The Fibroblastic Coconspirator in Cancer Progression (original) (raw)
. Author manuscript; available in PMC: 2008 Nov 7.
Published in final edited form as: Cold Spring Harb Symp Quant Biol. 2005;70:383–388. doi: 10.1101/sqb.2005.70.007
Abstract
A remarkable change has occurred in the thinking about epithelial-derived cancer in recent years: From almost entirely focusing on oncogenes and tumor suppressor genes has come the realization that the tumor microenvironment is a coconspirator in the carcinogenic process. Many types of stromal cells, including fibroblasts, adipocytes, macrophages, mast cells, and cells of the vascular system, are crucial contributors to epithelial carcinogenesis. Here, we focus on the fibroblast’s role in cancer progression and the molecules involved in the communications between the fibroblasts and the cancer cells, including fibroblast secreted protein 1 (FSP-1 or S100A4), transforming growth factor β (TGF-β), the chemokine CXCL-12 (stromal derived factor 1 α, SDF-1α), type I collagen, and matrix metalloproteinase 13 (MMP-13).
It is now accepted that the stromal microenvironment contributes to tumorigenesis in cancers of epithelial origin. The mutation that initiates the carcinoma occurs in the epithelium, but events that promote tumor progression involve the stroma. In fact, in some cases, the trigger for neoplastic progression may even come from signals within the stromal microenvironment (for review, see Radisky et al. 2001; Bhowmick et al. 2004a). The stromal microenvironment consists of several cell types (fibroblasts, macrophages, vascular components, and inflammatory cells of the innate and acquired immune response), as well as the extracellular matrix (ECM) that they elaborate and all the molecules that are concentrated and immobilized on it. All of these components communicate with each other and with the neoplastic cells to contribute to the aberrant tumor organ.
A classic example of a stromal signal that can trigger neoplasms is chronic inflammation. Epidemiological evidence supporting an association of inflammation with cancer comes from studies showing a relationship between inflammatory bowel disease and colon cancer, between Helicobacter pylori infection of the stomach and stomach cancer, and between hepatitis C infection and liver cancer (for review, see Coussens and Werb 2002). Experimental evidence for the link between inflammation and stromal promotion of cancer comes from the studies on two-stage carcinogenesis, in which mutagens do not produce tumors, but require the application of tumor promoters, such as phorbol esters, which can occur long after carcinogen exposure. The tumor promoters trigger an inflammatory response and generate an aberrant tumor-promoting stroma. Another process that can generate tumor-promoting stroma experimentally is irradiation. Irradiation of the mammary gland induces nonreversible changes in the stroma that contribute to neoplasia: Non-transformed mammary epithelial cells injected into irradiated mammary stromal fat pads have greatly increased tumor growth compared to those injected into the contralateral, nonirradiated mammary fat pads (Barcellos-Hoff and Ravani 2000). Similar results have been obtained when comparing nonirradiated fibroblasts with irradiated fibroblasts, where only the latter stimulates invasiveness of pancreatic cancer (Ohuchida et al. 2004).
Here we discuss the role of the fibroblastic component of the tumor stroma in stimulating neoplastic progression.
FIBROBLASTS INFLUENCE TUMOR PROGRESSION
The fibroblast is one of several cell types involved in stromal regulation of cancer. The fibroblast is capable of adapting to tissue injury, and during wound healing it changes its phenotype to become “reactive.” The reactive fibroblast is also known as a myofibroblast—a cell type that shares properties with both fibroblasts and the smooth muscle cells. In addition to wound healing, neoplasia represents another situation where reactive fibroblasts are observed. The reactive fibroblasts observed during neoplasia are often referred to as carcinoma-associated fibroblasts (CAFs). The CAFs differ from normal fibroblasts by abnormally high expression of smooth muscle actin and increased expression of proteolytic enzymes and ECM proteins, such as tenascin.
The importance of the CAFs in epithelial carcinogenesis has been established in recombination experiments: When immortalized, nontumorigenic human prostate epithelial cells were mixed with fibroblasts from human prostate carcinomas grafted to immune-deficient animals, the epithelial cells developed into large carcinomas. In contrast, mixing the epithelial cells with fibroblasts from a normal prostate gland did not result in carcinomas (Olumi et al. 1999).
