Role of NMDA Receptors in Dopamine Neurons for Plasticity and Addictive Behaviors (original) (raw)
. Author manuscript; available in PMC: 2008 Sep 29.
Summary
A single exposure to drugs of abuse produces an NMDA receptor (NMDAR)-dependent long-term potentiation (LTP) of AMPA receptor (AMPAR) currents in DA neurons; however, the importance of LTP for various aspects of drug addiction is unclear. To test the role of NMDAR-dependent plasticity in addictive behavior, we genetically inactivated functional NMDAR signaling exclusively in DA neurons (KO mice). Inactivation of NMDARs results in increased AMPAR-mediated transmission that is indistinguishable from the increases associated with a single cocaine exposure, yet locomotor responses to multiple drugs of abuse were unaltered in the KO mice. The initial phase of locomotor sensitization to cocaine is intact; however, the delayed sensitization that occurs with prolonged cocaine withdrawal did not occur. Conditioned behavioral responses for cocaine-testing environment were also absent in the KO mice. These findings provide evidence for a role of NMDAR signaling in DA neurons for specific behavioral modifications associated with drug seeking behaviors.
Introduction
The mesolimbic DA circuit, consisting of midbrain DA neurons in the ventral tegmental area (VTA), that project to the nucleus accumbens (NAc) and prefrontal cortex, plays a key role in reward and motivation and is a major target of abused drugs (Wise, 2004). Repeated exposure to drugs of abuse increases their psychomotor stimulant effects and elicits conditioned preferences for neutral stimuli in animal models of addiction. These behaviors are thought to reflect increased incentive motivation for drug acquisition and contextual or cue-evoked, drug-seeking behavior (Robinson and Berridge, 1993). Two classical models used to study these correlates of addiction are locomotor (or behavioral) sensitization and conditioned place preference (CPP) (Ettenberg, 1989; Kalivas and Stewart, 1991). The acquisition and expression of locomotor sensitization and CPP are attenuated by glutamate receptor antagonists, strongly implicating glutamate signaling and synaptic plasticity in the mesolimbic DA circuit as an underlying mechanism in addiction.
Plasticity at excitatory glutamatergic synapses plays a critical role in memory acquisition and consolidation. Signal transduction cascades, initiated by Ca2+ influx through NMDARs, mediate a rapid and sustained enhancement of glutamatergic synapses by regulating local synaptic strength through the modulation of AMPAR number and function (Malinow and Malenka, 2002). Drugs of abuse evoke an NMDAR-dependent, long-term potentiation (LTP) of AMPAR currents in DA neurons that may reflect an early memory trace in the acquisition of drug dependence (Borgland et al., 2004; Liu et al., 2005; Saal et al., 2003; Ungless et al., 2001). Functionally, LTP of AMPAR currents in VTA DA neurons following drug exposure is coincident with increased AMPA-evoked DA release that is thought to underlie the initiation of drug sensitization (Dunn et al., 2005; Kalivas and Alesdatter, 1993; Vezina and Queen, 2000; Zhang et al., 1997).
A dichotomy currently exists regarding the role of glutamate plasticity within VTA DA neurons as a critical neural adaptation underlying drug-seeking behavior (Tzschentke and Schmidt, 2000; Vanderschuren and Kalivas, 2000). Site-specific injection of glutamate antagonists into the VTA during repeated drug administration blocked behavioral sensitization (Dunn et al., 2005; Kalivas and Alesdatter, 1993; Vezina and Queen, 2000) and attenuated CPP (Harris and Aston-Jones, 2003; Harris et al., 2004). However, repeated infusion of NMDA directly into the VTA did not induce a sensitized response to a systemic cocaine injection (Schenk and Partridge, 1997). In addition, other groups have demonstrated day-to-day increases in locomotor responding to amphetamine or morphine when NMDAR antagonists were present during testing (Ranaldi et al., 2000). Disparities regarding the role of AMPARs also exist. Viral overexpression of the AMPAR subunit GluR1 produced a sensitized behavioral response to acute morphine treatment (Carlezon et al., 1997), and infusion of AMPAR antagonists into the VTA prevented sensitization (Dunn et al., 2005). However, mice with a genetic deletion of the AMPAR subunit GluR1 lacked cocaine-induced plasticity in DA neurons, yet behavioral sensitization was normal (Dong et al., 2004).
Significant differences in the methodologies used in these studies could explain the discrepancies observed, but they all share a lack of cell specificity. Although NMDAR antagonists were injected directly into the VTA, other cell types in this brain region likely express these receptors, such as GABAergic inter-neurons, GABAergic projection (Korotkova et al., 2004; Olson and Nestler, 2007), and glutamatergic neurons (Yamaguchi et al., 2007). Similarly, viral-mediated delivery of GluR1 likely infected both DA and non-DA neurons within the VTA, and the GluR1 knockout mice lacked this subunit in all cells.
To examine the cell-specific requirements of glutamate signaling in DA neurons for the long-term changes associated with drug exposure, we selectively inactivated NMDAR signaling in these cells. Absence of functional NMDAR increased synaptic AMPAR currents in DA neurons that were similar to the changes associated with a single cocaine exposure in vivo. Acute responses to the locomotor-stimulating effects of cocaine and other drugs of abuse were unaltered in mice lacking functional NMDAR in DA neurons, and the induction of behavioral sensitization progressed normally; however, cue-evoked drug seeking and the enhancement of drug craving following withdrawal were significantly impaired.
Results
Selective Inactivation of NMDARs in DA Neurons
NMDARs are heteromeric ion channels that are critically dependent on the NR1 subunit for proper channel function. Cell-specific, conditional inactivation of the NR1 subunit prevents NMDAR-mediated currents, blocks the induction of LTP, and impairs some forms of learning and memory (Tsien et al., 1996). To determine if NMDAR-dependent plasticity in DA neurons is critical for the neural adaptations associated with repeated drug administration, we used a genetic approach to selectively inactivate the NR1 subunit of the NMDARs in DA neurons. Cell-specific inactivation of the floxed NR1 (Grin1lox) allele (Tsien et al., 1996) was achieved by crossing these mice with mice expressing Cre recombinase under the control of the endogenous DA transporter gene (Slc6a3Cre) (Zhuang et al., 2005). For the experiments described here, Slc6a3+/Cre; Grin1Δ/lox (knockout, KO) mice and Slc6a3+/Cre; Grin1+/lox (control) mice were used (Figure 1A). Both KO and control mice have one functional allele for the DA transporter and one functional allele of NR1, except in DA neurons where Cre-mediated recombination should inactivate the remaining Grin1lox allele. KO mice were born at the expected Mendelian ratio and were visibly indistinguishable from control mice.
Figure 1. Generation and Histological Characterization of KO and Control Mice.
