1 T cells in B16 tumor treatment model. A. Tumor regression by adoptively transfer pmel-1 T cells. Each line represented the mean of measurements from 5 animals per group. Measurements were stopped at point where < 75% of originally treated mice were still surviving. The results shown were representative of three independent identical experiments. B. Population kinetics of transferred pmel-1 transgenic T cells. Cultured pmel-1 T cells were adoptively transferred into B16-bearing mice and given treatments as indicated as described in Materials and Methods. 2–5 mice per group were sacrificed at indicated time points and splenocytes from treated animals were harvested and analyzed for presence of transgenic TCR Vβ13 chain and secondary congenic marker thy1.1. Total transgenic splenocytes = (total average splenocyte count) × (% of Vβ13+/thy1.1+ double-positive splenocytes). Shown values were averaged from 3 experiments of identically treated groups of mice.
To study the kinetics of the tumor-reactive pmel-1 T cell population during the tumor treatment, we measured the donor T cell population over a time course of the first two weeks following adoptive transfer and immunization. Levels of transferred pmel-1 T cells were between 11,000 – 180,000 cells in the spleen 1 day following adoptive transfer. We found that the transgenic cell population increased in the spleen in all treatment groups during the next 4–7 days (Fig. 1B). Despite the observation that transgenic T cells increased in all treatment groups, immunization with the relevant antigen using FP-hgp100 and IL-2 cytokine administration induced the generation of a much larger population of total pmel-1 T cells. The peak of the pmel-1 T cell populations occurred 5 days following transfer and was >35-fold greater in number compared to the peak population when immunizing with an irrelevant antigen and giving IL-2. This peak was also >24-fold higher compared to the population peak when immunizing with FP-hgp100 without IL-2 cytokine, and >29-fold greater than expansion of adoptive transfer of cells alone (which peaked on 8 days following adoptive transfer). Following this period of cell expansion, the transgenic cell population dramatically contracted over a 24-hour period to approximately 30% of the peak population. The pmel-1 splenocyte population then gradually decreased after day 6 to reach a level of 3% of the peak value by day 11 and stabilizes at this level during the second week following adoptive transfer and immunization (data not shown). Kinetics from multiple experiments consistently followed a similar pattern of proliferation and contraction, peaking between days 4 and 5 following transfer and immunization (Fig. 1B).
The early expansion of transferred pmel-1 T cells upon immunization and IL-2 was not limited to the spleen. We have carefully examined multiple tissues and organs including peripheral blood, lymph node, lung, liver, brain, kidney, eye and tumor after the adoptive cell therapy. Kinetics of pmel-1 T cells in these tissues were found to be similar, which indicated that the proliferation of pmel-1 T cells with or without FPhgp100 and IL-2 immunization were not tissue specific (20).
Number of pmel-1 T cells during expansion correlates tumor treatment efficacy
To study the link between the number of pmel-1 T cells and their relationship to the efficacy of tumor treatment, we performed a dose titration experiment. When B16 melanoma-bearing mice were given increasing numbers of pmel-1 T cells (2×104, 2×105, 2×106, 2×107), the efficacy of tumor treatment was directly correlated with the increasing number of cells transferred (Fig. 2A). Although we have previously described the specificity of pmel-1 T cells to B16 tumor (20), we transferred culture transgenic T cells specific for a Kb restricted β-gal protein (called Bg-1) then immunized with hgp100 and IL-2. Even large numbers (2 × 106) of non-gp100-reactive Bg-1 cells did not cause B16 tumor regression (Fig. 2A).
Figure 2.