FIBROBLAST-SECRETED PROTEIN 1 STIMULATES EPITHELIAL CANCER PROGRESSION
The recombination experiments illustrate the effects of CAFs on epithelial carcinogenesis, but only recently have some of the molecules responsible for these effects been identified. However, most of these molecules are not exclusively expressed by the CAFs in the carcinomas. An example is fibroblast-secreted protein (FSP1, also called S100A4 or mts1), which is expressed in both CAFs and carcinoma cells during tumor progression (Ambartsumian et al. 1996), and possibly also by macrophages (Inoue et al. 2005). FSP1 is a calcium-binding protein with both intracellular and extracellular protein-binding partners. Intracellularly, it interacts with and possibly inactivates p53. FSP1 also interacts with non-muscle myosin heavy chain, actin filaments, and non-muscle tropomyosin, thereby potentially influencing the cytoskeleton and regulating cell motility (for review, see Helfman et al. 2005). The extracellular binding partners of FSP1 are largely unknown, with the exception of Annexin II. FSP1 binds to this coreceptor for the serine proteinase plasminogen, which results in increased activation of plasminogen (Semov et al. 2005). FSP1 is proangiogenic, and this is possibly mediated either by the activation of plasminogen or through the transcriptional up-regulation of matrix metalloproteinase (MMP) 13 (Schmidt-Hansen et al. 2004). Both of these proteinases are thought to play a role in endothelial cell invasion.
Compelling evidence exists for FSP1 as a crucial CAF-expressed factor regulating metastasis: Carcinoma cells that are metastatic when injected into wild-type mice are less likely to form tumors and do not metastasize at all when injected into _Fsp1_-/- mice. Coinjection of Fsp1+/+ fibroblasts with the tumor cells restores tumor development and metastasis in the _Fsp1_-/- animals, whereas coinjection with _Fsp1_-/- fibroblasts does not (Grum-Schwensen et al. 2005). This suggests that FSP1, when secreted by the fibroblasts, alters the stromal microenvironment, making it more favorable for tumor progression. This could be through the regulation of angiogenesis and inflammation: Tumors forming after coinjection of carcinoma cells with _Fsp1_-/- cells had significantly decreased numbers of infiltrating macrophages, smooth muscle actin-expressing myofibroblasts, and CD31-positive endothelial cells compared to tumors developing after coinjection with Fsp1+/+ cells (Grum-Schwensen et al. 2005).
As mentioned, FSP1 is also up-regulated in metastatic carcinoma cells, and this can result in the fibroblastic phenotype of the carcinoma cells known as epithelial-to-mesenchymal transition (EMT). EMT has been proposed to be the mechanism responsible for the metastatic phenotype induced by FSP1 (Xue et al. 2003). If FSP1 mainly exerts its tumor-promoting function as a secreted protein, the cellular source of its secretion might not be important. It is possible that FSP1 can be an important factor that induces angiogenesis, inflammation, and EMT, depending on which cell type it acts on rather than which cell type secretes it.
CXCL12 STIMULATES EPITHELIAL CANCER PROGRESSION THROUGH EFFECTS ON CANCER CELLS AND ENDOTHELIAL CELLS
The CXC chemokine CXCL12 (also known as stromal cell-derived factor 1 α, SDF-1α) is another important factor secreted by CAFs (Orimo et al. 2005). CXCL12 acts through several mechanisms: It acts directly on the mammary carcinoma cells stimulating proliferation through the CXCL12 receptor CXCR4. CXCL12 has also been proposed to stimulate metastasis to the lung and to lymph nodes through high expression of the chemokine at these organ sites, resulting in homing of the cancer cells, which express the CXCL12 receptor, to these organs (Muller et al. 2001). In addition to direct actions on the tumor cells, CXCL12 secretion by CAFs leads to recruitment of endothelial cell precursors to the growing tumor, thereby promoting angiogenesis (Orimo et al. 2005). As mentioned above, there is a strong link between stromal changes, inflammation, and carcinoma progression also when it comes to the effects mediated by the fibroblasts. So far, the functions of CAF-secreted CXCL12 have only been studied using xenograft models employing immune-compromised mice, which makes it impossible to address direct interactions between CAFs and leukocytes mediated by CXCL12. However, CXCL12 is a well-established chemoattractant for leukocytes, and it is thus very likely that CXCL12 would have additional effects acting through leukocytes if studied in the context of a full cellular immune response.
