ETV6-NTRK3 fusion oncogene initiates breast cancer from committed mammary progenitors via activation of AP1 complex (original) (raw)
. Author manuscript; available in PMC: 2008 Jun 1.
Published in final edited form as: Cancer Cell. 2007 Dec;12(6):542–558. doi: 10.1016/j.ccr.2007.11.012
SUMMARY
To better understand the cellular origin of breast cancer, we developed a mouse model that recapitulates expression of the ETV6-NTRK3 (EN) fusion oncoprotein, the product of the t(12;15)(p13;q25) translocation characteristic of human secretory breast carcinoma. Activation of EN expression in mammary tissues by Wap-Cre leads to fully penetrant, multifocal malignant breast cancer with short latency. We provide genetic evidence that in nulliparous Wap-Cre;EN females, committed alveolar bipotent or CD61+ luminal progenitors, are targets of tumorigenesis. Furthermore, EN transforms these otherwise transient progenitors through activation of the AP1 complex. Given increasing relevance of chromosomal translocations in epithelial cancers, such mice serve as a paradigm for the study of their genetic pathogenesis and cellular origins, and generation of novel preclinical models.
SIGNIFICANCE
For the largest class of human tumors, those of epithelial origin, little is known about their initiating genetic hits or cells of origin. Whether tissue stem cells or more committed progenitors are targets for transformation is uncertain. We developed a system in which epithelial tumorigenesis can be assessed from the initial event to frank malignancy. In this breast cancer model based on chromosomal translocation, we show through genetic marking that committed mammary progenitors, rather than mammary stem cells, are direct targets of transformation. We show that activation of the AP1 complex represents a critical downstream event of the ETV6-NTRK3 translocation. Further focus on this transcriptional complex as a target in human breast cancer is warranted.
INTRODUCTION
For most malignancies neither the initiating genetic event nor the cell of origin is known. Cancers are heterogeneous in composition and are organized in a hierarchy that includes cells competent to recreate the tumor on transplantation, designated tumor initiating cells (T-ICs) or cancer stem cells, and other cells comprising the bulk tumor mass (Reya et al., 2001). Following the first hit, whether it occurs within a stem or more differentiated cell, secondary events of genetic or epigenetic nature contribute to evolution of malignancy. Access to these early steps in cancer formation is impossible in patients. Animal models provide a window into this phase of cancer development, but are likely to be relevant to human biology only so far as the genetic events mirror those occurring in patients.
In considering these issues, we have sought to apply to a cancer of epithelial origin principles that have proved successful in the study of hematopoietic malignancies. For leukemias and childhood sarcomas, chromosomal translocations leading to the production of chimeric proteins serve as initiating genetic events (Rowley, 2001). Although some gene rearrangements associated with leukemia are rare, study of these infrequent events has defined transcription factors critical for normal differentiation and pathways more generally perturbed in malignancy.
Until lately, the contribution of chromosomal rearrangements to epithelial cancers has been viewed as minor. Recently, chromosomal rearrangements involving the ETS family transcription factors were identified in >50% cases of human prostate cancer (Tomlins et al., 2007; Tomlins et al., 2006; Tomlins et al., 2005). Furthermore, an EML4-ALK fusion gene was identified in 6.7% of non-small-cell lung cancer cases (Soda et al., 2007). These observations prompt reassessment of conclusions regarding the involvement of chromosomal rearrangements in epithelial cancer.
t(12;15)(p13;q25) is a unique recurrent chromosomal translocation associated with cancer of all germ layers, including human secretory breast carcinoma (SBC) (Tognon et al., 2002), congenital fibrosarcoma (Knezevich et al., 1998b), congenital mesoblastic nephroma (Knezevich et al., 1998a; Rubin et al., 1998), and acute myelogenous leukemia (Eguchi et al., 1999). It produces a fusion oncogene, ETV6-NTRK3 (EN), which encodes a chimeric protein made up of the oligomerization domain of ETV6 (also known as TEL, an ETS family transcription factor) and the protein tyrosine kinase (PTK) domain of NTRK3 (also known as TRKC, a TRK family tyrosine kinase receptor for neurotrophin-3). The consistent presence of this translocation in human SBC provides strong genetic epidemiological support for its role in the initiation of breast cancer (Tognon et al., 2002).
Breast cancer is representative of other epithelial malignancies in its heterogeneity, both genetically and clinically (Simpson et al., 2005). In part, phenotypic heterogeneity may reflect diverse cellular origins of different subtypes of breast cancer (Ince et al., 2007).
Here we report a murine model of human sporadic breast cancer based on the EN translocation. We demonstrate that two committed mammary progenitors in the normal mammary developmental hierarchy serve as target cells of breast cancer. In addition, by performing microarray analysis, we reveal that EN-initiated transformation is mediated largely through activation of the c-Jun/Fosl1 AP1 complex.
RESULTS
Generating the Etv6-NTRK3 (EN) conditional knockin allele
We generated a Cre-Lox EN knockin allele by introducing the portion of human NTRK3 cDNA encoding the PTK domain (as found in SBC patients) into exon 6 of the mouse Etv6 locus (Figure 1A). We rendered this fusion allele conditional by insertion of a “floxed” transcriptional terminator sequence [“stopper” (Mao et al., 1999), Figure 1A] into the intron upstream of the knockin NTRK3 cDNA. The resulting allele, once activated by Cre-mediated excision of the floxed “stopper”, produces a mouse Etv6-human NTRK3 hybrid protein, which transforms NIH 3T3 cells (data not shown). Therefore, the EN knockin allele recapitulates the chimeric protein seen in patients.
Figure 1. Generating the Etv6-NTRK3 (EN) conditional knockin allele.
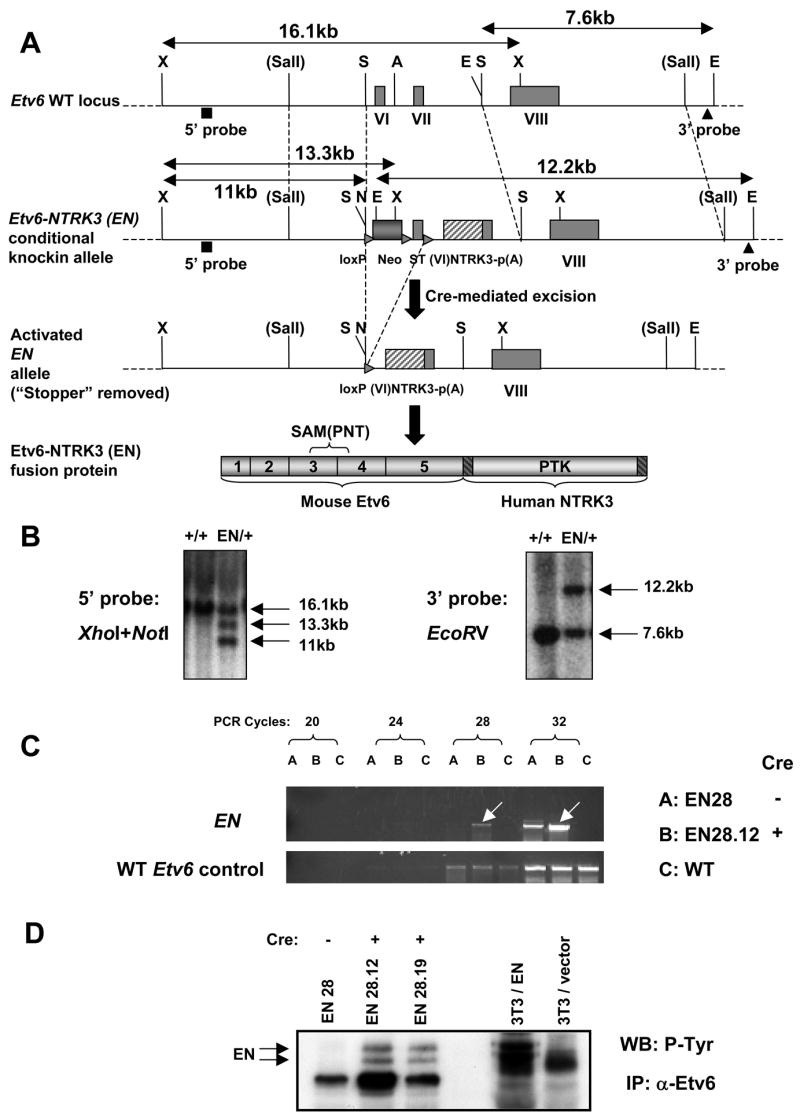
(A) Schematic diagrams of the endogenous WT Etv6 allele, the EN conditional knockin allele, the activated EN allele upon Cre-mediated excision of the floxed Neo-stopper cassette (ST: stopper), and the EN fusion protein produced from the activated EN allele. The 5′ and 3′ probes for southern blot are shown. X: _Xho_I, S: _Spe_I, A: _Apa_I, E: _EcoR_V, N: _Not_I, VI, VII, VIII: exon 6, 7, and 8 of Etv6, p(A): poly(A) signal, SAM(PNT): sterile alpha motif/pointed domain, PTK: protein tyrosine kinase domain.
(B) Southern blot screen and confirmation of correctly targeted ES cell clones. The 5′ probe recognizes a 16.1kb _Xho_I fragment from the WT Etv6 allele, a 13.3kb _Xho_I fragment (due to incomplete digestion of _Not_I) and an 11kb _Xho_I-_Not_I fragment (complete digestion) from the EN knockin allele. The 3′ probe recognizes a 7.6kb _EcoR_V fragment from the WT allele, and a 12.2kb _EcoR_V fragment from the EN allele.
(C) RT-PCR analysis showing greatly elevated expression of the EN fusion transcript from the “stopper”-excised ES cells (EN28.12). Note EN has slightly leaky expression in the parental EN28 ES cells (without excision of the “stopper”).
(D) Western blot showing detection of the EN fusion protein (tyrosine phosphorylated doublet) from the “stopper”-excised ES cells (EN28.12, EN28.19), but not from the parental EN28 ES cells. EN-3T3 cells were used as the positive control.
We identified correctly targeted ES cell clones using Southern blot (Figure 1B). By RT-PCR, we found EN ES cells without excision exhibited slightly leaky expression of the EN allele, but removal of the “stopper” greatly increased its expression (Figure 1C). At the protein level (Figure 1D), no EN fusion protein was detected in unexcised ES cells. However, its expression was readily visible from “stopper”-excised ES cells. Thus, the EN conditional knockin allele functions as designed.
The endogenous Etv6 locus is active in mammary epithelial cells
Because EN is under the control of the endogenous Etv6 promoter, we first examined where Etv6 is normally expressed in mammary glands (MGs) using a mouse strain we generated (TEA175) that carries an Etv6 locus having a β-Geo cassette inserted between its exons 2 and 3 and serves as a reporter for its expression.
Upon staining MGs from TEA175 heterozygous females for _lac_Z activity, we found Etv6 is expressed in both ductal and alveolar mammary epithelial cells (MECs) (Figure S1A in the Supplemental Data available with this article online). In addition, flow cytometry analysis revealed _lac_Z+ cells in all 4 major MEC subpopulations defined previously (Stingl et al., 2006) (Figure S1B).
