Bid-induced Conformational Change of Bax Is Responsible for Mitochondrial Cytochrome c Release during Apoptosis (original) (raw)
Abstract
Here we report that in staurosporine-induced apoptosis of HeLa cells, Bid, a BH3 domain containing protein, translocates from the cytosol to mitochondria. This event is associated with a change in conformation of Bax which leads to the unmasking of its NH2-terminal domain and is accompanied by the release of cytochrome c from mitochondria. A similar finding is reported for cerebellar granule cells undergoing apoptosis induced by serum and potassium deprivation. The Bax-conformational change is prevented by Bcl-2 and Bcl-xL but not by caspase inhibitors. Using isolated mitochondria and various BH3 mutants of Bid, we demonstrate that direct binding of Bid to Bax is a prerequisite for Bax structural change and cytochrome c release. Bcl-xL can inhibit the effect of Bid by interacting directly with Bax. Moreover, using mitochondria from Bax-deficient tumor cell lines, we show that Bid- induced release of cytochrome c is negligible when Bid is added alone, but dramatically increased when Bid and Bax are added together. Taken together, our results suggest that, during certain types of apoptosis, Bid translocates to mitochondria and binds to Bax, leading to a change in conformation of Bax and to cytochrome c release from mitochondria.
Keywords: apoptosis, Bax, Bid, cytochrome c, mitochondria
The Bcl-2 family is composed of proteins which play a pivotal role in controlling apoptosis (programmed cell death) induced by a variety of stimuli. Some proteins within this family such as Bcl-2, Bcl-xL, Bcl-w, Mcl-1, A1, adenovirus E1B19K, and Caenorhabditis elegans ced-9 inhibit apoptosis while others including Bax, Bak, Bok/Mtd, Bcl-xS, Bad, Bid, Bik/Nbk, Bim, HRK, Blk, and C. elegans Egl-1 promote apoptosis (for reviews see Yang and Korsmeyer, 1996; Kroemer, 1997; Reed, 1997; Kelekar and Thompson, 1998). These proteins can form both homo- and heterodimers and as a consequence of this, they can function either independently or in concert to regulate apoptosis (Knudson and Korsmeyer, 1997). Dimerization of Bcl-2 family members involves interactions between conserved amino acid sequences known as Bcl-2 homology (BH)1 domains. Four of these domains (BH1, BH2, BH3, and BH4) have now been identified and they appear to play a crucial role in specifying the pro- or antiapoptotic properties of a given family member (Yang and Korsmeyer, 1996; Kroemer, 1997; Reed, 1997; Kelekar and Thompson, 1998).
Antiapoptotic proteins such as Bcl-2 and Bcl-xL possess all four BH domains and both BH1 and BH2 appear necessary for their dimerization with Bax and for suppression of apoptosis (Yin et al., 1994; Chittenden et al., 1995; Sedlak et al., 1995). In contrast, the proapoptotic proteins Bax, Bak, and Bok lack a recognizable BH4 domain while Bid, Bad, Bik/Nbk, Hrk, Bim, Blk, and Egl-1 are characterized by the presence of a BH3 domain only (“BH3 only” proteins). Outside of this region these proteins display considerable sequence diversity.
The BH3 domain was first identified as a stretch of 16 amino acids required for Bak to heterodimerize with Bcl-xL and to promote cell death (Chittenden et al., 1995). Similarly, the ability of Bad to bind Bcl-xL through its BH3 domain is required to promote apoptosis (Kelekar et al., 1997; Zha et al., 1997). However, it has now been shown that the proapoptotic activity triggered by this domain was not always dependent on its interaction with antiapoptotic proteins. Thus, Bid BH3 mutants which lack the ability to bind Bcl-2, but which retain the ability to bind Bax, are still potent activators of apoptosis (Wang et al., 1996). These observations suggest that the mechanisms by which BH3 domains trigger apoptosis may vary from one family member to another and this may reflect involvement in multiple pathways leading to cell death.
Recent structural studies have shown that in monomeric form Bcl-xL consists of two central hydrophobic helices (α5 and α6) surrounded by five amphipathic helices, with a 60-residue flexible loop linking the BH4 and BH3 domains (Muchmore et al., 1996; Aritomi et al., 1997). The BH1, BH2, and BH3 domains lie in close proximity to each other and form an elongated hydrophobic cleft which can bind BH3-containing peptides (Sattler et al., 1997). The overall structure of Bcl-xL with the two central hydrophobic helices is reminiscent of the structure of the pore-forming domain of the bacterial toxins diphtheria toxin or colicins A and E1 which act as channels for ions or small proteins. Like these toxins, some members of the Bcl-2 family such as Bcl-xL (Minn et al., 1997), Bcl-2 (Schendel et al., 1997), and Bax (Antonsson et al., 1997; Schlesinger et al., 1997) were also shown to be capable of forming channels in synthetic lipid membranes. Furthermore, from amino acid sequence analysis and structure modeling using the Bcl-xL crystallographic coordinates, we can predict that Bak and Bok, like Bax, should also display pore-forming activities whereas the BH3 only proteins should not. Indeed, we found that Bid is not able to form pores in liposomes (Antonsson, B., unpublished data). The role which has been proposed for the BH3 only proteins is rather to act as intracellular ligands for membrane-bound Bcl-2, Bcl-xL, or Bax (Wang et al., 1996; Sattler et al., 1997), such that upon ligand binding, Bcl-2, Bcl-xL, or Bax would be activated and exert their function. Structural analysis of the Bcl-xL/Bak complex suggests that within the Bak protein, the BH3 region points toward the interior of the protein, making these residues unavailable for interaction with Bcl-xL (Sattler et al., 1997). Therefore, exposure of the BH3 domain of Bak requires a structural change of the protein. Consistent with such a model, we now show that direct binding of Bid to Bax triggers a change in the conformation of Bax which is associated with the release of cytochrome c from mitochondria.
Materials and Methods
Antibodies
Rabbit pAbs were raised against amino acids 1–21 of human Bax (06-499, lot 15670; Upstate Biotechnology), amino acids 11–30 of human Bax (Bax N-20 sc-493; Santa Cruz Biotechnology), amino acids 43–61 of murine Bax (13686E; PharMingen). Note that the antibodies raised against human Bax also recognize murine Bax. A mouse mAb raised against amino acids 3–16 of human Bax (clone YTH-2D2, 2282-MC; Genzyme) was also used for in vitro binding assays. Rabbit pAb raised against amino acids 4–21 of human Bcl-2 was from Santa Cruz Biotechnology (Bcl-2 N-19 sc-492). Mouse mAb raised against amino acids 41–54 of Bcl-2 was obtained from Genosys (OM-11-925A). Rabbit pAb against amino acids 18–233 of human Bcl-xS/L was from Transduction Laboratories (B22630). Rabbit pAb against the NH2 terminus of human Bcl-x was from Santa Cruz Biotechnology (Bcl-xS/L S-18 sc-634). Mouse mAb against mitochondrial heat shock protein 70 (mt-hsp-70) was from Affinity Bioreagents, Inc. (MA3-028). Anti–human Bak mAb raised against amino acids 1–52 was obtained from Calbiochem (AM03). Mouse mAb against bovine cytochrome oxidase subunit IV (COX-IV) which reacts specifically with the rat and human homologue of the protein was from Molecular Probes (A-6431). The mouse monoclonal anti–cytochrome c antibody that recognizes the native form of rat, mouse, and human cytochrome c was from PharMingen (65971A) and the rabbit polyclonal anti–cytochrome c antibody was generated against bovine cytochrome c. A rabbit pAb was generated against recombinant full-length Bid.
