A post-docking role for active zone protein Rim (original) (raw)
. Author manuscript; available in PMC: 2008 Nov 21.
Published in final edited form as: Nat Neurosci. 2001 Oct;4(10):997–1005. doi: 10.1038/nn732
Abstract
Rim1 was previously identified as a Rab3 effector localized to the presynaptic active zone in vertebrates. Here we demonstrate that C. elegans unc-10 mutants lacking Rim are viable, but exhibit behavioral and physiological defects that are more severe than those of Rab3 mutants. Rim is localized to synaptic sites in C. elegans, but the ultrastructure of the presynaptic densities is normal in Rim mutants. Moreover, normal levels of docked synaptic vesicles were observed in mutants, suggesting that Rim is not involved in the docking process. The level of fusion competent vesicles at release sites was reduced fivefold in Rim mutants, but calcium sensitivity of release events was unchanged. Furthermore, expression of a constitutively open form of syntaxin suppressed the physiological defects of Rim mutants, suggesting Rim normally acts to regulate conformational changes in syntaxin. These data suggest Rim acts after vesicle docking likely via regulating priming.
Docking, priming and fusion of synaptic vesicles occur in close proximity to the presynaptic specialization1–3. Hence, many molecular components regulating these events of the synaptic vesicle cycle are likely to be associated with this common ultrastructural feature of synapses. Only a limited set of proteins have been demonstrated to localize to the presynaptic density4. One such protein, Rim1 (Rab3 interacting molecule), is likely to be associated with the presynaptic specialization, based on immunostaining using light and electron microscopy5. Rim interacts with Rab3, cAMP-GEFII and Munc13-1 (refs. 5–7). These interactions suggest that Rim could regulate multiple steps of exocytosis, specifically, docking of synaptic vesicles, regulating cAMP-dependent plasticity or priming of synaptic vesicles for rapid release.
Rim1 was originally identified as a protein that interacts with the GTP-bound form of the synaptic vesicle-associated GTPase Rab3 (ref. 5). Rab3-GTP is bound to synaptic vesicles, and has been implicated in the transport and docking of synaptic vesicles at the presynaptic density8–11. Through Rab3 interactions, Rim could physically couple synaptic vesicles to release sites and regulate docking in response to Rab3 GTPase signaling. Such a potential involvement for Rim would parallel the function of EEA1, a Rab5 GTP binding protein, which acts to link Rab5 positive vesicles to target membranes12.
Rim1 and Rim2 also interact with cAMP-GEFII (ref. 6). This protein is a guanine nucleotide exchange factor for the small GTPase Rap1, whose activity is stimulated by cAMP6. Disruption of interactions between native Rim2 and cAMP-GEFII reduced cAMP-mediated secretion of growth hormone from PC12 cells. cAMP levels modulate neurotransmitter release in many preparations, including the crayfish neuromuscular junction13, the Drosophila neuromuscular junction14, Aplysia gill and siphon withdrawal15, and hippocampal mossy fiber LTP16. The interaction of Rim with cAMP-GEFII suggests that Rim may participate in cAMP-mediated plasticity and may, therefore, have functions independent of Rab3.
Vertebrate Rim also interacts with the N-terminal domain of Munc13-1 (ref. 7). Proteins of the UNC-13 family are required for priming of synaptic vesicles for release in mice, Drosophila and C. elegans, and thereby determine the size of the readily releasable pool of synaptic vesicles17–20. Furthermore, in cultured hippocampal neurons, overexpression of the N-terminal domain of Munc13-1 dramatically reduced the readily releasable pool of synaptic vesicles7. The interactions with Munc13 suggest that Rim may be involved in the post-docking step of vesicle priming at the nerve terminal.
What, then, is Rim’s function? One way of testing these models is to analyze mutants lacking the Rim protein. Here we demonstrate that in C. elegans, Rim is not required for synapse development, morphological docking of synaptic vesicles or calcium-regulated fusion. Rather, our studies indicate that a primary function of Rim is to regulate the efficiency of a post-docking step of the release pathway.
Results
unc-10 encodes a C. elegans homolog of Rim
unc-10 mutants were originally isolated based on their uncoordinated locomotion21. These mutants also exhibit resistance to the acetylcholine esterase inhibitor aldicarb22, which is a hallmark of many C. elegans synaptic transmission mutants23,24. unc-10 was positionally cloned and shown to encode the C. elegans Rim homolog by three criteria. First, lesions predicted to disrupt the Rim gene were identified in nine independently isolated unc-10 mutants (Fig. 1a). Second, Rim protein immunore-activity was absent in the unc-10(md1117) null mutant (see below; Fig. 2). Third, a plasmid containing the Rim coding region was capable of rescuing the behavioral and pharmacological defects associated with unc-10 mutants (Figs. 1a and 3b). BLAST searches of the C. elegans genome and protein databases revealed no other predicted Rim-like proteins; thus, unc-10 represents the only Rim-related gene expressed in C. elegans. The md1117 lesion lacks the entire Rim coding region (Fig. 1a; see Methods) and is used as the null mutation in this study.
Fig. 1.