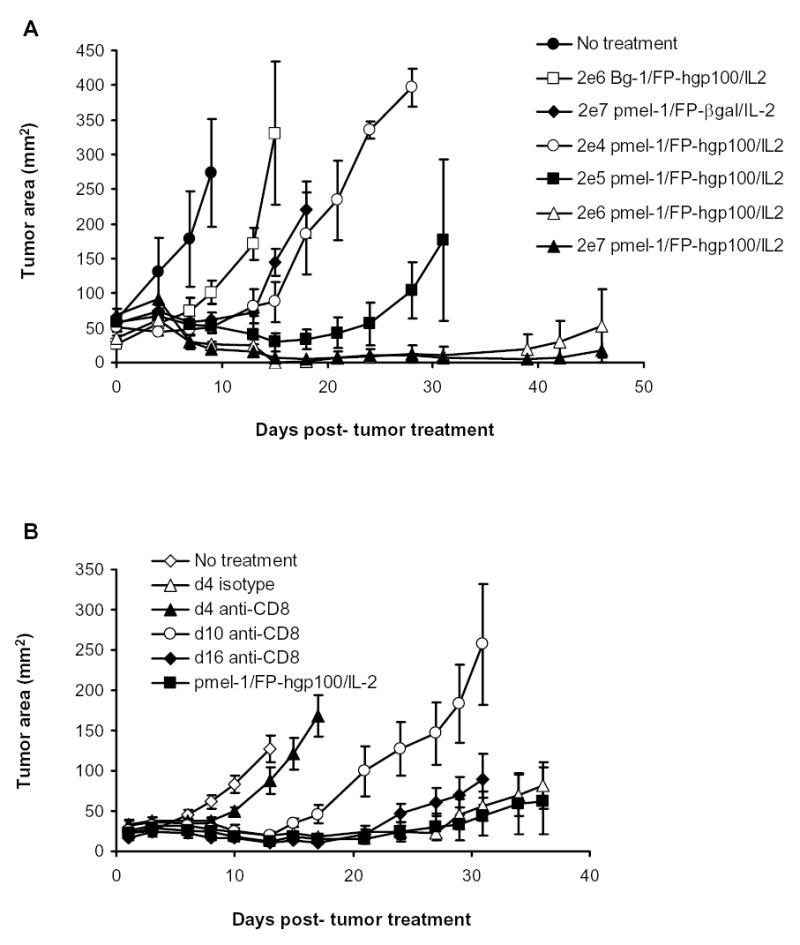
Requirement of pmel-1 cells during B16 tumor treatment. A. Pmel-1 specific B16 tumor treatment. B16 tumor treatment was performed as previously described. The numbers of cell transferred are indicated in the legend. Bg-1 cells that were in vitro stimulated with 1μM of β-gal peptide and 60 IU of IL-2 for 7 days were used as control. 4–5 mice were in each group. Tumor growth between mice given 2 × 104 pmel-1 cells and 2 × 105, 2 × 106 or 2 × 107 pmel-1 T cells were statistically significantly different (p = 0.034, by Wilcoxon Rank Sum Test) B. Depletion of pmel-1 T cells at early time points following adoptive transfer impaired tumor efficacy of full treatment. B16-bearing mice were irradiated and given standard adoptive transfer of pmel-1 T cells, immunization and IL-2 treatment. Mice were subsequently depleted of CD8+ T cells by a single i.p. injection of purified anti-mouse CD8α antibody (clone 53-6.7) or control isotype antibody at a dose of 150μg at indicated time points. Lines of measurements from 5 animals per group. Measurements were stopped at point where <75% of originally treated mice were still surviving. Measurements were representative of two identical independent experiments
To investigate how long the pmel-1 T cells were required for tumor regression, we performed experiments to deplete pmel-1 cells at different time points following adoptive transfer and treatment in irradiated recipients. Because greater than 95% of the transferred cultured pmel-1 cells were CD8+ T cells, we used an established CD8 depleting mAb in our experiments. Mice given pmel-1 T cells, immunization and IL-2 were subsequently depleted of their CD8+ cell populations at d4, d10, and d16 following adoptive transfer using an anti-mouse CD8 antibody. Depletion of the CD8+ pmel-1 cell population during the expansion phase of the response (4 days post-transfer) resulted in greatly abrogated tumor regression (Fig. 2B). However, depletion at later points (10 and 16 days post-transfer) resulted in progressively decreased interference of tumor treatment. We found that > 95% of the transgenic CD8+ cells were depleted within 2 days of antibody injection (data not shown). These results suggested that expansion of tumor-reactive cells was associated with the extended regression of established B16 melanoma.