Activation of EN in mammary glands by Wap-Cre leads to mammary tumors with complete penetrance
Without Cre-mediated activation of EN, heterozygotes were indistinguishable from their wild type (WT) littermates. Some EN heterozygous females (and very rarely, males) developed mammary tumors at advanced ages (>1 year), possibly due to the leakiness of the “stopper” and low level EN expression (Figure 1C).
To activate EN expression in MGs, we initially planned to use 2 commonly used MG-specific Cre mouse lines, MMTV-Cre and Wap-Cre (Wagner et al., 1997). Unfortunately, use of MMTV-Cre led to a lethal myeloproliferative disease within several weeks after birth, apparently due to expression of MMTV-Cre in the hematopoietic system (data not shown).
The more restricted expression of the endogenous Wap gene as well as that of the Wap-Cre transgene (Boulanger et al., 2005; Kordon et al., 1995; Robinson et al., 1995; Robinson et al., 1996; Wagner et al., 2002) afforded an approach for activation of EN in MGs without accompanying effects in other tissues. In maturing and mature nulliparous female mice, Wap+ cells are present only transiently as a minor subset at estrus. Wap expression is greatly elevated in differentiating MECs during late pregnancy and lactation, and then turned down following involution.
Based on the expression pattern of Wap, we initially predicted that Wap-Cre;EN (hereafter, WCEN) female mice might require rounds of pregnancies in order to express sufficient Cre to activate EN. Unexpectedly, however, all nulliparous WCEN females develop multifocal mammary tumors as early as 4-month of age (Figure 2A), with preceding lobuloalveolar hyperplasia (Figure 2B). Parous WCEN females develop similar multifocal mammary tumors with antecedent alveolar hyperplasia, and with no significant differences in tumor latency and histology. Some aged WCEN males also develop mammary tumors (Figure 2A and data not shown).
Figure 2. Wap-Cre;EN mice develop mammary tumors with antecedent alveolar hyperplasia.
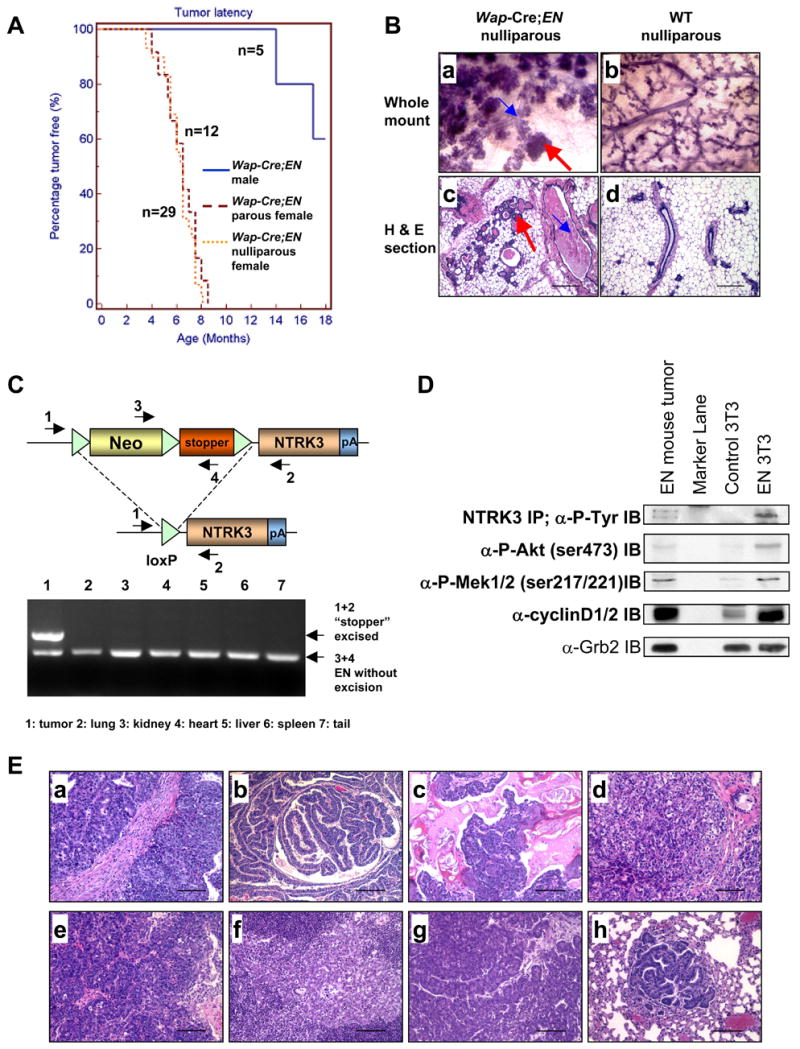
(A) Tumor free curves.
(B) Mammary glands (MGs) of WCEN females (a,c) exhibit extensive lobuloalveolar hyperplasia compared to WT (b,d). a–b: MG whole mounts stained with hematoxylin; c–d: hematoxylin & eosin (H&E) stained MG sections (scale bars=200μm). Red arrows: alveolar hyperplasia. Blue arrows: dilated ducts and accumulation of secretions within ducts.
(C) PCR analysis on genomic DNA shows excision of the Neo-stopper cassette only in the tumor. Locations of PCR primers (1–4) are indicated.
(D) Western blot analysis of the WCEN tumor. EN-3T3 cells were used as the positive control. IP/Western confirms EN expression in both the tumor and EN-3T3 cells.
(E) Histology (H&E stained sections, scale bars=100μm) of mammary tumors developed in WCEN mice. Panel c shows squamous metaplasia; Panels e and f were from a nulliparous female, e shows a primary mammary tumor and f shows metastasis in a lymph node; Panels g and h were from a male, g shows a primary mammary tumor and h shows metastasis in the lung.
To confirm that mammary tumors in WCEN animals resulted from activation of EN expression, we performed PCR analysis on genomic DNA prepared from tumors and other organs of WCEN animals and found that the “stopper” in the EN allele was excised only in tumor cells (Figure 2C). Consistent with this, we also detected EN protein in tumor tissues by Western blot (Figure 2D). Previous studies demonstrated that expression of the EN fusion protein in 3T3 cells led to constitutive phosphorylation of Mek1/2 and Akt as well as to a constitutive high level expression of cyclin D1/2 (Tognon et al., 2001). These features also characterized EN-initiated mammary tumors (hereafter, EN tumors) (Figure 2D).
EN tumors were heterogeneous both with respect to morphology and rate of tumor progression (Figure 2Ea–2Ed and data not shown). Most EN tumors were highly invasive and transplantable upon subcutaneous injection into immunodeficient mice. The rate of tumor regrowth following transplantation correlated with the apparent rate of progression of the corresponding primary tumor. Due to the relatively short latency of these tumors, most WCEN mice failed to show signs of metastasis. On occasion, metastases to lymph node and lung were observed (Figure 2Ef and 2Eh).
The transient Wap+ cells in nulliparous Wap-Cre;EN mammary glands are target cells of EN
In nulliparous female mice, Wap is activated in a small, transient population of MECs during estrus (Kordon et al., 1995; Robinson et al., 1995; Robinson et al., 1996). Further studies in _Wap-Cre;Rosa-Stop-lac_Z (hereafter, WCLZ) females showed that Wap-Cre+ (thus also _lac_Z+) MECs failed to persist in WCLZ mice during diestrus. Since _lac_Z+ cells do not accumulate in nulliparous WCLZ mice, it is inferred that the MECs in which Wap is first activated do not proliferate extensively and possibly die through apoptosis (Boulanger et al., 2005; Henry et al., 2004; Wagner et al., 2002). In addition, one can infer that Wap-Cre+ cells are not equivalent to mammary stem cells (MaSCs) in virgin mice.
Since WCEN nulliparous females develop multifocal mammary tumors with 100% penetrance and the transient Wap+ MECs are likely the only cells in virgins that express Cre and, therefore, activate EN expression, we conjectured that these cells might be direct targets of EN transformation (Figure 3A). To test this hypothesis, we interbred a _Rosa-Stop-lac_Z reporter (Mao et al., 1999) into the WCEN background and generated Wap-Cre;EN;Rosa-Stop-lacZ (hereafter, WCENLZ) females. We predicted the presence of _lac_Z+ hyperplastic MECs in MGs and subsequent development of _lac_Z+ mammary tumors (Figure 3A). Indeed _lac_Z staining was primarily restricted to hyperplastic alveolar cells in hyperplastic MGs (Figure 3B–3C). Moreover, _lac_Z+ cells were confined to tumor epithelial cells in mammary tumors (Figure 3D–3E). Thus, genetic marking suggests that activation of EN by Cre rescues an otherwise transient subpopulation of MECs and maintains them and their progeny for subsequent steps in progression to frank malignancy. Furthermore, we reasoned that T-ICs in WCENLZ tumors should also be marked by _lac_Z. To test this, we transplanted tumor cells into immunodeficient mice after limited dilutions. Without fractionation, typically ~104–105 EN tumor cells were needed to form new tumors in _Rag2_−/− mice. In contrast, only ~103–104 (sometimes even as low as 600) sorted _lac_Z+ tumor cells were sufficient to initiate new tumors in _Rag2_−/− hosts. This suggests TICs, evolved from EN target cells, are enriched in the population of _lac_Z-marked EN tumor cells.
Figure 3. Target cells of mammary tumors developed in Wap-Cre;EN virgins are transient mammary progenitors, rather than MaSCs.
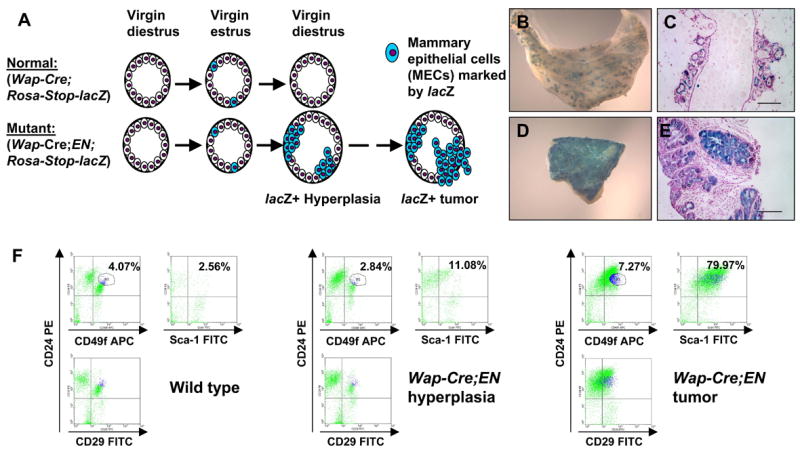
(A) Schematic diagram of the _lac_Z-marking experiment. Normal WCLZ virgin MGs contain a transient wave of _lac_Z+ MECs. In WCENLZ mutant MGs, the transient _lac_Z+ MECs appear to be rescued from death and lead to _lac_Z+ alveolar hyperplasia and eventually _lac_Z+ mammary tumors.