Organization of the C. elegans unc-10 gene. (a) Genomic region of the unc-10 gene including selected restriction sites. Exons are shown as colored boxes that define domains using the same color code as in (b). Zinc finger, PDZ and C2 domains were identified using the SMART program43. The glutamine- and asparagine-rich domain (Q/N) was delineated by visual inspection. The position of unc-10 point mutations (arrows) and deletions (|\#x02014;|) are depicted below the genomic map. Genomic regions included in plasmids used in this work are also illustrated. Open boxes in the plasmids indicate deletions of the corresponding unc-10 genomic regions. Green fluorescent protein (GFP) sequences inserted in plasmids are denoted as labeled boxes. (b) Comparison of the domain organization of the C. elegans UNC-10 and rat Rim1 proteins. The percentage sequence identity between shared domains and the positions of alternative splicing are depicted. The alanine and proline rich domain (A/P) and alternative splice forms of Rim1 were identified previously5. (c) Sequence alignments of selected domains of Rim homologs. Alignments were created and formatted using the clustalW and SeqVu programs.
Fig. 2.

Rim protein localizes to a subdomain of the synapse. Whole mounts of wild type (a–e) and unc-10(md1117) (f, g) C. elegans fixed and prepared for immunohistochemical detection of Rim (green) and RAB-3 (red) proteins. (a) A lateral view of the head region of an adult demonstrates that Rim protein is abundant in the ventral and dorsal nerve cords and in the nerve ring. Scale bar, 20 μm. (b, c) Closer inspection of the nerve ring (b) and ventral nerve cord (c) reveals that Rim is restricted to discrete puncta. (d) A view of the dorsal nerve cord illustrates that Rim is more discretely localized compared to the synaptic vesicle-associated protein RAB-3. (e) A view of an SAB neuron innervating head muscle demonstrates that one or two Rim punctum are localized within each RAB-3 staining varicosity. Scale bar (b–e), 2 μm. (f) Rim (green) but not RAB-3 (red) immunoreactivity is absent in unc-10(md1117) animals as visualized in the SAB neurons. (g) Isolated view of the Rim channel (green) of (f) confirms that Rim immunoreactivity is absent in the unc-10(md1117) mutant. Scale bar (f), (g) 20 μm.
Fig. 3.

Behavioral defects of Rim mutants. (a) Bright field micrographs of young adult wild type and unc-10 mutant animals. (b) Behavior of wild type, unc-10 mutant and unc-10 transgenic animals after 12 h in the presence of varying concentrations of aldicarb. pRIM6 was used as the source of Rim (+), pRIM19 at the source of RimΔZn, pRIM9 as the source of RimΔC2B and pRIM10 as the source of RimΔZnQ. RimΔZnQ 1 and 2 are two independent lines expressing this construct.
Analysis of unc-10 EST cDNA clones and RT-PCR products revealed that multiple related Rim proteins are produced from the locus. The largest product exhibits a domain organization similar to that of rat Rim1 and Rim2 (Fig. 1b). C. elegans Rim and rat Rim1 share 44% sequence identity in the Rab3 interacting zinc finger domain, 58% sequence identity in the PDZ domain and 37% and 53% sequence identity in the two C2 domains (Fig. 1b and c). No significant similarity between the C. elegans and rat proteins was detected in the remaining coding regions. An alanine- and proline-rich region identified in rat Rim1 was absent from the C. elegans protein. Instead, the analogous region of the C. elegans protein contains a large glutamine- and asparagine-rich domain. Alternatively spliced forms of C. elegans Rim either include or exclude intron 13 just N-terminal to the PDZ domain and intron 24 preceding the second C2 domain (Fig. 1a; see Methods). In the latter case, the alternative form leads to termination of the coding sequences before the second C2 domain.
Rim localizes to a subdomain of the presynaptic terminal
Vertebrate Rim is concentrated at the presynaptic active zone5. To localize the homologous protein in C. elegans, antibodies were raised against the N-terminal zinc finger of Rim and used for whole-mount immunostaining (see Methods). Rim staining was restricted to discrete puncta in synapse-rich regions of the nervous system including the nerve ring (Fig. 2a and b), the ventral nerve cord (Fig. 2a and c) and the dorsal nerve cord (Fig. 2a and d). The size of the puncta was uniform and comparable to the optical resolution limit of the microscope (~300 nm). Double labeling with antibodies directed against RAB-3, a synaptic vesicle protein, revealed that Rim was much more tightly clustered than synaptic vesicle markers (Fig. 2d and e). In the head of the worm, where individual synaptic varicosities of SAB neurons can be examined, one or two Rim puncta were localized in the middle of each RAB-3 staining synaptic varicosity (Fig. 2e). In unc-10(md1117) animals that lack the entire Rim coding region, no immunoreactivity was observed with the anti-Rim antibody, although RAB-3 was normally localized (Fig. 2f and g). These data also demonstrate the specificity of the anti-Rim antibody. Localized Rim protein was observed in all larval stages and was undetectable in cell bodies (data not shown). A Rim promoter-GFP fusion (pRIM5; Fig. 1a) was expressed in most if not all neurons, suggesting that Rim expression is pan-neuronal (data not shown). Thus, the immunohistochemistry indicates that Rim is restricted to a subdomain of the presynaptic terminal in C. elegans, a finding consistent with its localization to the presynaptic specialization in vertebrate synapses5.