Increase in transgenic T cell population is directly associated with cell proliferation
Because either transgenic cell proliferation or migration to the spleen in response to different treatment regimens could cause an increase in cell numbers, we used BrdU to assess cell proliferation at different time points following adoptive transfer. Proliferation of the transgenic population appeared to be the greatest at the earliest time point (d 2–3) when >50% of the transferred cells were positive for BrdU incorporation (Fig. 3). This proliferation level gradually decreases during the cell expansion phase of cell growth (3–5 days after transfer). After this rapid expansion, the proliferation of transgenic T cells comes to an abrupt halt (see Fig. 3, d5–6). This abrupt decrease in proliferation of the pmel-1 T cells corresponds to a marked cell contraction that begins after day 5 as shown by pmel-1 population measurements (Fig. 1B). Proliferation was primarily limited to transgenic cell population as analysis of non-transgenic cells showed that < 5% of non-transgenic cells incorporated BrdU at any given time point (data not shown). Proliferation at day 1 and day 2 following adoptive transfer were not measured as the total number of pmel-1 T cells was found to be insufficient for similar analysis.
Figure 3.
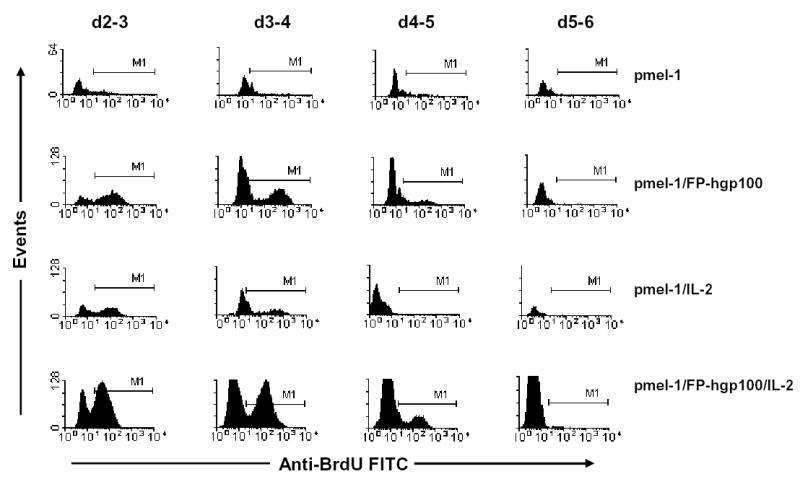
Proliferation profile of transgenic pmel-1 T cells following adoptive transfer and activation. Pmel-1 T cells were adoptively transferred and given treatments as indicated. A single injection of 1 mg BrdU was administered to recipient mice i.p. to label in vivo proliferating cells. Sixteen hours later, pmel-1 cells in spleens of recipient mice were then analyzed on lymphocyte and thy1.1+ cell populations for BrdU incorporation. M1 region represented positive detection of BrdU incorporation for in vitro cultured proliferating pmel-1 T cells following peptide stimulus and IL-2 support.
Removal of either IL-2 support or immunization resulted in much smaller levels of proliferation of the transgenic cell population compared to that of cells treated with full immunization and IL-2 at all the different time points measured (Fig. 3, row 1–3). Despite differences in the number of proliferating cells, all treatment groups exhibited a similar pattern of proliferation for the transgenic cell population that correlated with the pmel-1 population kinetics shown in Fig. 1B. Analysis of pmel-1 proliferation by BrdU incorporation in other organ systems indicated that there were similar levels of pmel-1 T cell proliferation occurring in lymph nodes, peripheral blood, and spleen during the first week following transfer (20). Thus, the observed increases in transgenic T cell population were directly associated with cell proliferation and not merely due to altered patterns of cellular migration.
Transferred pmel-1 T cells proliferate faster than B16 tumor cells during expansion
To determine whether there is a detrimental kinetic disparity between the growth of the malignancy and the accumulation of anti-tumor effector cells in our tumor treatment model, we compared the growth rates of B16 melanoma and that of tumor-reactive pmel-1 T cells.
We first calculated the net changes in the cell populations during the T cell expansion and early contraction (Table I). Assuming there was no cell death during the period of measurement, the net fold increase in cell population would be equal to 2n where n was equal to the number of divisions. Alternatively, the number of divisions could be determined as the log2 of the net fold increase in cell numbers. Therefore:
Table I.