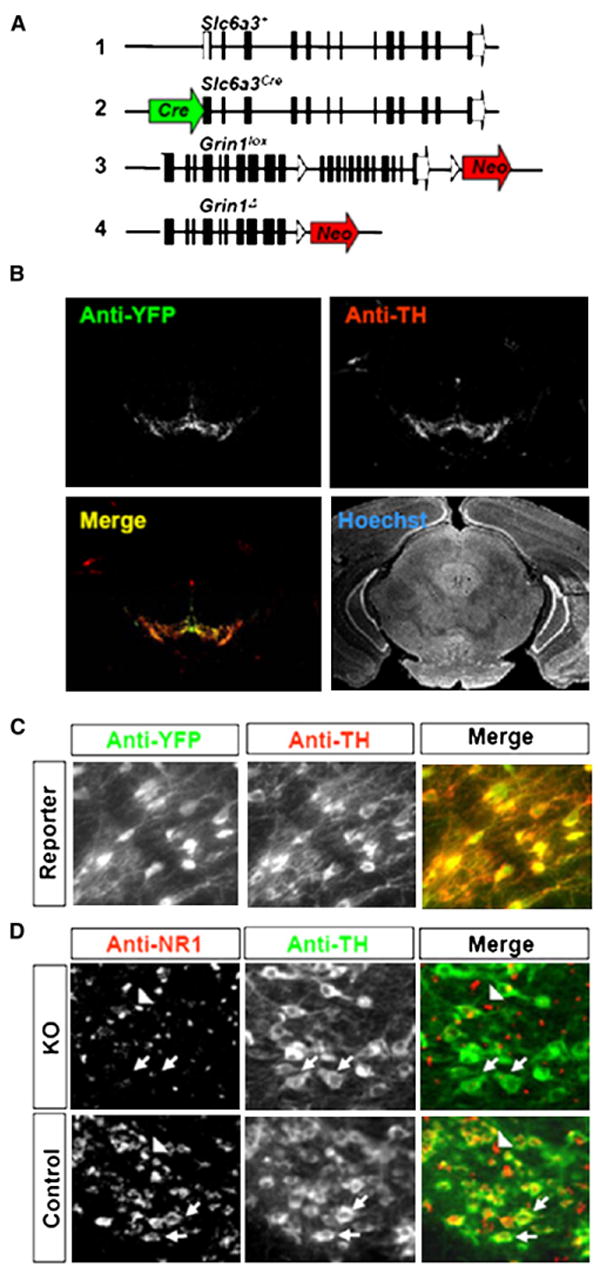
(A) Schematic of Slc6a3 and Grin1 alleles in control and KO mice. Wild-type Slc6a3 allele (1), Slc6a3Cre allele (2), Grin1lox allele (3), and Grin1Δ allele (4).
(B) Low-power (2.5×) image of immunofluoresence staining for YFP, TH, and Hoechst nuclear stain in the midbrain of Rosa26YFP reporter mice.
(C) High-power (20×) image of double immunofluoresence staining of VTA DA neurons for TH and YFP.
(D) High-power (20×) image of double immunofluoresence staining of VTA DA neurons for TH and NR1. NR1 expression was undetectable in TH-positive (arrows), but not TH-negative (arrow heads) neurons in the VTA of KO mice.
To confirm that Cre-mediated recombination in Sl6a3Cre mice is localized to DA neurons, we crossed these mice with Rosa26_YFP_ reporter mice (Srinivas et al., 2001). Double immunofluorescence labeling for yellow fluorescent protein (YFP) and tyrosine hydroxylase (TH), the rate-limiting enzyme in dopamine synthesis, revealed a high level of coexpression in the midbrain, but not in the overlying cortex or hippocampus (Figures 1B and 1C). In addition, we detected YFP in other cells known to express TH and DAT, such as the hypothalamus and olfactory bulb (data not shown), similar to that previously described (Coulter et al., 1996; Zhuang et al., 2005; Turiault et al., 2007). The extent of Grin1 gene inactivation in KO mice was examined by immunohistochemistry using antibodies against NR1. KO mice displayed no observable expression of NR1 in TH-expressing midbrain DA neurons compared to controls; however, NR1 expression in non-DA cells in the midbrain was normal (Figure 1D).
To confirm a lack of functional NMDARs, whole-cell, voltage-clamp recordings from DA and non-DA neurons were obtained from brain slices through the VTA of KO and control mice. DA neurons were identified by the presence of hyperpolarization-activated potassium currents (Ih) (Figure 2A) as previously described (Borgland et al., 2004; Saal et al., 2003; Schilstrom et al., 2006; Ungless et al., 2001). There was no NMDAR component of the excitatory postsynaptic current (EPSC) in DA neurons from KO mice (Figure 2B); however, NMDAR-mediated EPSCs were intact in non-DA neurons of the VTA of KO mice (Figure 2B).
Figure 2. Inactivation of Functional NMDAR in DA Neurons.
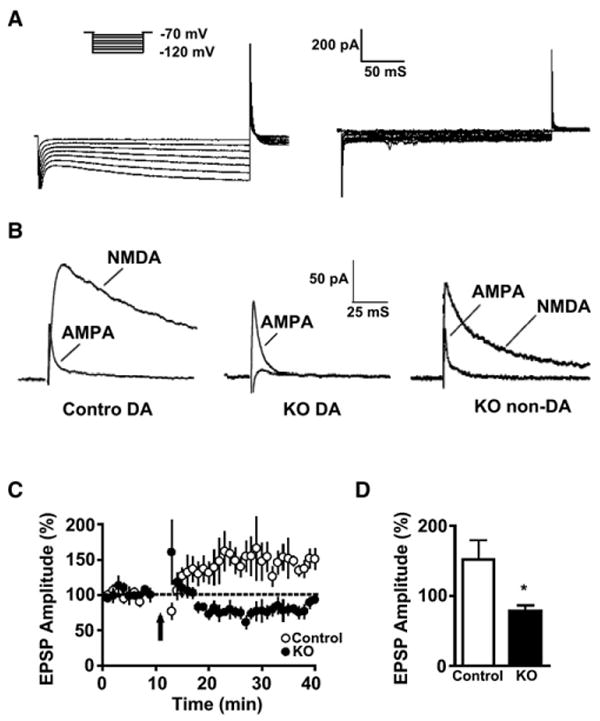
(A) Representative traces of Ih currents used to identify DA (left) and non-DA neurons (right).
(B) Representative traces of NMDAR and AMPAR EPSCs from DA and non-DA neurons recorded from VTA slices of control and KO mice.
(C) LTP evoked by high-frequency afferent stimulation was not induced in DA neurons from KO mice.
(D) Histogram representing the magnitude of percent increase in EPSP amplitude 25 min after induction (*p < 0.01, Student's t test). Data are from VTA slices from control (n = 7) and KO (n = 6) mice and represent means ± SEM.
NMDAR activation is thought to be critical for the induction of LTP in DA neurons (Borgland et al., 2004; Saal et al., 2003; Schilstrom et al., 2006; Ungless et al., 2001). We monitored evoked excitatory, postsynaptic potential (EPSP) amplitudes in DA neurons from VTA slices following high-frequency afferent stimulation using a spike-timing protocol of induction (Liu et al., 2005). As predicted, we observed robust LTP in DA neurons from control, but not KO mice (Figures 2C and 2D).
Inactivation of NMDARs and In Vivo Cocaine Exposure Evoke Similar Changes in Synaptic AMPAR Currents
Many drugs of abuse produce an LTP of AMPAR currents in DA neurons that is blocked by NMDAR antagonists (Borgland et al., 2004; Liu et al., 2005; Saal et al., 2003; Ungless et al., 2001). To determine if NMDAR activation in DA neurons is necessary for cocaine-evoked LTP of synaptic AMPAR currents, we monitored spontaneous miniature excitatory postsynaptic currents (mEPSCs) in drug-naive and cocaine-treated mice (Figure 3A) as previously described (Ungless et al., 2001). As predicted, a single cocaine exposure increased mEPSC amplitude and frequency in control mice (Figures 3B and 3C). Unexpectedly, drug-naive KO mice also showed a significant increase in mEPSC amplitude (16.3 ± 1.3 pA versus 10.8 ± 0.8 pA, p < 0.01 student's t test) and frequency (3.2 ± 0.3 Hz versus 1.6 ± 0.2 Hz, p < 0.01 student's t test) compared to drug-naive controls (Figures 3B and 3C). These changes were similar to the increases associated with a single cocaine exposure (Figures 3B and 3C).