Rim mutants display behavioral deficits
Rim mutants were originally identified based upon their uncoordinated phenotypes21. The nine sequenced mutations (Fig. 1a; see Methods) cause a similar spectrum of phenotypes (Fig. 3, Table 1 and data not shown). Rim mutants including those carrying the complete loss-of-function md1117 lesion are viable, fertile and can be easily distinguished from the wild type based on their small size and slow, jerky, loopy locomotion (Fig. 3a and data not shown). Rim mutants also exhibit defects in feeding, defecation, and mating behavior (Table 1 and data not shown). In addition, as previously demonstrated, Rim mutants show robust resistance to the acetyl-cholinesterase inhibitor aldicarb (Fig. 3b)22. This spectrum of behavioral and pharmacological defects is consistent with a defect in synaptic function. However, Rim mutant phenotypes are less severe than those of mutants disrupting essential components of the C. elegans synaptic release machinery, such as syntaxin, synaptobrevin and UNC-13 (refs. 25–27).
Table 1.
Behavioral phenotypes of Rim mutants.
| Genotype | Locomotion (μM/s; n = 60–90) | Pumping (pumps/min; n = 9–15) | Thrashing (body bends/min; n = 8–9) | Defecation period (s; n = 55–110) | EMC (n = 50–100) |
|---|---|---|---|---|---|
| wild type | 202 ± 5.7 | 240 ± 17 | 120 ± 5 | 44.3 ± 4.0 | 98% |
| unc-10(md1117) | 72 ± 6.4 | 145 ± 18 | 32 ± 12 | 60.8 ± 10 | 85% |
| unc-10(js244) | 82 ± 5.5 | 117 ± 25 | 25 ± 14 | n.d. | n.d. |
| unc-10(e102) | 87 ± 5.7 | 154 ± 31 | 35 ± 10 | 48.3 ± 6.9 | 85% |
| rab-3(js49) | 159 ± 7.1 | 189 ± 11 | 103 ± 12 | 46.2 ± 3.8 | 95% |
| aex-3(y255) | 168 ± 5.0 | 196 ± 13 | 104± 6.8 | 45.1 ± 4.5 | 46% |
| rbf-1(js232) | 192 ± 8.0 | 226 ± 15 | 117 ± 7.7 | 47.0 ± 4.1 | 96% |
| unc-10(md1117); aex-3(y255) | 75 ± 4.2 | 138 ±15 | 31 ± 7.9 | 57 ± 12 | 42% |
| unc-10(md1117); rab-3(js49) | 67.8 ± 7.4 | 144 ± 18 | 29 ± 8.6 | 62 ±7.5 | 83% |
| unc-10(md1117); rbf-1(js232) | 65.9 ± 6.8 | 137 ± 20 | 34 ± 10 | 59 ± 6.7 | 87% |
Interactions between Rim, RAB-3 and rabphilin
Vertebrate Rim was identified as a Rab3 effector5; hence, it is of interest to compare the defects of C. elegans Rim mutants with those lacking RAB-3 and other C. elegans homologs of Rab3 effectors. Mutations in three other components implicated in the Rab3 GTPase pathway have been isolated in C. elegans. Lesions in each of these genes result in milder phenotypes than those associated with the Rim md1117 deletion. For instance, both mutants in rab-3 and the C. elegans Rab3 guanine nucleotide exchange factor homolog aex-3 have mild locomotory phenotypes, mating abnormalities and exhibit weak resistance to aldicarb (Table 1)11,28. Furthermore, mutations in the C. elegans homolog of the Rab3 effector rabphilin29 do not result in defects in mating or stimulated locomotion resistance to aldicarb30. As these observations suggest that Rim might act in other capacities besides as a Rab3 effector, we examined double mutants between Rim and other C. elegans Rab3 signaling molecules. If Rab3 action is mediated through other effectors, then Rim RAB-3 double mutants or Rim rabphilin double mutants should exhibit more severe defects than should either single mutant. However, we observed that RAB-3 Rim and rabphilin Rim double mutants exhibited locomotory, defecation and pharyngeal pumping phenotypes indistinguishable from Rim mutants (Table 1). Aex-3 Rim double mutants, also behaved identically to Rim single mutants except that the double mutant displayed the more severe defecation behavior exhibited by aex-3 mutations. Furthermore, the aldicarb resistance profiles of all three double mutants were very similar to that of the unc-10 single mutant (data not shown). Thus, our data are consistent with Rab3 signaling operating primarily through Rim.
Zinc finger and C2 domains essential for Rim function
The zinc finger domain of vertebrate Rim interacts with both Rab3 and Munc13-1. Thus, we tested if deletions of the zinc finger domain disrupted Rim function. A genomic clone of Rim (pRIM6) fully restored aldicarb sensitivity (Fig. 3b) and rescued the behavioral deficits (data not shown) of Rim mutant animals. Transgenic animals expressing Rim in either a wild type or an unc-10 mutant background were hypersensitive to aldicarb (Fig. 3b and data not shown). These data suggest that Rim is a limiting regulator of synaptic strength. By contrast, introduction of constructs deleting either the zinc finger domain (pRIM19) or the second C2 domain (pRIM9) failed to either restore aldicarb sensitivity (Fig. 3b) or rescue behavioral deficits (data not shown). The dysfunction associated with the zinc finger lesion was not a result of decreased expression or mislocalization, as the RimΔZn protein was appropriately localized (Fig. 4b and data not shown). Although a zinc finger domain deletion did not rescue the Rim md1117 deletion, a larger N-terminal deletion clone (pRIM10) could restore function to the hypomorphic e102 Rim mutant but not the null allele (Fig. 3b). This finding suggests that a PDZ/C2A/C2B protein retains certain biological activities provided by Rim. Consistent with this finding, both vertebrate Rim1 and Rim2 loci also express products lacking the Zinc finger domain, called Nim1 and Nim2, respectively31.