(B–E) B and D show whole mounts of WCENLZ MG and tumor stained for _lac_Z activity. C and E are _lac_Z-stained tissue sections counterstained with nuclear fast red. Note in B and C, _lac_Z+ cells are mainly restricted to the alveolar compartment. Scale bars=100μm.
(F) Flow cytometry analysis of a WT MG, a WCEN hyperplastic MG, and a WCEN tumor. The profiles shown here have already been gated for lineage-negative (Lin−) cells. Percentages of positive cells are from a representative experiment.
Wap-Cre;EN hyperplastic mammary glands and tumors accumulate CD24+Sca-1+ cells
To characterize further the nature of the EN target cells, we examined surface markers that have previously been used to fractionate mammary tissues into enriched stem or progenitor populations (Shackleton et al., 2006; Stingl et al., 2006; Welm et al., 2002). By flow cytometry analysis most EN tumor cells were CD24+Sca-1+ (Figure 3F). Moreover, MGs from mature nulliparous WCEN females contained a greater number of CD24+Sca-1+ cells than WT controls even before they developed tumors (Figure 3F). In contrast, the MaSC-enriched Lin−CD29hiCD24+ [(Shackleton et al., 2006), and the similar Lin−CD49fhiCD24+, (Stingl et al., 2006)] subpopulation is not altered in WCEN hyperplastic MGs (Figure 3F). These data argue that mammary tumors in WCEN mice are unlikely to arise from transformation of MaSCs; instead, they may be derived from committed CD24+Sca-1+ mammary progenitors.
Wap-Cre;EN mammary tumors are derived from committed alveolar progenitors
In analogy to the hematopoietic system, a developmental hierarchy for MECs, including MaSCs, bipotent ductal or alveolar progenitors, single lineage-restricted progenitors, and mature luminal or myoepithelial cells, has been proposed (Asselin-Labat et al., 2007; Hennighausen and Robinson, 2005). To position target cells of EN in this hierarchy, we performed immunostaining for the luminal epithelial cell marker keratin 8 (K8), basal/myoepithelial cell markers keratin 5 (K5) and 14 (K14), p63, and α-smooth muscle actin (SMA), as well as the mammary progenitor marker keratin 6 (K6), and estrogen receptor (ERα) (Figure 4A and Figure S2). Overall, we have identified 2 broad tumor types in WCEN mice. The majority of EN tumors (type 1, ~90%) exhibit relatively well-differentiated glandular structures and contain K8+ luminal epithelial cells surrounded by basal/myoepithelial cells that are positive for K5, K14, p63, and sometimes, SMA. Occasionally there are K5+K14+p63+SMA+or− basal/myoepithelial cells “leaking” into the region of luminal epithelial cells (Figure 4A and Figure S2A and S2D). In the most extreme case, some type 1 tumors (type 1a, or regions within these tumors) contain a large number of K5+K14+p63+SMA+or− basal/myoepithelial cells intermixed with a smaller number of K8+ luminal epithelial cells in a less organized manner (Figure S2A). A second class of EN tumors (type 2, ~10%) exhibits no K5+K14+p63+ basal/myoepithelial cells, but contains predominantly K8+ luminal epithelial cells. Interestingly, a large number of K8+ cells in these tumors are also K14+ (but K5−) (Figure 4A and Figure S2A). In contrast, most type 1 tumors lack these abnormal K8+K14+ cells. However, some type 1a tumors do contain a small number of K8+K14+ cells (Figure S2A). In addition, we also identified an additional subtype of type 1 tumors (type 1b) that exhibit glandular structures, with K8+ luminal epithelial cells surrounded by K5+K14+ basal/myoepithelial cells. However, many of these K8+ luminal cells are also K14+ (Figure S2A), thus representing tumors with features of both types (1 and 2). A summary of the staining properties of cells in the different types of EN tumors is provided in Figure S2B. The majority of type 1 tumors contain K6-expressing cells, whereas very few or no K6+ cells are present in type 2 tumors (Figure S2C). However, tumors of both types express Sca-1 (data not shown) and are ERα+ (Figure S2E). Interestingly, upon careful examination by immunofluorescence co-localization, we observed a significant number of ERα+p63+ and ERα+SMA+ cells, in addition to the ERα+p63−SMA− luminal epithelial cells (Figure S2E).
Figure 4. Two major types of Wap-Cre;EN tumors characterized by immunostaining and microarray analysis.
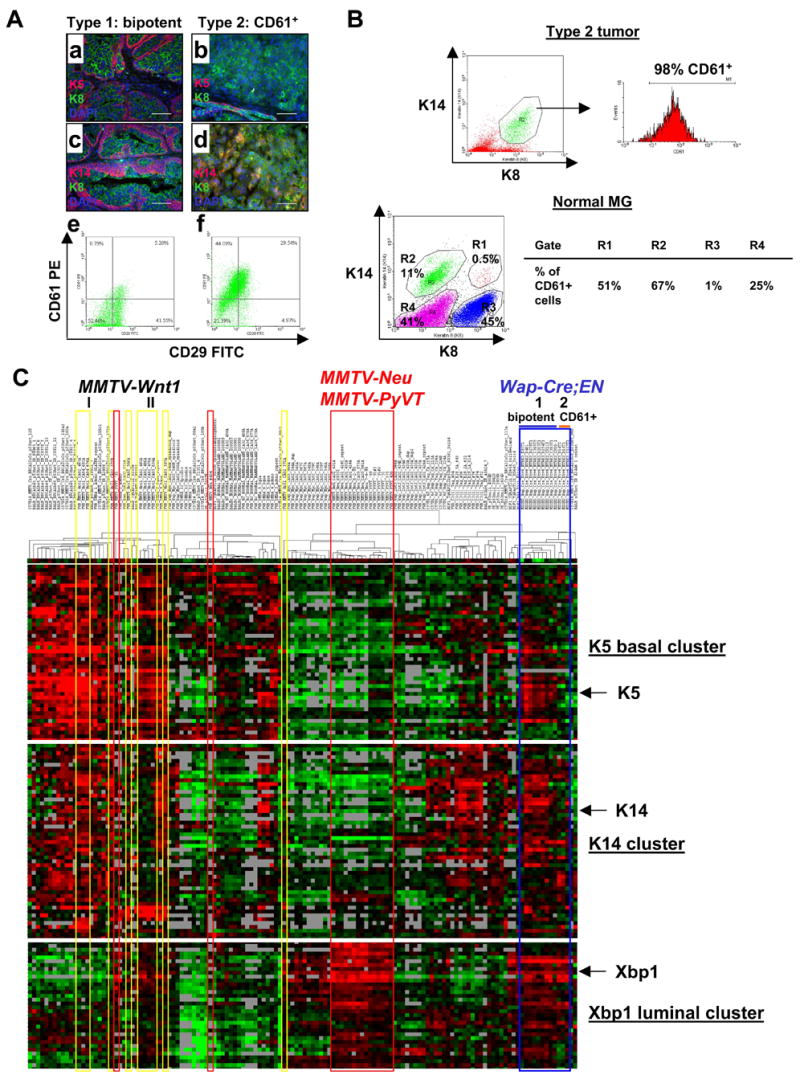
(A) EN type 1 (bipotent: a,c,e) and type 2 (CD61+: b,d,f) tumors based on K5/K8 (a–b), K14/K8 (c–d), and CD61/CD29 (e–f) staining patterns. Scale bars=50μm.
(B) Flow cytometry analysis showing that K8+K14+ cells in EN type 2 tumors are CD61+. In normal MGs, almost all CD61+ MECs (gates R1-3) are K14+. Gate R4 represents non-epithelial cells in MGs.
(C) Hierarchical cluster analysis of 12 WCEN tumors (Type 1 and 2, blue lines) together with 122 samples from other murine models of breast cancer and normal MGs. MMTV-Wnt1 tumors (clusters I and II) are highlighted by yellow lines. MMTV-Neu and MMTV-PyVT tumors are highlighted by red lines. Heat maps of 3 representative gene clusters (K5, K14, and Xbp1) are shown (heat maps showing all genes in the intrinsic gene list are shown in Figure S5). Gene names for each cluster are listed in Table S8. In the heat map, red, black, and green represent above average, average, and below average levels of expression, respectively. Gray indicates no data recorded.
Recently, CD61 (integrin β3) has been shown to mark the luminal progenitors in normal MGs (Asselin-Labat et al., 2007). Interestingly, we find that all type 2 tumors abundantly express CD61, whereas most of the type 1 tumors contain much fewer CD61+ cells (Figure 4Ae–4Af), with the exception of type 1b tumors. Microarray expression profiling confirms that type 2 tumors express the highest levels of CD61, followed by type 1b, and then the remaining type 1 tumors (data not shown). Since type 2 tumors exhibit “luminal” appearance and are characterized by large numbers of K8+K14+ cells, we asked whether these double positive cells might represent blocked CD61+ progenitors. In fact, flow cytometry analysis revealed that almost all K8+K14+ cells in these tumors are CD61+ (Figure 4B). In normal MGs, the majority of CD61+ cells are K8−K14+ cells, whereas K8+K14− luminal cells are CD61− (Figure 4B). We also detected a small population of K8+K14+ cells, a significant number of which also express CD61 (Figure 4B). These data suggest that CD61+ luminal progenitors are K14-expressing cells, and the abnormal K8+K14+ cells in EN type 2/type 1b tumors probably represent CD61+ luminal progenitors blocked in differentiation toward K8+K14− mature luminal epithelial cells.
In type 1 EN tumors, the K5+ basal/myoepithelial cells, in addition to the K8+ luminal cells, are part of the tumor epithelial cell population. When we compared microarray expression profiles generated from sorted tumor epithelial cells based on the above-described _lac_Z marker in WCENLZ females to those from unfractionated tumors, we observed slight enrichment (rather than loss) of both the K5 basal gene cluster and the K14 cluster (as defined in Figure 4C, discussed below) in sorted _lac_Z+ tumor cells, as determined by gene set enrichment assay (GSEA) (data not shown) (Subramanian et al., 2005). This confirms that basal/myoepithelial cells in EN tumors are part of the tumor epithelial cells, not normal cells recruited to tumors.
Due to the presence of mixed cell types in type 1 EN tumors, the cells from which tumors originate could be MaSCs, bipotent progenitors, or multiple lineage-committed progenitors. Since the above-described genetic marking experiment and flow cytometry analysis (Figure 3) both rule out MaSCs as targets of EN, we next asked whether tumors are derived from distinct lineage-restricted progenitors (i.e., multiclonal), or from bipotent progenitors (i.e., clonal).
