Molecular mechanisms of sulfasalazine-induced T-cell apoptosis (original) (raw)
Abstract
- Impaired apoptosis of T-lymphocytes is involved in the development of chronic inflammatory disorders. Previously we have shown that the anti-inflammatory drug sulfasalazine induces apoptosis in a murine T-lymphocyte cell line. The aims of the present study were to expand these observations to human systems and to analyse the molecular basis for sulfasalazine-induced apoptosis.
- Sulfasalazine induces apoptosis both in Jurkat cells, a human T-leukaemia cell line (ED50 value ∼1.0 mM), and in primary human peripheral blood T-lymphocytes (ED50 value ∼0.5 mM). In contrast SW620 colon carcinoma cells or primary human synoviocytes are not affected at these concentrations suggesting a cell type-specific sensitivity to sulfasalazine.
- Sulfasalazine triggers the mitochondrial accumulation of Bax and induces a collapse of the mitochondrial transmembrane potential (ΔΨm).
- Sulfasalazine causes cytochrome c release from mitochondria and subsequent activation of caspase-3 and downstream substrates. However, the pan-caspase inhibitor Z-VAD.fmk fails to inhibit sulfasalazine-induced apoptosis.
- Sulfasalazine stimulates mitochondrio-nuclear translocation of the novel apoptogenic factor _a_poptosis-_i_nducing _f_actor (AIF) and triggers large-scale DNA fragmentation, a characteristic feature of AIF-mediated apoptosis.
- Sulfasalazine-induced ΔΨm loss, AIF redistribution, and cell death are fully prevented by overexpression of Bcl-2.
- In conclusion, our data suggest that sulfasalazine-induced apoptosis of T-lymphocytes is mediated by mitochondrio-nuclear translocation of AIF and occurs in a caspase-independent fashion. Sulfasalazine-induced apoptosis by AIF and subsequent clearance of T-lymphocytes might thus provide the molecular basis for the beneficial therapeutic effects of sulfasalazine in the treatment of chronic inflammatory diseases.
Keywords: Sulfasalazine, chronic inflammatory diseases, rheumatoid arthritis, inflammatory bowel disease, AIF, caspase-independent apoptosis, mitochondria, Bax
Introduction
Homeostasis of the immune response requires tight regulation of proliferation and cell death. Useless or potentially autoreactive cells are deleted by programmed cell death (apoptosis) (Scaffidi et al., 1999). In the developing immune system autoreactive thymocytes undergo T-cell-receptor-mediated apoptosis upon encountering ‘self' antigens in the thymus (MacDonald & Lees, 1990). Apoptosis leads to safe clearance of unwanted cells during the resolution of inflammation by limiting the persistence of activated T-cells (Strasser et al., 1995), B-cells (McDonnell et al., 1989; Nisitani et al., 1993), granulocytes (Haslett, 1992), and macrophages (Munn et al., 1995). Accumulating evidence suggests that failure of apoptosis is a major mechanism responsible for progression of initially mild or self-limited diseases to more severe chronic inflammatory stages (Thompson, 1995; Anderson, 1996; Ravirajan et al., 1999). Chronic inflammatory diseases such as rheumatoid arthritis and inflammatory bowel disease have been shown to be associated with abnormally low levels of apoptosis of pathogenic leukocytes (Ina et al., 1995; Boirivant et al., 1999; Salmon et al., 1997; Mountz et al., 1994). Therefore, therapeutic induction of apoptosis holds the attraction to be effective in the treatment of these and other chronic inflammatory diseases (Thompson, 1995; Anderson, 1996; Ravirajan et al., 1999).
Cytotoxic drugs may induce apoptosis through different intracellular pathways such as death receptor systems and mitochondria dependent signals. Upon CD95-ligand/receptor interaction, caspase-8 is recruited to the receptor via the adapter molecule FADD, leading to autoactivation of caspase-8 and subsequent activation of downstream caspases (Nagata, 1997; Walczak & Krammer, 2000). Induction of CD95-ligand and CD95 expression after treatment with cytotoxic drugs such as doxorubicin have been described in a variety of tumour cell lines. Blockade of the CD95/CD95-ligand interaction by antagonistic antibodies may inhibit drug-induced apoptosis in some cases (Friesen et al., 1996; Fulda et al., 1997). However, drug-triggered apoptosis may also occur independently of the CD95-system (Eischen et al., 1997; Newton & Strasser, 2000).
Mitochondrial membrane permeabilization is considered to be one of the initial events of the apoptotic process, including cell death induced by chemotherapeutic drugs. Opening of the mitochondrial permeability transition pore, which is under the control of members of the Bcl-2 family, may culminate in the permeabilization of the outer mitochondrial membrane and the release of potentially apoptogenic proteins such as cytochrome c and AIF from the intermembrane space (Kroemer, 1997; Green & Reed, 1998; Gross et al., 1999). Cytosolic cytochrome c binds to Apaf-1 in a ternary complex with caspase-9, leading to activation of caspase-9, which in turn activates caspase-3 (Li et al., 1997). Caspase-3 substrates include poly(ADP-ribose)polymerase (PARP) (Nicholson et al., 1995; Tewari et al., 1995) and ICAD/DFF-45 (Enari et al., 1998; Liu et al., 1997). Cleavage of ICAD (_i_nhibitor of the _c_aspase-_a_ctivated _D_Nase) leads to activation of CAD and cleavage of DNA into characteristic oligonucleosomal-length fragments (Enari et al., 1998).
AIF (_a_poptosis-_i_nducing _f_actor) was more recently cloned and identified as a mitochondrial intermembrane space protein with homology to bacterial NADH oxidoreductases. In response to apoptotic stimuli AIF is released, migrates to the nucleus and participates in the induction of chromatin condensation, the exposure of phosphatidylserine in the outer leaf of the plasma membrane, and the dissipation of the mitochondrial transmembrane potential. These effects seem to be caspase-independent, since none of them are prevented by the broad spectrum caspase inhibitor Z-VAD.fmk and are independent of the apoptosome complex (Susin et al., 1999, 2000; Ferri et al., 2000).
Sulfasalazine was synthesized in 1942 to combine an antibiotic, sulfapyridine, and an anti-inflammatory agent, 5-aminosalicylic acid (5-ASA) (Svartz, 1941). Sulfasalazine was the first drug with proven efficacy for ulcerative colitis (Riis et al., 1973). Controlled trials demonstrated a significant therapeutic benefit also in the treatment of rheumatoid arthritis (van der heijde et al., 1989; Neumann et al., 1983). Sulfasalazine and other salicylates as well as corticosteroids are still mainstays in the therapy of inflammatory bowel disease and rheumatoid arthritis. How sulfasalazine achieves its therapeutic effect is still not completely understood. Previously, we have shown that sulfasalazine interferes with NF-κB/Rel activation, most likely by interference with the ATP-binding site of the IκB-inducing-kinases, IKKα and IKKβ (Wahl et al., 1998; Weber et al., 2000; Liptay et al., 1999). In addition, we observed that prolonged incubation with sulfasalazine or higher doses of sulfasalazine-induced apoptosis in a murine T-lymphocyte cell line (Liptay et al., 1999).
In the present study we extended these observations to human systems and analysed the molecular mechanisms for sulfasalazine-induced apoptosis. We found that sulfasalazine perturbs mitochondrial function, most likely by mitochondrial accumulation of Bax. Sulfasalazine treatment leads to reduction of the mitochondrial transmembrane potential (ΔΨm), cytochrome c release, activation of caspase-3 and cleavage of known caspase-3 substrates. However, caspase activity seems not to be required for sulfasalazine-induced cell death, since Z-VAD.fmk pre-treated cells die equally effective. Rather the effect of sulfasalazine appears to be mediated by AIF. Sulfasalazine induces AIF release from the mitochondrial intermembrane space and nuclear translocation of AIF, which is blocked by Bcl-2 overexpression, but not affected by caspase inhibition. These data indicate that sulfasalazine-induced apoptosis is mediated by mitochondrio-nuclear translocation of AIF. Sulfasalazine-induced apoptosis by AIF and subsequent clearance of T-lymphocytes might thus provide the molecular basis for the beneficial therapeutic effects of sulfasalazine in the treatment of chronic inflammatory diseases.