Fig. 4.
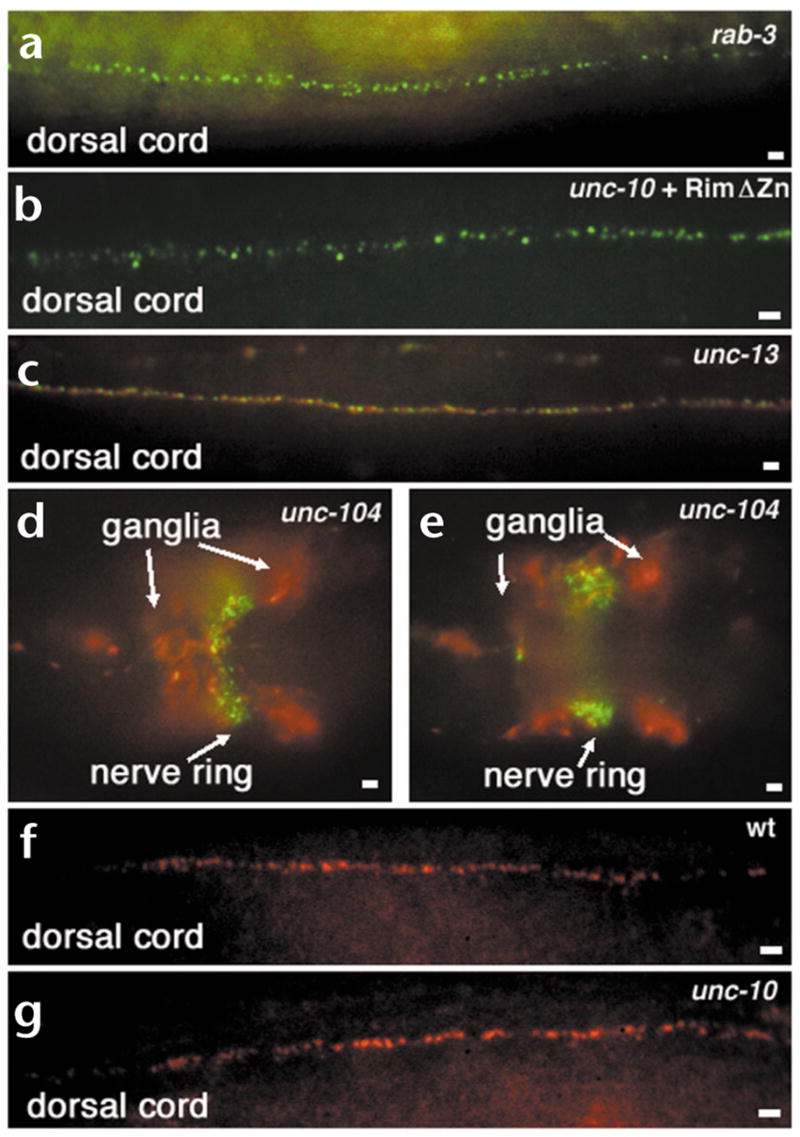
Localization of Rim, RAB-3 and UNC-13 in different mutant backgrounds. (a–e) Whole-mount mutant C. elegans animals fixed and prepared for immunohistochemical detection of Rim (green) and RAB-3 (red) proteins. (a) Rim protein is localized indistinguishably from the wild type despite the absence of RAB-3 in rab-3(js49) animals. Compare with Fig. 2a–c. (b) Rim protein localizes normally in a transgenic unc-10(md1117) Rim null animal expressing the RimΔZn construct pRIM19. (c) Both Rim and RAB-3 are normally localized in the severe unc-13(s69) mutant. (d, e) Images at two focal planes showing that Rim is localized in discrete puncta at the nerve ring despite the mislocalization and accumulation of RAB-3 in neuronal cell bodies (ganglia) in an unc-104(e1265) kinesin mutant. (f, g) Whole-mount mutant C. elegans animals fixed and prepared for immunohistochemical detection of long form of UNC-13 (red) in wild type (f) and unc-10(md1117) Rim null (g) mutant animals. UNC-13 localization in a Rim mutants is indistinguishable from wild type. Scale bars, 2 μm.
Rim localization independent of Rim-interacting proteins
Vertebrate Rim interacts with both Rab3 and Munc-13 (refs. 5, 7). These interactions may be responsible for either transporting or tethering Rim to specific structural zones at the synapse. Alternatively, Rim may be involved in localizing Rim-interacting proteins to the synapse. Hence, we addressed whether the localization of C. elegans Rim and Rim-interacting proteins were dependent on each other and on the transport of synaptic vesicle components to the synapse.
Vertebrate Rim interacts with Rab3 via its N-terminal zinc finger domain5. However, Rim localization was normal in a rab-3 null mutant (Fig. 4a) and a Rim mutant lacking the Rab3 interacting zinc finger domain also localized to distinct tightly clustered puncta (Fig. 4b). The zinc finger of Rim has also been shown to interact with the N-terminus of the Munc13-1 long form, the vertebrate homolog of unc-13 (ref. 7). In C. elegans, the long form of UNC-13 localizes to discrete puncta at synapses25. Both Rim and RAB-3 were localized normally in the severe unc-13(s69) mutant (Fig. 4c). Finally, transport of all known synaptic vesicle proteins in C. elegans depends upon the kinesin UNC-104. In unc-104(e1265) animals, although synaptic vesicle proteins and synaptic vesicles accumulate in neuronal cell bodies11,32,33, Rim was still concentrated in puncta in the nerve cord (Fig. 4d and e). Consistent with our finding, it was previously shown that presynaptic densities are still present in unc-104 mutants despite massive depletion of synaptic vesicle populations32. Thus, transport of Rim to the synapse and its synaptic localization are independent of both its known interacting partners and the synaptic vesicle transport motor.