We used a Wap-rtTA-Cre transgenic line that expressed Cre in Wap+ cells in the presence of doxycycline (Utomo et al., 1999). We generated animals with both Wap-rtTA-Cre and EN, some of which also carried a conditional luciferase reporter at the Rosa26 locus [_Rosa-Stop-Luc_, similar to the above-described _Rosa-Stop-lac_Z reporter, (Safran et al., 2003)]. Unexpectedly, we found that the Wap-rtTA-Cre transgene exhibits leaky Cre expression and Wap-rtTA-Cre;EN nulliparous females developed mammary tumors without doxycycline. These tumors also featured cells of both luminal and basal/myoepithelial lineages (Figure S3A). Since Cre should be expressed at a low level without doxycycline, sometimes it might excise the “stopper” at the EN allele but not that at the Rosa-Stop-Luc allele within the same cell. If a tumor arose from a cell with this genotype, tumor epithelial cells would all inherit the recombined EN allele and the unmodified Rosa-Stop-Luc allele. Indeed, in a tumor arising in a Wap-rtTA-Cre;EN;Rosa-Stop-Luc nulliparous female, we detected excision only at the EN locus, and not at the Rosa-Stop-Luc locus (Figure S3B). Since it is highly unlikely for multiple lineage-restricted progenitors to obtain the same partial excision pattern (as described above) and then give rise to a tumor with this pattern, our observation supports the clonal origin of type 1 EN tumors from bipotent progenitors.
Furthermore, microarray data reveal that compared to normal MGs, both type 1 and 2 EN tumors express high levels of the alveolar cell marker κ-casein, but low levels of the ductal cell marker NKCC1 (also known as “solute carrier family 12, member 2”) (Figure S4A), suggesting an alveolar cellular origin. In contrast, MMTV-Wnt1 tumors do not express (or express low levels of) κ-casein, but express NKCC1 in a fraction of tumor cells, consistent with MaSCs as potential target cells to give rise to both ductal and/or alveolar cells (Li and Rosen, 2005) (Figure S4B). Taken together, we conclude that target cells of mammary tumors arising in WCEN females are either committed bipotent alveolar progenitors (type 1) or luminal alveolar progenitors (type 2). These progenitors are within the transient Wap+ cells in nulliparous females.
Wap-Cre;EN mammary tumors express both luminal and basal gene clusters
We compared EN tumors to other murine breast cancer models by microarray expression profiling, using hierarchical clustering based on an intrinsic gene list developed for murine models of breast cancer (Herschkowitz et al., 2007). As shown in Figure 4C and Figure S5, in general, compared to other murine models, EN tumors are relatively homogeneous and cluster together. However, the 3 type 2 tumors (D14T1, D14T2, and C117T1) form their own sub-cluster and are separated from the type 1 tumors. In contrast, tumors from parous (CR115, CR90-1, D14T1, and D14T2) and nulliparous (the remaining samples) females are mixed together and do not form distinct sub-clusters. These data suggest that the initiating oncogenic event and cellular origin, but not the reproductive history, are more important in determining phenotypes of EN tumors
All type 1 EN tumors express 3 gene clusters, including a K5 basal gene cluster, a strong K14 cluster, and a strong Xbp1 luminal gene cluster, similar to MMTV-Wnt1 tumors (Figure 4C). Type 2 EN tumors express the K14 and Xbp1 clusters, but not the K5 cluster. In contrast, tumors from both MMTV-Neu and MMTV-PyVT mice only express the Xbp1 luminal cluster (Figure 4C). Of note, MMTV-Wnt1 tumors are more heterogeneous than EN tumors and they can also be divided into 2 major sub-clusters. One contains K5 and K14 clusters, but not the Xbp1 luminal cluster (Figure 4C, cluster I). The other contains the K5 basal cluster and the Xbp1 luminal cluster (though expressed at slightly lower levels), but not the K14 cluster (Figure 4C, cluster II). These differences may be due in part to their different cells of origin, with MaSCs as more likely targets of MMTV-Wnt1 tumors, and committed progenitors as targets of EN.
An AP1 signature associated with EN-mediated mammary tumorigenesis
To understand the mechanism of EN-mediated mammary tumorigenesis, we first performed GSEA on microarray expression profiles from unsorted EN tumors compared to those from normal MGs, using curated gene sets (c2 collection) for metabolic and signaling pathways from the GSEA molecular signature database website (http://www.broad.mit.edu/gsea/msigdb/msigdb_index.html). Unexpectedly, this analysis only revealed pathways that seem to be common for tumor cells in general (e.g., reflecting increased metabolic activities and proliferation, Table S1). Two possibilities may account for this result. First, pathways that are active in both normal MGs and tumors may not be revealed by this assay. Second, non-cancerous cells in these tumors could introduce significant “noise” in unsorted tumor samples.
In our WCENLZ animals, tumor epithelial cells are labeled by _lac_Z (Figure 3B–3E). Thus, we have the opportunity to mark tumor epithelial cells and separate them from stromal cells. In addition, target cells of EN in WCLZ virgin females as well as premalignant, hyperplastic MECs in WCENLZ females are also marked by _lac_Z, thus providing an opportunity to isolate these cells for analysis.
We generated microarray expression profiles for several _lac_Z-sorted samples from either WCENLZ hyperplastic MGs that had not developed visible tumors, or mammary tumors that arose in WCENLZ females. We then compared sorted tumor cells to sorted hyperplastic MECs by GSEA using the above-described c2 gene sets, in an attempt to identify pathways upregulated during tumor progression. The top gene sets enriched in sorted tumor cells derived from this comparison are more informative than the comparison using unsorted tumors (Table S2, compare to Table S1). Pathways related to the hypoxia response in tumors, WNT signaling, TRK/NGF signaling, TGFβ signaling, and genes regulated by MYC are evident. To further validate the effectiveness of this approach, we also compared unsorted tumors to sorted hyperplastic MECs (thus both comparisons have the same baseline). The top gene sets enriched in unsorted tumors revealed from this analysis are very similar to those compared to normal MGs (Table S3, compare to Table S1), suggesting GSEA using sorted tumor cells may reveal pathways with better specificity than using unsorted tumors.
We next compared sorted tumor cells to unsorted tumors by GSEA using c2 gene sets. We reasoned that genes/pathways upregulated specifically in tumor epithelial cells would be further enriched in this comparison. This would also potentially reveal an overall EN “signature” in the tumor epithelial cell compartment composed of pathways upregulated during the initial transformation, or tumor progression, or both. Among gene sets enriched in sorted tumor cells, we observed pathways related to genes regulated by C/EBP, IL6 response, HOXA5 targets, WNT signaling, JNK signaling, and TGFβ signaling (Table S4).
To determine what portion of the EN tumor signature is acquired during tumor initiation, ideally we would need to sort normal target cells of EN and compare their expression profiles to those from sorted hyperplastic MECs. Unfortunately, _lac_Z-staining of mammary tissues by FDG dye yields a significant amount of background staining (~1–3% even for _lac_Z− tissues). This background staining does not cause difficulty when sorting _lac_Z+ hyperplastic MECs or tumor cells, which typically constitute ~10–40% of total cells, but prohibits sorting the small population of transient EN target cells [0.8–4% as estimated by Wagner et al (Wagner and Smith, 2005)] with sufficient confidence. To overcome this technical limitation, we turned to an in vitro model of EN signaling. Because EN is a unique oncoprotein in that it transforms cells of all germ layers, we reasoned that the mechanism of EN-mediated transformation might be conserved in different cell types. EN-mediated signaling in NIH 3T3 cells has been studied previously (Tognon et al., 2001; Wai et al., 2000), many of which were also observed in EN tumors (Figure 2D). Thus, we generated microarray expression profiles from EN-transduced 3T3 cells (EN-3T3) and compared them to those of untransformed 3T3 cells. By performing GSEA using c2 gene sets, we identified several common gene sets that are enriched in both EN-3T3 cells and sorted EN tumor cells, including 2 IL6 pathways, “WNT_TARGETS”, “CHEN_HOXA5_TARGETS_UP”, and “TGF_BETA_SIGNALING_PATHWAY” (Table S5, compare to Table S4).
Since “WNT_TARGETS” arose among the top-enriched gene sets in all 3 analyses, we next focused on this gene set. By comparing the core enrichment genes from all 3 analyses, we observed striking similarity between sorted tumor cells (compared to unsorted tumors) and EN-3T3 cells (compared to control 3T3 cells) (Figure S6A–S6B), including genes such as JUN, FOSL1, PLAUR, and CD44. JUN and FOSL1 (also known as FRA1) encode components of the AP1 complex and are target genes of β-catenin in human colorectal carcinomas. PLAUR encodes uPAR and its transcription is activated through AP1 (Mann et al., 1999). In addition, CD44 can also be upregulated by AP1 activity (Lamb et al., 1997). Thus, this 4-gene set appears to represent an AP1 signature.
Consistent with the above data, we also observed enrichment of all gene sets containing genes with the AP1 binding motif in the c3 collection of GSEA database in sorted tumor cells and EN-3T3 cells (Table S6). More directly, we manually compiled a gene set for AP1 target genes based on published literature (plus JUN and FOSL1) (Eferl and Wagner, 2003) and observed its significant enrichment in both sorted EN tumor cells and EN-3T3 cells (Figure 5A–5B). In addition to the above-described 4-gene set, we identified additional AP1 target genes that are upregulated in both comparisons, including HBEGF (heparin-binding EGF-like growth factor), BCL2L11 (BIM), CDKN1A (p21), and CDKN2A (p16). p16INK4A and p21WAF1 typically function as tumor suppressors. However, both may also act as oncogenes. In fact, overexpression of p16 is found in breast cancer cases with a more malignant phenotype (Milde-Langosch et al., 2001). Of note, we did not observe significant enrichment of this AP1 signature in sorted EN tumor cells compared to sorted EN hyperplastic MECs (Figure 5C and Figure S6C), suggesting that at least part of this signature might have already been acquired in hyperplastic MECs during the initial stage of EN transformation.
Figure 5. Enrichment of c-Jun/Fosl1 and AP1 targets in EN-transformed cells.