Figure 3. Cocaine and NMDAR Inactivation Increase Synaptic AMPAR Currents in VTA DA Neurons.
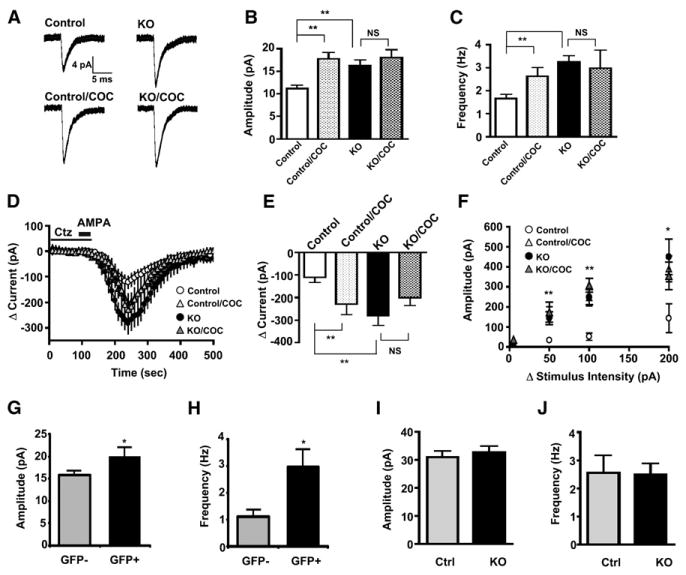
(A) Average AMPAR miniature EPSCs (mEPSCs) from DA neurons of drug-naive control and KO mice 24 hr after a single cocaine (20 mg/kg) injection (mEPSCs traces are the average of 60 consecutive events).
(B) Histogram of average mEPSC amplitude (**p < 0.01 compared to control, Student's t test).
(C) Histogram of average mEPSC frequency (**p < 0.01 compared to control, Student's t test). Data collected from VTA slices from naive control, n = 6; naive KO, n = 8; cocaine-treated control, n = 6; and cocaine-treated KO, n = 4 (B and C).
(D) Average AMPA-evoked (2.5 μM AMPA in 50 μM cyclothiazide, ctz) EPSCs in DA neurons from drug-naive control and KO mice 24 hr after a single cocaine (20 mg/kg) injection.
(E) Histogram of average change in AMPA-evoked currents 150 s after AMPA application (p < 0.01 compared to control, Student's t test). Data collected from VTA slices from naive control, n = 7; naive KO, n = 7; cocaine-treated control, n = 6; and cocaine-treated KO, n = 4 (D and E).
(F) Input-output curves from stimulus-evoked AMPAR EPSCs expressed as a fixed increase over threshold (*p < 0.05, **p < 0.01 compare to control, Student's t test). Data collected from VTA slices from naive control, n = 6; naive KO, n = 7; cocaine-treated control, n = 6; and cocaine-treated KO, n = 4.
(G) Histogram of average mEPSC amplitude from AAV-Cre-GFP transduced and control DA neurons.
(H) Histogram of average mIPSC frequency. Data collected from GFP-negative, n = 9, and GFP-positive, n = 8 cells (G and H).
(I) Histogram of average mIPSC amplitude.
(J) Histogram of average mIPSC frequency. Data collected from VTA slices from control, n = 8, and KO, n = 9 (I and J).
Data expressed as means ± SEM.
Cocaine increases postsynaptic AMPAR currents in DA neurons without affecting presynaptic transmitter release, as monitored by the paired-pulse ratio between two electrical stimuli applied at an interval of 50 ms (Ungless et al., 2001). Similarly, we did not observe a significant difference in paired-pulse ratio in DA neurons from control and KO mice (0.861 ± 0.056, n = 19, versus 0.941 ± 0.053, n = 25, respectively). To confirm that changes in mEPSC amplitude and frequency observed in KO mice is due to increased postsynaptic AMPAR currents similar to that evoked by cocaine, we monitored AMPA-evoked EPSCs using whole-cell voltage clamp in VTA slices from drug-naive and cocaine-treated control and KO mice as described (Borgland et al., 2004; Liu et al., 2005; Ungless et al., 2001). Consistent with our data from mEPSCs, AMPA-evoked currents from KO mice were significantly increased compared to controls (peak change in current, 282 ± 41 pA versus 110 ± 24 pA, p < 0.01, Student's t test) and was not different than cocaine-treated mice (Figures 3D and 3E). These findings were confirmed by monitoring the input-output relationship at various stimulus intensities. AMPAR EPSCs were significantly larger at each stimulus intensity tested (Figure 3F).
Because the DA transporter gene is expressed by embryonic day 14 (Fujita et al., 1993), it is possible that early developmental inactivation of NMDAR signaling engages compensatory mechanisms resulting in increased synaptic AMPAR currents (Turrigiano and Nelson, 2004). To determine whether increased synaptic AMPAR are restricted to early developmental inactivation of NMDAR, we injected an adenoassociated virus (AAV) expressing a Cre-GFP fusion protein directly into the VTA of 3-week-old mice, a time after which DA dependence has developed (Zhou and Palmiter, 1995). Synaptic AMPAR currents were monitored 3–5 days following viral injection. AAV-Cre-GFP transduction of DA neurons significantly decreased NMDAR EPSCs, increasing overall AMPAR/NMDAR ratios compared to neighboring GFP-negative DA neurons (2.71 ± 0.36 versus 1.24 ± 0.12, p < 0.01). Similar to KO mice, AMPAR mEPSCs amplitude and frequency were significantly increased in GFP-positive DA neurons compared to GFP-negative DA neurons (Figures 3G and 3H). These findings indicate that NMDAR inactivation -induced increases in synaptic AMPAR currents are not restricted to early development.
To determine whether genetic NMDAR inactivation also alters inhibitory synaptic transmission, we measured spontaneous miniature IPSCs (mIPSC) in DA neurons from control and KO mice as described (Liu et al., 2005). mIPSC amplitude (Figure 3I) and frequency (Figure 3J) were not different between the two groups, indicating that chronic NMDAR inactivation selectively modulates excitatory, but not inhibitory synaptic transmission.
Altered Glutamate Signaling in DA Neurons of KO Mice Does Not Affect Locomotor-Stimulating Effects of Drugs
Increased AMPAR signaling in DA neurons is correlated with elevated DA release and heightened responses to the psychomotor-stimulating effects of drugs of abuse (Carlezon et al., 1997; Zhang et al., 1997); thus, chronic absence of NMDARs and consequent enhancement of AMPAR currents might alter the excitability of these cells resulting in enhanced locomotor activity. To determine if KO mice have heightened locomotor activity in general, we monitored their exploratory behavior during a 90 min exposure to novel activity chambers. The time course of habituation to novelty (Figure 4A) and cumulative locomotor activity (Figure 4G) did not differ between the two groups. Locomotor activity during a single day-night cycle was also monitored. Both control and KO mice were relatively inactive during the light phase (day) and demonstrated increased activity shortly after dark phase (night) onset that persisted for approximately 12 hr (Figure 4B).
Figure 4. Novelty and Drug-Induced Locomotor Activity.

(A) Locomotor response to novel environment (control, n = 12, and KO, n = 13).
(B) Day-night locomotion (control, n = 9, and KO, n = 7).
(C) Locomotor response to morphine (25 mg/kg) (control, n = 7, and KO, n = 8).
(D) Locomotor response to amphetamine (2.5 mg/kg) (control, n = 12, and KO, n = 13).
(E) Locomotor response to cocaine (20 mg/kg) (control, n = 15, and KO, n = 12).
(F) Locomotor response to D1/D5 receptor agonist SKF 81297 (7.5 mg/kg) (control, n = 7, and KO, n = 8).