Although Rim is not transported or localized by its interacting partners, Rim may regulate the localization of these proteins and thereby modulate synaptic transmission. However, UNC-13 is still distributed in discrete clusters at synapses (Fig. 4f and g) in unc-10 mutants. Similarly, RAB-3 is concentrated at synapse-rich sites (Fig. 2f and data not shown) in unc-10 mutants. These experiments suggest that Rim is not a scaffold protein orchestrating the organization of the presynaptic nerve terminal.
Synaptic organization in Rim mutants
To further address whether defects in synaptic organization or development could account for the behavioral defects in Rim mutants, we examined neuronal architecture and synaptic morphology by both light and electron microscopy. At the level of the light microscope, development of the nervous system appeared normal as assessed by the distribution of the neuronal plasma membrane protein syntaxin and DiO staining of chemosensory neurons. Nerve cords, commissures, the nerve ring and neuronal soma were appropriately positioned and displayed no gross morphological abnormalities (data not shown). As discussed above, the distribution of the synapse-associated proteins synaptotagmin, RAB-3 and UNC-13 also appeared indistinguishable from that of the wild type (Figs. 2d and f, 4f and g, and data not shown). Thus, the general architecture of the nervous system appeared intact in Rim mutants.
The ultrastructural organization of neuromuscular junctions was also relatively normal. Presynaptic specializations were present and of the same size as in the wild type (Fig. 5; Table 2). These data suggest that Rim is not essential for the organization of the presynaptic density, although it is likely to be a component of this sub-structure. Synaptic vesicles were clustered at active zones; however, the total number of synaptic vesicles was significantly reduced in both Rim mutant alleles analyzed (Table 2). Despite the decrease in total number of synaptic vesicles, the number of docked vesicles was similar to the wild type in both mutant alleles examined (Table 2). Thus, the synaptic defect in Rim mutants is subtle and is similar in scope to the reduction in synaptic vesicles observed at rab-3 mutant synapses11.
Fig. 5.

Structure of neuromuscu-lar junctions in Rim mutants. Electron micrographs of neuro-muscular junctions from young adult wild type and Rim mutants. The presynaptic specialization (blue arrowhead), and representative synaptic vesicles (red arrowhead) and docked synaptic vesicles (green arrowhead) are labeled. Scale bar, 200 nm.
Table 2.
Summary of characterization of unc-10 synapses using electron microscopy.
| Genotype | Average number SVs/synapse | Docked vesicles/synapse | Length of active zone/synapse (nm) | Average length of synapse (nm) | SVs/mid-synaptic profile |
|---|---|---|---|---|---|
| N2 (n = 30) | 104.8 ± 6.7a,b | 2.8 ± 0.2 | 138 ± 11.2 | 334 ± 12.1c | 25.2 ± 1.6a,d |
| unc-10(md1117) (n = 30) | 51.5 ± 4.7a | 3.1 ± 0.3 | 130 ± 8.2 | 274 ± 16.7c | 14.2 ± 1.0a |
| unc-10(ox120) (n = 19) | 62.6 ± 6.1b | 3.0 ± 0.2 | 133 ± 12.6 | 278 ± 23.0c | 16.8 ± 1.2d |
Release defects in Rim mutants
The presence of morphologically docked synaptic vesicles suggests that Rim is required at a post-docking step in the synaptic vesicle cycle. To characterize the defect in synaptic transmission, we examined the physiology of neuromuscular transmission using intracellular recording techniques34. We measured postsynaptic currents elicited by stimulation with an extracellular electrode placed in the ventral nerve cord near motor neuron cell bodies. Evoked responses from the Rim null mutant were threefold smaller than responses of the wild type (Fig. 6a and b). We also examined the frequency and amplitude of miniature postsynaptic currents. These currents, called minis, result from neurotransmitter released by the fusion of one or a few synaptic vesicles. The frequency of endogenous minis was reduced fivefold in the presence of calcium in unc-10 mutants compared to that of the wild type (Fig. 6c and d). The average amplitude of the miniature synaptic currents was unchanged (28.16 ± 2.1 pA for the wild type and 24.7 ± 1.5 pA for md1117). Because the amplitude of miniature events in Rim mutants was normal, it is unlikely that the Rim synaptic defect is a consequence of an alteration in the sensitivity of the postsynaptic receptor field. Moreover, the presence of minis indicates that Rim is not essential for synaptic vesicle fusion itself.
Fig. 6.