Methods
Cell culture and treatments
Jurkat cells, a human acute T-cell leukaemia cell line, and Bcl-2-Jurkats, Jurkat cells stably transfected with Bcl-2 (a kind gift from Georg Häcker, Department of Microbiology, Technical University of Munich, Germany), OKT-3-sensitive Jurkat cells (a kind gift from Gudrun Strauß, Dept. of Paediatrics, University of Ulm, Germany) were grown under standard conditions in RPMI 1640 medium supplemented with 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 2 mM L-Glutamin (all GIBCO Life technologies, Eggenstein, Germany) and 10% FCS (Biochrom, Berlin, Germany). SW620 cells, a human colon carcinoma cell line, and primary synoviocytes (a kind gift from Rolf Brenner, Department of Orthopaedics, University of Ulm, Germany) were grown in DMEM (GIBCO Life technologies) supplemented as described above. Sulfasalazine (Sigma, Deisenhofen, Germany) was freshly dissolved in culture media and added to the cultures at the indicated concentrations and for the indicated time periods. The following apoptosis inducers or inhibitors were added: TNFα (150 U ml−1, Sigma), PHA M (5 μg ml−1, Difco, Detroit, MI, USA), staurosporin (1 μM, Sigma), OKT-3 monoclonal antibody (a kind gift from Gudrun Strauß, Department of Paediatrics, University of Ulm, Germany), neutralizing mouse anti-CD95L monoclonal antibody NOK-1 (100 μg ml−1, Becton Dickinson, Heidelberg, Germany), activating mouse anti-CD95 monoclonal antibody (100 ng ml−1, Upstate Tech.), bongkrekic acid (50 μM, kindly provided by J.A. Duine, University of Delft, Delft, The Netherlands), Z-VAD.fmk (100 μM, Bachem, Heidelberg, Germany).
Isolation and culture of human primary peripheral blood T-lymphocytes
Primary peripheral mononuclear cells of four healthy donors were isolated using Ficoll gradient centrifugation (Pharmacia, Freiburg, Germany). T-lymphocytes were isolated by rosetting with neuraminidase (Boehringer, Mannheim, Germany) treated sheep erythrocytes. The percentage of T-cells was greater than 90% as determined by flow cytometry after staining for CD-3 (FACSCalibur, Becton Dickinson). Cells were seeded at a density of 106 cells per ml in RPMI 1640 media supplemented with 10% FCS.
Determination of apoptosis
For quantitative determination of apoptosis, cells were treated as indicated and analysed by flow cytometry using Cell Quest software (Becton Dickinson). Apoptotic cells were identified on the basis of their characteristic change in the FSC/SSC profile as described (Friesen et al., 1996; Carbonari et al., 1994).
For analysis of chromatin condensation Jurkat T-cells were treated as indicated. Five × 104 cells were seeded on poly-lysine coated cover slips. Cells were fixed with 4% paraformaldehyde for 10 min at 37°C, washed in 100 mM NaCl, 10 mM EDTA, 10 mM Tris pH 7.0 and stained for 1 h at 37°C in washing solution containing 0.1 μg ml−1 4,6-diamidino-2-phenylindole (DAPI). Chromatin condensation was assayed under a fluorescence microscope mounted on an inverted microscope with an 10×63 oil objective (Carl Zeiss, Inc., Thornwood, NY, U.S.A.).
Hypodiploid DNA was determined as described (Liptay et al., 1999). For analysis of oligosomal DNA fragmentation, 2×107 cells were treated for 24 h as indicated. Cells were washed, centrifuged and resuspended in 10 mM Tris pH 8.0, 400 mM NaCl, 2 mM EDTA, 1% SDS. 200 μg ml−1 Proteinase K was added and cells were incubated at 50°C over night. After protein precipitation with NaCl, DNA was precipitated. Twenty μg DNA were run on a 2% agarose gel. DNA was visualized by ethidium bromide staining and UV illumination.
For quantitative analysis of phosphatidylserine exposure Jurkat cells were treated as indicated. Cells were washed once in PBS and resuspended in annexin V binding buffer (10 mM HEPES pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Cells were adjusted to 106 cells per 100 μl binding buffer and Fluorescein isothiocyanate (FITC)-conjugated annexin V (Becton Dickenson) was added to a final concentration of 25 mg ml−1. After 15 min incubation cells were analysed by flow cytometry.
Preparation of whole cell, mitochondrial, cytoplasmic and nuclear protein extracts
For whole cell extracts cells were lysed for 10 min at 4°C in 150 mM NaCl, 50 mM Tris pH 7.5, 0.05% SDS, 1% NP40 and 0.4 mM PMSF. Isolation of mitochondria by ultracentrifugation and extraction of mitochondrial proteins was performed as described previously (Fulda et al., 1999). Cytoplasmic (Gross et al., 1998) and nuclear protein extracts (Schmid et al., 1991) were prepared as described. Protein concentration was determined by the Bradford method.
Western blot analysis
Western blot analyses were done as previously described (Wahl et al., 1998; Fulda et al., 1999). Equal protein load was controlled by Ponceau red staining of membranes. The following antibodies were used: anti-CD95L (1 : 250, Becton Dickenson), anti-TRAIL (1 : 1000, Becton Dickenson), anti-β-actin (1 : 5000, Sigma), anti-cytochrome c (1 : 3000, Becton Dickenson), anti-Bcl-2 (1 : 500, Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), anti-Caspase-3 (1 : 1000, Becton Dickenson), anti-PARP (1 : 1000, Becton Dickenson), anti-ICAD/DFF-45 (1 : 1000, Upstate Biotechnology, Lake Placid, NY, U.S.A.), anti-AIF (1 : 2000, was previously described in Susin et al., 1999 and anti-Bax (1 : 1000, Calbiochem, Bad Soden, Germany), goat anti-mouse IgG (1 : 5000, Amersham), and goat anti-rabbit IgG (1 : 5000, Amersham). ECL (Amersham) or Lumilight (Boehringer Mannheim) was used for detection.
Determination of mitochondrial membrane potential
For determination of mitochondrial potential 5×105 per ml cells were incubated with 3,3′-dihexyloxacarbocyanide iodine (DiOC6(3)), 40 nM, Molecular Probes, Inc., Eugene, OR, U.S.A.) for 15 min at 37°C and analysed by flow cytometry (Becton Dickenson).
Preparation of agarose plugs and pulse field gel electrophoresis
For detection of large-scale DNA fragmentation 2.5×105 cells were resuspended in 40 μl PBS and mixed with 40 μl 1.2% InCert Agarose, FMC. The plugs were digested twice for 24 h in 0.5 M EDTA pH 9.5, 1% SLS with 2 mg/ml proteinase K (Boehringer Mannheim) and washed in 10T1E. Pulse field gel electrophoresis was carried out in a Biometra Rotaphor System for 23 h at 13°C in 0.25× TBE with 30–5 s log, 120–110° lin, 180–120 V log, Rotor Speed 7.1, 1% Seakem GTG Agarose, FMC. Molecular weight standards were from Gibco (25 kb-ladder) and New England Biolabs (Yeast molecular weight standard).
Immunofluorescence staining
Five times 104 cells were seeded on poly-lysine coated cover slips and treated as indicated. Cells were fixed in 4% paraformaldehyde for 60 min, permeabilized with 0.1% SDS for 10 min, and blocked with 3% BSA, 0.1% Tween 40 for 20 min. Rabbit anti-AIF polyclonal antibody (as previously described in Susin et al., 1999) was added (1 : 250) for 30 min and revealed by a Cy3-conjugated goat anti-rabbit IgG antibody (1 : 1000, Dianova-Immunotech., Hamburg, Germany). Slides were analysed using a confocal laser scanning microscope mounted on an inverted microscope with a 10×63 oil objective (Carl Zeiss, Inc., Thornwood, NY, U.S.A.). Control experiments without the first or without the secondary antibody confirmed that all detectable fluorescence was specific (not shown).
Results
Sulfasalazine induces apoptosis in human Jurkat T-lymphocytes
To test whether sulfasalazine induces apoptosis in human T-lymphocytes, Jurkat T-cells were incubated with increasing doses of sulfasalazine for 24 h and analysed by forward scatter/sideward scatter (FSC/SSC) analysis in a fluorescence-activated cell sorter (FACS). Apoptotic lymphocytes were identified on the basis of their characteristic forward/sideward scatter changes (Carbonari et al., 1994). A concentration dependent induction of apoptosis was found with 45% dead cells at 1.0 mM and over 80% dead cells at 2.0 mM sulfasalazine (Figure 1A). Jurkat cells were incubated with 2.0 mM sulfasalazine for varying time periods (0, 1, 2, 4, 8 and 24 h) followed by FACS analysis. As shown in Figure 1B induction of apoptosis by sulfasalazine is fast leading to more than 60% apoptotic cells after 8 h. Jurkat T-cells treated with sulfasalazine displayed typical morphological and biochemical features of apoptotic cells. Treatment with 2.0 mM sulfasalazine for 24 h induced chromatin condensation and formation of nuclear bodies in over 80% of the cells as determined by staining with the intercalating dye DAPI (4,6-diamidino-2-phenylindole) (Figure 1C). Sulfasalazine treatment (2.0 mM) was associated with a loss of DNA with a sub-G1 peak in 61% of Jurkat cells after 24 h (Figure 1D). Oligonucleosomal DNA fragmentation was detected in sulfasalazine-treated cells (1.0 and 2.0 mM), but not in untreated or TNFα (150 U ml−1) treated cells (Figure 1E). Phosphatidylserine exposure was induced by sulfasalazine (2.0 mM) with 36% annexin V positive cells after 6 h, and 85% positive cells after 24 h treatment (Figure 1F). Taken together, these results clearly demonstrate that sulfasalazine induces apoptosis in Jurkat T-lymphocytes. Interestingly, incubation of a human colon carcinoma epithelial cell line, SW620 cells (Figure 1G), or primary human synoviocytes (Figure 1H) with 2.0 mM sulfasalazine for 24 h did not result in any significant induction of cell death (data not shown), suggesting a cell type-specific sensitivity to sulfasalazine.