TGF-β HAS BOTH CANCER-PROMOTING AND -INHIBITING EFFECTS, DEPENDING ON WHICH CELL TYPE IT ACTS ON
TGF-β is one of the key players involved in the communications between CAFs and carcinoma cells, but again is expressed by multiple cell types including the stromal fibroblasts, the inflammatory cells, and carcinoma cells (Bhowmick and Moses 2005). Whereas FSP1 and CXCL12 clearly are CAF-secreted promoters of carcinogenesis, TGF-β is a factor with much more complicated effects on tumorigenesis. TGF-β is immune-suppressive when acting on inflammatory cells, thereby promoting carcinogenesis through inhibition of the immune response against the neoplasm. However, when acting on the epithelium, TGF-β is growth-inhibiting and thus inhibits carcinogenesis until the carcinoma cells overcome the growth-suppressive effects of TGF-β, and TGF-β becomes a stimulator of metastasis (for review, see Bhowmick and Moses 2005). Several groups have shown that TGF-β1 can induce differentiation of resting fibroblasts into myofibroblasts in culture (for review, see Elenbaas and Weinberg 2001). Furthermore, increased secretion of TGF-β in irradiated mammary stroma has been suggested as part of the mechanism by which irradiated stroma stimulates tumorigenesis (Barcellos-Hoff and Ravani 2000). Finally, overexpression of TGF-β by fibroblasts stimulates neoplastic growth of human breast epithelium in vivo (Kuperwasser et al. 2004). Thus, TGF-β is a key player in both the generation of a reactive stroma and its action. The actions of TGF-β are clearly cell-type-specific: When the TGF-β receptor II is genetically removed from fibroblasts in mice, rendering these fibroblasts unresponsive to TGF-β signaling, the mice develop neoplasias and carcinomas without any further genetic manipulations of the epithelium (Bhowmick et al. 2004b). However, ablation of the TGF-β receptor II in epithelial cells inhibits tumor progression (Forrester et al. 2005). These results suggest that TGF-β acting on the fibroblasts normally protects the epithelium from developing into carcinomas, whereas TGF-β secreted by CAFs and acting on the epithelium promotes carcinogenesis.
TYPE I COLLAGEN AND CANCER PROGRESSION
In carcinoma, the CAFs are largely responsible for the desmoplastic response, which is a strong stromal response characterized by pronounced changes in the ECM, including increased amounts of collagens, fibronectins, proteoglycans, and glycosaminoglycans (Elenbaas and Weinberg 2001). Many carcinomas, including human breast cancer, show a remarkable up-regulation of fibrillar collagen and collagen-associated proteins. In fact, some of the changes in the composition of the ECM may occur before the carcinoma evolves: High mammographic breast density is a strong predisposing factor for the development of sporadic breast cancer and confers a risk of about 4 relative to women with fatty breasts (Vacek and Geller 2004). Mammographic density is reflective of a changed stromal microenvironment, including increased amounts of collagen (Guo et al. 2001). Since many stromal effects on normal development and tumor development of the mammary gland are shared (Wiseman and Werb 2002), we have undertaken studies to determine the role of type I collagen metabolism in the developing mammary gland. Type I collagen is one of the classic substrates of the MMPs, a family of proteolytic enzymes identified as modifiers of mammary carcinogenesis (for review, see Egeblad and Werb 2002). We found that cleavage of collagen by MMPs is an important step in normal development of the mammary gland (M. Egeblad et al., in prep.). Whether type I collagen metabolism also plays a role in mammary carcinoma remains to be determined. However, it is noteworthy that the presence of collagen-dense fibrotic foci within mammary carcinomas is correlated with an adverse prognosis (Hasebe et al. 2001) and that increased expression of collagen type I is associated with increased risk of metastasis and decreased survival in many human cancers, including breast, lung, and prostate cancers (Ramaswamy et al. 2003).