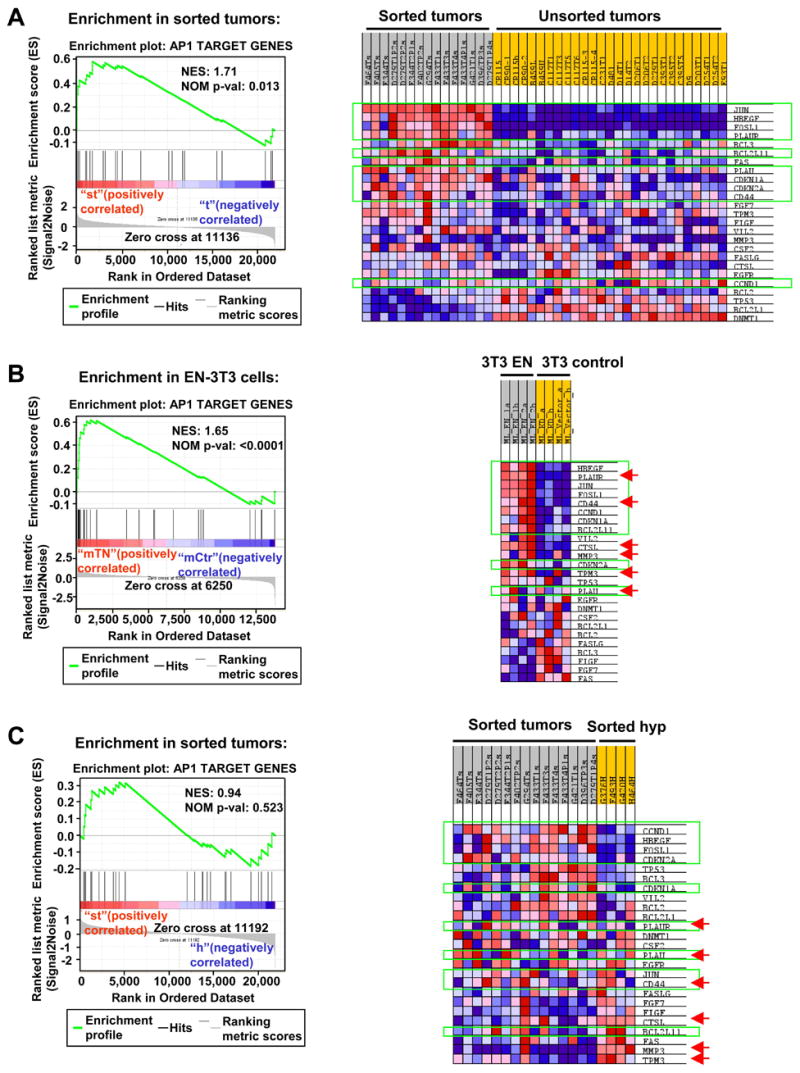
(A–C) GSEAs show enrichment of c-Jun/Fosl1 and AP1 target genes in sorted EN tumor cells compared to unsorted tumors (A) and sorted hyperplastic cells (C), and in EN-3T3 cells (B). Genes expressed at higher levels in both sorted tumors and EN-3T3 cells are highlighted with green lines. Genes expressed at higher levels in both sorted hyperplastic MECs and EN-3T3 cells are indicated by red arrows.
To confirm involvement of the c-Jun/Fosl1 AP1 complex in EN-mediated transformation, we performed electrophoretic mobility shift assays (EMSAs) on nuclear lysates from 3T3 cells and observed a significantly larger and specific bandshift created by AP1 complex formation in EN-3T3 cells (Figure 6A). This AP1 complex was composed of c-Jun and Fosl1 or Fosl2 (Figure 6B). Western blot analysis confirmed significantly elevated levels of total c-Jun, phosphorylated c-Jun, and Fosl1 only in EN-3T3 cells (Figure 6C), suggesting c-Jun/Fosl1 is indeed the major AP1 complex formed upon EN-mediated transformation.
Figure 6. Upregulation and activation of the c-Jun/Fosl1 AP1 complex is associated with EN-mediated transformation.
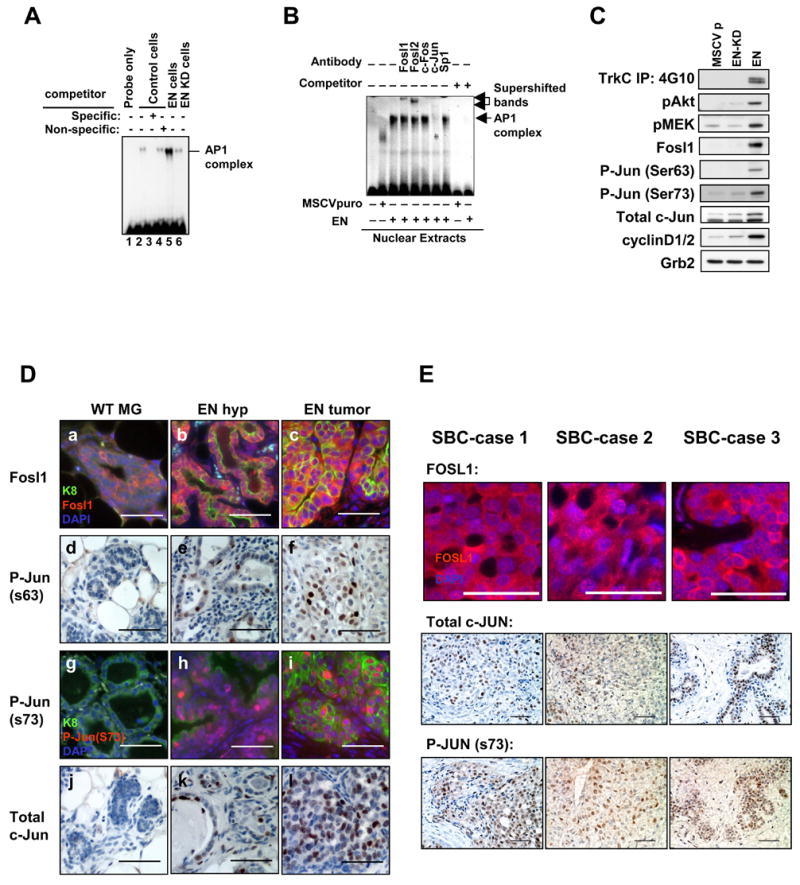
(A) AP1 EMSAs on nuclear lysates from control, EN, or EN kinase dead (KD) transduced 3T3 cells. Incubation with a specific unlabelled competitor blocks bandshift (lane 3). EN-3T3 nuclear lysates produced a significantly larger bandshift compared to control or EN KD-3T3 lysates (Compare Lane 5 to Lane 2 and 6).
(B) Antibodies specific to Fosl1, Fosl2, and c-Jun bandshifted or destroyed the AP1 bandshift produced by EN nuclear extracts whereas antibodies to c-Fos and Sp1 had no effect.
(C) Western blot analysis performed on lysates from serum starved control (MSCVp), kinase dead EN (EN-KD), and EN 3T3 cells. EN cells possessed a tyrosine phosphorylated oncoprotein doublet and elevated levels of phosphorylated Akt, MEK, and c-Jun (ser 63 and 73) as well as elevated levels of Fosl1, total c-Jun and cyclin D1/2. Loading control: Grb2.
(D) Immunofluorescence (a–c, g–i) and immunohistochemical (d–f, j–l) staining of WT MGs (a, d, g, j), EN hyperplastic MGs (b, e, h, k), and EN mammary tumors (c, f, i, l) with antibodies against Fosl1 (a–c), phosphorylated c-Jun at Ser63 (d–f) and at Ser73 (g–i), and total c-Jun (j–l). WT MGs in a, d, and j were from nulliparous female mice. WT MG in g was from a lactating female. Scale bars=50μm.
(E) Immunostaining of 3 SBC cases confirms presence/activation of the c-JUN/FOSL1 AP1 complex. Scale bars=50μm.
To validate upregulation and activation of the c-Jun/Fosl1 complex in EN tumors, we performed immunostaining using antibodies against Fosl1, phosphorylated c-Jun, and total c-Jun. In WT MGs, Fosl1 is mainly localized in cytoplasm (Figure 6Da). In EN hyperplastic MECs and tumor cells, the overall intensity of Fosl1 staining is increased, and more importantly, significant Fosl1 nuclear staining is detected (Figure 6Db–6Dc). In addition, upregulation and phosphorylation of c-Jun is also evident in EN hyperplastic MECs and tumor cells, but not in WT MGs (Figure 6Dd–6Dl), suggesting activation of this AP1 complex is an early event (i.e., present in hyperplastic MECs) and persists in EN-mediated tumorigenesis.
To determine the c-Jun/Fosl1 status in human SBC, we stained 3 SBC cases with c-JUN and FOSL1 antibodies. As shown in Figure 6E, in 3 of 3 SBC samples studied, we detected nuclear staining of FOSL1, and expression and activation (phosphorylation) of c-JUN, suggesting that activation of the c-JUN/FOSL1 complex is indeed associated with human SBC.
Expression of a dominant negative c-Jun blocks EN-mediated transformation
To determine if AP1 activation is necessary for EN-mediated transformation, we employed the dominant negative (DN) c-Jun TAM67, which lacks its transactivation domain, to block the AP1 activity (Ludes-Meyers et al., 2001) in several different systems. Co-expression of DN c-Jun in EN-3T3 cells blocked EN-mediated transformation both morphologically and molecularly (Figure 7A–7D). Ectopic expression of EN in the murine EpH4 cells and human MCF10A cells led to larger spheroids in 3-D Matrigel cultures, which was reverted by co-expression of DN c-Jun (Figure 7E–7F). We have shown previously that EN-transduced EpH4 (EN-EpH4) cells formed tumors upon subcutaneous injection in immunodeficient mice (Tognon et al., 2002). We now show that expression of DN c-Jun in EN-EpH4 cells significantly reduces their tumorigenic properties in vivo (Figure 7G–7H). We compared microarray expression profiles between EN-EpH4 tumors and DN c-Jun/EN-EpH4 tumors by GSEA. As expected, many gene sets enriched in sorted EN tumor cells (compared to unsorted EN tumors) are downregulated in DN c-Jun/EN-EpH4 tumors (Table S7). Importantly, one of them is “JNK_UP”, a gene set directly related to JNK-JUN signaling.
Figure 7. Expression of a dominant negative (DN) c-Jun blocks EN-mediated transformation.
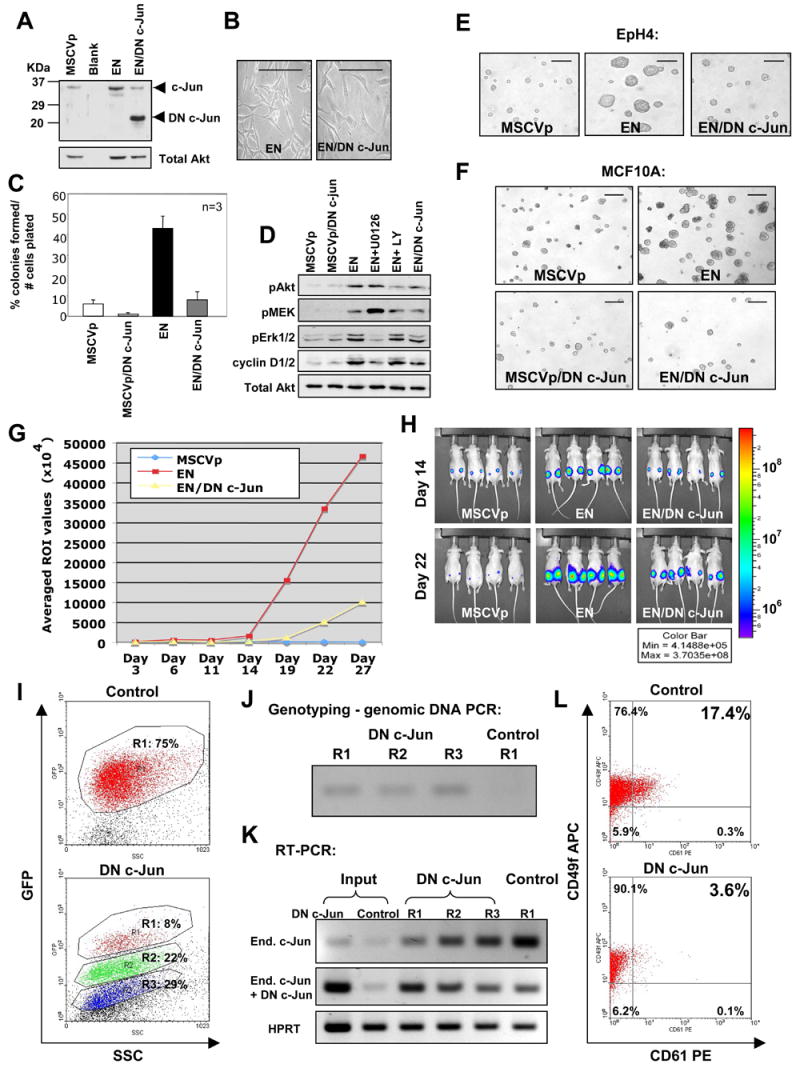
(A) Expression of DN c-Jun in EN expressing cells confirmed by western blot analysis. Loading control: total Akt.