Population/volume change of transferred T cells or tumor*
| Treatment group or tumor | d1–3 | d3–4 | d4–5 | d5–6 | d6–7 | d7–8 | d8–11 |
|---|---|---|---|---|---|---|---|
| pmel-1 | 2.6 | 1.2 | −1.2 | 1.5 | 1.3 | 2.8 | −0.3 |
| pmel-1/FP-hgp100 | 30.2 | 1.9 | 1.1 | −1.5 | −1.4 | 1.0† | −1.4 |
| pmel-1/FP-βgal/IL-2 | 9.6 | 2.7 | 1.5 | −2.3 | −0.8 | 1.1 | −2.3 |
| pmel-1/FP-hgp100/IL-2 | 502.5 | 7.4 | 1.9 | −3.6 | −1.3 | −1.7 | −4.2 |
| B16.F10‡ | 3.8 |
Doubling time = Time of Proliferationlog2(fold increase in cell population)
The assumption of no cell death for the calculations of the net population change, although convenient for our calculations, was an untested hypothesis. However, attempts at measuring in vivo apoptosis utilizing standard annexin V staining and TdT-based assays to detect early- or late-stage apoptotic events for adoptively transferred pmel-1 T cells were, in our experience, unsuccessful (data not shown). This was possibly due to very efficient in vivo clearance mechanisms of dying lymphocytes triggered by very early apoptotic events (21). As this calculation did not consider cell death, which would necessitate compensation of cell loss to achieve the observed net population changes, the derived doubling times were most likely conservative estimates and would be even faster if cell death were taken into consideration.
For pmel-1 T cells transferred in combination with immunization and IL-2, cells underwent a net 502.5-fold increase (approximately 9 divisions) during the 48-hour period from days 1–3 following transfer. Therefore, at the height of cell proliferation between 1–3 days following adoptive transfer, fully activated pmel-1 T cell division occurred at a rate of approximately 5.3 hours/cell division. We performed similar calculations from other groups (shown in Fig. 2B and Table I) and summarized in Table II. During the period from 1 to 3 days after adoptive cell transfer, pmel-1 T cells expanded approximately 30-fold and at a calculated rate of 9.8 hours/cell division in response to immunization without IL-2 support. Measurement of the kinetics of transgenic cells in response to IL-2 cytokine without immunization resulted in a more modest 9.6-fold increase in population with a doubling time of 14.7 hours. Normal homeostatic proliferation of the transgenic cell population in irradiated recipients resulted in a slight 2.6-fold increase in cell population during this 48-hour period, resulting in a doubling time of 34.8 hours.
Table II.
Calculated doubling time for transferred cells and B16-tumor cells *
| Treatment group or tumor | d1–3 | d3–5 |
|---|---|---|
| pmel-1 | 34.8 | N/A† |
| pmel-1/FP-hgp100 | 9.8 | 42.7 |
| pmel-1/FP-bgal/IL-2 | 14.7 | 24.4 |
| pmel-1/FP-hgp100/IL-2 | 5.3 | 12.7 |
| B16.F10‡ | 24.9 |
We then calculated proliferation rates for a subcutaneously established B16 tumor by measuring growth of untreated tumor from multiple experiments. We determined the volume of tumors as that of a half-sphere = (4/3πr3)/2, where the radius of the sphere was the half of the average of perpendicular diameters. Because the average change in tumor volume was approximately proportional to the number of tumor cells comprising the growth, we calculated that the greatest change in tumor growth for naïve mice was approximately 3.8-fold over a 48 hour period (between days 12–14 following tumor challenge) (Fig. 4A). B16 tumor growth in naïve mice was thus at an approximate rate of 24.9 hours/cell division during optimal growth (Table II).
Figure 4.