(G) Cumulative ambulations for 90 min following exposure to a novel environment (NOV), cocaine (COC, 20 mg/kg i.p.), amphetamine (AMP, 2.5 mg/kg i.p.), morphine (MOR, 25 mg/kg s.c.), or D1/D5 receptor agonist SKF 81297 (SKF, 7.5 mg/kg i.p.).
Data are presented as mean ± SEM.
Psychomotor-stimulating effects of drugs of abuse are mediated by increasing synaptic DA levels in the NAc. Morphine enhances DA release by decreasing inhibitory inputs onto DA neurons, cocaine blocks DA reuptake, and amphetamine purges vesicular DA pools and blocks reuptake (Vanderschuren and Kalivas, 2000). Cumulative locomotor responses to morphine (25 mg/kg), cocaine (20 mg/kg), or amphetamine (2.5 mg/kg) during the 90 min after drug exposure did not differ between the two groups (Figure 4G). In addition, the kinetics of drug-induced locomotor activity was similar in both groups of mice (Figures 4C–4E).
To determine if response to DA signaling is altered in KO mice, we monitored locomotor activity in response to the D1/D5 receptor agonist SKF81297 (7.5 mg/kg i.p.). We did not observe a significant difference between KO and control mice (Figures 4F and 4G). Thus, loss of functional NMDAR in DA cells does not significantly affect locomotor responses associated with activation of DA signaling, suggesting that the increased AMPAR currents in KO mice do not affect DA release, reuptake, or signaling to postsynaptic cells.
The Initial Phase of Behavioral Sensitization to Cocaine Does Not Require NMDAR Activation
NMDAR-dependent LTP of AMPAR currents following a single cocaine injection is thought to underlie the acquisition of behavioral sensitization. To determine if behavioral sensitization to cocaine is dependent on NMDAR signaling in DA neurons, we monitored locomotor activity in response to cocaine administration for 5 consecutive days in an activity chamber that was distinct from the home cage. Repeated cocaine injections (20 mg/kg) produced a significant increase in activity that did not differ between control and KO mice (Figures 5A and 5B). We also monitored locomotor activity in response to repeated injections of 15 mg/kg cocaine but did not observe robust behavioral sensitization in either group of mice (data not shown). Because our behavioral analysis was conducted on a genetic background in which mice were heterozygous for Grin1 and Slc6a3, it is possible that the results were skewed by an altered baseline response to cocaine. To eliminate this possibility, we monitored sensitization to 20 mg/kg cocaine in wild-type mice. Locomotor activity in response to the first cocaine injection and the level of sensitization was not different between wild-type (Slc6a3+/+; Grin1+/+) and control (Slc6a3+lox; Grin1Cre/+) mice (day1 ambulations, 297 ± 68 versus 245 ± 72; and day 5 ambulations, 894 ± 213 versus 835 ± 136, respectively).
Figure 5. Behavioral Sensitization to Cocaine.

(A) Cumulative 90 min locomotor response to cocaine (20 mg/kg, i.p.) from control (n = 15) and KO (n = 12) mice across days; DW, days withdrawal (*p < 0.05 compared to control treatment day 5 and KO 21 DW).
(B) Ambulations per 5 min bin in response to cocaine on treatment days 1 and 5.
(C) Ambulations per 5 min bin in response to cocaine after 21 days of withdrawal (21 DW).
(D) Locomotor activity per 5 min bin on treatment days 1 and 5 during 90 min habituation to activity chambers.
(E) Average anticipatory locomotor activity during the first 15 min of the habituation phase (*p < 0.05 compared to treatment day 1).
Data are presented as means ± SEM.
Locomotor sensitization continues to increase after cocaine administration stops (Heidbreder et al., 1996); thus, we also monitored locomotor response to cocaine (20 mg/kg) in control and KO mice after 3 and 21 days of withdrawal. Locomotor responses after 3 days of cocaine withdrawal were not significantly different in the two groups; however, after 21 days of withdrawal there was a significant enhancement of behavioral sensitization in control mice that was not observed in KO mice (Figures 5A and 5C; repeated-measures ANOVA, F(7,175) = 3.93, p < 0.05).
Conditioned Behavioral Responses to Cocaine Depend on Functional NMDAR in DA Neurons
After drug sensitization, locomotor activity increases when a rodent is placed in the conditioning chamber; this is thought to reflect cue- (or environment-) induced incentive motivation (Berridge and Robinson, 1998). To determine if functional NMDAR signaling in DA neurons is necessary for this context-dependent anticipatory locomotor activity, we monitored the initial locomotor responses of mice during the daily habituation phase that preceded cocaine administration in the sensitization experiment. The highest level of locomotor activity occurred during the first 15 min of re-exposure to the conditioning chamber (Figure 5D). Analysis of cumulative locomotor responses during the first 15 min revealed significantly enhanced anticipatory locomotor responses on the fifth day of training and after 3 or 21 days of cocaine withdrawal by control but not by KO mice (Figure 5E; repeated-measures ANOVA: F(6,150) = 3.88, p = 0.001).
To determine if CPP for cocaine is also attenuated in KO mice, we used a balanced, unbiased CPP paradigm. Baseline preference for the conditioning chambers was determined during a 20 min pretest; we did not observe a significant chamber preference in either group (data not shown). The next day, control and KO mice were given either cocaine (10 mg/kg or 20 mg/kg) or saline in contextually distinct compartments of a three-compartment CPP box. Following one, two, or three exposures to cocaine, control mice, but not KO mice, displayed a significant preference for the drug-paired side at both 10 mg/kg (Figure 6A; repeated-measures ANOVA, test day F(3,60) = 3.02, p = 0.036; post hoc analysis control, all test days versus baseline p < 0.05; KO, p > 0.4, no significant genotype × day interaction was detected) and 20 mg/kg (Figures 6C and 6E; repeated-measures ANOVA, day F(3,60) = 2.83, p = 0.045; post hoc analysis control, all test days versus baseline p < 0.05; KO, p > 0.6, no significant genotype × day interaction was detected). Locomotor responses to cocaine during pairing sessions did not differ between the two groups (Figures 6B and 6D).
Figure 6. Conditioned Place Preference for Cocaine.
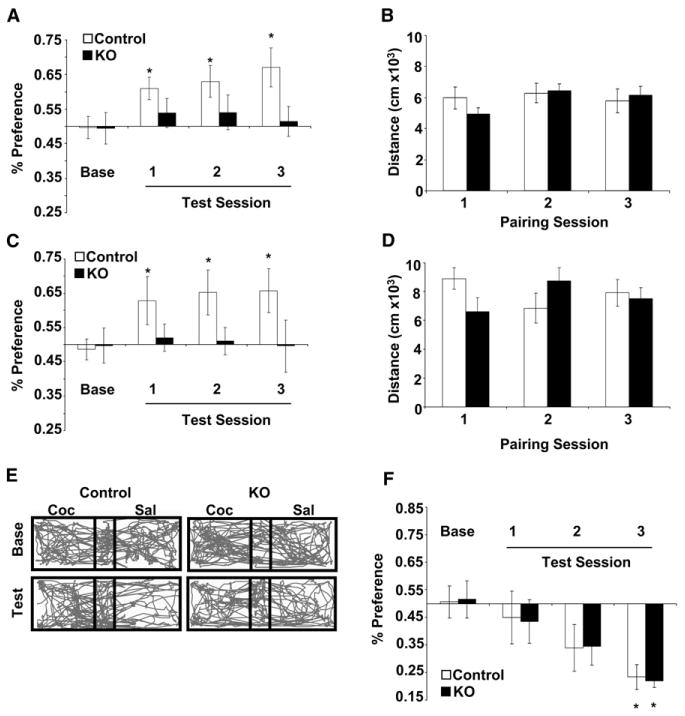
(A) Percent preference for cocaine-paired compartment during baseline (Base) and following 1, 2, or 3 pairings with cocaine at 10 mg/kg; n = 12 control mice and n = 10 KO mice (*p < 0.05 compared to baseline).