Cell Cultures
Primary cultures of cerebellar granule cells (CGC) were prepared from 8-d-old rat pups according to Villalba et al. (1997) with slight modifications. In brief, freshly dissected cerebella were incubated with 0.25 mg/ml trypsin for 15 min at 37°C and trypsin inhibitor (0.5 mg/ml) was added to stop the reaction. Then, digested cerebella were mechanically dissociated with a flame-narrowed Pasteur pipette in HBSS in the presence of DNase I (0.1 mg/ml) and trypsin inhibitor. Cells were seeded at a density of 0.25 × 106 cells/cm2 in Falcon dishes previously coated with poly-d-lysine hydrobromide (10 μg/ml) in basal medium Eagle (BME; GIBCO BRL) supplemented with 10% FCS, 20 mM KCl, 50 IU penicillin, 50 μg/ml streptomycin, 10 mM Hepes, 2 mM l-glutamine, and 1 mM sodium pyruvate. After 24 h, 10 μM cytosine β-d-arabinofuranoside was added to the culture medium. Cultures were incubated at 37°C in a humidified atmosphere of 5% CO2. Neurons were used after 7 d in culture.
HeLa cells and the stable HeLa cell line that constitutively overexpresses Bcl-2 (HeLa-Bcl-2) (Estoppey et al., 1997) were cultured in a 1:1 mixture of basal Iscove's medium and Ham's F12 medium (Seromed) supplemented with 10% FCS and 2 mM l-glutamine. HEK cells were cultured in DME-F12 medium (GIBCO BRL) supplemented with 10% FCS and 2 mM l-glutamine. LoVo (colon, adenocarcinoma, human), DU145 (prostate, carcinoma, human), and LS180 (colon, adenocarcinoma, human) were from the American Type Culture Collection and cultured according to the instructions provided.
Immunocytochemistry
For immunocytochemistry analysis, cells were seeded onto glass coverslips. Apoptosis was induced in primary cultures of CGC by washing twice and placing the neurons in a culture medium containing a normal concentration of KCl (5 mM) and lacking serum (basal medium Eagle– Glutamax-I supplemented with 50 IU penicillin, 50 μg/ml streptomycin, 10 mM Hepes, 2 mM l-glutamine, and 1 mM sodium pyruvate). Apoptosis in HeLa cells was induced by addition of 1 μM staurosporine in the presence or absence of 100 μM z-VAD-fmk. Stock solutions of the drugs (×1,000) were made in DMSO. Control cultures received solvent alone. Cells were fixed with 4% paraformaldehyde in PBS for 15 min and permeabilized with 0.2% Triton X-100 in PBS at room temperature. After washing, the cells were incubated for 2 h with anti-Bax pAbs (all diluted 1:100 in PBS + 5% normal goat serum) and anti–cytochrome c mAb (dilution 1:15 in PBS + 5% normal goat serum) or anti–mt-hsp-70 (dilution 1:500 in PBS + 5% normal goat serum), washed twice in PBS, and developed with fluorescein and Texas red–labeled goat anti–rabbit and goat anti–mouse antibodies, respectively. In the last wash, 1 μg/ml of Hoechst 33258 was added to the cells. Coverslips were placed on a glass slide in Vectashield mounting medium and observed by conventional or confocal fluorescence microscopy.
Subcellular Fractionation
At different times after induction of apoptosis, HeLa cells and CGC were harvested in isotonic mitochondrial buffer (MB: 210 mM mannitol, 70 mM sucrose, 1 mM EDTA, and 10 mM Hepes, pH 7.5) supplemented with protease inhibitor cocktail Complete (Boehringer Mannheim), and homogenized for 30–40 strokes with a Dounce homogenizer. Samples were transferred to Eppendorf centrifuge tubes and centrifuged at 500 g for 5 min at 4°C to eliminate nuclei and unbroken cells. The resulting supernatant was centrifuged at 10,000 g for 30 min at 4°C to obtain the heavy membrane pellet (HM) enriched for mitochondria. This supernatant was further centrifuged at 100,000 g for 1 h at 4°C to yield the light membrane pellet (not analyzed) and the final soluble fraction (S). The HM material was resuspended in MB supplemented with 1% Triton X-100. Soluble and HM fractions (30 and 15 μg, respectively) were separated by SDS-PAGE (4–20% Tris-Glycine gels; NOVEX) and transferred to a nitrocellulose membrane (NOVEX). After blocking nonspecific sites for 1 h at room temperature with 5% nonfat milk in PBS supplemented with 0.2% Tween 20, the membrane was incubated overnight at 4°C with the antibody raised against amino acids 1–21 of human Bax (Upstate Biotechnology) diluted 1:500 in PBS supplemented with 2.5% nonfat milk. To confirm equal loading and transfer, the membrane was subsequently stripped and reprobed for COX-IV. The immunoreactive proteins were visualized using horseradish peroxidase–linked goat anti–mouse antibody (Jackson ImmunoResearch Laboratories Inc.) and enhanced chemiluminescence (Amersham Life Sciences).
cDNA Cloning and Site-directed Mutagenesis of Bid and Bcl-xL
Single-stranded random-primed cDNA prepared from mouse thymus total RNA was used as a template for PCR amplification of Bid cDNA using sense primer 5′ ATGGATCCCCATGGACTCTGAGGT 3′ and antisense primer 5′ CACCTCGAGCCTCAGTCCATCTCGTTTC 3′. The PCR product was digested with BamH1/Xho1 and subcloned in the BamH1/Xho1 site of pBluescript II KS (Stratagene). The sequence of Bid was confirmed by sequencing. This construct was used as template to prepare NH2-terminal His-tagged Bid using as sense primer 5′ ATACCATGGCTCACCACCACCACCACCACATGGACTCTGAGGTCAGC 3′ and the same antisense primer as above. The PCR product was digested with Nco1/Xho1 and subcloned in the Nco1/Xho1 site of pET23d (Novagen).
BidmIII-1 and BidmIII-3 were generated in three steps. First, the 5′ portion of Bid was amplified using the 6-His sense primer, and mutant antisense primers 5′ CTGGATGTTGTGGGCGGCCTCATCG 3′ for BidmIII-1 and 5′ GTCCATCTCATCGGCTATTTGGGCGA 3′ for BidmIII-3. The 3′ portion of Bid was amplified using the sense 5′ CGATGAGGCCGCCCACAACATCCAG 3′ for BidmIII-1, 5′ TCGCCCAAATAGCCGATGAGATGGA 3′ for BidmIII-3, together with the antisense Xho1 containing primer as above. Second, the PCR products of each corresponding mutant were mixed and reamplified by PCR with the sense His primer and the antisense Xho1 primer. Third, the PCR products were digested with Nco1/Xho1 and subcloned in the Nco1/Xho1 site of pET23d. Mutations were confirmed by DNA sequencing.
The constructs pET23dHisBid, pET23dHisBidmIII-1, and pET23dHisBidmIII-3 were transformed in BL21 (DE3) Escherichia coli and protein expression was induced by 100 μM of isopropyl β-d-thiogalactopyranoside (IPTG).
Bcl-xLm: G138→ A was generated by PCR site-directed mutagenesis on the template pET23dHisBcl-xL, using Pwo DNA polymerase, sense primer 5′ GGGGTAAACTGGGCCCGCATTGTGGCC 3′, and antisense primer 5′ GGCCACAATGCGGGCCCAGTTTACCCC 3′. The PCR product was then digested with Dpn, purified using QIAquick (QIAquick PCR purification kit; Qiagen), and 2.5 μl purified PCR DNA transformed in competent E. coli XL1-Blue. Mutation was confirmed by DNA sequencing.
Production of Recombinant Proteins
His-tagged Bid and Bid mutants were expressed in the pET23d vector in E. coli. The recombinant proteins were recovered in the soluble bacteria fraction and purified by chromatography on Ni-NTA-Agarose followed by Q-Sepharose. The purified proteins which were >95% pure were stored in 25 mM Tris-HCl, 0.1 mM DTT, 30% glycerol, pH 7.5, at −80°C.
His-tagged human Bcl-xL and His-tagged human mutant Bcl-xL (Bcl-xLm: G138→ A) both lacking 24 amino acids at the COOH terminus were expressed in the pET23d vector in E. coli. The recombinant proteins were recovered in the soluble bacteria fraction and purified by chromatography on Ni-NTA-Agarose followed by Q-Sepharose. The purified proteins which were >95% pure were stored in 25 mM Tris-HCl, 0.2 mM DTT, 30% glycerol, pH 7.5, at −80°C.