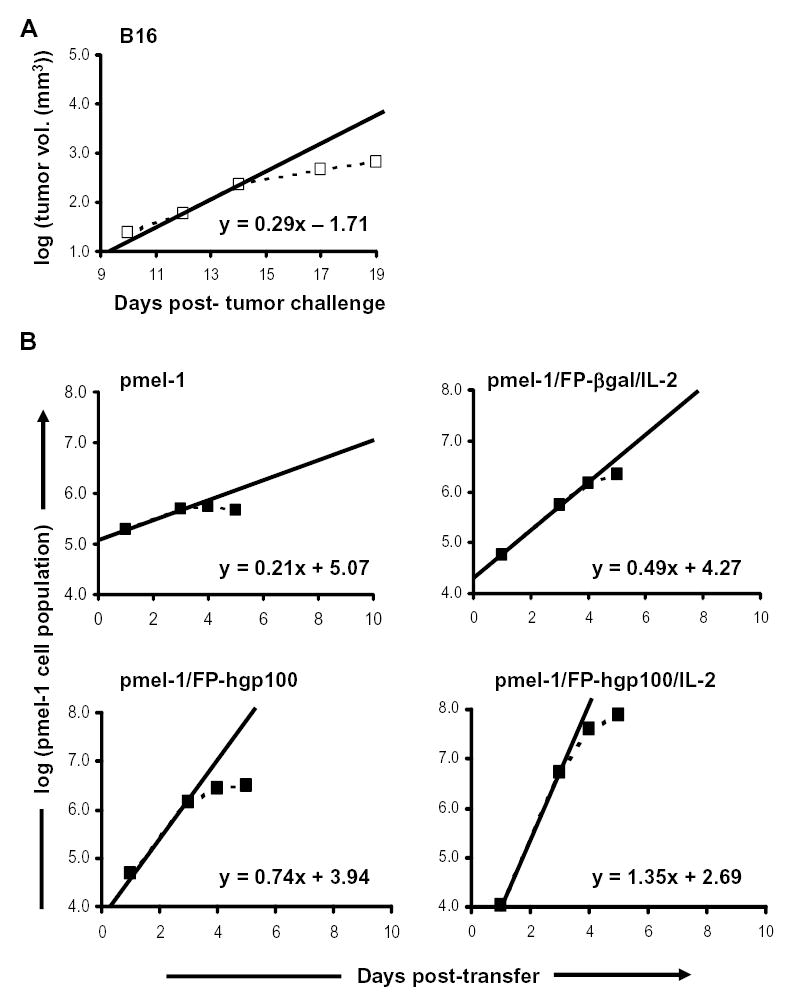
Growth rates of pmel-1 T cells and B16 tumor cells. A. B16 tumor growth in naïve C57BL/6 mice. Mice were injected s.c. with 1×105 B16.F10 cells. Tumors were measured and tumor volumes were calculated as that of a half-sphere = (4/3πr3)/2, where the radius of the sphere was determined as half the average of perpendicular diameters. Tumor volumes were shown as the average of 5 independent experiments with 5 mice per experiment and plotted as dotted lines. The solid line represented the linear function of tumor growth during optimal B16 growth period (12–14 days post-challenge) in naïve mice. Equation shown was the formula describing volume change during period of optimal B16 growth. B. Pmel-1 T cell growth during expansion phase following adoptive transfer. Recipient mice were given cultured pmel-1 T cells and treated as indicated. Total pmel-1 splenocyte populations at different time points were determined as the average of 3 experiments combining 2–5 mice/time point as described in Materials and Methods and were plotted as dotted lines. The solid line represented the linear function describing cell growth during optimal pmel-1 T cell expansion (1–3 days post-transfer). Equations shown are the formulae describing net population changes during optimal period of pmel-1 T cell growth.
We sought to compare the relative rates of T cells and tumor cells and plotted the growth of T cells or tumor on a logarithmic scale as a linear function of time (Fig. 4). We derived the rate at which the cells grew by using the slopes of population changes during different periods. Comparisons of the maximum slopes (1–3 days for pmel-1 T cells and 12–14 days for B16 tumor) provide clear evidence that the rate of change of the pmel-1 population in combination with immunization and IL-2 was much greater than that of B16 tumor growth. Additional analysis from doubling time calculations also provided further evidence that the expansion of pmel-1 T cells with full treatment was approximately 4.7-fold faster than that of growing B16 tumor and resulted in an approximately 130-fold larger increase in total cell population compared to B16 growth over a 48-hour period of time (Tables I and II). Thus, the expansion rate of cytokine and antigen-stimulated adoptively transferred CD8+ T cells exceeds that of the aggressively growing mouse tumor, B16.
Discussion
In this study, we report on the kinetic response of adoptively transferred transgenic CD8+ T cells following immunization and IL-2 cytokine administration in the context of the treatment of an established subcutaneous B16 melanoma in recipient mice. The full activation of transferred cells by immunization with FP-hgp100 and IL-2 cytokine resulted in a >30-fold larger peak population than when transferring cells alone, a >35-fold greater population compared to immunizing with an irrelevant antigen and IL-2 support, and a >24-fold higher population peak compared to immunization alone without IL-2 cytokine support. Depletion of the cells at the peak of expansion significantly diminished the efficacy of B16 tumor treatment. In addition, variation in the number of pmel-1 cells used with vaccination and IL-2 directly correlated with anti-tumor efficacy.