Figure 1.
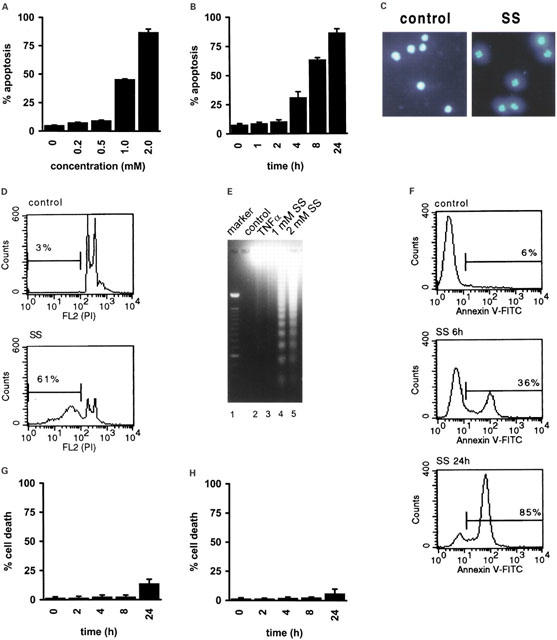
Sulfasalazine induces apoptosis in Jurkat T-lymphocytes. (A) Jurkat cells were incubated for 24 h with increasing concentrations of sulfasalazine, (B) or with 2.0 mM sulfasalazine for the indicated time periods. The percentage of apoptotic cells was determined by FACS analysis on the basis of their characteristic forward/sideward light scatter changes. Data are obtained from three independent experiments done in duplicate and are presented as mean and standard error of the mean (s.e.mean). (C) Sulfasalazine induces chromatin condensation. Jurkat cells were treated for 24 h with medium (control) or 2.0 mM sulfasalazine (SS), and examined under a fluorescence microscope after DAPI staining. Representative fields of one out of three independent experiments are shown. (D) Sulfasalazine induces DNA loss. Jurkat cells were treated for 24 h with (SS) or without (control) 2.0 mM sulfasalazine, stained with propidium iodide (PI), and analysed by flow cytometry. Numbers above the histogram markers indicate the percentage of apoptotic nuclei (broad hypodiploid peak) in a representative experiment out of four. (E) Sulfasalazine induces oligonucleosomal DNA fragmentation. Jurkat cells were treated for 24 h with medium (lane 2), 150 U ml−1 TNFα (lane 3), 1 mM (lane 4) or 2 mM sulfasalazine (lane 5). Genomic DNA was extracted and analysed on an ethidium bromide stained agarose gel. As molecular-weight marker a 100 bp DNA marker was used (lane 1). (F) Sulfasalazine induces phosphatidylserine exposure. Jurkat cells were treated with medium (control), or 2.0 mM sulfasalazine for 6 or 24 h as indicated. Cells were stained with FITC-conjugated annexin V and analysed by FACS. Numbers above the histogram markers indicate the percentage of apoptotic cells in a representative experiment out of four. Viability of (G) SW620 cells, a human colon carcinoma cell line, and (H) human primary synoviozytes is not affected by sulfasalazine. Cells were incubated with 2 mM sulfasalazine for the indicated time periods. Cell death was determined by trypan blue exclusion.
Sulfasalazine induces apoptosis in human primary peripheral blood T-lymphocytes
Resting peripheral blood T-lymphocytes are relatively resistant to apoptosis induced by physiological regulators such as CD95 triggering. However, upon mitogenic activation and extended culture in vitro, human peripheral blood T-lymphocytes become sensitive to CD95-, TCR- or PHA-mediated apoptosis (Klas et al., 1993; Wesselborg et al., 1993). Since sulfasalazine-induced apoptosis of Jurkat T-lymphocytes did not require additional T-cell activation we were interested to see whether sulfasalazine would be able to induce apoptosis in primary T-lymphocytes. Peripheral blood T-lymphocytes were isolated and a purity of over 90% was determined by CD-3 staining and FACS analysis. After 24 h purified T-cells were incubated with increasing doses of sulfasalazine for an additional 24 h. Twenty per cent of peripheral blood T-lymphocytes underwent spontaneous apoptosis. Addition of sulfasalazine resulted in over 50% apoptotic cells at 0.5 mM, and over 95% apoptotic cells at 1 mM sulfasalazine, indicating that primary T-lymphocytes are even more sensitive to sulfasalazine than Jurkat T-cells (compare Figure 2A,B to Figure 1A,B). Additional activation of primary T-lymphocytes with PHA (5 μg ml−1) or TNFα (150 U ml−1) did not result in an increased susceptibility to apoptosis induction (data not shown).
Figure 2.
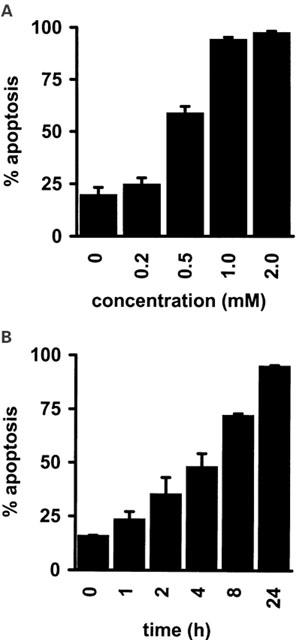
Sulfasalazine induces apoptosis in human primary peripheral blood T-lymphocytes. Peripheral blood T-lymphocytes were isolated (purity over 90%) and (A) incubated for 24 h with the indicated concentrations of sulfasalazine, or (B) with 2.0 mM sulfasalazine for the indicated time periods. The percentage of apoptotic cells was determined as described in Figure 1A. Data were obtained from three independent experiments done in duplicate and are presented as mean and standard error of the mean (s.e.mean).
Sulfasalazine induces apoptosis independently of the CD95-system
Drug-induced apoptosis can be mediated by the CD95-ligand/receptor-system in many cell types (Friesen et al., 1996; Fulda et al., 1997). Cytotoxic drugs such as doxorubicin, cisplatinum, and VP-16 were found to induce CD95L expression, thereby leading to apoptosis via CD95 triggering (Friesen et al., 1996; Fulda et al., 1997). More recently, it was demonstrated that doxorubicin also activates expression of TRAIL (Herr et al., 1999), another member of the CD95/TNFα superfamily, which mediates apoptosis in various tumour cell lines (Griffith & Lynch, 1998). Therefore, we were interested whether sulfasalazine-induced apoptosis involves the CD95- or TRAIL-ligand/receptor-systems. Jurkat T-lymphocytes were treated with 2.0 mM sulfasalazine for varying times and whole cell protein extracts were subjected to Western blot analysis. Sulfasalazine treatment did not induce TRAIL or CD95L expression (Figure 3A). Receptor/ligand interaction and activation of the CD95-system can be blocked by antagonistic antibodies as previously shown for CD95-mediated drug-induced apoptosis (Friesen et al., 1996). Therefore, Jurkat T-cells were preincubated with a neutralizing antibody (NOK-1) for 1 h followed by treatment with 2.0 mM sulfasalazine for an additional 24 h. As shown in Figure 3B this neutralizing antibody did not inhibit sulfasalazine-induced apoptosis. As a control for its efficacy, NOK-1 greatly reduced activation-induced cell death of Jurkat cells triggered by CD-3 crosslinking (Figure 3B). These data suggest that sulfasalazine-induced apoptosis is independent of the CD95-system.
Figure 3.