How collagen contributes to the development and progression of cancer is not known. However, it is known that cancer cells are influenced by the ECM. For example, the sensitivity of cancer cells to apoptotic stimuli is regulated by interactions between integrin receptors on the cancer cells and proteins in the ECM (Weaver et al. 2002). Furthermore, malignant transformation of the breast is associated with dramatic changes in gland tension that include increased ECM stiffness, elevated compression forces, and high tensional resistance stresses. These changes perturb tissue morphogenesis and facilitate tumor invasion (Paszek and Weaver 2004). Interestingly, overexpression of lysyl oxidase-related protein-1 (LOR-1), a protein that is involved in the cross-linking and thereby stabilization of the collagen fibers, results in the formation of very dense collagen fibers surrounding the tumors (Akiri et al. 2003). However, rather than preventing invasion through the encapsulation of the carcinoma, the LOR-1-overexpressing cells become highly invasive (Akiri et al. 2003).
In addition to any direct effects on the cancer cells, collagen may play a role in the regulation of leukocyte behavior within tumors. In mouse mammary tumors, we found high expression levels of collagen in areas of leukocytes. Indeed, the literature suggests cross-talk between the collagenous stroma and the infiltrating leukocytes in tumors: Macrophages and dendritic cells become activated and secrete chemokines in response to binding to type I collagen (Matsuyama et al. 2004). Vice versa, leukocytes produce the ECM protein, SPARC, which determines stroma and collagen deposition in carcinomas. In the absence of leukocyte-produced SPARC, tumor growth is reduced and large areas of necrosis and impaired vascularization are observed (Sangaletti et al. 2003).
REMODELING OF THE TUMOR MICROENVIRONMENT BY MMPs
The remodeling of the stromal microenvironment (e.g., the cleavage of type I collagen) is mediated in part by secreted proteinases, including the MMPs (Egeblad and Werb 2002). Among the more than 24 MMPs, MMP-13 caught our attention because of the cells that express it and because of its substrates: MMP-13 is expressed by CAF-like cells in human breast cancer (Nielsen et al. 2001), and in vitro, breast cancer cells can stimulate fibroblasts to secrete MMP-13 (Uria et al. 1997). However, MMP-13 may also be expressed by lymphocytes (Willmroth et al. 1998; Wahlgren et al. 2001). Several proteins are known to be substrates for MMP-13 in vitro, including TGF-β, CXCL12, and type I collagen (Egeblad and Werb 2002). Latent TGF-β is cleaved and activated by MMP-13 (D’Angelo et al. 2001), CXCL12 is cleaved and inactivated by MMP-13 (McQuibban et al. 2001), and type I collagen is cleaved into specific fragments by MMP-13. Thus, MMP-13 is a factor secreted by CAFs that may regulate the activity of other factors secreted by or acting on the CAFs, complicating the interpretation of the role of the individual factors in carcinogenesis. In addition to the examples with MMP-13 and its CAF-secreted substrates, TGF-β1 can up-regulate the CXCL12 receptor CXCR4 (Chen et al. 2005), TGF-β1 and type I collagen can stimulate FSP-1 protein expression (Okada et al. 1997), and FSP1 can stimulate endothelial cells to up-regulate the expression of MMP-13 (Schmidt-Hansen et al. 2004). Thus, stromal factors and stromal cells are coconspirators and may act both additively and synergistically, or may repress each other’s functions (Fig. 1).
Figure 1.