(B–C) DN c-Jun expression reverts EN-mediated morphological transformation (B, scale bars =50μm) and soft agar colony formation (C, error bars=mean +/− SD) in 3T3 cells.
(D) Western blot analysis of serum starved control cells (MSCVp) and EN cells [alone; co-expressing DN c-Jun; or treated with either MEK inhibitor U0126 or PI3K inhibitor LY294002 (LY) (25μM)]. Both U0126 treatment and co-expression of DN c-Jun had a significant effect upon cyclin D1/2 levels in EN cells. Loading control: total Akt.
(E) Effects of EN and DN c-Jun expression on EpH4 cell growth after 4 days in 3-D Matrigel cultures. Scale bars=500μm.
(F) Effects of EN and DN c-Jun expression on human MCF10A cell growth in 3-D Matrigel cultures. Scale bars=1000μm.
(G) Tumor growth curves of luciferase expressing cells injected subcutaneously into nude mice. ROI= regions of interest values in photons/sec/cm2/sr.
(H) Comparison of bioluminescent images of mice at day 14 and day 22.
(I) Top panel shows Gfp+ cells in an empty-vector transduced EN tumor developed in the _Rag2_−/− host. Lower panel shows 3 distinct populations of Gfp+ cells (R1–R3: express high, medium, and low levels of Gfp, respectively) in a DN c-Jun transduced EN tumor developed in the _Rag2_−/−host.
(J) PCR genotyping (using a forward primer in the DN c-Jun and a reverse primer in the PGK promoter of the viral vector) confirms integration of DN c-Jun viruses in all 3 populations (R1–R3) of the DN c-Jun/EN tumor. This primer set does not detect the control viruses (vector only).
(K) Expression levels of DN c-Jun are estimated indirectly by comparing expression levels of the endogenous c-Jun to those of total c-Jun (endogenous+DN c-Jun). Expression of the endogenous c-Jun is detected by a PCR primer set located in the region deleted in DN c-Jun. Expression of total c-Jun is detected by a second primer set located in the common region.
(L) Flow cytometry analysis shows reduced number of CD61+ cells in the DN c-Jun/EN tumor compared to the control tumor. A representative experiment is shown.
Lastly, we asked whether forced expression of DN c-Jun in primary EN tumor cells impairs their capacity to form new tumors upon transplantation. We developed a protocol to retrovirally transduce EN tumor cells ex vivo quickly, sort virally infected Gfp+ cells, and then transplant them into _Rag2_−/− mice immediately. In 3 independent experiments performed, 5 of 5 _Rag2_−/− mice transplanted with Gfp+ EN tumor cells transduced by the control virus (empty vector, LPIG) developed tumors in 2~3 months, whereas only 3 of 5 _Rag2_−/− mice transplanted with same numbers of Gfp+, DN c-Jun-transduced EN tumor cells developed tumors within the same time window. In one tumor derived from DN c-Jun-transduced EN tumor cells, we detected 3 distinct populations of tumor cells expressing high, medium, and low levels of Gfp (Figure 7I). Intriguingly, the Gfphi population was significantly smaller than the other two. In contrast, the majority of tumor cells in the control (LPIG) tumor were Gfp+ cells expressing high levels of Gfp. We individually sorted these distinct populations of Gfp+ cells and confirmed they were all derived from virally transduced cells (Figure 7J). We then quantitated DN c-Jun expression levels and compared these to virally transduced cells before transplantation (i.e., input). Consistent with their Gfp expression levels, the Gfphi, Gfpmid, and Gfplow cells in the DN c-Jun-transduced tumor also expressed high, medium, and low levels of DN c-Jun, respectively. In addition, even the Gfphi tumor cells expressed much lower levels of DN c-Jun than those of the input (Figure 7K). These findings suggest that selection against EN tumor cells expressing high levels of DN c-Jun occurs in vivo. Upon staining with antibodies for stem/progenitor markers CD61 and CD49f, we found the DN c-Jun-transduced tumors contained fewer CD61+CD49f+ cells than the control tumors (Figure 7L). Since CD61+CD49f+ MECs may represent stem/progenitor cells in MGs (Asselin-Labat et al., 2007), we hypothesize that forced expression of DN c-Jun in EN tumor cells may reduce the number of tumor cells capable of initiating new tumors upon transplantation. Furthermore, in one _Rag2_−/− mouse transplanted with DN c-Jun-transduced EN tumor cells, 2 physically separated tumors developed. One tumor expressed high levels of Gfp and DN c-Jun; the other expressed lower levels of Gfp and DN c-Jun (data not shown). We sorted Gfp+ cells from both tumors and serially transplanted same numbers of sorted cells (ranging from 500 to 5×105) into _Rag2_−/− mice. 1.5 months after transplantation, _Rag2_−/− mice injected with 1×105 and 5×105 Gfplow (thus also DN c-Junlow) cells have already developed new tumors, whereas none of the _Rag2_−/− mice transplanted with Gfphi (thus also DN c-Junhi) cells have developed tumors. This again suggests high levels of DN c-Jun expression in EN tumor cells reduce the number of tumor cells capable of forming new tumors upon transplantation.
DISCUSSION
Committed mammary progenitors as cells of origin for breast cancer
A central question in cancer biology is the cellular origin of cancers. Do cancers originate from normal stem cells that lose normal growth control, or do they initiate from progenitors or more differentiated cells after acquisition of stem cell attributes through mutation(s) (Lobo et al., 2007)? Current views are derived largely from studies of leukemias in the hematopoietic system. Since hematopoietic stem cells (HSCs) are endowed with self-renewal, it has been argued that transformation of HSCs provides a simple means to generate leukemic cells. Paradoxically, cellular phenotypes in diverse leukemias mirror those of progenitors, rather than HSCs (Lobo et al., 2007). In fact, evidence in both patients and mouse models favors a progenitor cell origin for many leukemias (e.g., see Jamieson et al., 2004; Krivtsov et al., 2006).
Similarly, although MaSCs are often proposed as cells of origin for breast tumors, cellular phenotypes of human breast cancer are not easily reconciled with this view. For instance, since MaSCs can give rise to both luminal and myoepithelial cells, one would expect to see mixed cell types in breast cancer if MaSCs represent the predominant cellular origin, yet most human breast tumors exhibit phenotypes of luminal epithelial cells (Sorlie et al., 2001). This suggests that more differentiated cells in MGs may serve as cells of origin for breast cancer. However, the existence of such committed progenitors in normal MGs remains to be directly demonstrated. Likewise, direct evidence is also lacking to show such cells as targets of transformation leading to breast cancer.
The cellular origin of human breast cancer is difficult to establish. Murine models, therefore, represent a tractable alternative for analysis. Previous studies using murine models have proposed both MaSCs and more differentiated cells, including mammary progenitors, as targets of breast cancer (Li and Rosen, 2005). Moreover, in murine models created by overexpression of oncogenes driven by the same promoter (e.g., MMTV), different oncogenes (e.g., Wnt1/β-catenin vs. Neu or H-Ras or PyMT) appear to preferentially transform distinct populations of MECs. However, due to use of exogenous promoters (thus oncogenes often expressed at non-physiological levels), and in the absence of genetic marking and thorough characterization of tumor cell types, these models cannot definitively assign target cells within the normal MEC developmental hierarchy.
In WCEN mice, EN is under the transcriptional control of the endogenous Etv6 promoter therefore should be expressed at a physiological level. Since mammary tumors develop in nulliparous WCEN females at 100% penetrance and transient Wap+ cells are the only cells in which EN expression is activated, this unique combination and the genetic marking experiment described above provide direct evidence to support committed mammary progenitors (within the transient Wap+ cells) as targets of EN.
Previous studies using genetic marking have identified a population of multipotent MECs (termed parity-induced MECs, or PI-MECs) that originate from _Wap-Cre_-expressing differentiating cells during the first pregnancy/lactating cycle, and survive through postlactational remodeling, and persist throughout the remainder of life (Boulanger et al., 2005; Henry et al., 2004; Wagner et al., 2002; Wagner and Smith, 2005). Recently, a population of alveolar progenitor cells, similar to PI-MECs, was identified in nulliparous mouse MGs (Booth et al., 2007). These cells represent lobule-restricted multipotent progenitors capable of proliferation and differentiation to both luminal and myoepithelial cells upon transplantation. Apparently, these progenitor cells are targets of EN in WCEN virgin females, as described in this study.
Our study also supports the existence of an epithelial cell hierarchy in both normal MGs and in mammary tumors. We have described at least 2 types of committed alveolar progenitors in the transient population of Wap+ MECs in nulliparous MGs: the bipotent alveolar progenitors as targets of type 1 tumors and the lineage-committed CD61+ luminal alveolar progenitors as targets of type 2 tumors. However, we cannot exclude the possibility that type 2 tumors are also derived from bipotent progenitors that are blocked in differentiation and only give rise to CD61+ immature luminal cells. We favor lineage-committed CD61+ luminal progenitors, rather than bipotent progenitors, as their target cells. In support of this conclusion, knockout of Gata3 in MGs led to an expansion of CD61+ luminal progenitors and a concomitant block in differentiation (Asselin-Labat et al., 2007), suggesting the existence of such luminal-restricted progenitors in the normal MEC hierarchy. In some type 1 EN tumors, we observed regions with extensive K5+K14+p63+ basal epithelial cells (type 1a), and regions with K8+K14+K5− immature luminal epithelial cells surrounded by K5+K14+p63+ basal/myoepithelial cells (type 1b). This can be best explained by the existence of an epithelial cell hierarchy in EN tumors. The committed bipotent alveolar progenitors are transformed by EN and become oncogenic bipotent progenitors, which give rise to abnormal luminal-restricted progenitors and basal/myoepithelial-committed progenitors. In most cases, these progenitors differentiate into well-differentiated type 1 EN bipotent tumors. Occasionally, either under the influence of the microenvironment, or after acquiring additional mutations, some of the abnormal luminal or basal/myoepithelial-committed progenitors become further transformed, and give rise to clones of immature luminal or basal epithelial cells (due to block in differentiation), respectively (Figure 8).