(B) Distance traveled during conditioning to cocaine (10 mg/kg).
(C) Percent preference for cocaine-paired compartment during baseline (Base) and following 1, 2, or 3 pairings with cocaine at 20 mg/kg; n = 10 control mice and n = 12 KO mice (*p < 0.05 compared to baseline).
(D) Distance traveled during conditioning to cocaine (20 mg/kg).
(E) Representative trace of locomotor activity during baseline (Base) and test day 3 (test) in a control and KO mouse.
(F) Percent preference for naloxone-paired compartment during baseline (Base) and following 1, 2, or 3 pairings with naloxone at 100 mg/kg (control n = 8 and KO n = 8) (*p < 0.05 compared to baseline).
Data are presented as mean ± SEM.
To examine whether KO mice can learn a context-dependent association, we monitored conditioned place aversion (CPA) to a noxious stimulus. Similar to cocaine CPP, mice were given three pairing sessions of saline in one compartment and naloxone (100 mg/kg s.c.) in the other compartment. On intermittent days, mice were tested for CPA. Both KO and control mice demonstrated a statistically significant CPA to naloxone during the third test session (Figure 6F).
Conditioned Behavioral Responses to Cocaine Depend on Functional NMDAR in the VTA
To confirm that the deficits observed in contextual reward association are due to a disruption of mesolimbic DA neurons and not other DAT-expressing cells, we injected AAV-Cre-GFP directly into the VTA. Although AAV-Cre-GFP viral transduction is not restricted to DA neurons in the VTA, it does provide anatomical specificity. Two weeks following AAV-Cre-GFP injections, mice were assessed for cocaine CPP. Following completion of the CPP paradigm, viral transduction of DA neurons was determined by counting the number of TH-positive cells in the VTA that were colabeled with nuclear GFP (Figure 7A). We did not observe a significant effect of viral injection on the overall number or health of TH-positive neurons (Figure 7A and data not shown). Because CPP scores for mice with <50% TH/GFP colabeling (average % TH/GFP, 19% ± 8.9%) were not different than heterozygous control-injected mice, we combined them into a single control group. Average CPP scores across days revealed a significant difference between mice with >50% GFP/TH colabeling (VTA KO, average % TH/GFP, 72% ± 5.3%) and controls (repeated-measures ANOVA, day F(3,45) = 9.2496, p < 0.001; post hoc analysis controls, all test days versus baseline p < 0.01; VTA KO, p > 0.2 all test days versus baseline; Figure 7B). Locomotor responses to cocaine did not differ between groups (Figure 7C). VTA KO mice showed elevated locomotor activity during baseline testing compared to controls (7,102 ± 680 cm versus 10,537 ± 1,149 cm; p < 0.01, Students t test); however, we did not detect differences on subsequent test days (4460 ± 424 cm versus 6164 ± 916 cm, test 1; 5242 ± 610 cm versus 5132 ± 838 cm, test 2; 5211 ± 726 cm versus 5402 ± 701). These findings indicate that functional NMDARs are necessary in the VTA for contextual reward association.
Figure 7. Conditioned Place Preference for Cocaine Following NMDAR Inactivation in the VTA.
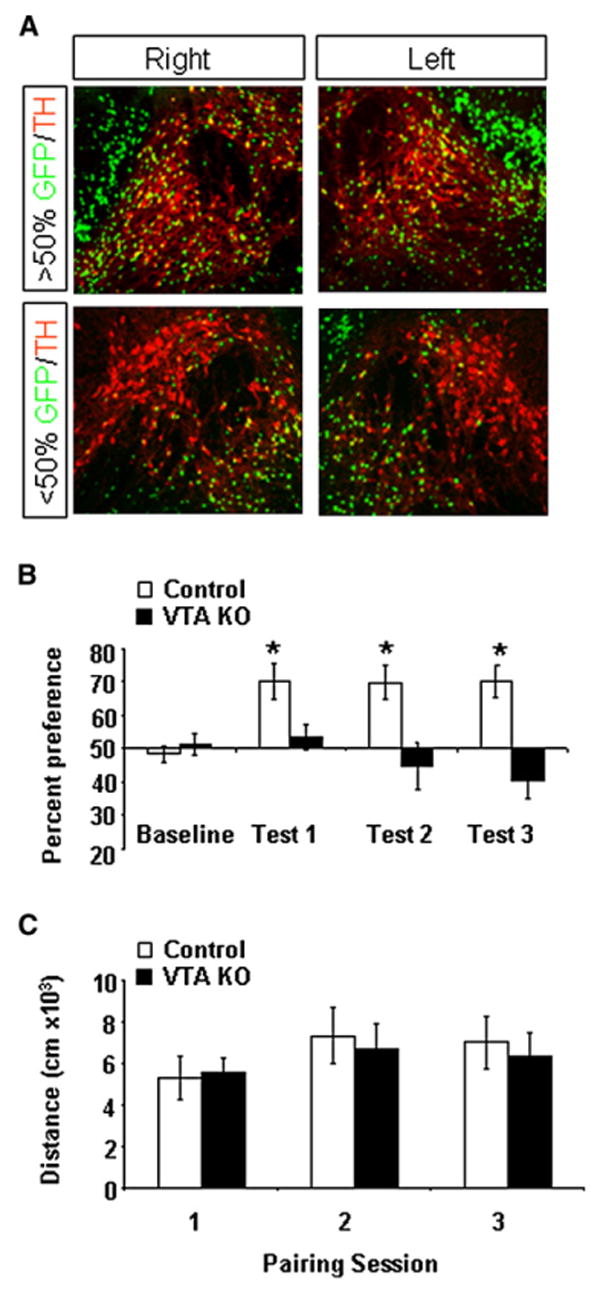
(A) Bilateral distribution of GFP-positive cells in the VTA labeled with TH immunofluoresence from a mouse with >50% colabeling (top) and <50% colabeling (bottom).
(B) Average percent preference score during baseline and across test days in VTA KO (n = 8) and control (n = 9) mice.
(C) Distance traveled during 20 min cocaine-pairing sessions.
Data are presented as mean ± SEM.
Discussion
Selective genetic inactivation of NMDARs in DA neurons increased synaptic AMPAR currents, and the magnitude of the increase was similar to that observed after a single cocaine exposure in vivo. The increase in AMPAR current in KO mice was unexpected because it had not been reported following genetic inactivation of NMDAR in other cell types (Iwasato et al., 2000; Nakazawa et al., 2002; Tsien et al., 1996); however, since the submission of this manuscript, two other studies have reported increased synaptic AMPAR currents similar to those described here (Ultanir et al., 2007; Adesnik et al., 2008). Increased synaptic AMPAR currents following pharmacological blockade of NMDAR has been suggested to be a process of synaptic scaling restricted to a critical period of synaptic development (Turrigiano and Nelson, 2004); however, the changes we observed in synaptic AMPAR currents following viral-mediated NMDAR inactivation in DA neurons from 3-week-old mice suggest that NMDAR-mediated synaptic scaling of AMPAR is likely not restricted to a critical period of DA neuron development because these changes were observed after DA dependence develops in mice (Zhou and Palmiter, 1995; Kim et al., 2002).