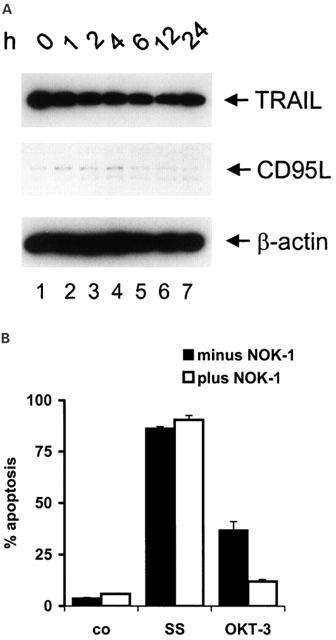
Sulfasalazine induces apoptosis independent of the CD95-system. (A) Sulfasalazine does not increase TRAIL or CD95L expression. Jurkat T-lymphocytes were treated with 2.0 mM sulfasalazine for varying periods as indicated. Expression of TRAIL and CD95L was determined by Western blot analysis of whole cell protein extracts using specific monoclonal antibodies. Detection of β-actin expression was used to control equal gel loading. (B) Lack of inhibition of sulfasalazine-induced apoptosis by a CD95L neutralizing antibody (NOK-1). Jurkat cells were treated for 24 h with medium (co), 2.0 mM sulfasalazine (SS) or 100 μg ml−1 OKT-3, in the presence or absence of NOK-1 (100 μg ml−1), which was added 1 h before SS. The percentage of apoptotic cells was determined as described in Figure 1A. Data are obtained from two independent experiments done in duplicates and are presented as mean and standard error of the mean (s.e.mean).
Sulfasalazine perturbs mitochondrial function
Since mitochondria have been implicated in various apoptotic pathways we next asked whether sulfasalazine-induced apoptosis is accompanied by alterations of mitochondrial functions. After treatment with 2.0 mM sulfasalazine for 0, 1, 2, 6, 12 or 24 h Jurkat T-cells were incubated with the potential-sensitive fluorochrome DiOC6(3) and analysed by FACS. A reduction in the mitochondrial transmembrane potential (ΔΨm) identified by reduced DiOC6(3) staining, was noted in a time dependent manner in sulfasalazine-treated cells (Figure 4A). To test if these mitochondrial alterations involve opening of the mitochondrial permeability transition (PT) pore, we next tested the effect of bongkrekic acid (BA), a specific inhibitor of the PT pore (Susin et al., 1997). Addition of BA inhibited the sulfasalazine triggered ΔΨm loss, indicating that mitochondrial alterations by sulfasalazine involved opening of PT pores (Figure 4B). Upon permeability transition, mitochondria have been shown to release apoptogenic factors such as cytochrome c from the mitochondrial intermembrane space into the cytoplasm. Therefore, Jurkat cells were treated for varying times with 2.0 mM sulfasalazine and mitochondrial or cytoplasmic protein extracts were used in Western blot experiments to analyse the subcellular distribution of cytochrome c. Following incubation with sulfasalazine a decrease in mitochondrial cytochrome c levels could be detected after 6 h, while an increase of cytoplasmic cytochrome c was already detectable after 2 h treatment (Figure 4C).
Figure 4.
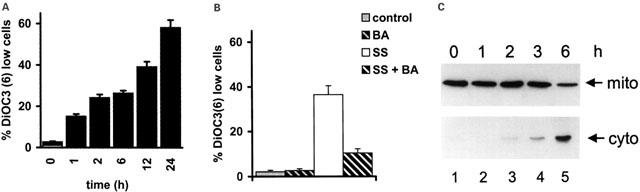
Sulfasalazine perturbs mitochondrial function. (A) Sulfasalazine induces a reduction of mitochondrial transmembrane potential (ΔΨm). Jurkat T-lymphocytes were incubated with 2.0 mM sulfasalazine for the indicated periods. ΔΨm was determined by staining with the potential-sensitive fluorochrome DiOC6(3) and FACS analysis. Data are given as mean and standard deviation of triplicates. Similar results were obtained in three independent experiments. (B) Loss of ΔΨm is inhibited by bongkrekic acid (BA). Jurkat cells were treated for 12 h with medium as control (grey bars) or 2.0 mM sulfasalazine (SS) (white bars), in the presence (hatched bars) or absence (plain bars) of 50 μM BA. ΔΨm was determined by staining with the potential-sensitive fluorochrome DiOC6(3) and FACS analysis. Data are given as mean and standard deviation of triplicates. Similar results were obtained in two independent experiments. (C) Sulfasalazine induces cytochrome c release from the mitochondria. Jurkat cells were treated with 2.0 mM sulfasalazine for the indicated times and mitochondrial (mito) and cytoplasmic (cyto) protein extracts were prepared. The subcellular distribution of cytochrome c was determined by Western blot analysis using a specific monoclonal antibody against cytochrome c.
Overexpression of Bcl-2 inhibits sulfasalazine-induced apoptosis
Overexpression of Bcl-2 has been found to confer resistance to chemotherapy-induced apoptosis (Dole et al., 1994). Bcl-2 inhibits opening of mitochondrial permeability transition pores (Susin et al., 1996) and/or stabilizes the barrier function of the outer mitochondrial membrane (Kluck et al., 1997; Yang et al., 1997). Therefore, we were interested in determining whether Bcl-2 is able to inhibit sulfasalazine-induced cell death. Wild-type or stably Bcl-2 transfected Jurkat T-cells were incubated with increasing doses of sulfasalazine for 24 h. Expression of Bcl-2 was confirmed by Western blot analysis (Figure 5A). Bcl-2 overexpression significantly reduced induction of apoptosis by sulfasalazine (Figure 5B). Inhibition of apoptosis seems to occur by stabilizing mitochondrial function, since overexpression of Bcl-2 strongly inhibited disruption of the mitochondrial transmembrane potential (ΔΨm) as determined by staining of sulfasalazine-treated Jurkat T-cells with DiOC6(3) and FACS analysis (Figure 5C).
Figure 5.
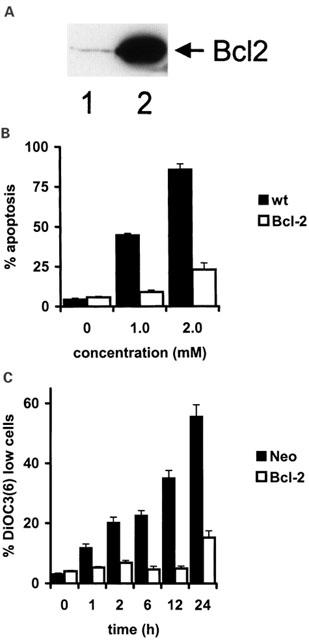
Overexpression of Bcl-2 inhibits sulfasalazine induced apoptosis. (A) Bcl-2 expression of wild-type (lane 1) and stably Bcl-2 transfected Jurkat cells (lane 2) was determined by Western blot analysis using a Bcl-2 specific polyclonal antibody. (B) Wild-type (black bars) and Bcl-2 transfected (white bars) Jurkat cells were incubated for 24 h with increasing doses of sulfasalazine as indicated. The percentage of apoptotic cells was determined as described in Figure 1A. Data are obtained from three independent experiments done in duplicate and are presented as mean and standard error of the mean (s.e.mean). (C) Bcl-2 inhibits sulfasalazine-induced loss of ΔΨm. Jurkat cells transfected with Bcl-2 (white bars) or vector only (black bars) were treated with 2.0 mM sulfasalazine for the indicated periods. ΔΨm was determined by staining with the potential-sensitive fluorochrome DiOC6(3) and FACS analysis. Data are given as mean and standard deviation of triplicates. Similar results were obtained in three independent experiments.
Sulfasalazine induces caspase activation, but caspase activity is not required for sulfasalazine induced apoptosis
Cytochrome c released from the mitochondria leads to binding of Apaf-1 in the cytoplasm and sequential activation of caspase-9 and caspase-3 (Li et al., 1997). Activated caspase-3 in turn cleaves downstream substrates such as poly(ADP-ribose)polymerase (PARP) (Nicholson et al., 1995; Tewari et al., 1995) and the more recently identified DNase inhibitor ICAD/DFF-45 (Enari et al., 1998; Liu et al., 1997). Since sulfasalazine treatment leads to ΔΨm breakdown and cytochrome c release we wondered whether sulfasalazine would induce the activation of caspase-3 and the cleavage of caspase-3 substrates. Jurkat T-cells were incubated for 3 and 6 h with 2.0 mM sulfasalazine and whole cell extracts were analysed by Western blotting. As a control, protein extracts of staurosporin treated Jurkat cells were used. After 3 h incubation with sulfasalazine caspase-3 (32 kDa) cleavage into an intermediate (20 kDa) and its active product (17 kDa) was observed (Figure 6A). In addition, sulfasalazine treatment resulted in proteolytic cleavage of two caspase-3 substrates. Full-length PARP (116 kDa) was cleaved into an 85 kDa fragment (Figure 6B), while ICAD/DFF-45 yielded a canonical 11 kDa product (Figure 6C).
Figure 6.