Figure 8. The mammary epithelial cell hierarchy in normal mammary glands and in mammary tumors from Wap-Cre;EN females.
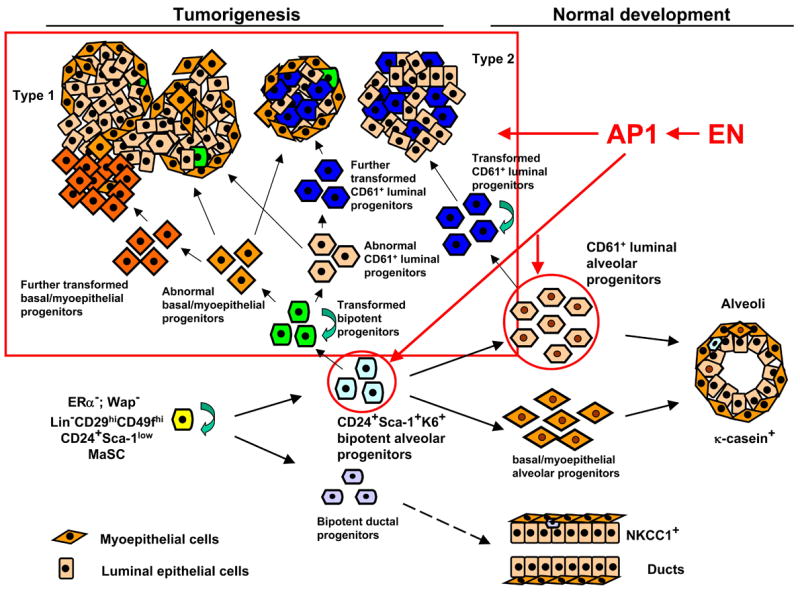
Proposed target cells of EN in WCEN females are indicated (red circles). Red arrows indicate EN-initiated tumorigenesis (both tumor initiation and progression) is mediated through AP1.
K14 is a type II keratin that pairs with the type I keratin, K5, to form heterodimers in basal cells of stratified epithelia. Thus, both K5 and K14 have been considered as basal cell markers and have been used to distinguish basal/myoepithelial cells from K8+ luminal epithelial cells. However, in this study, we show that K14 also stains a subset of K8+ luminal epithelial cells, in addition to K5+ basal/myoepithelial cells (Figure 4A and Figure S2A and S7). In fact, these K14+K8+ luminal cells represent blocked CD61+ luminal progenitors in EN tumors. Of note, in normal MGs, a population of K14+K8+ (Figure 4B, this study) or K14+K18+ luminal cells is also detected (Shackleton et al., 2006). In addition, our microarray analysis of murine breast cancer models also shows that the K14 gene cluster is distinct from the K5 basal gene cluster [Figure 4C, also (Herschkowitz et al., 2007)]. Thus, K14 is distinct from K5 as a marker, and a combination of both markers together with other markers (e.g., p63, SMA, K8) can be used to define subpopulations of cells in murine mammary tumors.
Although we show that EN transforms committed alveolar progenitors, we do not dismiss the possibility that EN may also transform MaSCs. Interestingly, as we described above, due to the leaky expression of the EN allele (without Cre-mediated excision of the “stopper”, thus independent of the Wap promoter), a small number of EN-only heterozygous mice develop mammary tumors at advanced ages. Most of these tumors fall within one of the 4 subtypes described above in WCEN females (Figure S7), suggesting target cells of EN in these 2 types of mice are probably similar. Although under this genetic setting, one cannot be certain if bipotent tumors with type 1 features are derived from committed bipotent progenitors (similar to WCEN) or from MaSCs, since they probably give rise to tumors with a similar appearance. In addition to tumors with features of these 4 subtypes, we also observed tumors either containing extensively K5+ basal cells, or containing mainly K8+ (but K14−) luminal cells (Figure S7). This is most likely due to the long latency of tumors that develop in _EN_-only mice (>1year compared to several months for WCEN females). More differentiated, single lineage committed cells may be transformed by EN after they accumulate additional mutations over an extended time period.
Transformation by EN oncoprotein is mediated through the AP1 complex
The AP1 transcriptional complex is composed of heterodimeric Jun/Fos family proteins (Eferl and Wagner, 2003). The AP1 pathway integrates multiple growth signals at the transcriptional level and regulates several cellular processes (Shen et al., 2007). In normal MGs, previous studies have shown that AP1 is a pivotal regulator of postnatal MG growth and development (Shen et al., 2006). In human breast cancer, c-JUN activation is associated with proliferation and angiogenesis in invasive breast cancer (Vleugel et al., 2006). Overexpression of the DN c-Jun in breast cancer cells induces a G1 cell cycle block and inhibits their growth both in vitro and in vivo (Liu et al., 2002). Fosl1 (Fra1) regulates proliferation and invasiveness of breast cancer cells (Belguise et al., 2005; Milde-Langosch et al., 2004). Overexpression of FOSL1 (FRA-I) protein has been observed in both hyperplastic and neoplastic human breast disorders (Chiappetta et al., 2007). In addition, Fosl1 was also used as an effective target for a DNA vaccine to protect mice against breast cancer (Luo et al., 2003).
Our findings demonstrate that EN expression leads to upregulation and activation of the c-Jun/Fosl1 AP1 complex. Several AP1 target genes are upregulated in both EN-3T3 cells and EN tumor cells (Figure 5A–5B and Figure S6A–S6B), including HBEGF and possibly CCND1 (cyclin D1), which stimulate proliferation. Cyclin D1 is the major positive regulator of cell-cycle progression induced by AP1 (Bakiri et al., 2000). Although we did not observe increased levels of cyclin D1 in sorted tumor cells directly compared to unsorted tumors (possibly due to its expression in stromal cells), we observed its upregulation in EN-3T3 cells both by microarray and Western blot (Figure 2D and 5B). We demonstrated its upregulation during tumor progression (Figure 5C). We also detected high levels of cyclin D1/2 expression in EN tumors by Western blot (Figure 2D). Intriguingly, our microarray analysis shows that several AP1 target genes known to promote angiogenesis and invasiveness, including PLAUR, PLAU, CD44, CTSL (cathepsin L), MMP3 (matrix metallopeptidase 3), and TPM3 (tropomyosin 3) are upregulated in both EN-3T3 cells and EN hyperplastic MECs (Figure 5B–5C), suggesting an EN/AP1-induced invasiveness program may be established early in EN-mediated transformation. This observations may partially account for the short latency and aggressiveness of EN tumors in mice, and the appearance of SBC in children as young as 3-years of age (Euhus et al., 2002).
Together with previous observations in human breast cancer cells, our study supports critical roles of AP1 in breast tumorigenesis and invasiveness. Further focus on this transcriptional complex as a target in human breast cancer is warranted.
Modeling chromosomal rearrangements in human epithelial tumors can provide novel insights into their pathogenesis and therapy
The breast cancer model described here is based on the t(12;15)(p13;q25) translocation in human cancer. Our data demonstrate that the translocation generated EN fusion oncoprotein is sufficient to initiate mammary tumorigenesis. This complements genetic evidence in humans suggesting this translocation is the primary event in the disorder (Tognon et al., 2002). In our model, EN is induced only in a very small number of cells in mammary tissues and the tumor cells emerge within the environment of normal cells, thus closely mimicking human disease initiated by somatic mutation. The recent findings of recurrent gene fusions in prostate cancer and lung cancer suggest that, balanced, disease-specific chromosomal rearrangements in epithelial cancers, may be more common and important than previously believed (Meyerson, 2007; Tomlins et al., 2005). As demonstrated in hematopoietic malignancies, study of gene rearrangements has contributed immeasurably to an understanding of both normal blood cell development and malignancy. In addition, study of gene fusions with kinase activities has revolutionized targeted therapies for cancer (e.g., imatinib for BCR-ABL fusion). We hope that the success in modeling an infrequent translocation seen in human epithelial cancer described here will inspire the generation of sophisticated models of other chromosomal arrangements that may occur more commonly in other epithelial tumors. Such engineered mice permit experimental access to the earliest target cells and steps in the transformation process, and may serve as effective models for preclinical testing.
EXPERIMENTAL PROCEDURES
Mice
Etv6-NTRK3 conditional knockin mice were generated by gene targeting (details are provided in supplemental methods). Wap-Cre, MMTV-Cre, and Wap-rtTA-Cre transgenic mice were acquired from MMHCC repository at NCI-Frederick. Rosa-Stop-lacZ reporter mice and _Rag2_−/−immunodeficient mice are maintained in our mouse colony. The Rosa-Stop-Luc reporter was kindly provided by Dr. William G. Kaelin, Jr. All studies involving mice were approved by the Children’s Hospital Boston Institutional Animal Care and Use Committee and performed in accordance with the relevant protocol.
Human SBC samples
The SBC cases were accrued from Pathology departments at Children’s and Women’s Health Centre of British Columbia, Instituto de Patologia e Imunologia Molecular da Universidade do Porto, and from Burnaby General Hospital, Burnaby, Canada. Informed consent was obtained for all patient samples used in this study and ethics approval was received from the Research Ethic Board (REB) at the University of British Columbia.
Pathology, immunostaining, _lac_Z staining, and flow cytometry
Standard protocols were followed. Detailed procedures are provided in supplemental methods.
Biochemical and cellular assays
Standard protocols were followed. Details about EMSAs, Western blot, immunoprecipitation studies, and soft agar assays are provided in supplemental methods.
Microarray data collection and analysis
Microarray expression profiles were collected using Affymetrix or Agilent chips and analyzed using dChip (Li and Wong, 2001) and GSEA (Subramanian et al., 2005) as described. Details are provided in supplemental methods. All microarray data have been deposited in NCBI’s Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) with GEO Series accession numbers GSE9355 (EN tumors using Affymetrix chips), GSE9354 (3T3 cells), GSE9353 (EpH4-derived tumors), and GSE9343 (selected EN tumors using Agilent chips).