Human Bax-α lacking 20 amino acids at the COOH terminus and human Bcl-2 lacking 34 amino acids at the COOH terminus were produced as described previously (Antonsson et al., 1997).
In Vitro Binding Assay
To characterize the interactions of Bid and the BH3 mutants BidmIII-1 and BidmIII-3 with Bax, Bcl-2, and Bcl-xL, recombinant proteins were mixed in 50 μl PBS (100 nM each, or 1 μM Bcl-2 or Bcl-xL with 100 nM Bid or BidmIII-1). After 30 min of incubation on ice, 450 μl of NP-40 buffer (142.5 mM KCl, 5 mM MgCl2, 1 mM EDTA, 0.25% NP-40, and 10 mM Hepes, pH 7.5) supplemented with protease inhibitor cocktail Complete (Boehringer Mannheim) was added to each binding mixture. Samples were rotated overnight at 4°C in the presence of 4 μg antibody. Then, 50 μl protein A–Agarose (for pAbs) or protein G–Agarose (for mAbs) (Boeh– ringer Mannheim) was added to each sample, rotated for 3 h at 4°C, recovered by centrifugation at 10,000 g for 1 min, and washed three times in 1 ml of NP-40 buffer. Materials bound to the beads were eluted by the addition of 50 μl of 3× SDS-PAGE sample buffer, thorough vortexing, and boiling for 5 min. Immunoprecipitated proteins were analyzed by SDS-PAGE and Western blotting, as described above.
Isolation of Mitochondria
Mitochondria were isolated from HeLa, HEK, HeLa-Bcl-2, LoVo, LS180, and DU145 cell lines by sucrose density gradient centrifugation. In brief, cells were harvested with PBS containing 1 mM EDTA, centrifuged at 750 g for 10 min, washed, and resuspended in isotonic MB supplemented with protease inhibitors. Cells were broken by five passages through a 25G1 0.5 × 25 needle fitted on a 2-ml syringe and the suspension was centrifuged at 2,000 g in an Eppendorf centrifuge at 4°C. This procedure was repeated until almost all of the cells were broken. Supernatants from each step were pooled before centrifugation at 13,000 g at 4°C for 10 min. The pellet was resuspended in 1 ml of MB and layered on top of a discontinuous sucrose gradient consisting of 20 ml of 1.2 M sucrose, 10 mM Hepes, pH 7.5, 1 mM EDTA, 0.1% BSA on top of 17 ml of 1.6 M sucrose, 10 mM Hepes, pH 7.5, 1 mM EDTA, 0.1% BSA. The samples were centrifuged at 27,000 rpm for 2 h at 4°C in a Beckman SW28 rotor. Mitochondria were recovered at the 1.6–1.2 M sucrose interface, washed, and resuspended in MB. Protein concentration was estimated by the method of Bradford (1976) with BSA as the standard. Electron microscopy studies revealed that most of the organelles recovered by this procedure are mitochondria with negligible contamination by endoplasmic reticulum membranes (not shown).
Analysis of Mitochondrial Bax Immunostaining by Flow Cytometry
Mitochondria (50–70 μg of proteins) were incubated with recombinant Bid proteins for 15 min at 30°C in 200 μl of KCl buffer (125 mM KCl, 4 mM MgCl2, 5 mM NaHPO4, 5 mM succinate, 0.5 mM EGTA, 15 mM Hepes-KOH, pH 7.4, 5 μM rotenone). Then mitochondria were fixed with PBS 4% paraformaldehyde, washed twice in PBS, incubated for 1 h at room temperature with the anti-Bax (amino acids 1–21) antibody or with the anti–Bcl-2 (amino acids 4–21) antibody both diluted 1:100 in PBS + 5% normal goat serum, washed twice in PBS, and incubated for 30 min at room temperature with fluorescein-labeled goat anti–rabbit IgG antibody (dilution 1:100 in PBS + 5% normal goat serum). After the final wash, fluorescence was analyzed by flow cytometry using a Facscalibur® flow cytometer (Becton Dickinson). Only mitochondria exhibiting a light scatter profile typical of intact mitochondria were analyzed and debris or nonmitochondrial membranes were excluded from the study by electronic gating.
In Vitro Assay for Cytochrome c Release
Mitochondria (30 μg) were incubated in the presence or absence of various recombinant proteins in 200 μl of KCl buffer for 15 min at 30°C and then centrifuged for 5 min at 13,000 g at 4°C. Mitochondrial pellets corresponding to 1.5-μg proteins and the corresponding volume of the supernatant fractions were separated by SDS-PAGE using 4–20% Tris-Gly gels (NOVEX) and their respective contents of cytochrome c were estimated by Western blotting using polyclonal anti–cytochrome c antibody (dilution 1:2,500). Equal loading of the mitochondrial pellet was verified using either an antibody against COX-IV or an antibody against mt-hsp-70. Antigen–antibody complexes were detected using a horseradish peroxidase– conjugated goat anti–rabbit IgG and enhanced chemiluminescence detection reagents.
Results
Bax Immunostaining in Cells Undergoing Apoptosis
We have investigated the subcellular distribution of Bax during apoptosis in HeLa cells treated with 1 μM staurosporine. Immunocytochemical studies with a pAb raised to amino acids 1–21 of Bax are shown in Fig. 1. Untreated HeLa cells displayed staining of the nucleolus but no cytosolic immunoreactivity (Fig. 1 A). The nucleolar staining does not appear to involve Bax since the same pattern was observed in two Bax-deficient tumor cell lines, LoVo and LS180 (Rampino et al., 1997) (not shown).
Figure 1.

Bax and cytochrome c immunostaining in cells undergoing apoptosis. (A–C) Control HeLa cells; (D–I) HeLa cells treated with 1 μM staurosporine for 4 h in the absence (D–F) or presence (G–I) of 100 μM z-VAD-fmk; (J–L) CGC cultured in a medium with low KCl (5 mM) and without serum for 7 h. Cells were double immunostained for Bax and cytochrome c and nuclei were visualized by Hoechst 33258 staining. A Bax pAb against amino acids 1–21 of human Bax (Upstate Biotechnology) was used in all cases. Arrows indicate Bax positive cells. Note the punctate Bax staining specifically in cells containing cytochrome c–depleted mitochondria. In CGC, in contrast to HeLa cells, the Bax immunostaining is seen only in neurons with a marginalized chromatin and not in neurons in the terminal phase of apoptosis (fragmented nucleus).
In contrast to untreated cells, in cultures exposed to staurosporine for 4 h, ∼30% of the cells displayed a punctate cytosolic pattern of Bax immunostaining suggestive of an association with the mitochondria (Fig. 1 D). No mitochondrial staining was observed with this Bax antibody in staurosporine-treated LoVo and LS180 cell lines, confirming the specificity of the Bax immunostaining in these experiments (not shown). Similar results were also obtained with another pAb raised against amino acids 11–30 of Bax (data not shown, but see Fig. 2). The mitochondrial localization of Bax in these cells was confirmed by confocal microscopy and double immunostaining with an antibody to mt-hsp-70 (Fig. 2). This protein, closely related to but distinct from the cytosolic hsp-70, is a 75-kD protein which resides constitutively in the mitochondrial matrix (Lill and Neupert, 1996).
Figure 2.

Bax staining appears in mitochondria. HeLa cells treated with 1 μM staurosporine for 2 h were double immunostained with antibodies against amino acids 11–30 of human Bax (Bax N-20 sc-493; Santa Cruz Biotechnology) and mt-hsp-70. Fluorescence microscopy was performed using a LSM 410 confocal microscope (Zeiss). The two channels are shown separately (A, anti-Bax; B, anti–mt-hsp-70) and merged (C).
It is interesting to note that the mitochondria revealed by Bax immunostaining displayed a small round shape and had a tendency to aggregate (Fig. 2), a finding previously observed in cells overexpressing Bax (Eskes et al., 1998; Rossé et al., 1998) or Bid (Li et al., 1998).