From both the population kinetics and proliferation analysis using BrdU incorporation, it is clear that a majority of the cell expansion occurs during the first three days following adoptive transfer. However, net changes in the pmel-1 population during this period must also take in consideration cell redistribution and migration to the spleen. Immunization and IL-2 could alter the migration and survival of adoptively transferred pmel-1 T cells, especially in the period just after adoptive transfer. Studies on the early kinetics of endogenous lymphocytes in response to superantigen SEB have shown that CD8+ cell populations were transiently decreased in the blood and spleen and were increased in the lymph nodes during antigen presentation and cell activation within the first 24 hours (22). Following the first 24 hours, there was rapid equilibration of the cells amongst the three compartments. In our own studies, we started measurements of T cell distribution after the first 24 hours – we did not see evidence of selective distribution of the pmel-1 cells(23).
When describing the T cell kinetic response, we must draw comparisons to previously described model systems utilizing similar TCR transgenic T cells to follow their progress in response to viral or non-self antigenic stimulation. Utilizing transgenic P14 T cells that were specific for a Db LCMV epitope, several studies from Ahmed et. al. have defined both endogenous (24) and adoptive transfer (16) responses to LCMV infection. The endogenous response measured in the Blatmann study showed an explosive expansion of antigen-reactive T cells following LCMV infection resulting in a calculated >50,000-fold net increase in the reactive cell population during the 8-day expansion phase. Their study further concluded that the endogenous and donor T cell responses were similar as evidenced by results that naïve donor P14 cells could similarly expand >1,000-fold (16). A separate study utilizing transgenic T cells reactive to the male antigen (17) also showed a 51-fold net increase of the transferred cell population following immunization. More recently, Van Stipdonk et al. revealed that adoptively transferred ova-specific T cells went rapid proliferation upon brief antigen stimulation (25). The expansion observed in our B16 tumor treatment model also reflected a very rapid expansion in response to immunization. Despite the differences in total numbers and expansion potential, these models all clearly show that the T cell response to viral infection and immunization occurs at an extremely rapid pace.
Our studies with the addition of IL-2 administration in combination with immunization showed significant improvement on overall T cell kinetics and tumor treatment efficacy compared to cells alone. Blatmann et al has studied the endogenous T cell response to LCMV infection in combination with IL-2 administration. The conclusion of these studies was that administration of IL-2 during the expansion phase of an antiviral immune response was detrimental to the CD8+ T cell response resulting in an increase in cell death during the contraction phase and had no effect on the overall cell expansion (24). Our own results, utilizing a 80-times higher dose of IL-2 (600,000IU twice daily), suggested that exposure to the cytokine following immunization resulted in a much greater level of proliferation resulting in a population peak that is >24-fold higher than without cytokine. In terms of cell depletion, our study also showed an increase in the rate of cell death during the contraction phase with IL-2 administration (Table I). However, the final transgenic cell count at day 11 was found to be greater in recipients receiving IL-2, presumably as a result of the differences in the total peak population at day 5 (Fig. 1B). Previous studies have also suggested that IL-2 is quickly excreted from the body within 2 hours following i.p. injection in mice (26). This rapid cytokine depletion may render an exogenously administered IL-2 at the dose of 15,000 IU per injection ineffectual to influence the overall T cell proliferative response. Our own investigation into dosing of IL-2 has suggested that even at 150,000 IU (1/4th our standard dose) the efficacy of tumor treatment is unaffected (data not shown).
While many investigators have previously reported on the efficacy of adoptive transfer models to treat tumors in mice, most of these studies have only addressed the efficacy of the transferred T cells (8, 11, 12, 27–29). Arguments for the use of adoptive cell transfer alone have suggested that it alleviates potential concerns of detrimental and complicating use of adjuvant, cytokines, or immunization. However, in the final analysis, the most compelling argument for utilizing other factors to complement cell transfer is its clear ability to increase the efficacy of tumor treatment. Results from this study clearly showed that complementation of adoptive cell therapy with immunization and IL-2 resulted in a profound difference in treatment of well-established B16 melanoma, which was mediated by the generation of a greatly increased tumor-reactive cell population.