Although the mechanism and function of synaptic scaling are not fully resolved, increasing evidence indicates that activation of NMDARs and its downstream targets play an important role in establishing synaptic AMPAR levels beyond LTP and LTD (Isaac et al., 2007). Inhibition NMDAR-mediated miniature synaptic events during chronic action potential blockade leads to rapid (<60 min) changes in synaptic AMPARs that is dependent on local protein synthesis (Sutton et al., 2006). These changes are likely to involve downstream targets of NMDAR activation, such as the immediate-early gene Arc/Arg3.1. Consistent with this idea, genetic inactivation of Arc/Arg3.1 leads to similar increases synaptic AMPAR currents (Shepherd et al., 2006).
Our observations that NMDAR inactivation leads to synaptic scaling of AMPAR currents that resembled the LTP induced by cocaine treatment allowed us to ask whether increased AMPAR currents in DA neurons is sufficient to elicit a sensitized phenotype. If elevated AMPAR currents observed in the KO mice increased the overall excitability of DA neurons, then we predicted that basal activity and locomotor responses to acute drug exposure would be augmented. Instead, we found that acute responses to drugs of abuse and behavioral sensitization to repeated cocaine were normal. These findings indicate that increased AMPAR currents are not sufficient to evoke a sensitized response and that the induction of sensitization occurs without further increasing the amplitude or frequency of AMPAR currents.
Our results related to drug sensitization appear to be at odds with some, but not all, pharmacological and electrophysiological studies implicating NMDAR-dependent LTP of AMPAR currents in DA neurons for the induction of behavioral sensitization. These differences may not be mutually exclusive. Repeated intra-VTA infusion of an NMDAR agonist does not evoke a sensitized response to a subsequent systemic cocaine injection (Schenk and Partridge, 1997); however, repeated intra-VTA infusion of an NMDAR antagonist blocks the induction of behavioral sensitization (Kalivas and Alesdatter, 1993; Vezina and Queen, 2000). These findings suggest that NMDAR-dependent LTP of AMPAR currents following acute drug exposure are necessary for the induction of behavioral sensitization but are not sufficient to evoke a sensitized response. Consistent with this idea, a single exposure to cocaine increases the magnitude of AMPAR LTP in DA neurons that directly correlates to the acute increase of locomotor activity; however, after repeated exposure to cocaine, the correlation between synaptic strength and sensitization is lost (Borgland et al., 2004). Transient LTP of AMPAR currents may be necessary to initiate long-term changes in DA neurons that are critical for sensitization to be expressed. Numerous changes in DA neurons following cocaine exposure have been linked to sensitization involving activation of neurotrophin signal transduction cascades (Berhow et al., 1995, 1996; Messer et al., 2000), changes in gene expression regulated by cAMP response-element binding protein (Olson et al., 2005), metabotropic glutamate signaling (Chiamulera et al., 2001; Bellone and Luscher, 2006; Mameli et al., 2007), and GABA singling (Liu et al., 2005). A mechanism in which NMDAR-dependent LTP of AMPAR currents is necessary, but not sufficient for behavioral sensitization would explain why increased AMPAR currents in these KO mice does not evoke a sensitized response to acute drug exposure and why sensitization develops normally. Nonetheless, our current findings cannot rule out the possibility that NMDAR on other cells in the VTA, such as GABA neurons, are critical for sensitization. Infusion of NMDAR antagonists increases locomotor activity that is blocked by a GABA agonist (Narayanan et al., 1996). In addition, we observed increased locomotor activity in response to novelty when NMDARs were indiscriminately inactivated in the VTA that was not observed when NMDARs were selectively inactivated in DA neurons, suggesting that NMDAR signaling in non-DA neurons may be important for regulating locomotor activity.
Behavioral sensitization after cocaine withdrawal is impaired in KO mice. This is consistent with the observation that NR1 expression increases following chronic cocaine administration (Fitzgerald et al., 1996) and following prolonged cocaine withdrawal (Loftis and Janowsky, 2000). In addition, analysis of human cocaine-overdose victims revealed significantly increased NR1 mRNA levels in the VTA (Tang et al., 2003). These delayed increases in NR1 expression may account for the increase in behavioral sensitization that occurs during withdrawal. Prolonged drug abstinence increases the probability of cue-induced drug craving in humans and rats (Gawin and Kleber, 1986; Grimm et al., 2001), suggesting that changes in NMDAR signaling in VTA DA neurons may be a critical molecular substrate for the enhanced drug craving that escalates during withdrawal (Grimm et al., 2001).
The conditioned-locomotor response to cocaine-testing environment (without cocaine administration) and the CPP for the chamber where cocaine was administered are thought to require learning to associate the hedonistic effect of the drug with the environment in which it was experienced. Thus, the lack of anticipatory locomotor activity and CPP for the environment in which cocaine was administered to KO mice suggests that NMDAR in DA neurons plays a critical role in contextual reward association. It is possible that a lack of NMDAR in DA neurons might impair burst firing by these cells (Overton and Clark, 1997). Bursts of DA are thought to imprint salient information in forebrain regions (Nicola et al., 2000); thus, a lack of cocaine CPP could be due to compromised NMDAR-dependent burst firing by DA neurons.
In conclusion, the present study demonstrates that inactivation of NMDARs exclusively in DA neurons impairs contextual reward association for cocaine and withdrawal-induced increases in sensitization. Together, these findings support a role for NMDAR-dependent modulation of DA neurons in cue-induced relapse to drug seeking.
Experimental Procedures
Animals
Generation of Slc6a3+/cre, Grin1Δ/lox Mice
Due to potential transient expression of Slc6a-Cre in germ cells, one copy of the floxed Grin1 allele was inactivated (Grin1Δ/+) by a genetic cross with Mox2-Cre mice. Male Slc6a3+/Cre, Grin1Δ/+ mice were bred with female Grin1lox/lox mice. DNA of offspring was used to monitor ectopic recombination of the Grinlox allele in non-DA cells by PCR. The Grinlox allele was genotyped by PCR primers (forward, 5′-AGATACAAGACCCTGACT; reverse, 5′-AGATGGTTGTGTTGTGAG) flanking the 5′ loxP site. The Grin1Δ allele was genotyped using the same Grinlox forward primer and a reverse primer (5′-CACTTGAGTAGCGCCAAGTGC) in the Pgk promoter of the Pgk-Neomycin cassette.
Behavior
All animal protocols were approved by the University of Washington, and University of California, San Francisco, Institutional Animal Care and Use Committees.
Locomotor activity was measured as consecutive beam breaks in activity chambers (San Diego Instruments, San Diego, CA). Prior to drug administration, animals were habituated to saline injections for 2 consecutive days in locomotor chamber; on the third day, animals were habituated for 90 min prior to drug injection.
CPP was measured as percent of time spent in cocaine-paired compartments, activity was monitored with a video acquisition system (Canopus MediaCruise, Tokyo, Japan), and data were analyzed using Ethovision software (Noldus Information Technology, Leesburg, VA). Mice were paired using an unbiased conditioning paradigm, such that an equal number of mice received cocaine in the preferred and nonpreferred chamber (Heusner and Palmiter, 2005).
Viral Injections
AAV-Cre-GFP virus contains an open reading frame encoding Cre-EGFP fusion protein with a myc tag and nuclear localization signal at the N terminus. It is driven by the cytomegalovirus-chicken beta-actin (CBA) promoter and is followed by a woodchuck postregulatory element (WPRE) and bovine growth hormone (bGH) polyadenylation sequence. It was prepared by transfection of HEK cells with helper plasmids and a plasmid expressing the AAV1 coat protein. The virus was purified by iodixanol and Q column procedures and titered at 1.2 × 1012 particles/ml. For viral injection experiments, mice were anesthetized with ketamine 100 mg/kg, xylazine 20 mg/kg, and acepromazine 0.6 mg/ ml and injected with 0.5 μl AAV-Cre-GFP bilaterally into the VTA using stereotaxic coordinates (x = 0.5, y = 3.5, and z = 4.5; Paxinos and Franklin, 2001). Mice were allowed recover for 2 weeks prior to behavioral testing. Slice electrophysiology was performed 3 to 5 days after viral injections.