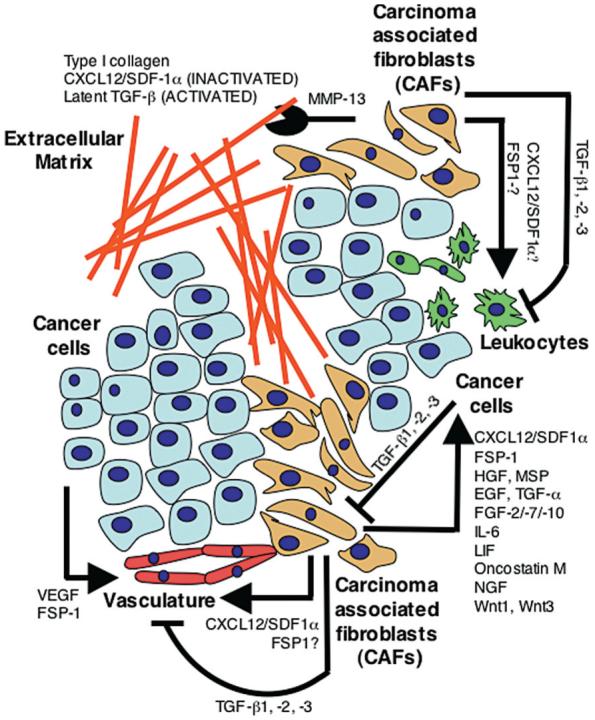
Molecular coconspirators of stromal-epithelial interactions during tumorigenesis. The cells in the tumor tissue communicate during tumor progression through the secretion of growth factors, chemokines, and cytokines. Shown here are examples of modulators of tumorigenesis that are secreted by one cell type and act on another through activation (arrows), inactivation (blocked lines), or proteolytic cleavage (chewing symbol).
WHAT IS A CAF?
Although we are now starting to understand what molecules are secreted by CAFs and what their functions are, we know surprisingly little about what the tumor-promoting CAFs themselves are and what distinguishes them from the normal fibroblasts. Like true fibroblasts, CAFs express vimentin. However, they also express α-smooth muscle actin and can contract collagen gels in vitro, thereby resembling myofibroblasts. Thus, the origin of these cells is unclear. They could be derived from fibroblasts or fibroblast precursors and change into CAFs through stimulation by the carcinoma cells. Indeed, it has been shown that when normal fibroblasts are cocultured with carcinoma cells, the fibroblasts undergo a myofibroblastic conversion (Ronnov-Jessen et al. 1995).
Interestingly, the CAFs maintain their ability to stimulate tumor progression through several cell passages but show no evidence of genetic alterations and senesce normally in culture (Orimo et al. 2005). Thus, this would suggest a nonreversible epigenetic change of the fibroblasts. However, the CAFs’ ability to stimulate carcinogenesis, even after having been cultured for several generations without further stimulation from the cancer cells, makes it tempting to speculate that the CAFs are an expanded population of an early developmental precursor initially present in the normal precancerous mammary gland, a population that first expands in response to signals from the cancer cells. Data from Schor and coworkers (Schor et al. 1994) suggest that cells with CAF-like properties are present before tumors evolve: When fibroblasts were isolated from breast tissue from patients undergoing surgery for either benign mammary gland lesions or cancer, it was found that the cancer patients’ fibroblasts were more motile than the fibroblasts from the patients with benign lesions, even though the fibroblasts were isolated in normal tissue, away from the cancer or the benign lesion. In a subset of the breast cancer patients, they even isolated fibroblasts from the skin that were abnormally motile.
These data therefore suggest that the presence of fibroblasts with CAF-like properties predisposes for the development of cancer, and they raise the question as to whether this is through germ-line mutations that determine fibroblast behavior. Indeed, the premalignant hamartomatous lesions of juvenile polyposis coli, which arise from germ-line mutations in the SMAD4 gene, are largely fibroblastic in nature, and thus a mutation in the stromal compartment initiates the development of the premalignant lesions that eventually lead to colon cancer (Howe et al. 1998; Kinzler and Vogelstein 1998; Vogelstein and Kinzler 2004). Data suggest that the stroma also can be the target of somatic mutations and that, at least in some cases, the mutations found in the fibroblastic cells and the carcinoma cells are different (Moinfar et al. 2000; Kurose et al. 2002). This strongly suggests that the CAFs are not derived from the carcinoma cells, e.g., by EMT, but rather that the mutations have arisen independently in the two cell populations. However, in other cases, CAFs may in fact be derived from carcinoma cells that have undergone EMT. Indeed, immortal fibroblast-like cells that had the same X-inactivation pattern as the epithelial carcinoma cells in the tumor have been isolated from human breast cancer (Petersen et al. 2003). The cells were not tumorigenic by themselves but behaved like CAFs, stimulating epithelial carcinoma cell activation of MMPs in vitro and tumor growth in vivo (Petersen et al. 2003).