Supplementary Material
01
02
Acknowledgments
We thank Dr. Yuko Fujiwara, Aimee Williams and Nicole Stokes for help with transgenic mice; Carol Browne for assistance with ES cell targeting; Melanie Hamblen for general support; Grigoriy Losyev for cell sorting; Dr. Leonard Zon for discussion; Dr. Tim Triche for help with microarray of 3T3 cells. This work was supported by grants to S.H.O. from the NCI Mouse Models of Human Cancer Consortium and from the Department of Defense, by grants to P.H.S. from the Canadian Institutes for Health Research (CIHR), by funds to C.M.P. from NCI (RO1-CA-101227-01) and HHSN-261200433008C (N01-CN43308), and by K99 Pathway-to-Independence award to Z.L. from NCI. S.H.O. is an Investigator of the Howard Hughes Medical Institute. C.E.T. was supported by a ReThink Breast Cancer Career Development Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asselin-Labat ML, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC, Hartley L, Robb L, Grosveld FG, van der Wees J, et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol. 2007;9:201–209. doi: 10.1038/ncb1530. [DOI] [PubMed] [Google Scholar]
- Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. Embo J. 2000;19:2056–2068. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belguise K, Kersual N, Galtier F, Chalbos D. FRA-1 expression level regulates proliferation and invasiveness of breast cancer cells. Oncogene. 2005;24:1434–1444. doi: 10.1038/sj.onc.1208312. [DOI] [PubMed] [Google Scholar]
- Booth BW, Boulanger CA, Smith GH. Alveolar progenitor cells develop in mouse mammary glands independent of pregnancy and lactation. J Cell Physiol. 2007;212:729–736. doi: 10.1002/jcp.21071. [DOI] [PubMed] [Google Scholar]
- Boulanger CA, Wagner KU, Smith GH. Parity-induced mouse mammary epithelial cells are pluripotent, self-renewing and sensitive to TGF-beta1 expression. Oncogene. 2005;24:552–560. doi: 10.1038/sj.onc.1208185. [DOI] [PubMed] [Google Scholar]
- Chiappetta G, Ferraro A, Botti G, Monaco M, Pasquinelli R, Vuttariello E, Arnaldi L, Di Bonito M, D’Aiuto G, Pierantoni GM, Fusco A. FRA-1 protein overexpression is a feature of hyperplastic and neoplastic breast disorders. BMC Cancer. 2007;7:17. doi: 10.1186/1471-2407-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- Eguchi M, Eguchi-Ishimae M, Tojo A, Morishita K, Suzuki K, Sato Y, Kudoh S, Tanaka K, Setoyama M, Nagamura F, et al. Fusion of ETV6 to neurotrophin-3 receptor TRKC in acute myeloid leukemia with t(12;15)(p13;q25) Blood. 1999;93:1355–1363. [PubMed] [Google Scholar]
- Euhus DM, Timmons CF, Tomlinson GE. ETV6-NTRK3--Trk-ing the primary event in human secretory breast cancer. Cancer Cell. 2002;2:347–348. doi: 10.1016/s1535-6108(02)00184-8. [DOI] [PubMed] [Google Scholar]
- Hennighausen L, Robinson GW. Information networks in the mammary gland. Nat Rev Mol Cell Biol. 2005;6:715–725. doi: 10.1038/nrm1714. [DOI] [PubMed] [Google Scholar]
- Henry MD, Triplett AA, Oh KB, Smith GH, Wagner KU. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene. 2004;23:6980–6985. doi: 10.1038/sj.onc.1207827. [DOI] [PubMed] [Google Scholar]
- Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince TA, Richardson AL, Bell GW, Saitoh M, Godar S, Karnoub AE, Iglehart JD, Weinberg RA. Transformation of Different Human Breast Epithelial Cell Types Leads to Distinct Tumor Phenotypes. Cancer Cell. 2007;12:160–170. doi: 10.1016/j.ccr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- Knezevich SR, Garnett MJ, Pysher TJ, Beckwith JB, Grundy PE, Sorensen PH. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 1998a;58:5046–5048. [PubMed] [Google Scholar]
- Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998b;18:184–187. doi: 10.1038/ng0298-184. [DOI] [PubMed] [Google Scholar]
- Kordon EC, McKnight RA, Jhappan C, Hennighausen L, Merlino G, Smith GH. Ectopic TGF beta 1 expression in the secretory mammary epithelium induces early senescence of the epithelial stem cell population. Dev Biol. 1995;168:47–61. doi: 10.1006/dbio.1995.1060. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Lamb RF, Hennigan RF, Turnbull K, Katsanakis KD, MacKenzie ED, Birnie GD, Ozanne BW. AP-1-mediated invasion requires increased expression of the hyaluronan receptor CD44. Mol Cell Biol. 1997;17:963–976. doi: 10.1128/mcb.17.2.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Rosen JM. Stem/progenitor cells in mouse mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia. 2005;10:17–24. doi: 10.1007/s10911-005-2537-2. [DOI] [PubMed] [Google Scholar]
- Liu Y, Ludes-Meyers J, Zhang Y, Munoz-Medellin D, Kim HT, Lu C, Ge G, Schiff R, Hilsenbeck SG, Osborne CK, Brown PH. Inhibition of AP-1 transcription factor causes blockade of multiple signal transduction pathways and inhibits breast cancer growth. Oncogene. 2002;21:7680–7689. doi: 10.1038/sj.onc.1205883. [DOI] [PubMed] [Google Scholar]
- Lobo NA, Shimono Y, Qian D, Clarke MF. The Biology of Cancer Stem Cells. Annu Rev Cell Dev Biol. 2007 doi: 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- Ludes-Meyers JH, Liu Y, Munoz-Medellin D, Hilsenbeck SG, Brown PH. AP-1 blockade inhibits the growth of normal and malignant breast cells. Oncogene. 2001;20:2771–2780. doi: 10.1038/sj.onc.1204377. [DOI] [PubMed] [Google Scholar]
- Luo Y, Zhou H, Mizutani M, Mizutani N, Reisfeld RA, Xiang R. Transcription factor Fos-related antigen 1 is an effective target for a breast cancer vaccine. Proc Natl Acad Sci U S A. 2003;100:8850–8855. doi: 10.1073/pnas.1033132100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci U S A. 1999;96:1603–1608. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Orkin SH. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci U S A. 1999;96:5037–5042. doi: 10.1073/pnas.96.9.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M. Cancer: broken genes in solid tumours. Nature. 2007;448:545–546. doi: 10.1038/448545a. [DOI] [PubMed] [Google Scholar]
- Milde-Langosch K, Bamberger AM, Rieck G, Kelp B, Loning T. Overexpression of the p16 cell cycle inhibitor in breast cancer is associated with a more malignant phenotype. Breast Cancer Res Treat. 2001;67:61–70. doi: 10.1023/a:1010623308275. [DOI] [PubMed] [Google Scholar]
- Milde-Langosch K, Roder H, Andritzky B, Aslan B, Hemminger G, Brinkmann A, Bamberger CM, Loning T, Bamberger AM. The role of the AP-1 transcription factors c-Fos, FosB, Fra-1 and Fra-2 in the invasion process of mammary carcinomas. Breast Cancer Res Treat. 2004;86:139–152. doi: 10.1023/B:BREA.0000032982.49024.71. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Robinson GW, McKnight RA, Smith GH, Hennighausen L. Mammary epithelial cells undergo secretory differentiation in cycling virgins but require pregnancy for the establishment of terminal differentiation. Development. 1995;121:2079–2090. doi: 10.1242/dev.121.7.2079. [DOI] [PubMed] [Google Scholar]
- Robinson GW, Smith GH, Gallahan D, Zimmer A, Furth PA, Hennighausen L. Understanding mammary gland development through the imbalanced expression of growth regulators. Dev Dyn. 1996;206:159–168. doi: 10.1002/(SICI)1097-0177(199606)206:2<159::AID-AJA5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Rowley JD. Chromosome translocations: dangerous liaisons revisited. Nat Rev Cancer. 2001;1:245–250. doi: 10.1038/35106108. [DOI] [PubMed] [Google Scholar]
- Rubin BP, Chen CJ, Morgan TW, Xiao S, Grier HE, Kozakewich HP, Perez-Atayde AR, Fletcher JA. Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion: cytogenetic and molecular relationship to congenital (infantile) fibrosarcoma. Am J Pathol. 1998;153:1451–1458. doi: 10.1016/S0002-9440(10)65732-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safran M, Kim WY, Kung AL, Horner JW, DePinho RA, Kaelin WG., Jr Mouse reporter strain for noninvasive bioluminescent imaging of cells that have undergone Cre-mediated recombination. Mol Imaging. 2003;2:297–302. doi: 10.1162/15353500200303154. [DOI] [PubMed] [Google Scholar]
- Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- Shen Q, Uray IP, Li Y, Krisko TI, Strecker TE, Kim HT, Brown PH. The AP-1 transcription factor regulates breast cancer cell growth via cyclins and E2F factors. Oncogene. 2007 doi: 10.1038/sj.onc.1210643. [DOI] [PubMed] [Google Scholar]
- Shen Q, Zhang Y, Uray IP, Hill JL, Kim HT, Lu C, Young MR, Gunther EJ, Hilsenbeck SG, Chodosh LA, et al. The AP-1 transcription factor regulates postnatal mammary gland development. Dev Biol. 2006;295:589–603. doi: 10.1016/j.ydbio.2006.03.042. [DOI] [PubMed] [Google Scholar]
- Simpson PT, Reis-Filho JS, Gale T, Lakhani SR. Molecular evolution of breast cancer. J Pathol. 2005;205:248–254. doi: 10.1002/path.1691. [DOI] [PubMed] [Google Scholar]
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara SI, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, Eaves CJ. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognon C, Garnett M, Kenward E, Kay R, Morrison K, Sorensen PH. The chimeric protein tyrosine kinase ETV6-NTRK3 requires both Ras-Erk1/2 and PI3-kinase-Akt signaling for fibroblast transformation. Cancer Res. 2001;61:8909–8916. [PubMed] [Google Scholar]
- Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, Becker L, Carneiro F, MacPherson N, Horsman D, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2:367–376. doi: 10.1016/s1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, Cao X, Wei JT, Rubin MA, Shah RB, Chinnaiyan AM. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–3400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Utomo AR, Nikitin AY, Lee WH. Temporal, spatial, and cell type-specific control of Cre-mediated DNA recombination in transgenic mice. Nat Biotechnol. 1999;17:1091–1096. doi: 10.1038/15073. [DOI] [PubMed] [Google Scholar]
- Vleugel MM, Greijer AE, Bos R, van der Wall E, van Diest PJ. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum Pathol. 2006;37:668–674. doi: 10.1016/j.humpath.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Boulanger CA, Henry MD, Sgagias M, Hennighausen L, Smith GH. An adjunct mammary epithelial cell population in parous females: its role in functional adaptation and tissue renewal. Development. 2002;129:1377–1386. doi: 10.1242/dev.129.6.1377. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Smith GH. Pregnancy and stem cell behavior. J Mammary Gland Biol Neoplasia. 2005;10:25–36. doi: 10.1007/s10911-005-2538-1. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, Li M, Furth PA, Hennighausen L. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai DH, Knezevich SR, Lucas T, Jansen B, Kay RJ, Sorensen PH. The ETV6-NTRK3 gene fusion encodes a chimeric protein tyrosine kinase that transforms NIH3T3 cells. Oncogene. 2000;19:906–915. doi: 10.1038/sj.onc.1203396. [DOI] [PubMed] [Google Scholar]
- Welm BE, Tepera SB, Venezia T, Graubert TA, Rosen JM, Goodell MA. Sca-1(pos) cells in the mouse mammary gland represent an enriched progenitor cell population. Dev Biol. 2002;245:42–56. doi: 10.1006/dbio.2002.0625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
02