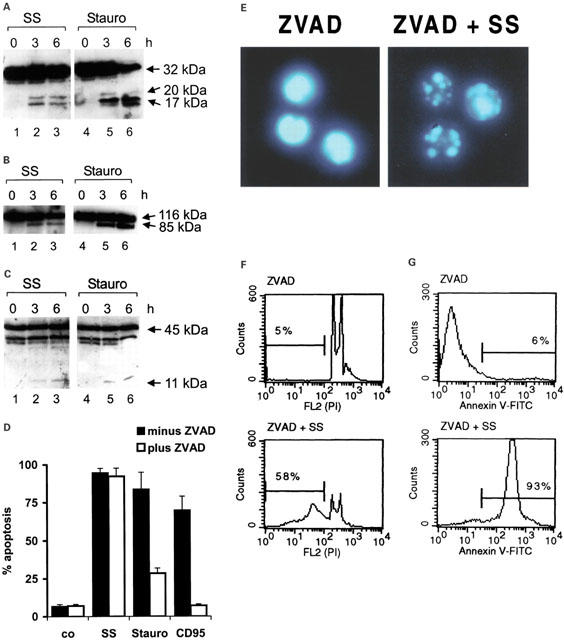
Sulfasalazine induces caspase activation, but caspase activity is not required for sulfasalazine-induced apoptosis. (A, B, C) Sulfasalazine induces cleavage of caspase-3, PARP and ICAD/DFF-45. Jurkat cells were incubated with medium or 2.0 mM sulfasalazine (SS) for 3 and 6 h. As control staurosporin (Stauro, 1 μM) treated cells were processed in parallel. Whole cell protein extracts were subjected to Western blot analysis for caspase-3, PARP and ICAD/DFF-45 processing using specific antibodies. (A) Cleavage of caspase-3 (32 kDa) resulted in a 20 kDa intermediate and a 17 kDa active subunit as indicated. Processing of (B) PARP (116 kDa) and (C) ICAD/DFF-45 (45 kDa) yielded 85 and 11 kDa products, respectively. (D) Inhibition of caspase activity does not inhibit sulfasalazine-induced apoptosis. Jurkat cells were pre-treated with (white bars) or without (black bars) Z-VAD.fmk (100 μM) for 1 h, followed by 24 h incubation with medium as control (co), 2.0 mM sulfasalazine (SS), 1 μM staurosporin (Stauro), and 100 ng ml−1 activating anti-CD95 antibody (CD95). The percentage of apoptotic cells was determined as described in Figure 1A. Data are obtained from three independent experiments done in duplicate and are presented as mean and standard error of the mean (s.e.mean). (E) Inhibition of caspase activity does not inhibit sulfasalazine-induced chromatin condensation. Jurkat cells were pre-treated for 1 h with Z-VAD.fmk (100 μM), followed by 24 h incubation with medium (ZVAD) or 2.0 mM sulfasalazine (ZVAD+SS). Cells were stained with DAPI and examined under a fluorescence microscope. Representative fields of one out of three independent experiments are shown. (F) Inhibition of caspase activity does not inhibit sulfasalazine-induced loss of DNA. Jurkat cells were treated as described under (E). Cells were stained with propidium iodide (PI) and analysed by flow cytometry. Numbers above the histogram markers indicate the percentage of apoptotic nuclei in a representative experiment out of four. (G) Inhibition of caspase activity does not inhibit sulfasalazine-induced phosphatidylserine exposure. Jurkat cells were treated as described under E. Cells were stained with FITC-conjugated annexin V and analysed by FACS. Numbers above the histogram markers indicate the percentage of apoptotic cells in a representative experiment out of three (compare to Figure 1C,D and F.)
To test whether caspase activity is required for sulfasalazine-induced apoptosis Jurkat T-cells were preincubated for 1 h with or without the wide-range caspase inhibitor Z-VAD.fmk, followed by 24 h treatment with sulfasalazine (2.0 mM), staurosporin (1 μM), or activating anti-CD95 antibody (100 ng ml−1) as control. As expected, pre-treatment with Z-VAD.fmk inhibited CD95- and staurosporin-induced apoptosis, while sulfasalazine-mediated cell death was not reduced by preincubation with Z-VAD.fmk (Figure 6D). In addition, Z-VAD.fmk did not prevent sulfasalazine-induced chromatin condensation, loss of DNA, or phosphatidylserine exposure (Figure 6E–G compare to Figure 1C, D and F).
These results indicate that sulfasalazine-triggered cytochrome c release leads to activation of caspase-3 and subsequent cleavage of known caspase-3 substrates. However, inhibition of caspases by Z-VAD.fmk did not prevent sulfasalazine-induced apoptosis, suggesting that other caspase-independent mechanisms are involved in sulfasalazine-mediated apoptosis.
Sulfasalazine leads to mitochondrio-nuclear translocation of AIF
AIF (_a_poptosis-_i_nducing _f_actor) was recently cloned as a mitochondrial intersperse membrane protein. In response to apoptotic stimuli AIF is liberated, migrates to the nucleus and participates in the induction of chromatin condensation, dissipation of the mitochondrial membrane potential, and phosphatidylserine exposure in a Z-VAD.fmk insensitive manner (Susin et al., 1999). Since sulfasalazine-induced apoptosis remarkably resembles these characteristics we next tested whether sulfasalazine leads to mitochondrial release and nuclear translocation of AIF. Jurkat T-cells were treated with medium as control or 2.0 mM sulfasalazine for varying times as indicated. To investigate the subcellular distribution of AIF mitochondrial, cytoplasmic and nuclear protein extracts were prepared and used for Western blot analysis. A specific AIF antibody recognizes two different transcription/translation products of ∼57 and ∼67 kDa (Susin et al., 1999). A decrease of mitochondrial AIF can be detected after 6 h, while an increase of cytoplasmic and nuclear AIF levels was observed after 3 h incubation with sulfasalazine (Figure 7A).
Figure 7.
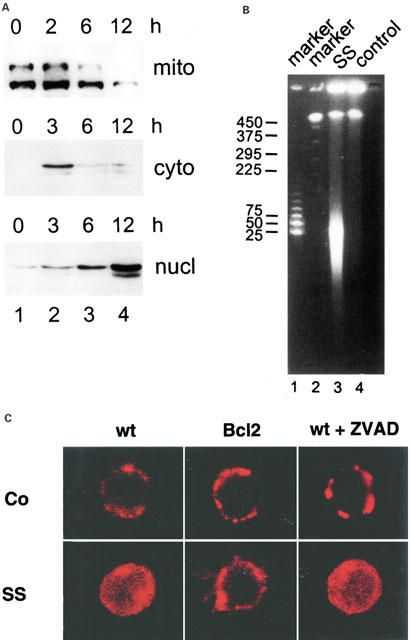
Sulfasalazine leads to mitochondrio-nuclear translocation of AIF. (A) Jurkat T-lymphocytes were incubated with 2.0 mM sulfasalazine for the indicated periods. Mitochondrial (mito), cytoplasmic (cyto) and nuclear extracts (nucl) were prepared. The subcellular distribution of AIF was determined by Western blot analysis using a specific polyclonal antibody against AIF which detects two different transcription/translation products of ∼57 and ∼67 kDa. (B) Sulfasalazine induces large-scale DNA fragmentation. High molecular-weight DNA of Jurkat cells treated for 24 h with 2.0 mM sulfasalazine (SS) or medium as control was analysed by pulse-field gel electrophoresis. Sizes of the molecular-weight markers are indicated in kbp. (C) Sulfasalazine-induced translocation of AIF is caspase-independent and inhibited by Bcl-2. Wild-type (wt) or Bcl-2 transfected (Bcl-2) Jurkat cells were incubated for 24 h with sulfasalazine (SS) or medium alone as control (co). Wild-type cells were incubated with SS in the presence (+ZVAD) or absence of Z-VAD.fmk (100 μM, added 1 h before SS). The subcellular distribution of AIF was determined by immunofluorescence microscopy using an AIF specific polyclonal antibody and a Cy-3 conjugated secondary antibody. The presented images reflect the dominant (>80%) phenotype of subcellular AIF distribution obtained under each condition.
A characteristic feature of AIF is its ability to induce large-scale DNA fragmentation, probably by activating a sessile nuclear DNase (Susin et al., 1999). Therefore, we analysed high-molecular weight DNA from control or sulfasalazine treated Jurkat T-cells by pulse-field gel electrophoresis. Sulfasalazine treated cells but not controls exhibited a strong signal at ∼50 kbp, indicative of large-scale DNA fragmentation (Figure 7B).
Sulfasalazine-induced mitochondrio-nuclear translocation of AIF was confirmed by immunofluorescence microscopy using an AIF specific polyclonal antibody and a Cy3-conjugated secondary antibody (red fluorescence). In untreated, wild-type Jurkat T-cells immunofluorescence detection of AIF revealed a punctuate cytoplasmic staining typical for mitochondrial localisation (Figure 7C). The same staining pattern was found in untreated Jurkat cells stably transfected with Bcl-2, or in wild-type Jurkat cells pre-treated with the caspase inhibitor Z-VAD.fmk. Treatment of wild-type Jurkat cells with 2.0 mM sulfasalazine for 24 h resulted in a diffuse distribution of AIF in the cytoplasm and within the nucleus. This AIF translocation was not prevented by pre-treatment with Z-VAD.fmk, yet was fully suppressed by Bcl-2 overexpression.