Overcoming the continuous growth of an established tumor mass is the primary goal for adoptive immunotherapy. Speculation by Hanson, et. al. has suggested that for treatment of large tumors there “may be an insufficient tumor-specific T cell response, which results in a detrimental kinetic disparity between the growth of the malignancy and the accumulation of host anti-tumor effector cells.” (8) Considering the results from our study, the rate of expansion for adoptive transfer of cells alone was approximated to be slightly slower than the growth of untreated B16 tumor. Thus, it is not surprising that only small tumors could be successfully treated in studies investigating cell transfer alone (8). The rate of T cell expansion after adoptive cell transfer with immunization and IL-2 far exceeded he rate at that even the highly aggressive B16 tumor could grow. Our analysis of growth rates indicated that the expansion differential of pmel-1 T cell proliferation with full treatment was occurring approximately 4.7-fold faster than that of growing B16 tumor and resulted in an approximately 130-fold larger increase in total cell population compared to B16 growth over a 48-hour period. Although this initial rate of expansion by T cells was sufficient to destroy tumors in these settings, high levels of anti-tumor T cells were not sustained after the first week. We therefore believe that one of the keys to the improvement of adoptive therapy for cancer will depend on finding methods to sustain or prolong the proliferation and activity of the tumor-reactive cells.
We have observed greater than a 50% response rate in patients with metastatic melanoma who received cultured tumor-reactive lymphocytes along with high dose of IL-2 in the lymphodepleted setting (14, 15). In this study, we have clearly shown that the successful use of adoptively transferred T cells requires proper in vivo activation of the transferred T cells (19). However, in clinical trials where bulk TIL are transferred, it would be difficult to measure how a heterogeneous population with varieties of antigen specificities would respond to effective stimulation. Interestingly, in some of the responders, lymphocytosis was observed and presumably induced by endogenous tumor antigen.
In summary, the observations in this report clearly indicate that adoptive cell transfer accompanied by immunization and IL-2 can result in a dramatic increase in proliferation of the transferred T cell population. These large numbers of anti-tumor T cells can mediate the extended regression of well-established B16 melanoma. Our results also suggest a need for additional measures to sustain the high level of tumor-reactive T cells following the initial proliferation response. One possibility to improve the sustenance of transferred T cells is the substitution of IL-2 with other growth cytokines, such as IL-15 that may not induce increased cell death or the induction of T regulatory cells that is attributed to IL-2 during cell contraction (30). Alternatively, continuous immunization against the targeted tumor-associated antigens and cytokine exposure may serve to reactivate the contracting T cell population. Analysis of T cell kinetics is but one aspect correlating to the efficacy of the adoptive cell therapy and provides us with a model by which we may be able to rationally design and assess future strategies in clinical adoptive cell therapies.
Footnotes
This research was supported by the Intramural Research Program of the NIH, NCI and was performed in partial fulfillment of a Ph.D. in Biochemistry (to D.C.P.) at the George Washington University, Washington, D.C.