Defects in evoked and spontaneous release at the neuromuscular junction. (a) Representative evoked synaptic responses of voltage-clamped muscle (holding potential, −60 mV) in the wild type and in Rim mutants in response to a 2-ms stimulus to motor neurons in the ventral nerve cord in the presence of 3 mM calcium. (b) Mean amplitudes of evoked responses from the wild type (n = 22) and unc-10(md1117) Rim mutants (n = 11). Errors bars, s.e.m. (c) Representative traces of endogenous currents in muscles from the wild type and unc-10(md1117) mutants in 3 mM calcium. (d) Average frequencies of endogenous synaptic currents in the wild type (n = 27) and unc-10(md1117) (n = 14) muscles. (e) Representative evoked synaptic responses of voltage-clamped muscle (holding potential, −60 mV) in the wild type and in Rim mutants in response to consecutive 2-ms stimuli to motor neurons in the ventral nerve cord in 0.4 mM and 5.0 mM calcium. (f) Ratio of the amplitude of evoked responses recorded in muscle in 0.4 versus 5.0 mM calcium in the wild type (n = 14) and unc-10(md1117) (n = 7). (g) Representative trace of spontaneous calcium-free miniature postsynaptic currents in the wild-type and unc-10(md1117) muscles. (h) Average frequencies of spontaneous calcium-free miniature post-synaptic events in wild type (n = 6) and unc-10(md1117) (n = 7) muscles.
Rim contains C2 domains similar to those found in synaptotagmin, which have been implicated in calcium regulation of transmitter release35. Therefore, we tested if evoked release in Rim mutants exhibited altered calcium dependence. We examined evoked release at both 0.4 mM and 5 mM calcium in both the wild type and in unc-10 mutants. Though release was decreased under both conditions in Rim mutant animals, the ratio of release at the two calcium levels was indistinguishable from that of the wild type (Fig. 6e and f). Thus, the dependence of release on calcium was not changed, which indicates that Rim does not have a major role in regulating the calcium sensitivity of fusion.
Priming has been defined as those steps required for docked vesicles to transition to competence for calcium-triggered fusion36. To determine if there are defects in priming in the unc-10 mutants, we examined the pool size of fusion-competent vesicles. Primed vesicles will spontaneously fuse with the plasma membrane even in the absence of calcium. The frequency of miniature events was reduced in 0-mM calcium compared to the wild type (Fig. 6g and h). Again, miniature amplitudes were similar in the wild type and in mutants (23.6 ± 1.4 pA and 32.3 ± 4.1 pA, respectively). Moreover, there was a four- to fivefold reduction in mini frequency in the mutants compared to the wild type in both high and low calcium. Because the calcium sensitivity for release is unaltered, these data are most consistent with a decrease in the pool of fusion-competent vesicles at release sites.
Open syntaxin bypasses Rim mutant phenotypes
At a molecular level, one step in priming is believed to correspond to the formation of SNARE complexes35,37. The priming step may be regulated, for example, by the unfolding of the SNARE protein syntaxin from the closed to the open state to initiate SNARE complex formation. A constitutively open form of syntaxin can be generated by introducing two amino acid substitutions into the protein38. The L166A, E167A mutant form of syntaxin can fully rescue C. elegans syntaxin null mutants20. If Rim acts at priming, it could function to promote the conformational change of syntaxin from the closed to the open state. If that were true, then open syntaxin should bypass the requirement for Rim. To test this model, transgenes carrying wild-type and open forms of syntaxin were crossed into unc-10 mutants. Open syntaxin fully suppressed the physiological defects of unc-10 mutants (Fig. 7a). In contrast, introducing wild-type syntaxin had no effect on the size (Fig. 7) or the calcium sensitivity (data not shown) of evoked responses of unc-10 mutants. These data suggest that Rim acts at the priming step of exocytosis rather than fusion.
Fig. 7.

Open syntaxin suppresses Rim-evoked release defect. (a) Representative evoked synaptic responses of voltage-clamped muscle (holding potential, −60 mV) in the wild type, Rim mutant with and without syntaxin constructs in response to a 2-ms stimulus to motor neurons in the ventral nerve cord in the presence of 5.0 mM calcium. The complete genotype of the Rim mutant syntaxin(+) strain is unc-10(md1117) oxIs33; unc-64(js115) and Rim syntaxin(open) strain is unc-10(md1117); unc-64(js115) oxIs34. (b) Mean amplitude of responses ± s.e.m. (wild type, n = 14; unc-10(md1117), n = 7; unc-10(md1117); syx(wt), n = 4; unc-10(md1117); syx(open), n = 10).
Discussion
Rim is a conserved component of release sites
We showed that the C. elegans unc-10 gene encodes a homolog of the vertebrate RIM family of active zone proteins. C. elegans and vertebrate RIM proteins all contain highly conserved zinc finger, PDZ, C2A and C2B domains arranged in a similar topological order. Whereas the Rim family in rodents consists of at least two genes, UNC-10 is the only protein that shares the Rim domain structure in the worm. Our data suggest that C. elegans Rim, like the vertebrate protein, is associated with the presynaptic specialization. However, analysis of the unc-10 phenotype indicates that Rim is not required for the development or the structural organization of synapses.