Sulfasalazine induces mitochondrial accumulation of Bax
Bax and Bax-like proteins are pro-apoptotic homologs of the Bcl-2 family known to form ion channels on isolated mitochondria (Schendel et al., 1998). Bax is both a membrane and cytoplasmic protein and there is increasing evidence that the cytoplasmic form undergoes a conformational change and translocates to mitochondrial membranes in response to apoptotic stimuli (Gross et al., 1998; Hsu et al., 1997). To investigate whether sulfasalazine induces mitochondrial translocation of Bax, Jurkat T-cells were treated for varying time periods with 2.0 mM sulfasalazine and mitochondrial protein extracts (the same as used in Figure 7A) were analysed by Western blotting. As shown in Figure 8 sulfasalazine treatment for 2 h induced an increase of mitochondrial Bax. These results suggest Bax translocation to be an initial event in sulfasalazine induced apoptosis.
Figure 8.
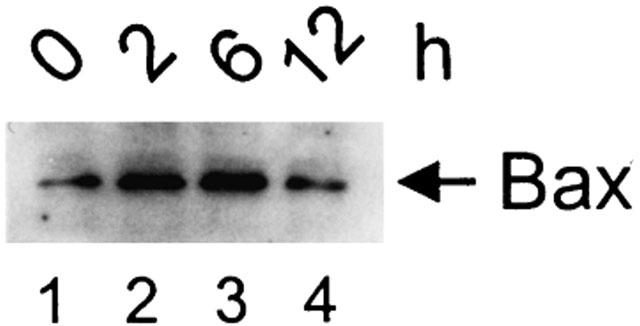
Sulfasalazine induces mitochondrial accumulation of Bax. Jurkat T-lymphocytes were incubated with 2.0 mM sulfasalazine for the indicated periods. The same mitochondrial extracts as in Figure 7A were used for Western blot analysis using a specific polyclonal antibody against Bax.
Discussion
Here we show, that sulfasalazine, a therapeutic agent widely involved in the treatment of inflammatory bowel disease and rheumatoid arthritis, induces apoptosis in a human T-lymphocyte cell line and in human primary peripheral blood T-lymphocytes in a concentration- and time dependent manner. Sulfasalazine-induced cell death shows typical morphological and biochemical signs of apoptosis such as cell shrinkage, chromatin condensation, formation of nuclear bodies, loss of DNA, oligonucleosomal DNA fragmentation and translocation of phosphatidylserine to the outer leaflet of the plasma membrane. This process does not involve activation of the CD95-ligand/receptor system as proposed for a variety of cytotoxic drugs (Friesen et al., 1996; Fulda et al., 1997). Sulfasalazine induces a bongkrekic acid-sensitive collapse of the mitochondrial transmembrane potential (ΔΨm), which is prevented by bongkrekic acid, an inhibitor of the permeability transition pore, as well as by Bcl-2. Sulfasalazine releases the apoptogenic factors cytochrome c and AIF from mitochondria and stimulates the activation of caspase-3 as well as the caspase-mediated cleavage of PARP and ICAD. It is not clear whether cytochrome c, AIF, both, or yet another pro-apoptotic mitochondrial factor such as the recently identified Smac/DIABLO (Du et al., 2000; Verhagen et al., 2000) leads to caspase-3 activation by sulfasalazine. Cytochrome c is a good candidate, since it is released from the mitochondria as early as 2 h after sulfasalazine treatment. It is well established that cytochrome c after release from the mitochondria binds to Apaf-1 in a ternary complex leading to activation of caspase-9, which in turn activates caspase-3 (Li et al., 1997). AIF might also be involved since it was shown that AIF in combination with a cytosolic factor releases caspase-9 from isolated mitochondria and cleaves caspase-9 as well as the caspase substrate Z-VAD.afc through an indirect, mitochondrion-dependent mechanism (Susin et al., 1999). In addition, AIF induces ΔΨm dissipation and subsequent cytochrome c release in a positive autocrine loop (Susin et al., 1999), leading to an increase of caspase-3 activation and amplification of caspase-3-dependent apoptosis.
Nonetheless, caspase activity seems not to be required for sulfasalazine-induced cell death, since cells treated with the wide ranging caspase inhibitor Z-VAD.fmk display typical signs of apoptosis such as cell shrinkage, chromatin condensation, loss of DNA and phosphatidylserine exposure. These features remarkably resemble those described for AIF mediated apoptosis (Susin et al., 1999, 2000; Daugas et al., 2000). Indeed, sulfasalazine causes a mitochondrio-nuclear translocation of AIF, which is caspase-independent, but can be blocked by overexpression of Bcl-2. In addition, sulfasalazine-treated cells show large-scale DNA fragmentation, which is another characteristic of AIF-mediated apoptosis (Susin et al., 1999; Daugas et al., 2000). These data strongly indicate that sulfasalazine-induced apoptosis is, at least in part, mediated by AIF, a caspase-independent, mitochondrial effector of apoptotic cell death.
The molecular mechanisms which lead to mitochondrial membrane permeabilization and release of apoptogenic factors are a matter of debate. Physical disruption of the outer mitochondrial membrane due to swelling of the matrix (Vander-Heiden et al., 1997; Petit et al., 1998), as well as the existence of non-specific, protein permeable pores in the outer membrane, have been proposed (Kluck et al., 1997; Yang et al., 1997; Shimizu et al., 1999). Since sulfasalazine does not induce ΔΨm loss of isolated mitochondria (Fulda & Liptay, unpublished data) we analysed the involvement of known pro-apoptotic members of the Bcl-2 family. Sulfasalazine treatment for 2 h causes an early accumulation of Bax to the mitochondria. It is known that in response to apoptotic stimuli, cytoplasmic Bax undergoes a conformational change and translocates to mitochondrial membranes (Gross et al., 1998; Hsu et al., 1997; Wolter et al., 1997; Goping et al., 1998). There it may ‘puncture' the outer membrane releasing cytochrome c, AIF and other apoptogenic factors (Jurgensmeier et al., 1998) or promote the opening of the permeability transition (PT) pore through association with the adenine nucleotide translocator (ANT), a crucial component of the PT pore (Marzo et al., 1998). Taken together, our data suggest that sulfasalazine-induced apoptosis might be initiated by translocation of Bax to the mitochondria.
Lack of apoptosis is seen in patients with rheumatoid arthritis and inflammatory bowel disease, although the cause of impaired apoptosis is unknown. It has been suggested that in rheumatoid arthritis the inflammatory T-cell infiltrate persists because apoptosis is actively suppressed by the synovial microenvironment (Salmon et al., 1997). High expression of the anti-apoptotic protein Bcl-xl was detected in synovial T-lymphocytes from patients with rheumatoid arthritis compared to healthy controls (Salmon et al., 1997). Lamina propria T-cells from patients with Crohn's disease and ulcerative colitis exhibit increased proliferation, cytokine production and decreased rates of apoptosis (Boirivant et al., 1999). In addition, mucosal T-cells of patients with Crohn's disease are resistant to apoptosis induced by deprivation of IL-2 in vitro (Ina et al., 1995). Since apoptosis is emerging as a major mechanism for safe clearance of unwanted cells during physiological resolution of inflammation, pharmacological induction of apoptosis emerges as an attractive therapeutic goal. Data presented in this paper show that sulfasalazine may well induce apoptosis of T-lymphocytes at doses which do not affect viability of primary synoviocytes or colon epithelial cells. The concentration of sulfasalazine achieved in inflamed tissue is not known. However, it is reported that stool concentrations after an average oral dose of 3–6 g per day in patients with inflammatory bowel disease are in the order of 1.25 to 2.0 mM and interstitial concentrations may be as high as 0.5–1.0 mM (Peppercorn & Goldman, 1973). These local concentrations are comparable to those used in our study. In contrast, reported serum levels of sulfasalazine are 10–15 μg ml−1, equivalent to 0.025–0.038 mM (Das et al., 1973). These lower concentrations explain that lymphopenia is not a common side effect seen in patients treated with sulfasalazine.
Previously, we have demonstrated that sulfasalazine is an inhibitor of NF-κB/Rel activation, most likely due to a direct interference with the ATP-binding site of the IκB kinases, IKKα and IKKβ. However, this effect is achieved at lower concentrations (Wahl et al., 1998; Weber et al., 2000). During the last few years evidence has accumulated showing that NF-κB plays a predominant role in the pathogenesis of rheumatoid arthritis and inflammatory bowel disease. Overexpression of NF-κB regulated cytokines as well as increased nuclear level of NF-κB was demonstrated in the mucosa of patients with active Crohn's disease and ulcerative colitis (Schmid & Adler, 2000).