CONCLUSIONS AND PERSPECTIVE
It appears that tumors have developed multiple different ways to ensure that CAF-like cells are present in the tumor organ: CAFs may be an expanded precursor cell population, epigenetically changed fibroblasts, mutated fibroblasts, or even epithelial cells that have undergone EMT. Why have the tumors developed so many different ways of recruiting CAFs? Do the CAFs arise as a part of a defense mechanism against the tumor—an attempt to encapsulate the tumors—or do the cancer cells recruit the CAFs (by either of the mechanisms mentioned above) because the communications between the cancer cells and the CAFs are crucial for the tumor progression? These are some of the important questions to be addressed in the future.
The molecular basis for the influence of fibroblast-like cells, the CAFs, on epithelial cancers is emerging, and genes expressed by the stromal cells in the tumors are promising prognostic predictors in human breast cancer (West et al. 2005). Whereas a normal stroma may protect the epithelium from tumorigenesis, an aberrant stroma can initiate tumorigenesis (Olumi et al. 1999; Bhowmick et al. 2004b; Kuperwasser et al. 2004; Ohuchida et al. 2004). Stunningly, restoration of normal microenvironmental signaling can reverse the malignant phenotype in vitro even though the tumor cells retain all their mutations (Weaver et al. 1997). In vivo, malignant mouse teratocarcinoma cells, propagated for 8 years as ascites tumors, when injected into blastocysts, remarkably contributed to multiple cell types in the mice that developed normally with no teratocarcinomas (Mintz and Illmensee 1975).
The ability of the CAFs to regulate epithelial carcinogenesis makes them potential drug targets. However, we are still far from developing strategies to restore aberrant signaling between the fibroblasts and the epithelium in carcinomas. Nevertheless, a few drugs that target the stromal influence on carcinogenesis (e.g., angiogenesis inhibitors) are showing significant effects on cancer patient survival, proving that targeting the stroma is a feasible direction for cancer treatment (Joyce 2005). The complex signaling network within the cancer cells has long been studied and targeted for drug development. The complex signaling between the cells in the tumor tissue is now taking off.
ACKNOWLEDGMENTS
This work was supported by grants from the National Cancer Institute (CA57621, CA58207, and CA10537), a fellowship from the Danish Medical Research Council to M.E., and a Ruth L. Kirschstein National Research Service Award from the National Institutes of Health (F32 CA103534) to L.E.L.