During apoptosis induced by diverse stimuli, cytochrome c is released from mitochondria into the cytosol (Kharbanda et al., 1997; Kluck et al., 1997; Kroemer et al., 1997; Yang et al., 1997; Bossy-Wetzel et al., 1998), and this is thought to be a crucial event in the apoptotic pathway. Recent evidence suggests that Bax may be involved in the pathway leading to cytochrome c release (Eskes et al., 1998; Jürgensmeier et al., 1998). Therefore, the appearance of Bax in mitochondria after staurosporine-induced apoptosis prompted us to study the cytochrome c distribution in these cells. A double immunostaining showed that while normal HeLa cells displayed a reticulate staining for cytochrome c (Fig. 1 B), the Bax positive cells almost invariably (>95%) displayed a diffuse cytosolic pattern of cytochrome c staining (Fig. 1 E). In all cases, Bax positive cells displayed a fragmented nucleus as assessed by Hoechst 33258 staining (Fig. 1 F). Therefore, detection of mitochondrial Bax is closely correlated with both depletion of cytochrome c from mitochondria and cell death as revealed by apoptotic nuclei.
We next decided to test whether blocking cell death, either by Bcl-2 or by using inhibitors of caspase activity, could influence the appearance of Bax staining during apoptosis. In HeLa cells constitutively overexpressing Bcl-2 (HeLa-Bcl-2) which we have shown previously to be resistant to staurosporine-induced apoptosis (Estoppey et al., 1997), we found that <0.1% of the cells became Bax positive after a 4-h treatment with 1 μM staurosporine and no release of cytochrome c was detected. The results of immunofluorescence in the Bcl-2 overexpressing strain were essentially indistinguishable from those obtained using untreated cells (Fig. 1, A–C). In contrast, the presence of 100 μM of the caspase peptide inhibitor z-VAD-fmk did not prevent the appearance of Bax immunoreactivity and cytochrome c release from mitochondria, despite total blockade of nuclear fragmentation (Fig. 1, G–I). This is consistent with recent observations that cytochrome c release is an upstream event in the pathway leading to caspase activation (Bossy-Wetzel et al., 1998; Eskes et al., 1998).
We next tested whether the appearance of a mitochondrial Bax staining during apoptosis also occurs in other cell types. We used primary cultures of CGC undergoing apoptosis after KCl and serum deprivation (D'Mello et al., 1993). We show that, as with HeLa cells exposed to staurosporine, these neurons displayed a mitochondrial staining with the antibody raised against amino acids 1–21 of Bax, cytochrome c–depleted mitochondria, and a condensed nucleus (Fig. 1, J–L). Similar results were obtained with another pAb raised against amino acids 43–61 of murine Bax. Interestingly, in CGC, unlike HeLa cells, only the neurons with a marginalized chromatin, representing an early stage in apoptosis, displayed a Bax staining, whereas those in the terminal phase of apoptosis, with a highly condensed nucleus and a shrunken cell body, appeared Bax negative (Fig. 1, J–L). This suggests that in these neurons the Bax immunoreactivity is a transient event in the apoptotic pathway.
Bax Mitochondrial Levels Do Not Increase during Apoptosis
Two hypotheses could explain the appearance of Bax mitochondrial staining during apoptosis. Either Bax could translocate to mitochondria as recently reported (Wolter et al., 1997; Gross et al., 1998) or alternatively, Bax may be present continuously in mitochondria but may undergo a conformational change rendering it immunoreactive towards the NH2-terminal antibodies. To distinguish between these two possibilities, we compared Bax levels by Western blot analysis in mitochondria from control and apoptotic HeLa cells and CGC (Fig. 3). These studies showed that in control cultures of both cell types Bax is distributed in both the soluble fraction (S) and HM fraction enriched in mitochondria and that the level of mitochondrial Bax does not increase during apoptosis (see also Fig. 4 A). Therefore, Bax translocation to the mitochondria is unlikely to account for the appearance of Bax immunoreactivity. It is interesting to note that the level of Bax detected by the antibody raised against amino acids 1–21 of Bax decreases in the soluble fraction during apoptosis as noted earlier (Wolter et al., 1997; Gross et al., 1998). However, the concomitant appearance of smaller immunoreactive fragments, at least as seen in CGC, suggests that this may be the result of Bax proteolysis. Therefore, the results in Fig. 3 suggest that the punctate pattern of Bax immunostaining observed in apoptotic cells is not due to an increased level of mitochondrial Bax but rather to a structural change of the protein which leads to exposure of the NH2-terminal domain.
Figure 3.

Subcellular distribution of Bax in HeLa cells and CGC undergoing apoptosis. HeLa cells treated with 1 μM staurosporine for 3, 6, and 9 h and CGC cultured in a medium with a low KCl concentration (5 mM) and without serum for 6, 14, and 24 h were homogenized in isotonic buffer and separated into a soluble fraction (S) and an HM fraction enriched in mitochondria. The fractions (respectively, 30- and 15-μg proteins) were analyzed by Western blot with the pAb raised against amino acids 1–21 of human Bax (Upstate Biotechnology) and the monoclonal anti– COX-IV antibody.
Figure 4.
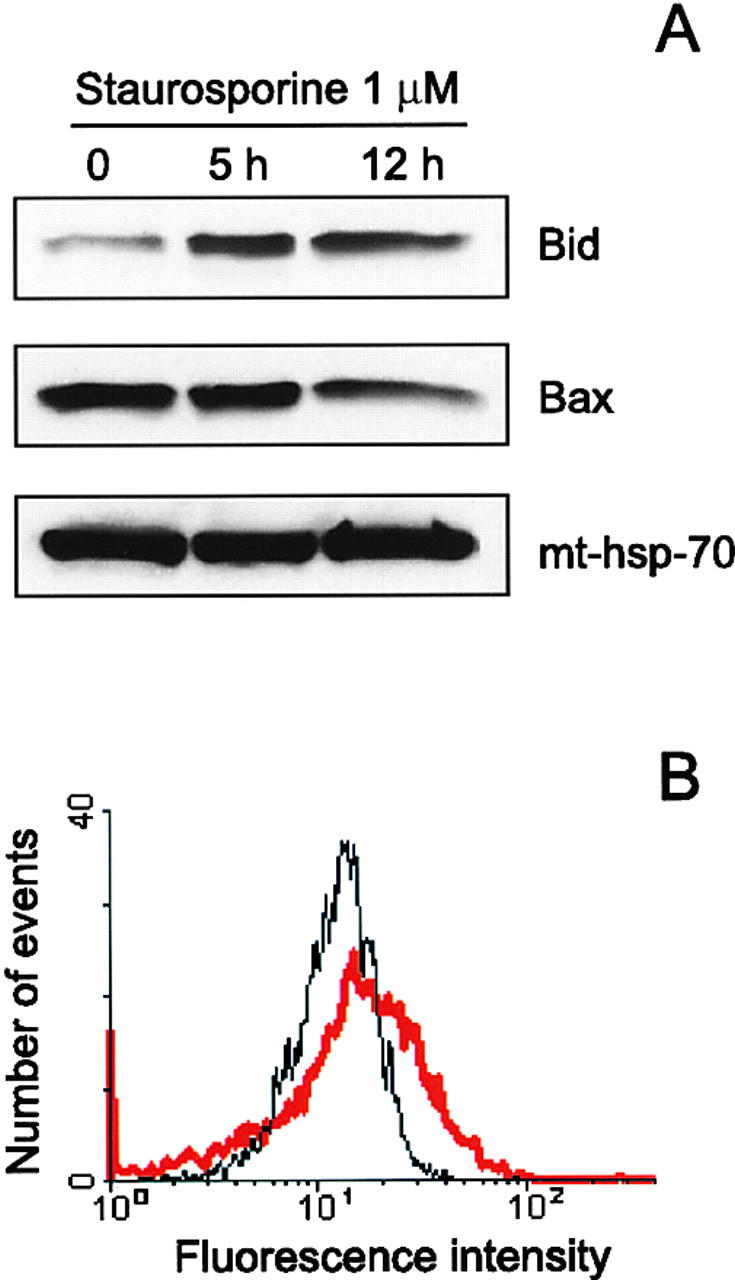
Translocation of Bid to mitochondria during staurosporine-induced apoptosis of HeLa cells. (A) Mitochondria from HeLa cells before and after treatment with 1 μM staurosporine for 5 and 12 h were isolated on a sucrose gradient and analyzed for the presence of Bid and Bax by Western blotting. pAbs against full-length recombinant Bid (top) and to amino acids 1–21 of human Bax (middle) were used. Level of mt-hsp-70 (bottom) was used as a gel loading control. (B) Mitochondria from control HeLa cells (black curve) and HeLa cells treated with 1 μM staurosporine for 9 h (red curve) were isolated on a sucrose gradient, fixed, immunostained for Bid, and analyzed by flow cytometry.