In addition to the pivotal role of NF-κB/Rel proteins in the regulation of the immune response, there is increasing evidence that NF-κB plays also an important role in the regulation of apoptosis. Most reports demonstrated an anti-apoptotic effect of NF-κB, most likely by increased transcription of anti-apoptotic gene products. In contrast, there is also strong evidence for a pro-apoptotic role of NF-κB (Lin et al., 1999). However, sulfasalazine-induced apoptosis of T-lymphocytes can not be explained by inhibition of NF-κB activation, since it was not overcome by overexpression of the NF-κB subunit RelA (Liptay & Schmid, unpublished data). Nevertheless, it cannot be excluded that inhibition of NF-κB activation plays an additional role in sulfasalazine mediated apoptosis.
In conclusion, our data strongly indicate that sulfasalazine-induced T-lymphocyte apoptosis is independent of caspase activity and involves the mitochondrio-nuclear translocation of AIF. AIF release might be initiated by mitochondrial accumulation of Bax. These findings elucidate why treatment with sulfasalazine leads to clearance of inflammatory cells and therefore can break the cycle of unrelenting cellular activation and tissue damage in chronic inflammation.
Acknowledgments
We thank Sabine Schirmer for excellent technical assistance, Iris Rueß for preparing the manuscript. Supported by grants from Novartis-Stiftung für Therapeutische Forschung (to R.M. Schmid) and Deutsche Krebshilfe (to S. Liptay and R.M. Schmid), the Ligue Nationale contre le Cancer (to G. Kroemer) and the European Community (to G. Kroemer and K.-M. Debatin).
Abbreviations
AIF
apoptosis inducing factor
5-ASA
5-aminosalicylic acid
SS
sulfasalazine
ΔΨm
mitochondrial transmembrane potential
References
- ANDERSON G.P. Resolution of chronic inflammation by therapeutic induction of apoptosis. Trends Pharmacol. Sci. 1996;17:438–442. doi: 10.1016/s0165-6147(96)01004-8. [DOI] [PubMed] [Google Scholar]
- BOIRIVANT M., MARINI M., DI FELICE G., PRONIO A.M., MONTESANI C., TERSIGNI R., STROBER W. Lamina propria T cells in Crohn's disease and other gastrointestinal inflammation show defective CD2 pathway-induced apoptosis. Gastroenterology. 1999;116:557–565. doi: 10.1016/s0016-5085(99)70177-0. [DOI] [PubMed] [Google Scholar]
- CARBONARI M., CIBATI M., CHERCHI M., SBARIGIA D., PESCE A.M., DELL'ANNA L., MODICA A., FIORILLI M. Detection and characterization of apoptotic peripheral blood lymphocytes in human immunodeficiency virus infection and cancer chemotherapy by a novel flow immunocytometric method. Blood. 1994;83:1268–1277. [PubMed] [Google Scholar]
- DAS K.M., EASTWOOD M.A., MCMANUS J.P., SIRCUS W. The metabolism of salicylazosulphapyridine in ulcerative colitis. I. The relationship between metabolites and the response to treatment in inpatients. Gut. 1973;14:631–641. doi: 10.1136/gut.14.8.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAUGAS E., NOCHY D., RAVAGNAN L., LOEFFLER M., SUSIN S.A., ZAMZAMI N., KROEMER G. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000;476:118–123. doi: 10.1016/s0014-5793(00)01731-2. [DOI] [PubMed] [Google Scholar]
- DOLE M., NUNEZ G., MERCHANT A.K., MAYBAUM J., RODE C.K., BLOCH C.A., CASTLE V.P. Bcl-2 inhibits chemotherapy-induced apoptosis in neuroblastoma. Cancer Res. 1994;54:3253–3259. [PubMed] [Google Scholar]
- DU C., FANG M., LI Y., LI L., WANG X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- EISCHEN C.M., KOTTKE T.J., MARTINS L.M., BASI G.S., TUNG J.S., EARNSHAW W.C., LEIBSON P.J., KAUFMANN S.H. Comparison of apoptosis in wild-type and Fas-resistant cells: chemotherapy-induced apoptosis is not dependent on Fas/Fas ligand interactions. Blood. 1997;90:935–943. [PubMed] [Google Scholar]
- ENARI M., SAKAHIRA H., YOKOYAMA H., OKAWA K., IWAMATSU A., NAGATA S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- FERRI K.F., JACOTO E., BLANCO J., ESTE J.A., ZAMZAMI N., SUSIN S.A., XIE Z., BROTHERS G., REED J.C., PFENNINGER J.M., KREOMER G. Apoptosis control in syncytia induced by the HIV envelope glycoprotein complex. Role of mitochondria and caspases. J. Exp. Med. 2000;192:1081–1092. doi: 10.1084/jem.192.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRIESEN C., HERR I., KRAMMER P.H., DEBATIN K.M. Involvement of the CD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat. Med. 1996;2:574–577. doi: 10.1038/nm0596-574. [DOI] [PubMed] [Google Scholar]
- FULDA S., LUTZ W., SCHWAB M., DEBATIN K.M. MycN sensitizes neuroblastoma cells for drug-induced apoptosis. Oncogene. 1999;18:1479–1486. doi: 10.1038/sj.onc.1202435. [DOI] [PubMed] [Google Scholar]
- FULDA S., SIEVERTS H., FRIESEN C., HERR I., DEBATIN K.M. The CD95 (APO-1/Fas) system mediates drug-induced apoptosis in neuroblastoma cells. Cancer Res. 1997;57:3823–3829. [PubMed] [Google Scholar]
- GOPING I.S., GROSS A., LAVOIE J.N., NGUYEN M., JEMMERSON R., ROTH K., KORSMEYER S.J., SHORE G.C. Regulated targeting of Bax to mitochondria. J. Cell. Biol. 1998;143:207–215. doi: 10.1083/jcb.143.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GREEN D.R., REED J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- GRIFFITH T.S., LYNCH D.H. TRAIL: a molecule with multiple receptors and control mechanisms. Curr. Opin. Immunol. 1998;10:559–563. doi: 10.1016/s0952-7915(98)80224-0. [DOI] [PubMed] [Google Scholar]
- GROSS A., JOCKEL J., WIE M.C., KORSMEYER S.J. Enforced dimerization of Bax results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROSS A., MCDONNELL J.M., KORSMEYER S.J. BCL-2 family members and the mitochondria in apoptosis. Genes. Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- HASLETT C. Resolution of acute inflammation and the role of apoptosis in the tissue fate of granulocytes. Clin. Sci. Colch. 1992;83:639–648. doi: 10.1042/cs0830639. [DOI] [PubMed] [Google Scholar]
- HERR I., WILHELM D., BOHLER T., ANGEL P., DEBATIN K.M. JNK/SAPK activity is not sufficient for anticancer therapy-induced apoptosis involving CD95-L, TRAIL and TNF-alpha. Int. J. Cancer. 1999;80:417–424. doi: 10.1002/(sici)1097-0215(19990129)80:3<417::aid-ijc14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- HSU Y.T., WOLTER K.G., YOULE R.J. Cytosol-to-membrane redistribution of Bax and Bcl-xl during apoptosis. Proc. Natl. Acad. Sci. U.S.A. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INA K., BINION D.G., WEST G.A., DOBREA G.M., FIOCCHI C. Secretion of soluble factors and phagocytosis by intestinal fibroblasts regulate T-cell apoptosis. Gastroenterology. 1995;108:A841. [Google Scholar]
- JURGENSMEIER J.M., XIE Z., DEVERAUX Q., ELLERBY L., BREDESEN D., REED J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. U.S.A. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLAS C., DEBATIN K.M., JONKER R.R., KRAMMER P.H. Activation interferes with the APO-1 pathway in mature human T cells. Int. Immunol. 1993;5:625–630. doi: 10.1093/intimm/5.6.625. [DOI] [PubMed] [Google Scholar]
- KLUCK R.M., BOSSY W.E., GREEN D.R., NEWMEYER D.D. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- KROEMER G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- LI P., NIJHAWAN D., BUDIHARDJO I., SRINIVASULA S.M., AHMAD M., ALNEMRI E.S., WANG X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- LIN B., WILLIAMS S.C., TAO Y., SCHLEICHER M.S., CANO L.L., DUKE R.C., SCHEINMAN R.I. NF-kappaB functions as both a proapoptotic and antiapoptotic regulatory factor within a single cell type. Cell. Death Differ. 1999;6:570–582. doi: 10.1038/sj.cdd.4400528. [DOI] [PubMed] [Google Scholar]