Genetic interactions between Rab3 and Rab3 effectors
Rim was originally isolated as a Rab3 effector, binding to it through its zinc finger domain5. These initial biochemical properties of Rim suggested a potential role for Rim in regulating the docking or tethering of synaptic vesicles. However, the behavioral and pharmacological defects observed in unc-10 mutants are more severe than those of mutants in Rab3 (ref. 11), the Rab3 nucleotide exchange factor AEX-3 (ref. 28), or the Rab3 effector rabphilin30. Moreover, the ultrastructural defects of unc-10 mutants, although mild, are more severe than those of Rab3 mutants. Both Rim and Rab3 mutants have about half as many synaptic vesicles at the center of the synapse compared to the wild type. However, in Rab3 mutants, the missing vesicles are found dispersed at the edges of the varicosity11, whereas in Rim mutants, there is a clear deficit of vesicles. Though the deficit in synaptic vesicles would be consistent with a partial disruption of endocytosis, we detected no defects suggestive of an endocytosis block. In identified C. elegans endocytosis mutants, synaptobrevin accumulates on the plasma membrane39–41. In contrast, synaptobrevin-GFP was not diffusely distributed in unc-10 mutants (data not shown). Our genetic analysis and previous biochemical studies of Rim suggest that Rim plays wider roles than that of a Rab3 effector. The severity of the both the behavioral and ultrastructural phenotypes of Rim mutants likely reflects these other functions.
Rim’s function at the synapse
Electrophysiological analyses of neuromuscular transmission in Rim mutants reveal a presynaptic defect in exocytosis. Our data indicate that the fusion-competent vesicle pool is decreased fivefold without altering the calcium sensitivity of release. Furthermore, Rim neuromuscular junctions still contain normal levels of morphologically docked vesicles. Hence, Rim does not seem to modulate the size of the fusion-competent vesicle pool by disrupting vesicle docking, but rather by regulating the subsequent priming step of vesicle fusion. Priming is thought to consist of multiple biochemical steps including formation of SNARE protein complexes35,37. We demonstrated that constitutively open syntaxin can bypass the requirement for Rim. These data suggest that Rim acts before the SNARE complex is formed and eliminates the possibility that Rim participates in fusion itself. Consistent with a role in priming, the calcium sensitivity of release in Rim mutants is normal.
Together these data suggest that Rim likely regulates fusion competence by promoting conformational changes in syntaxin. The Rim-interacting protein UNC-13 also appears to act at this step. UNC-13 mutants disrupt priming completely, but this physiological defect can also be rescued by open syntaxin20. Rim null mutants are very similar in both behavior and physiological properties to hypomorphic UNC-13 mutants consistent with RIM acting to modulate UNC-13 activity through direct interaction via its zinc finger domain7.
Rim’s possible function in priming
Biochemical studies also indicate that Rim regulates the priming step of the synaptic vesicle cycle and that the interaction with UNC-13 is essential for this function7. Moreover, UNC-13 mutants disrupt priming completely, but this physiological defect can also be rescued by open syntaxin20. Thus, Rim may coordinate docking and priming by regulating UNC-13 priming activity. Binding of vesicles via Rab3-GTP to Rim may signal the presence of a docked synaptic vesicle. Rim may then signal to UNC-13 to change the conformation of syntaxin from the closed to the open state. Syntaxin could then engage synaptobrevin on the docked vesicle to form SNARE complexes and to prime the vesicle for release.
Methods
Genetic analysis
All strains were maintained as described previously42. The following mutations were used in the study: aex-3(sa5), dpy-7(e88), rab-3(js49), rbf-1(js232), unc-6(n102), unc-10(e102, js244, js318, md293, md330, md1117, md1995, md2122, ox110, ox120, unc-13(s69) and unc-104(e1265). unc-10(md330) and unc-10(e102) were positioned by three-factor mapping. From dpy-7 unc-6/unc-10(md330), 2 of 3 Unc-6 non-Dpy recombinants and 6 of 16 Dpy non-Unc-6 recombinants carried unc-10. From dpy-7 unc-6/unc-10(e102), 6 of 10 Unc-6 non-Dpy recombinants and 6 of 16 Dpy non-Unc-6 recombinants carried unc-10. The genotype of double mutants among unc-10(md1117), rab-3(js49) and rbf-1(js232) were confirmed by molecular criteria. The aex-3(sa5); unc-10(md1117) double mutant genotype was confirmed by non-complementation in test crosses.