Translocation of Bid to Mitochondria during Apoptosis
Resolution of the three-dimensional structure of Bcl-xL in its non–membrane-integrated conformation revealed the existence of a hydrophobic cleft on the surface of the protein formed by the BH1, BH2, and BH3 domains which provides a likely binding site for BH3 containing peptides (Muchmore et al., 1996; Sattler et al., 1997). This suggested to us that the binding of a BH3 domain protein to Bax could induce the structural changes which we postulate to explain the appearance of Bax immunostaining during apoptosis. A likely candidate for such a protein was Bid, because this protein had been shown previously to strongly interact with Bax (Wang et al., 1996). As Bid is expected to be mainly cytosolic (Wang et al., 1996), we reasoned that in order to induce a change in the Bax structure, Bid might translocate to mitochondria during apoptosis. This hypothesis was tested using HeLa cells undergoing apoptosis. Mitochondria from these cells were purified on a sucrose gradient and the respective levels of Bid and Bax were determined by Western blotting at 5 and 12 h after addition of 1 μM staurosporine (Fig. 4 A). These results first of all confirmed our earlier observation that the levels of Bax in mitochondria do not change during apoptosis and in addition showed a significant increase in the level of Bid. The increased level of Bid in these mitochondria was confirmed by flow cytometry (Fig. 4 B). Thus, the observed translocation of Bid to mitochondria during apoptosis is consistent with the idea that Bid could be responsible for the change in Bax conformation, and this hypothesis was tested using isolated mitochondria from HeLa cells.
Bid Triggers Bax Conformational Change
In a first series of experiments, mitochondria from HeLa cells were isolated on a sucrose gradient and incubated for 15 min with or without 1 μM Bid. Bax immunostaining was then assessed by flow cytometry (Fig. 5). When an antibody to the NH2-terminal sequence (amino acids 1–21) of Bax was used (Fig. 5 A), the mean fluorescence intensity of mitochondria from HeLa cells exposed to 1 μM Bid was about fivefold higher than that of untreated mitochondria (Fig. 5 A). A similar effect was observed with mitochondria from HEK cells (Fig. 5 B). Dose–response analysis (see Fig. 7 A) using Bid concentrations in the range of 1 nM to 10 μM indicated that half the maximal increase in Bax-immunofluorescence intensity (EC50) in mitochondria from HeLa cells was obtained at a concentration of 5 nM Bid, with saturation reached at 1 μM. In contrast, using mitochondria from the three Bax-deficient tumor cell lines LoVo, DU145, and LS180, no increase in fluorescence was detected after addition of 8 μM Bid (Fig. 5, D–F).
Figure 5.

Flow cytometric analysis of Bax immunofluorescence in mitochondria after exposure to Bid. Mitochondria from HeLa cells (A), HEK cells (B), Bcl-2–overexpressing HeLa cells (C), as well as from LoVo (D), DU145 (E), and LS180 (F) tumor cell lines, were isolated on a sucrose gradient and incubated for 15 min at 30°C in the presence or absence of 1 μM (A and C) or 8 μM recombinant Bid (B and D–F). After incubation, mitochondria were fixed and immunostained with the antibody to amino acids 1–21 of Bax (A and B, D–F) or with the antibody to amino acids 4–21 of human Bcl-2 (C). Immunostained mitochondria were analyzed by flow cytometry. Black curve: control. Red curve: after treatment with Bid.
Figure 7.
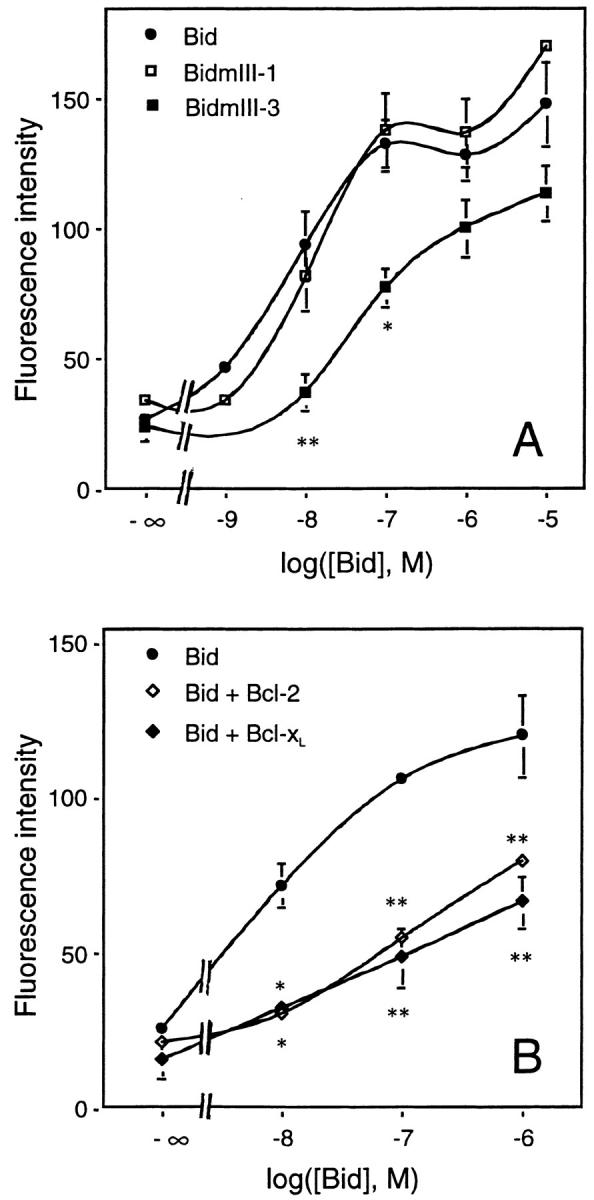
Dose–response curve of Bid-induced Bax immunoreactivity. (A) Effects of BH3 mutations in Bid. Mitochondria from HeLa cells were isolated on a sucrose gradient and incubated for 15 min at 30°C with increasing concentrations of recombinant wild-type Bid or BidmIII-1 and BidmIII-3 mutants. Mitochondria were then fixed, immunostained with the antibody raised against amino acids 1–21 of Bax, and analyzed by flow cytometry. (B) Inhibition of Bid effect by Bcl-2 and Bcl-xL. Mitochondria isolated from HeLa cells were incubated with increasing concentrations of recombinant Bid for 15 min at 30°C in the presence or absence of 1 μM recombinant Bcl-2 or Bcl-xL before fixation, Bax immunostaining, and FACS® analysis. Results are mean ± SEM of three independent experiments, each performed in duplicate. *P < 0.05; **P < 0.01; significantly different from corresponding values obtained with wild-type Bid (ANOVA followed by Bonferroni's test).
Using mitochondria isolated from HeLa-Bcl-2 cells and an antibody raised against the NH2 terminus of Bcl-2 (amino acids 4–21), we found that, in contrast to Bax, the Bcl-2 fluorescence intensity did not change after incubation with 8 μM Bid (Fig. 5 C).