- LIPTAY S., BACHEM M., HACKER G., ADLER G., DEBATIN K.M., SCHMID R.M. Inhibition of nuclear factor kappa B and induction of apoptosis in T-lymphocytes by sulfasalazine. Br. J. Pharmacol. 1999;128:1361–1369. doi: 10.1038/sj.bjp.0702937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU X., ZOU H., SLAUGHTER C., WANG X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- MACDONALD H.R., LEES R.K. Programmed death of autoreactive thymocytes. Nature. 1990;343:642–644. doi: 10.1038/343642a0. [DOI] [PubMed] [Google Scholar]
- MARZO I., BRENNER C., ZAMZAMI N., JURGENSMEIER J.M., SUSIN S.A., VIEIRA H.L., PREVOST M.C., XIE Z., MATSUYAMA S., REED J., KROEMER G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- MCDONNELL T.J., DEANE N., PLATT F.M., NUNEZ G., JAEGER U., MCKEARN J.P., KORSMEYER S.J. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57:79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- MOUNTZ J.D., WU J., CHENG J., ZHOU T. Autoimmune disease. A problem of defective apoptosis. Arthritis Rheum. 1994;37:1415–1420. doi: 10.1002/art.1780371002. [DOI] [PubMed] [Google Scholar]
- MUNN D.H., BEALL A.C., SONG D., WRENN R.W., THROCKMORTON D.C. Activation-induced apoptosis in human macrophages: developmental regulation of a novel cell death pathway by macrophage colony-stimulating factor and interferon gamma. J. Exp. Med. 1995;181:127–136. doi: 10.1084/jem.181.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGATA S. Apoptosis by death factor. Cell. 1997;8:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- NEUMANN V.C., GRINDULIS K.A., HUBBALL S., MCCONKEY B., WRIGHT V. Comparison between penicillamine and sulphasalazine in rheumatoid arthritis: Leeds-Birmingham trial. Br. Med. J. Clin. Res. Ed. 1983;287:1099–1102. doi: 10.1136/bmj.287.6399.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEWTON K., STRASSER A. Ionizing radiation and chemotherapeutic drugs induce apoptosis in lymphocytes in the absence of Fas or FADD/MORT1 signaling. Implications for cancer therapy. J. Exp. Med. 2000;191:195–200. doi: 10.1084/jem.191.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLSON D.W., ALI A., THORNBERRY N.A., VAILLANCOURT J.P., DING C.K., GALLANT M., GAREAU Y., GRIFFIN P.R., LABELLE M., LAZEBNIK Y.A. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- NISITANI S., TSUBATA T., MURAKAMI M., OKAMOTO M., HONJO T. The bcl-2 gene product inhibits clonal deletion of self-reactive B lymphocytes in the periphery but not in the bone marrow. J. Exp. Med. 1993;178:1247–1254. doi: 10.1084/jem.178.4.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEPPERCORN M.A., GOLDMAN P. Distribution studies of salicylazosulfapyridine and its metabolites. Gastroenterology. 1973;64:240–245. [PubMed] [Google Scholar]
- PETIT P.X., GOUBERN M., DIOLEZ P., SUSIN S.A., ZAMZAMI N., KROEMER G. Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: the impact of irreversible permeability transition. FEBS Lett. 1998;426:111–116. doi: 10.1016/s0014-5793(98)00318-4. [DOI] [PubMed] [Google Scholar]
- RAVIRAJAN C.T., PITTONI V., ISENBERG D.A. Apoptosis in human autoimmune diseases. Int. Rev. Immunol. 1999;18:563–589. doi: 10.3109/08830189909088499. [DOI] [PubMed] [Google Scholar]
- RIIS P., ANTHONISEN P., WULFF H.R., FOLKENBORG O., BONNEVIE O., BINDER V. The prophylactic effect of salazosulphapyridine in ulcerative colitis during long-term treatment. A double-blind trial on patients asymptomatic for one year. Scand. J. Gastroenterol. 1973;8:71–74. [PubMed] [Google Scholar]
- SALMON M., SCHEEL T.D., HUISSOON A.P., PILLING D., SHAMSADEEN N., HYDE H., D'ANGEAC A.D., BACON P.A., EMERY P., AKBAR A.N. Inhibition of T cell apoptosis in the rheumatoid synovium. J. Clin. Invest. 1997;99:439–446. doi: 10.1172/JCI119178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCAFFIDI C., KIRCHHOFF S., KRAMMER P.H., PETER M.E. Apoptosis signaling in lymphocytes. Curr. Opin. Immunol. 1999;11:277–285. doi: 10.1016/s0952-7915(99)80045-4. [DOI] [PubMed] [Google Scholar]
- SCHENDEL S.L., MONTAL M., REED J.C. Bcl-2 family proteins as ion-channels. Cell. Death. Differ. 1998;5:372–380. doi: 10.1038/sj.cdd.4400365. [DOI] [PubMed] [Google Scholar]
- SCHMID R.M., ADLER G. NF-κB/Rel/IκB: Implications in gastrointestinal diseases. Gastroenterology. 2000;118:1208–1228. doi: 10.1016/s0016-5085(00)70374-x. [DOI] [PubMed] [Google Scholar]
- SCHMID R.M., PERKINS N.D., DUCKETT C.S., ANDREWS P.C., NABEL G.J. Cloning of an NF-kappa B subunit which stimulates HIV transcription in synergy with p65. Nature. 1991;352:733–736. doi: 10.1038/352733a0. [DOI] [PubMed] [Google Scholar]
- SHIMIZU S., NARITA M., TSUJIMOTO Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- STRASSER A., HARRIS A.W., HUANG D.C., KRAMMER P.H., CORY S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 1995;14:6136–6147. doi: 10.1002/j.1460-2075.1995.tb00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUSIN S.A., DAUGAS E., RAVAGNAN L., SAMEJIMA K., ZAMZAMI N., LOEFFLER M., COSTANTINI P., FERRI K.F., IRINOPOULOU T., PREVOST M.C., BROTHERS G., MAK T.W., PENNINGER J., EARNSHAW W.C., KROEMER G. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUSIN S.A., LORENZO H.K., ZAMZAMI N., MARZO I., SNOW B.E., BROTHERS G.M., MANGION J., JACOTOT E., COSTANTINI P., LOEFFLER M., LAROCHETTE N., GOODLETT D.R., AEBERSOLD R., SIDEROVSKI D.P., PENNINGER J.M., KROEMER G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- SUSIN S.A., ZAMZAMI N., CASTEDO M., DAUGAS E., WANG H.G., GELEY S., FASSY F., REED J.C., KROEMER G. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J. Exp. Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUSIN S.A., ZAMZAMI N., CASTEDO M., HIRSCH T., MARCHETTI P., MACHO A., DAUGAS E., GEUSKENS M., KROEMER G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SVARTZ N. Ett nytt sulfonamidpreparat. Forelopande meddelande. Nord. Med. 1941;9:544–. [Google Scholar]
- TEWARI M., QUAN L.T., O'ROURKE K., DESNOYERS S., ZENG Z., BEIDLER D.R., POIRIER G.G., SALVESEN G.S., DIXIT V.M. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- THOMPSON C.B. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- VAN DER HEIJDE D.M., VANRIEL P.L., NUVER-ZWART I., GRIBNAU F.W., VAD-DE-PUTTE L.B. Effects of hydroxychloroquine and sulphasalazine on progression of joint damage in rheumatoid arthritis. Lancet. 1989;1:1036–1038. doi: 10.1016/s0140-6736(89)92442-2. [DOI] [PubMed] [Google Scholar]
- VANDER-HEIDEN M.G., CHANDEL N.S., WILLIAMSON E.K., SCHUMACKER P.T., THOMPSON C.B. Bcl-xl regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- VERHAGEN A.M., EKERT P.G., PAKUSCH M., SILKE J., CONNOLLY L.M., REID G.E., MORITZ R.L., SIMPSON R.J., VAUX D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- WAHL C., LIPTAY S., ADLER G., SCHMID R.M. Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J. Clin. Invest. 1998;101:1163–1174. doi: 10.1172/JCI992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALCZAK H., KRAMMER P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp. Cell. Res. 2000;56:58–66. doi: 10.1006/excr.2000.4840. [DOI] [PubMed] [Google Scholar]
- WEBER C.K., LIPTAY S., WIRTH T., ADLER G., SCHMID R.M. Suppression of NFkappaB activity by sulfasalazine is mediated by direct inhibition of IkappaB kinases alpha and beta. Gastroenterology. 2000;119:1209–1218. doi: 10.1053/gast.2000.19458. [DOI] [PubMed] [Google Scholar]
- WESSELBORG S., JANSSEN O., KABELITZ D. Induction of activation-driven death (apoptosis) in activated but not resting peripheral blood T cells. J. Immunol. 1993;150:4338–4345. [PubMed] [Google Scholar]
- WOLTER K.G., HSU Y.T., SMITH C.L., NECHUSHTAN A., XI X.G., YOULE R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell. Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG J., LIU X., BHALLA K., KIM C.N., IBRADO A.M., CAI J., PENG T.I., JONES D.P., WANG X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]