REFERENCES
- Akiri G, Sabo E, Dafni H, Vadasz Z, Kartvelishvily Y, Gan N, Kessler O, Cohen T, Resnick M, Neeman M, Neufeld G. Lysyl oxidase-related protein-1 promotes tumor fibrosis and tumor progression in vivo. Cancer Res. 2003;63:1657. [PubMed] [Google Scholar]
- Ambartsumian NS, Grigorian MS, Larsen IF, Karlstrom O, Sidenius N, Rygaard J, Georgiev G, Lukanidin E. Metastasis of mammary carcinomas in GRS/A hybrid mice transgenic for the mts1 gene. Oncogene. 1996;13:1621. [PubMed] [Google Scholar]
- Barcellos-Hoff MH, Ravani SA. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000;60:1254. [PubMed] [Google Scholar]
- Bhowmick NA, Moses HL. Tumor-stroma interactions. Curr. Opin. Genet. Dev. 2005;15:97. doi: 10.1016/j.gde.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004a;432:332. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004b;303:848. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- Chen S, Tuttle DL, Oshier JT, Knot HJ, Streit WJ, Goodenow MM, Harrison JK. Transforming growth factor-beta1 increases CXCR4 expression, stromal-derived factor-1alpha-stimulated signalling and human immunodeficiency virus-1 entry in human monocyte-derived macrophages. Immunology. 2005;114:565. doi: 10.1111/j.1365-2567.2004.02110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo M, Billings PC, Pacifici M, Leboy PS, Kirsch T. Authentic matrix vesicles contain active metalloproteases (MMP). A role for matrix vesicle-associated MMP-13 in activation of transforming growth factor-beta. J. Biol. Chem. 2001;276:11347. doi: 10.1074/jbc.M009725200. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2002;2:161. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell Res. 2001;264:169. doi: 10.1006/excr.2000.5133. [DOI] [PubMed] [Google Scholar]
- Forrester E, Chytil A, Bierie B, Aakre M, Gorska AE, Sharif-Afshar AR, Muller WJ, Moses HL. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005;65:2296. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- Grum-Schwensen B, Klingelhofer J, Berg CH, El-Naaman C, Grigorian M, Lukanidin E, Ambartsumian N. Suppression of tumor development and metastasis formation in mice lacking the S100A4(mts1) gene. Cancer Res. 2005;65:3772. doi: 10.1158/0008-5472.CAN-04-4510. [DOI] [PubMed] [Google Scholar]
- Guo YP, Martin LJ, Hanna W, Banerjee D, Miller N, Fishell E, Khokha R, Boyd NF. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer Epidemiol. Biomark. Prev. 2001;10:243. [PubMed] [Google Scholar]
- Hasebe T, Sasaki S, Imoto S, Ochiai A. Highly proliferative fibroblasts forming fibrotic focus govern metastasis of invasive ductal carcinoma of the breast. Mod. Pathol. 2001;14:325. doi: 10.1038/modpathol.3880310. [DOI] [PubMed] [Google Scholar]
- Helfman DM, Kim EJ, Lukanidin E, Grigorian M. The metastasis associated protein S100A4: Role in tumour progression and metastasis. Br. J. Cancer. 2005;92:1955. doi: 10.1038/sj.bjc.6602613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, Tomlinson IP, Houlston RS, Bevan S, Mitros FA, Stone EM, Aaltonen LA. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- Inoue T, Plieth D, Venkov CD, Xu C, Neilson EG. Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int. 2005;67:2488. doi: 10.1111/j.1523-1755.2005.00358.x. [DOI] [PubMed] [Google Scholar]
- Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280:1036. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, Richardson A, Weinberg RA. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc. Natl. Acad. Sci. 2004;101:4966. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat. Genet. 2002;32:355. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- Matsuyama W, Wang L, Farrar WL, Faure M, Yoshimura T. Activation of discoidin domain receptor 1 isoform b with collagen up-regulates chemokine production in human macrophages: Role of p38 mitogen-activated protein kinase and NF-kappa B. J. Immunol. 2004;172:2332. doi: 10.4049/jimmunol.172.4.2332. [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, Overall CM. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J. Biol. Chem. 2001;276:43503. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc. Natl. Acad. Sci. 1975;72:3585. doi: 10.1073/pnas.72.9.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: Implications for tumorigenesis. Cancer Res. 2000;60:2562. [PubMed] [Google Scholar]
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- Nielsen BS, Rank F, Lopez JM, Balbin M, Vizoso F, Lund LR, Dano K, Lopez-Otin C. Collagenase-3 expression in breast myofibroblasts as a molecular marker of transition of ductal carcinoma in situ lesions to invasive ductal carcinomas. Cancer Res. 2001;61:7091. [PubMed] [Google Scholar]