To investigate the role of the BH3 domain of Bid in the stimulation of Bax immunoreactivity, we produced two different recombinant BH3 mutants of Bid, BidmIII-1 (M97,D98→ A,A) and BidmIII-3 (G94→ A) described previously by Wang et al. (1996). First, we tested these mutants for binding to Bax, Bcl-2, and Bcl-xL in vitro (Fig. 6 A). As shown by Wang et al. (1996), the mutant BidmIII-3 had a lowered affinity for Bax but still bound Bcl-2 or Bcl-xL, whereas BidmIII-1 showed a lowered affinity for Bcl-2 or Bcl-xL but was still able to interact strongly with Bax (Fig. 6 A). When tested for their ability to stimulate Bax immunoreactivity, BidmIII-3 was 10-fold less efficient (EC50 < 50 nM) than wild-type Bid, whereas BidmIII-1 was as effective as wild-type Bid (Fig. 7 A). Importantly, the effect of Bid on Bax immunoreactivity was dramatically reduced by at least two orders of magnitude when 1 μM Bcl-2 or Bcl-xL was added to the mitochondrial preparation (Fig. 7 B).
Figure 6.

Interactions between BH3 mutants of Bid and Bcl-2 family proteins in vitro. (A) 100 nM of Bid, BidmIII-1, or BidmIII-3 was incubated in PBS with 100 nM of Bax, Bcl-xL, Bcl-xLm, or Bcl-2 for 30 min on ice. Recombinant proteins were then immunoprecipitated (IP) with the pAb against Bid or a mAb against Bcl-2 (Genosys) and separated by SDS-PAGE. Immunoblottings (IB) were performed using the pAb against Bid, an mAb against Bax (Genzyme), a pAb against Bcl-x (Transduction Laboratories), or a pAb against Bcl-2 (sc-492). (B) 100 nM of Bid or BidmIII-1 was incubated with 1 μM of Bcl-xL or Bcl-2 for 30 min on ice, followed by steps described in A. The same antibodies were used for immunoprecipitation and immunoblotting. In addition, the polyclonal anti–Bcl-x antibody from Santa Cruz Biotechnology (sc-634) was used to precipitate Bcl-xL.
Bax Conformational Change Is Accompanied by a Release of Cytochrome c from Mitochondria
Overexpression of Bax in cells or addition of recombinant Bax directly to mitochondria results in the release of cytochrome c (Xiang et al., 1996; Eskes et al., 1998; Jürgensmeier et al., 1998). This Bax-induced cytochrome c release from mitochondria could be related to a change in conformation of the protein triggered by Bid. To test this hypothesis, mitochondria were incubated with Bid (1–100 nM) for 15 min and levels of cytochrome c were assessed by Western blotting in both mitochondrial pellets and supernatants (Fig. 8 A). As with Bax (Eskes et al., 1998), we found that Bid induced the release of cytochrome c in a concentration-dependent manner. A maximal effect was observed at 10 nM Bid which corresponds to >90% loss of mitochondrial cytochrome c. BidmIII-1 was as active as wild-type Bid, whereas BidmIII-3 was ∼10-fold less active (Fig. 8 A).
Figure 8.

Bid-induced cytochrome c release from mitochondria. (A) Mitochondria from HeLa cells were isolated on a sucrose gradient, incubated for 15 min at 30°C with increasing concentrations of recombinant Bid as indicated. The incubation mix was centrifuged and the supernatants and pellets were analyzed for cytochrome c content by Western blot. mt-hsp-70 was used as a gel loading control. (B) Mitochondria from HeLa cells isolated on a sucrose gradient were incubated for 15 min at 30°C with 100 nM of wild-type Bid in the presence or absence of 1 μM of Bcl-2, Bcl-xL, or a mutant of Bcl-xL in which Gly 138 was replaced by an alanine (Bcl-xLm). Mitochondria were then centrifuged and the cytochrome c content of both the pellet and the supernatant of each sample was estimated by Western blotting. Equal loading of the mitochondrial pellet was confirmed with the antibody to mt-hsp-70.
Altogether, these results demonstrate a tight correlation between the effect of Bid on mitochondrial cytochrome c release and on Bax NH2-terminal immunoreactivity (see Fig. 7).
Bcl-xL and Bcl-2 Inhibit Bid-induced Cytochrome c Release
To test whether Bcl-xL and Bcl-2 were able to inhibit Bid-induced cytochrome c release, mitochondria were incubated with 100 nM Bid together with 1 μM recombinant Bcl-xL or Bcl-2 (Fig. 8 B). Consistent with the reduced Bax immunofluorescence reported in Fig. 7 B, we found that both proteins were able to inhibit Bid-induced cytochrome c release. Furthermore, Bcl-xL and Bcl-2 were also able to abrogate the effect of BidmIII-1 (Fig. 8 B). However, even under these conditions in which they are in excess (1 μM), Bcl-xL and Bcl-2 do not bind BidmIII-1 (100 nM) sufficiently to explain their inhibitory effect on cytochrome c release (Fig. 6 B). Therefore, these data indicate that the inhibition of cytochrome c release from mitochondria by Bcl-2 and Bcl-xL does not require interaction with Bid.
The inhibitory effect of Bcl-2 on Bid-induced cytochrome c release was further confirmed using mitochondria isolated from Bcl-2–overexpressing HeLa cells. Fig. 9 A shows that Bid was 10-fold less efficient in triggering the release of cytochrome c from HeLa-Bcl-2 mitochondria compared with mitochondria from control HeLa cells.
Figure 9.

Involvement of Bax and Bcl-2 in the Bid-induced release of cytochrome c from mitochondria. (A) Mitochondria were isolated on a sucrose gradient from HeLa cells, Bcl-2–overexpressing HeLa cells, and from the Bax-deficient colon tumor cells LS180. They were incubated for 15 min at 30°C in the presence of increasing concentrations of recombinant Bid and the contents of cytochrome c of both the mitochondrial pellets and the supernatants were estimated by Western blot. Equal loading of the mitochondrial pellet was confirmed with the monoclonal anti–COX-IV antibody. (B) Mitochondria from LS180 cells were incubated for 15 min at 30°C with 5 μM recombinant Bax lacking 20 amino acids at the COOH terminus in the presence or absence of 50 nM recombinant Bid. Cytochrome c was detected by Western blot in the mitochondrial pellets and the supernatants. Equal loading of the mitochondrial pellet was confirmed with the antibody against mt-hsp-70. (C) Flow cytometric analysis of Bak immunostaining of mitochondria isolated from HeLa and LS180 cells. Mitochondria from both cell types were isolated on a sucrose gradient and incubated with 1 μM recombinant Bid for 15 min at 30°C before fixation, immunostaining with an antibody raised against NH2-terminal domain of Bak (amino acids 1–52; Calbiochem), and FACS® analysis. Black curve: control. Red curve: 1 μM Bid.
To test whether the inhibition of Bid-induced cytochrome c release by Bcl-xL required its binding to Bax, we used a Bcl-xL mutant in which Gly 138 was replaced by an alanine (Bcl-xLm: G138→ A). This mutant does not bind to Bax and is unable to rescue cells from apoptosis (Muchmore et al., 1996; Ottilie et al., 1997). In contrast to wild-type Bcl-xL, we found that this Bcl-xL mutant was unable to inhibit the Bid effect (Fig. 8 B). However, in vitro binding experiments showed that Bcl-xLm was still able to bind Bid (Fig. 6 A). Altogether, these results indicate that Bcl-xL prevents Bid-induced cytochrome c release through its interaction with Bax and not through its ability to bind Bid.
Bid-induced Release of Cytochrome c from Mitochondria from Bax-deficient Cells
To demonstrate that Bid-induced cytochrome c release was mediated by Bax, we tested the ability of Bid to trigger cytochrome c release from mitochondria isolated from the Bax-deficient tumor cell line LS180. The results of Western blot analysis showed that, as expected, Bax was not detectable in this cell line, whereas the levels of Bcl-xL and Bak found associated with mitochondria were similar to levels in mitochondria from HeLa cells (not shown). The results in Fig. 9 A show that Bid was at least 10-fold less efficient in triggering cytochrome c release from mitochondria from LS180 cells, compared with mitochondria from HeLa cells. In contrast, if mitochondria from LS180 cells were incubated with both Bid and Bax together, the mitochondria were completely depleted of cytochrome c within 15 min (Fig. 9 B), confirming that the major effect of Bid in these mitochondria was indeed mediated by Bax.