Immunohistochemistry
Proteins were detected with primary antibodies to tyrosine hydroxylase (monoclonal antibody 1:1000, Chemicon, Temecula, CA), NR1 (polyclonal antibody 1:100; Chemicon, Temecula, CA), and YFP (polyclonal antibody 1:1000; Molecular Probes, Eugene, OR). Primary antibodies were detected using CY2-or CY3-conjugated, goat anti-mouse and goat anti-rabbit antibodies (1:200, Jackson Immunolabs, West Grove, PA).
VTA Slice Preparations
VTA slices from young adult mice (p25 to p35) were prepared as previously described (Ungless et al., 2001). In brief, mice were anesthetized with halothane and killed. A block of tissue containing midbrain was sliced in the horizontal plane (190 μm) in ice-cold low Ca2+ artificial cerebrospinal fluid (ACSF). Slices were then transferred to a holding chamber containing ACSF, in mM, 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2, 18 NaHCO3 and 11 glucose and equilibriated at 31°C–34°C for at least 1 hr. All solutions were bubbled with 95% O2/5% CO2 and perfused over the slice at a rate of 2.5 ml/min.
Electrophysiology
Whole-cell recordings were performed using an Axopatch 1D amplifier (Axon Instruments, Foster City, CA). For EPSCs recordings, electrodes (2–6 MΩ) contained in mM: 120 cesium methansulfonic acid, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 TEA-Cl, 2.5 MgATP, 0.25 MgGTP (pH 7.2–7.4). Picrotoxin (100 μM) was added to the ACSF for recording, to block GABAA receptor-mediated IPSCs.
NMDAR or AMPAR traces were constructed by averaging 15 EPSCs elicited at +40 mV or at −70 mV. NMDAR responses were calculated by subtracting the average response in the presence of 50 μM D-APV (AMPAR only) from that recorded in its absence. AMPA mEPSCs were collected using Clampex (Axon Instruments) from cells voltage-clamped at −70 mV in the presence of lidocaine (500 μM) and of D-APV (50 μM) (120 sweeps for each cell, 1 s per sweep) and analyzed using Mini Analysis program (Synaptosoft, Decatur, GA). Detection limit was set to include only events >7 pA, <1 ms rise time, and <3 ms decay time.
For mIPSCs recordings, electrodes (2–6 MΩ) contained in mM: KCl 128, NaCl 20, MgCl2 1, EGTA 1, CaCl2 0.3, Mg-ATP 2, GTP 0.25, buffered with HEPES 10 (pH 7.2–7.4). Neurons were voltage-clamped at −70 mV. D-APV (50 μM) and CNQX (100 μM) were added to the ACSF for recording to block NMDAR- and AMPAR-mediated synaptic currents and lidocaine (500 μM), to abolish action potential-driven IPSCs. mIPSCs were measured as previously described (Bonci and Williams, 1997). For evoked excitatory postsynaptic potentials (EPSPs), recordings electrodes (2–6 MΩ) contained 0.95% KOH (v/v), 0.76% methanesulfonic acid (v/v), 0.18% hydrochloric acid (v/v), 20 mM HEPES, 0.2 mM EGTA, 2.8 mM NaCl, 2.5 mg/ml MgATP, and 0.25 mg/ml GTP (pH 7.2–7.4). Neurons were current-clamped at −70 mV, and LTP was induced by using a spike-timing protocol as previously described (Liu et al., 2005). Twenty bursts of EPSP spike pairs were delivered, with each burst consisting of five paired stimuli delivered at 10 Hz (interburst interval of 5 s). The postsynaptic spikes were evoked ∼5 ms after the onset of EPSPs by injecting depolarizing current pulses (1–2 nA, 3 ms). The magnitude of LTP was computed by averaging 30 consecutive EPSPs 5 min before and 25 min after the end of the induction protocol.
Acknowledgments
We thank Dr. Joe Tsien for the Grin1lox mice, Dr. Xiaoxi Zhuang for the Slc6a3Cre mice, Dr. Matt During for the AAV-_Cre_-GFP, Glenda Froelick for technical assistance, and members of the Palmiter and Bonci labs for helpful discussions during these studies. This work was supported by grants from the National Institute on Drug Abuse through the National Institutes of Health (to R.D.P. and L.S.Z. F32DA022829 and A.B. 1RO1DA15096-01); the state of California for medical research on alcohol and substance abuse through the University of California, San Francisco and the Wheeler Center for the Neurobiology of Addiction (A.B. and E.A.).
References
- Adesnik H, Guangnan L, During MJ, Pleasure SJ, Nicoll RA. NMDA receptors inhibit synapse unsilincing during brain development. Proc Natl Acad Sci USA. 2008;105:5597–5602. doi: 10.1073/pnas.0800946105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Luscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- Berhow MT, Russell DS, Terwilliger RZ, Beitner-Johnson D, Self DW, Lindsay RM, Nestler EJ. Influence of neurotrophic factors on morphine- and cocaine-induced biochemical changes in the mesolimbic dopamine system. Neuroscience. 1995;68:969–979. doi: 10.1016/0306-4522(95)00207-y. [DOI] [PubMed] [Google Scholar]
- Berhow MT, Hiroi N, Nestler EJ. Regulation of ERK (extracellular signal regulated kinase), part of the neurotrophin signal transduction cascade, in the rat mesolimbic dopamine system by chronic exposure to morphine or cocaine. J Neurosci. 1996;16:4707–4715. doi: 10.1523/JNEUROSCI.16-15-04707.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev. 1998;28:309–369. doi: 10.1016/s0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Boundy VA, Haile CN, Lane SB, Kalb RG, Neve RL, Nestler EJ. Sensitization to morphine induced by viral-mediated gene transfer. Science. 1997;277:812–814. doi: 10.1126/science.277.5327.812. [DOI] [PubMed] [Google Scholar]
- Chiamulera C, Epping-Jordan MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, Corsi M, Orzi F, Conquet F. Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat Neurosci. 2001;4:873–874. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- Coulter CL, Happe HK, Murrin LC. Postnatal development of the dopamine transporter: a quantitative autoradiographic study. Brain Res Dev Brain Res. 1996;92:172–181. doi: 10.1016/0165-3806(96)00004-1. [DOI] [PubMed] [Google Scholar]
- Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, Malenka RC. Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(−/−) mice. Proc Natl Acad Sci USA. 2004;101:14282–14287. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JM, Inderwies BR, Licata SC, Pierce RC. Repeated administration of AMPA or a metabotropic glutamate receptor agonist into the rat ventral tegmental area augments the subsequent behavioral hyperactivity induced by cocaine. Psychopharmacology (Berl) 2005;179:172–180. doi: 10.1007/s00213-004-2054-9. [DOI] [PubMed] [Google Scholar]
- Ettenberg A. Dopamine, neuroleptics and reinforced behavior. Neurosci Biobehav Rev. 1989;13:105–111. doi: 10.1016/s0149-7634(89)80018-1. [DOI] [PubMed] [Google Scholar]
- Fitzgerald LW, Ortiz J, Hamedani AG, Nestler EJ. Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J Neurosci. 1996;16:274–282. doi: 10.1523/JNEUROSCI.16-01-00274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, Shimada S, Nishimura T, Uhl GR, Tohyama M. Ontogeny of dopamine transporter mRNA expression in the rat brain. Brain Res Mol Brain Res. 1993;19:222–226. doi: 10.1016/0169-328x(93)90031-j. [DOI] [PubMed] [Google Scholar]
- Gawin FH, Kleber HD. Abstinence symptomatology and psychiatric diagnosis in cocaine abusers. Clinical observations Arch Gen Psychiatry. 1986;43:107–113. doi: 10.1001/archpsyc.1986.01800020013003. [DOI] [PubMed] [Google Scholar]