- Ohuchida K, Mizumoto K, Murakami M, Qian LW, Sato N, Nagai E, Matsumoto K, Nakamura T, Tanaka M. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004;64:3215. doi: 10.1158/0008-5472.can-03-2464. [DOI] [PubMed] [Google Scholar]
- Okada H, Danoff TM, Kalluri R, Neilson EG. Early role of Fsp1 in epithelial-mesenchymal transformation. Am. J. Physiol. 1997;273:F563. doi: 10.1152/ajprenal.1997.273.4.F563. [DOI] [PubMed] [Google Scholar]
- Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, Weaver VM. The tension mounts: Mechanics meets morphogenesis and malignancy. J. Mammary Gland Biol. Neoplasia. 2004;9:325. doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- Petersen OW, Nielsen HL, Gudjonsson T, Villadsen R, Rank F, Niebuhr E, Bissell MJ, Ronnov-Jessen L. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am. J. Pathol. 2003;162:391. doi: 10.1016/S0002-9440(10)63834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky D, Hagios C, Bissell MJ. Tumors are unique organs defined by abnormal signaling and context. Semin. Cancer Biol. 2001;11:87. doi: 10.1006/scbi.2000.0360. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003;33:49. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Ronnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J. Clin. Invest. 1995;95:859. doi: 10.1172/JCI117736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangaletti S, Stoppacciaro A, Guiducci C, Torrisi MR, Colombo MP. Leukocyte, rather than tumor-produced SPARC, determines stroma and collagen type IV deposition in mammary carcinoma. J. Exp. Med. 2003;198:1475. doi: 10.1084/jem.20030202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Hansen B, Ornas D, Grigorian M, Klingelhofer J, Tulchinsky E, Lukanidin E, Ambartsumian N. Extracellular S100A4(mts1) stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrix metalloproteinase activity. Oncogene. 2004;23:5487. doi: 10.1038/sj.onc.1207720. [DOI] [PubMed] [Google Scholar]
- Schor AM, Rushton G, Ferguson JE, Howell A, Redford J, Schor SL. Phenotypic heterogeneity in breast fibroblasts: Functional anomaly in fibroblasts from histologically normal tissue adjacent to carcinoma. Int. J. Cancer. 1994;59:25. doi: 10.1002/ijc.2910590107. [DOI] [PubMed] [Google Scholar]
- Semov A, Moreno MJ, Onichtchenko A, Abulrob A, Ball M, Ekiel I, Pietrzynski G, Stanimirovic D, Alakhov V. Metastasis-associated protein S100A4 induces angiogenesis through interaction with Annexin II and accelerated plasmin formation. J. Biol. Chem. 2005;280:20833. doi: 10.1074/jbc.M412653200. [DOI] [PubMed] [Google Scholar]
- Uria JA, Stahle-Backdahl M, Seiki M, Fueyo A, Lopez-Otin C. Regulation of collagenase-3 expression in human breast carcinomas is mediated by stromal-epithelial cell interactions. Cancer Res. 1997;57:4882. [PubMed] [Google Scholar]
- Vacek PM, Geller BM. A prospective study of breast cancer risk using routine mammographic breast density measurements. Cancer Epidemiol. Biomark. Prev. 2004;13:715. [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat. Med. 2004;10:789. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- Wahlgren J, Maisi P, Sorsa T, Sutinen M, Tervahartiala T, Pirila E, Teronen O, Hietanen J, Tjaderhane L, Salo T. Expression and induction of collagenases (MMP-8 and -13) in plasma cells associated with bone-destructive lesions. J. Pathol. 2001;194:217. doi: 10.1002/path.854. [DOI] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 1997;137:231. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F, Werb Z, Bissell MJ. β4 Integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2:205. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RB, Nuyten DS, Subramanian S, Nielsen TO, Corless CL, Rubin BP, Montgomery K, Zhu S, Patel R, Hernandez-Boussard T, Goldblum JR, Brown PO, van de Vijver M, van de Rijn M. Determination of stromal signatures in breast carcinoma. PLoS Biol. 2005;3:e187. doi: 10.1371/journal.pbio.0030187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmroth F, Peter HH, Conca W. A matrix metalloproteinase gene expressed in human T lymphocytes is identical with collagenase 3 from breast carcinomas. Immunobiology. 1998;198:375. doi: 10.1016/S0171-2985(98)80046-6. [DOI] [PubMed] [Google Scholar]
- Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer. Science. 2002;296:1046. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C, Plieth D, Venkov C, Xu C, Neilson EG. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res. 2003;63:3386. [PubMed] [Google Scholar]