However, it should also be noted that even in LS180 mitochondria without exogenous Bax, Bid was nevertheless able to induce release of cytochrome c at higher concentrations (Fig. 9 A), suggesting that there may be other proapoptotic targets for Bid in the mitochondrial membrane. Since we showed that Bak is present at about the same level in mitochondria from HeLa cells and LS180 cells, we therefore decided to test whether Bid was also able to induce a conformational change in Bak. Results in Fig. 9 C show that exposure of mitochondria from both HeLa cells and LS180 cells to 1 μM Bid, followed by immunostaining with an NH2-terminal antibody to Bak (amino acids 1–52), resulted in a threefold increase in Bak immunoreactivity in both cell types as judged by subsequent FACS® analysis. Thus, a component of the Bid-induced cytochrome c release from these mitochondria is also likely to be mediated via Bak.
Discussion
Recent reports have shown that Bid is responsible for the release of cytochrome c from mitochondria during Fas- and TNF-mediated apoptosis (Li et al., 1998; Luo et al., 1998). However, the mechanisms by which Bid triggers this event are unclear. We now report, in a different system, that Bax mediates Bid-induced cytochrome c release from mitochondria. During staurosporine-induced apoptosis of HeLa cells, Bid translocates from the cytosol to mitochondria, an event associated with a change in conformation of Bax and a release of cytochrome c. Using isolated mitochondria, we demonstrate that direct binding of Bid to Bax is a prerequisite for Bax structural change and cytochrome c release.
Bid but Not Bax Translocates from the Cytosol to Mitochondria during Staurosporine-induced Apoptosis
It has been reported recently that, during Fas- and TNF-mediated apoptosis, Bid becomes cleaved by caspase 8 and its COOH-terminal domain translocates to mitochondria (Li et al., 1998; Luo et al., 1998). Using staurosporine-treated HeLa cells, we found that full-length Bid can also translocate to mitochondria during apoptosis. However, we have been unable to detect the cleaved form of Bid attached to mitochondria. Our findings suggest that caspase-induced Bid cleavage is not an essential requirement for its movement to mitochondria. Translocation of full-length Bid may thus occur in types of apoptosis in which activation of caspases is not a primary event in the apoptotic pathway.
In contrast to Bid, we did not detect any significant translocation of Bax to mitochondria, although we did observe a decrease in Bax cytosolic levels in both HeLa cells and CGC undergoing apoptosis. At least in the case of CGC, the appearance of several smaller Bax immunoreactive bands by Western blot suggests that this decrease in the level of Bax in the cytosol may be the result of proteolytic degradation. The characterization of these lower molecular weight species and the involvement of caspases or, as reported recently, calpain (Wood et al., 1998) in Bax proteolysis is currently ongoing.
The absence of Bax translocation to mitochondria during apoptosis has also been observed by Goping et al. (1998) in some of their experiments with FL5.12 cells undergoing apoptosis after IL-3 deprivation. In contrast, others have reported a translocation of Bax to mitochondria in IL-3–deprived FL5.12 cells (Gross et al., 1998) in staurosporine-treated HL-60 (Hsu et al., 1997) and in GFP-Bax–overexpressing Cos-7 kidney epithelial cells (Wolter et al., 1997). These apparent conflicting results may be due to the type of cells or cell death stimuli used in these various studies. In all cultured cells that we examined so far, including HeLa, Cos-7, and HEK cell lines as well as cultured primary neurons from superior cervical ganglia and cerebellar granule neurons (data presented here; Nichols, A., and J.-C. Martinou, unpublished results), we have observed high levels of Bax associated with mitochondria in addition to cytosolic Bax. In clear contrast, in cells from freshly dissected tissues but not exposed to culture conditions, including thymus, spleen, or brain, Bax was practically cytosolic and hardly detectable in mitochondria (Antonsson, B., unpublished data), a finding which confirms previous results from Hsu et al. (1997). Thus, it seems that simply maintaining cells in culture leads to redistribution of a significant part of cytosolic Bax to mitochondria. The important question which remains to be understood is why these cells, despite significant levels of mitochondrial-associated Bax, do not spontaneously undergo apoptosis. Does Bax remain inactive in cultured cells because of possible association with antiapoptotic proteins such as Bcl-xL or Bcl-2? or another protein? and/ or because of an inert configuration? The data presented here provide a possible explanation to this question.
Bax Undergoes a Bid-dependent Conformational Change
During apoptosis of HeLa cells and cerebellar granule neurons, Bax immunoreactivity was found to increase despite the apparent absence of Bax translocation to mitochondria. Moreover, addition of Bid directly to isolated mitochondria was sufficient to stimulate the appearance of Bax immunoreactivity towards NH2-terminal antibodies. Altogether, these results suggest that Bax immunoreactivity increases because the NH2-terminal region of the protein undergoes a conformational change making it accessible to antibodies. We presume that this domain, which may include the BH3 domain, must be localized on the external face of the outer mitochondrial membrane since it was accessible to antibodies without the need for membrane permeabilization. Alteration of the structure of Bax was inhibited by Bcl-2 or Bcl-xL, but not by the caspase inhibitor z-VAD-fmk, suggesting that, at least in the models of apoptosis described here, exposure of this domain is not a caspase-dependent event.
Bid-induced exposure of the NH2-terminal domain of Bax could reflect either a conformational change in Bax or a displacement of another protein serving to mask critical immunoreactive residues. For example, interaction of Bid with antiapoptotic proteins may be responsible for the dissociation of Bax heterodimers and the unmasking of Bax epitopes. However, BidmIII-3, with reduced affinity for Bax but which retained a high affinity for Bcl-2 and Bcl-xL, was much less active in enhancing Bax immunoreactivity and resulted in a 10-fold lower release of cytochrome c than wild-type Bid. On the other hand, BidmIII-1, with a reduced affinity for Bcl-2 and Bcl-xL but which retained high affinity for Bax, behaved as wild-type Bid. In addition, in ELISA competitive binding studies, Bid was found unable to dissociate Bax/Bcl-2 and Bax/Bcl-xL complexes (not shown), confirming previous data demonstrating that Bid failed to bind Bax/Bcl-2 heterodimers (Wang et al., 1996). Altogether, these data suggest that Bid-induced unmasking of the NH2-terminal domain of Bax results from a direct binding of Bid to Bax and not from a loss of Bax interaction with Bcl-2 or Bcl-xL.
Our data also suggest that Bcl-xL and Bcl-2 inhibit the effect of Bid by interacting directly with Bax and not by preventing the interaction between Bid and Bax. First, Bcl-xL and Bcl-2 were able to inhibit Bid as well as BidmIII-1, which has a low affinity for Bcl-xL and Bcl-2, indicating that their mechanism of action was independent of their affinity for Bid. Second, a mutant of Bcl-xL (Bcl-xLm: G138→ A) which has lost its ability to interact with Bax (Ottilie et al., 1997) but which is still able to bind Bid was unable to block Bid effect. This finding shows that Bcl-xL must bind Bax to prevent its activation but that its inhibitory effect is independent of its binding to Bid.
On the other hand, it has been reported that heterodimerization of Bcl-xL with Bax was not always essential for its death-repressing action (Cheng et al., 1996) since several mutations that disrupted the ability of Bcl-xL to interact with Bax still retained an antiapoptotic activity (Cheng et al., 1996). Therefore, our data suggest that these Bcl-xL mutants exerted their activity either upstream of Bax conformational change or downstream of Bax activation and cytochrome c release, as recently reported for Bcl-2 (Rossé et al., 1998; Zhivotovsky et al., 1998).