References
- 1.Blattman JN, Greenberg PD. Cancer immunotherapy: a treatment for the masses. Science. 2004;305:200–5. doi: 10.1126/science.1100369. [DOI] [PubMed] [Google Scholar]
- 2.Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3:630–41. doi: 10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- 3.Zhou X, Jun dY, Thomas AM, et al. Diverse CD8+ T-cell responses to renal cell carcinoma antigens in patients treated with an autologous granulocyte-macrophage colony-stimulating factor gene-transduced renal tumor cell vaccine. Cancer Res. 2005;65:1079–88. [PubMed] [Google Scholar]
- 4.Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65:3437–46. doi: 10.1158/0008-5472.CAN-04-4262. [DOI] [PubMed] [Google Scholar]
- 5.Niethammer AG, Xiang R, Becker JC, et al. A DNA vaccine against VEGF receptor 2 prevents effective angiogenesis and inhibits tumor growth. Nat Med. 2002;8:1369–75. doi: 10.1038/nm1202-794. [DOI] [PubMed] [Google Scholar]
- 6.Cassetti MC, McElhiney SP, Shahabi V, et al. Antitumor efficacy of Venezuelan equine encephalitis virus replicon particles encoding mutated HPV16 E6 and E7 genes. Vaccine. 2004;22:520–7. doi: 10.1016/j.vaccine.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–75. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanson HL, Donermeyer DL, Ikeda H, et al. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 2000;13:265–76. doi: 10.1016/s1074-7613(00)00026-1. [DOI] [PubMed] [Google Scholar]
- 9.Marzo AL, Lake RA, Robinson BW, Scott B. T-cell receptor transgenic analysis of tumor-specific CD8 and CD4 responses in the eradication of solid tumors. Cancer Res. 1999;59:1071–9. [PubMed] [Google Scholar]
- 10.Shrikant P, Mescher MF. Control of syngeneic tumor growth by activation of CD8+ T cells: efficacy is limited by migration away from the site and induction of nonresponsiveness. J Immunol. 1999;162:2858–66. [PubMed] [Google Scholar]
- 11.Cordaro TA, de Visser KE, Tirion FH, et al. Tumor size at the time of adoptive transfer determines whether tumor rejection occurs. Eur J Immunol. 2000;30:1297–307. doi: 10.1002/(SICI)1521-4141(200005)30:5<1297::AID-IMMU1297>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 12.Ryan MH, Bristol JA, McDuffie E, Abrams SI. Regression of extensive pulmonary metastases in mice by adoptive transfer of antigen-specific CD8(+) CTL reactive against tumor cells expressing a naturally occurring rejection epitope. J Immunol. 2001;167:4286–92. doi: 10.4049/jimmunol.167.8.4286. [DOI] [PubMed] [Google Scholar]
- 13.Overwijk WW, Tsung A, Irvine KR, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–86. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blattman JN, Grayson JM, Wherry EJ, et al. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat Med. 2003;9:540–7. doi: 10.1038/nm866. [DOI] [PubMed] [Google Scholar]
- 17.Veiga-Fernandes H, Walter U, Bourgeois C, McLean A, Rocha B. Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nat Immunol. 2000;1:47–53. doi: 10.1038/76907. [DOI] [PubMed] [Google Scholar]
- 18.Zimmerman C, Brduscha-Riem K, Blaser C, Zinkernagel RM, Pircher H. Visualization, characterization, and turnover of CD8+ memory T cells in virus-infected hosts. J Exp Med. 1996;183:1367–75. doi: 10.1084/jem.183.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer DC, Balasubramaniam C, Hanada K, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–16. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurosaka K, Takahashi M, Watanabe N, Kobayashi Y. Silent cleanup of very early apoptotic cells by macrophages. J Immunol. 2003;171:4672–9. doi: 10.4049/jimmunol.171.9.4672. [DOI] [PubMed] [Google Scholar]
- 22.Vasseur F, Le Campion A, Pavlovitch JH, Penit C. Distribution of cycling T lymphocytes in blood and lymphoid organs during immune responses. J Immunol. 1999;162:5164–72. [PubMed] [Google Scholar]
- 23.Palmer DC, Balasubramaniam S, Hanada K, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–16. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blattman JN, Antia R, Sourdive DJ, et al. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195:657–64. doi: 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. 2001;2:423–9. doi: 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 26.Cheever MA, Thompson JA, Kern DE, Greenberg PD. Interleukin 2 (IL 2) administered in vivo: influence of IL 2 route and timing on T cell growth. J Immunol. 1985;134:3895–900. [PubMed] [Google Scholar]
- 27.Granziero L, Krajewski S, Farness P, et al. Adoptive immunotherapy prevents prostate cancer in a transgenic animal model. Eur J Immunol. 1999;29:1127–38. doi: 10.1002/(SICI)1521-4141(199904)29:04<1127::AID-IMMU1127>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 28.Peng L, Shu S, Krauss JC. Treatment of subcutaneous tumor with adoptively transferred T cells. Cell Immunol. 1997;178:24–32. doi: 10.1006/cimm.1997.1124. [DOI] [PubMed] [Google Scholar]
- 29.Zimmermann C, Prevost-Blondel A, Blaser C, Pircher H. Kinetics of the response of naive and memory CD8 T cells to antigen: similarities and differences. Eur J Immunol. 1999;29:284–90. doi: 10.1002/(SICI)1521-4141(199901)29:01<284::AID-IMMU284>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 30.Klebanoff CA, Finkelstein SE, Surman DR, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci U S A. 2004;101:1969–74. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]