- Grimm JW, Hope BT, Wise RA, Shaham Y. Neuroadaptation. Incubation of cocaine craving after withdrawal. Nature. 2001;412:141–142. doi: 10.1038/35084134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris GC, Aston-Jones G. Critical role for ventral tegmental glutamate in preference for a cocaine-conditioned environment. Neuropsycho-pharmacology. 2003;28:73–76. doi: 10.1038/sj.npp.1300011. [DOI] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, Byrne R, Aston-Jones G. Glutamate-associated plasticity in the ventral tegmental area is necessary for conditioning environmental stimuli with morphine. Neuroscience. 2004;129:841–847. doi: 10.1016/j.neuroscience.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Heidbreder CA, Thompson AC, Shippenberg TS. Role of extracellular dopamine in the initiation and long-term expression of behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1996;278:490–502. [PubMed] [Google Scholar]
- Heusner CL, Palmiter RD. Expression of mutant NMDA receptors in dopamine D1 receptor-containing cells prevents cocaine sensitization and decreases cocaine preference. J Neurosci. 2005;25:6651–6657. doi: 10.1523/JNEUROSCI.1474-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Ashby M, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–731. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Alesdatter JE. Involvement of N-methyl-D-aspartate receptor stimulation in the ventral tegmental area and amygdala in behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1993;267:486–495. [PubMed] [Google Scholar]
- Kim DS, Froelick GJ, Palmiter RD. Dopamine-dependent desensitization of dopaminergic signaling in the developing mouse striatum. J Neurosci. 2002;22:9841–9849. doi: 10.1523/JNEUROSCI.22-22-09841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova TM, Ponomarenko AA, Brown RE, Haas HL. Functional diversity of ventral midbrain dopamine and GABAergic neurons. Mol Neurobiol. 2004;29:243–259. doi: 10.1385/MN:29:3:243. [DOI] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftis JM, Janowsky A. Regulation of NMDA receptor subunits and nitric oxide synthase expression during cocaine withdrawal. J Neurochem. 2000;75:2040–2050. doi: 10.1046/j.1471-4159.2000.0752040.x. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mameli M, Balland B, Lujan R, Luscher C. Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science. 2007;317:530–533. doi: 10.1126/science.1142365. [DOI] [PubMed] [Google Scholar]
- Messer CJ, Eisch AJ, Carlezon WA, Jr, Whisler K, Shen L, Wolf DH, Westphal H, Collins F, Russell DS, Nestler EJ. Role for GDNF in biochemical and behavioral adaptations to drugs of abuse. Neuron. 2000;26:247–257. doi: 10.1016/s0896-6273(00)81154-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Quirk MC, Chitwood RA, Watanabe M, Yeckel MF, Sun LD, Kato A, Carr CA, Johnston D, Wilson MA, Tonegawa S. Requirement for hippocampal CA3 NMDA receptors in associative memory recall. Science. 2002;297:211–218. doi: 10.1126/science.1071795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan S, Willins D, Dalia A, Wallace L, Uretsky N. Role of dopaminergic mechanisms in the stimulatory effects of MK-801 injected into the ventral tegmental area and the nucleus accumbens. Pharmacol Biochem Behav. 1996;54:565–573. doi: 10.1016/0091-3057(95)02220-1. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- Olson VG, Nestler EJ. Topographical organization of GABAergic neurons within the ventral tegmental area of the rat. Synapse. 2007;61:87–95. doi: 10.1002/syn.20345. [DOI] [PubMed] [Google Scholar]
- Olson VG, Zabetian CP, Bolanos CA, Edwards S, Barrot M, Eisch AJ, Hughes T, Self DW, Neve RL, Nestler EJ. Regulation of drug reward by cAMP response element-binding protein: evidence for two functionally distinct subregions of the ventral tegmental area. J Neurosci. 2005;25:5553–5562. doi: 10.1523/JNEUROSCI.0345-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overton PG, Clark D. Burst firing in midbrain dopaminergic neurons. Brain Res Brain Res Rev. 1997;25:312–334. doi: 10.1016/s0165-0173(97)00039-8. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press; 2001. [Google Scholar]
- Ranaldi R, Munn E, Neklesa T, Wise RA. Morphine and amphetamine sensitization in rats demonstrated under moderate- and high-dose NMDA receptor blockade with MK-801 (dizocilpine) Psychopharmacology (Berl) 2000;151:192–201. doi: 10.1007/s002130000480. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Schenk S, Partridge B. Effects of acute and repeated administration of N-methyl-D-aspartate (NMDA) into the ventral tegmental area: locomotor activating effects of NMDA and cocaine. Brain Res. 1997;769:225–232. doi: 10.1016/s0006-8993(97)00712-9. [DOI] [PubMed] [Google Scholar]
- Schilstrom B, Yaka R, Argilli E, Suvarna N, Schumann J, Chen BT, Carman M, Singh V, Mailliard WS, Ron D, Bonci A. Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J Neurosci. 2006;26:8549–8558. doi: 10.1523/JNEUROSCI.5179-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Tang WX, Fasulo WH, Mash DC, Hemby SE. Molecular profiling of midbrain dopamine regions in cocaine overdose victims. J Neurochem. 2003;85:911–924. doi: 10.1046/j.1471-4159.2003.01740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Turiault M, Parnaudeau S, Milet A, Parlato R, Rouzeau JD, Lazar M, Tronche F. Analysis of dopamine transporter gene expression pattern—generation of DAT-iCre transgenic mice. FEBS J. 2007;274:3568–3577. doi: 10.1111/j.1742-4658.2007.05886.x. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Schmidt WJ. Blockade of behavioral sensitization by MK-801: fact or artifact? A review of preclinical data. Psychopharmacology (Berl) 2000;151:142–151. doi: 10.1007/s002130000395. [DOI] [PubMed] [Google Scholar]
- Ultanir SK, Kim JE, Hall BJ, Deernick T, Ellisman M, Ghosh A. Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc Natl Acad Sci USA. 2007;104:19553–19558. doi: 10.1073/pnas.0704031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- Vezina P, Queen AL. Induction of locomotor sensitization by amphetamine requires the activation of NMDA receptors in the rat ventral tegmental area. Psychopharmacology (Berl) 2000;151:184–191. doi: 10.1007/s002130000463. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Sheen W, Morales M. Glutamatergic neurons are present in the rat ventral tegmental area. Eur J Neurosci. 2007;25:106–118. doi: 10.1111/j.1460-9568.2006.05263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Hu XT, White FJ, Wolf ME. Increased responsiveness of ventral tegmental area dopamine neurons to glutamate after repeated administration of cocaine or amphetamine is transient and selectively involves AMPA receptors. J Pharmacol Exp Ther. 1997;281:699–706. [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell. 1995;83:1197–1209. doi: 10.1016/0092-8674(95)90145-0. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Masson J, Gingrich JA, Rayport S, Hen R. Targeted gene expression in dopamine and serotonin neurons of the mouse brain. J Neurosci Methods. 2005;143:27–32. doi: 10.1016/j.jneumeth.2004.09.020. [DOI] [PubMed] [Google Scholar]