Bax Plays a Key Role in the Bid-induced Release of Cytochrome c from Mitochondria
Addition of Bid to mitochondria is sufficient to trigger cytochrome c release (Li et al., 1998; Luo et al., 1998; this study). Here, we extend this observation and provide evidence for a mechanism by which Bax may mediate the Bid effect. Consistent with this, we have shown that Bid was at least 10-fold less efficient in triggering cytochrome c release from mitochondria from LS180 tumor cells which possess no detectable Bax (Rampino et al., 1997) compared with mitochondria from HeLa cells. This difference seems to be due to the deficiency in Bax and not to an intrinsic property of mitochondria from LS180 cells acquired during tumorigenicity, since in the presence of recombinant Bax, mitochondria from these cells respond to Bid in the same way as mitochondria from HeLa cells. Importantly, in these complementation studies, addition of Bid was found to potentiate Bax effect. This potentiation may be explained by a change in conformation of Bax induced by Bid. However, Bax does not seem to be acting alone in this process since a low level of cytochrome c was released when mitochondria from LS180 cells were exposed to high concentrations of Bid. This may be due to another Bax-like protein present on their mitochondria. Bak (which is present in similar amounts in the mitochondria of LS180 and HeLa cells) may represent such an alternative protein and was also found to undergo a conformational change in Bid-treated mitochondria.
In conclusion, we have shown that upon binding to Bid, mitochondrially associated Bax undergoes a conformational change, leading to release of cytochrome c in the cytosol. A change in Bax conformation leading to exposure of the NH2-terminal domain had been observed already by Hsu and Youle (1997) when the protein was in a solution containing nonionic detergents. This change in conformation may expose the BH3 domain of Bax which, as proposed for Bak (Sattler et al., 1997), may normally be oriented toward the interior of the protein and thus be unavailable for homo- or heterodimerization. As a dimer, Bax may then be able to insert within mitochondrial membranes leading to the observed changes in mitochondrial activity (Gross et al., 1998). A recent report indicates that, during apoptosis, the association of Bax with mitochondrial membranes changes from a weak to a strong insertion (Goping et al., 1998), a process which is regulated by the NH2-terminal domain of Bax. As a working hypothesis, we propose that Bax remains weakly attached to mitochondria in the absence of apoptotic stimulus, and that BH3 only proteins such as Bid may promote apoptosis by modifying the structure of Bax leading to its insertion into mitochondrial membranes.
Acknowledgments
We thank Dr. Steve Arkinstall for critical reading of the manuscript, Marc Herzlich for technical assistance, Christopher Hebert for artwork, Claude Berney for help with the flow cytometer and confocal microscope, and Dr. Tim Wells for encouraging support.
Abbreviations used in this paper
BH
Bcl-2 homology
CGC
cerebellar granule cells
COX-IV
cytochrome oxidase subunit IV
HM
heavy membrane
MB
mitochondrial buffer
mt-hsp-70
mitochondrial heat shock protein 70
Footnotes
Part of this work was funded by the European Community (Biotech grant BIO4CT96 0774 to Jean-Claude Martinou).
References
- Antonsson B, Conti F, Ciavatta AM, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod J-J, Mazzei G, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- Aritomi M, Kunishima N, Inohara N, Ishibashi Y, Ohta S, Morikawa K. Crystal structure of rat Bcl-xL . J Biol Chem. 1997;272:27886–27892. doi: 10.1074/jbc.272.44.27886. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO (Eur Mol Biol Organ) J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Cheng EH-Y, Levine B, Boise LH, Thompson CB, Hardwick JM. Bax-independent inhibition of apoptosis by Bcl-xL . Nature. 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- Chittenden T, Flemington C, Houghton AB, Ebb RG, Gallo GJ, Elangovan B, Chinnadurai G, Lutz RJ. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. EMBO (Eur Mol Biol Organ) J. 1995;14:5589–5596. doi: 10.1002/j.1460-2075.1995.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskes R, Antonsson B, Osen-Sand A, Montessuit S, Richter C, Sadoul R, Mazzei G, Nichols A, Martinou J-C. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ions. J Cell Biol. 1998;143:217–224. doi: 10.1083/jcb.143.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estoppey S, Rodriguez I, Sadoul R, Martinou J-C. Bcl-2 prevents activation of CPP32 cysteine protease and cleavage of poly (ADP-ribose) polymerase and U1-70 kD proteins in staurosporine-mediated apoptosis. Cell Death Differ. 1997;4:34–38. doi: 10.1038/sj.cdd.4400205. [DOI] [PubMed] [Google Scholar]
- Goping IS, Gross A, Lavoie JN, Nguyen M, Jemmerson R, Roth K, Korsmeyer SJ, Shore GC. Regulated targeting of BAX to mitochondria. J Cell Biol. 1998;143:207–215. doi: 10.1083/jcb.143.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO (Eur Mol Biol Organ) J. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y-T, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- Hsu Y-T, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-XLduring apoptosis. Proc Natl Acad Sci USA. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelekar A, Thompson CB. Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol. 1998;8:324–330. doi: 10.1016/s0962-8924(98)01321-x. [DOI] [PubMed] [Google Scholar]
- Kelekar A, Chang BS, Harlan JE, Fesik SW, Thompson CB. Bad is a BH3 domain-containing protein that forms an inactivating dimer with Bcl-XL. Mol Cell Biol. 1997;17:7040–7046. doi: 10.1128/mcb.17.12.7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharbanda S, Pandey P, Schofield L, Isreals S, Roncinske R, Yoshida K, Bharti A, Yuan Z-M, Saxena S, Weichselbaum R, et al. Role for Bcl-xLas an inhibitor of cytosolic cytochrome c accumulation in DNA damage-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:6939–6942. doi: 10.1073/pnas.94.13.6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Knudson CM, Korsmeyer SJ. Bcl-2 and Bax function independently to regulate cell death. Nat Genet. 1997;16:358–363. doi: 10.1038/ng0897-358. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- Kroemer GC. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu C, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Lill R, Neupert W. Mechanisms of protein import across the mitochondrial outer membrane. Trends Cell Biol. 1996;6:56–61. doi: 10.1016/0962-8924(96)81015-4. [DOI] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl-2 interacting protein, mediates cytochrome c release in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Vélez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. Bcl-xLforms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong S-L, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- Ottilie S, Diaz J-L, Chang J, Wilson G, Tuffo KM, Weeks S, McConnell M, Wang Y, Oltersdorf T, Fritz LC. Structural and functional complementation of an inactive Bcl-2 mutant by Bax truncation. J Biol Chem. 1997;272:16955–16961. doi: 10.1074/jbc.272.27.16955. [DOI] [PubMed] [Google Scholar]
- Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M. Somatic frameshift mutations in the BAXgene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- Rossé T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC. Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA. 1997;94:5113–5118. doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger PH, Gross A, Yin X-M, Yamamoto K, Saito M, Waksman G, Korsmeyer SJ. Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic BCL-2. Proc Natl Acad Sci USA. 1997;94:11357–11362. doi: 10.1073/pnas.94.21.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, Korsmeyer SJ. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sci USA. 1995;92:7834–7838. doi: 10.1073/pnas.92.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba M, Bockaert J, Journot L. Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J Neurosci. 1997;17:83–90. doi: 10.1523/JNEUROSCI.17-01-00083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Yian X-M, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- Wolter KG, Hsu Y-T, Smith CL, Nechushtan A, Xi X-G, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DE, Thomas A, Devi LA, Berman Y, Beavis RC, Reed JC, Newcomb EW. Bax cleavage is mediated by calpain during drug-induced apoptosis. Oncogene. 1998;17:1069–1078. doi: 10.1038/sj.onc.1202034. [DOI] [PubMed] [Google Scholar]
- Xiang J, Chao DT, Korsmeyer ST. Bax-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proc Natl Acad Sci USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, Korsmeyer SJ. Molecular thanatopsis: a discourse on the BCL2 family and cell death. Blood. 1996;88:386–401. [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yin X-M, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Osipov K, Jockel J, Waksman G, Korsmeyer SJ. BH3 domain of BAD is required for heterodimerization with BCL-XLand pro-apoptotic activity. J Biol Chem. 1997;272:24101–24104. doi: 10.1074/jbc.272.39.24101. [DOI] [PubMed] [Google Scholar]
- Zhivotovsky B, Orrenius S, Brustugun OT, Doskeland SO. Injected cytochrome c induces apoptosis. Nature. 1998;391:449–450. doi: 10.1038/35060. [DOI] [PubMed] [Google Scholar]