Molecular analysis of unc-10
cDNAs yk445g5 and yk253c4 (donated by Y. Kohara) were sequenced and cDNAs yk153f9 and yk76f3 and yk219h6 were restriction mapped and partially sequenced to identify potential alternative splice sites. Alternatively spliced intron 13 was present in yk445g5 and absent from yk253c4 and yk219h6. Two alternative splice forms of intron 25 were detected. The long form of the intron leads to a termination codon before the coding sequence for the second C2 domain and was represented in yk76f3, yk153f9 and yk253c4. The short form of the intron that produces a Rim protein extending through the second C2 domain was identified by RT-PCR. Intron 24 was unspliced from cDNAs yk445g5 and yk219h6. This message also leads to termination of the translation before the second C2 domain. The 5′ sequence of the unc-10 transcript was assembled by RT-PCR, as previously described27. Trans-splicing to the SL1 leader occurred at cttcag/20 bp 5′ of the initiator methionine. Plasmid pRIM4 contains the 17.5 kb _Bam_HI/Asp718 genomic fragment from cosmid C34H12 inserted into pBluescript KS(−). pRIM4 rescues the behavioral and aldicarb resistance phenotypes of both unc-10(md1117) and unc-10(e102). pRIM5 consists of a 4.0-kb _Hind_III/_Pst_I fragment inserted into the GFP(S65C) vector pPD95.69. pRim6 deletes 4 kb of sequences from pRIM4 3′ of the polyadenylation signal between Asp718 and SwaI restriction sites. pRIM6 rescues the behavioral and aldicarb resistance phenotypes of both unc-10(md1117) and unc-10(e102). pRIM9 deletes the BlpI/SphI fragment encompassing the C2B coding region. pRIM10 replaces the 4-kb PstI fragment of pRIM6 that codes for the zinc finger domain, Q/N rich domain and a portion of the PDZ domain with GFP(S65C) and the missing PDZ sequences. The construct was built by overlap PCR using pPD95.81 as a source of GFP. pRIM10 plasmid is predicted to express a Rim product encoding codons UNC-10(1–13):GFP:UNC-10(590–1563). Assayed by GFP fluorescence, this product appears localized to puncta in synaptic regions, but less discretely than the full-length protein detected with antibodies. pRIM19 deletes a 900 bp ApaLI fragment containing exons 2–4 (codons 24 to 108). The sequence of lesions in the unc-10 gene are as follows: for e102, EMS induced G to A in last base of the fifteenth exon disrupting the splice acceptor site; for js244, EMS induced C to T in the first base of Q201 codon yielding a stop; for js318, a tripartite lesion of C to T of the second base of S295, deletion of sequences coding from A315 to R1172 and deletion of sequences coding from P1255 to 1446 bp past the polyadenylation site; for md293, deletion of ggataatttcagAAATGAATGAACAACGGAC spanning the intron 7 and exon 8 boundary; md1117, deletion of 8601 bp resulting in a tttttgtcaattca/atttaaatattaacgt junction sequence that removes the entire Rim coding region and spans from 591 bp 5′ of the translation initiation codon to 29 bp 3′ of the polyadenylation site; md1995, G to A in the third base changing W907 codon to a stop; md2122, C to T in the first base of Q707 codon yielding a stop; ox110, a 571-bp deletion starting in intron 13 and ending in exon 16 resulting in the gggtcttaccaaggg/atacttctgtcgag junction sequence; ox120, C to T in the first base of Q287 codon yielding a stop. The md330 lesion has not been precisely defined but deletes all of the coding sequences of unc-10 and extends further 5′ and 3′ than the md1117 lesion.
Behavioral and pharmacological analysis
Locomotion assays were performed as described30. Briefly, a group of 25 young adults were imaged every 5 seconds before and after application of a stimulus of 5 taps of a specified intensity. Locomotion rates were quantified by summing the distances traveled by all worms in 5-s intervals of the movie using Scion Image (Scion Corporation, Frederick, Maryland). Thrashing, pharyngeal pumping and defecation assays were done as previously described23. The criterion for aldicarb resistance was failure to respond to prodding with a platinum wire. Aldicarb resistance was scored at 12–14 h.
Immunocytochemistry
Antibodies were raised against a His6-tagged fusion to amino acids 1–142 of UNC-10. The fusion protein was purified on Ni-NTA agarose (Qiagen, Valencia, California) in 8 M urea, and renatured by dialysis against phosphate buffered saline. Rabbit 5431 antiserum was used at 1:12,500 for immunohistochemistry. Mouse polyclonal ascities directed against RAB-3 (ref. 11) and rabbit antisera against synaptotagmin33 and long form UNC-13 (ref. 25) have been previously described. Animals were fixed and prepared for immunocytochemistry using a modified Bouin’s fixative11. Mouse and rabbit antisera were detected with Alexa 488 and Alexa 568 conjugated species-specific IgG (Molecular Probes, Eugene, Oregon).
Electron microscopy
L4 worms from all genotypes were aged at 22°C between 24–48 h. They were subsequently fixed in 0.67% gluteraldehyde and 0.67% Osmium in 10 mM HEPES buffer at 4°C for 1 h. After this the worms were cut around the vulva and fixed at 4°C for 3 h in 2% Osmium in 10 mM HEPES. The remaining processing was done as previously described39. 50–60 nm sections were cut and stained using standard procedures. The dorsal and ventral cords of each section was photographed at either 15,000× or 20,000×. The developed negatives were digitized using a Kodak digital camera (Eastman Kodak, Rochester, New York). Neuromuscular junction synapses were identified by the presence of a presynaptic density (AZ) and synaptic vesicles. A note was made of the number of sections with the presynaptic density and the total number of sections in the synapse. The total length of the presynaptic specialization and synapse was calculated by multiplying the number of sections with 60. The number of synaptic vesicles was counted in every section spanning the presynaptic specialization until the number dropped below six to nine vesicles or the cell could not be followed. Only 25–40 nm regular sized vesicles were counted as synaptic vesicles. Vesicles were considered as docked if they touched the plasma membrane within a distance of approximately 80 nm on either side of the presynaptic specialization and if they touched the plasma membrane on single adjacent sections in the region of the presynaptic specialization. Number of synaptic vesicles at the mid-synaptic profile was calculated as an average of all synaptic vesicles in well-developed AZ sections for a given synapse. Five, two and three animals were analyzed for N2, unc-10(md1117) and unc-10(ox120), respectively. Mean, standard error of the mean (s.e.m.) and calculation of _p_-value for the _t_-test with unequal variance was done using Microsoft Excel.
Electrophysiology
Electrophysiological methods were essentially as previously described19. Briefly, adult worms were immobilized in saline with a cyanoacrylic glue (Histoacryl Blue, B. Braun, Melsungen, Germany). A lateral incision was made to expose the neuromuscular junctions along the ventral nerve cord. The basement membrane overlying the muscles was enzymatically removed using 0.23 mg/ml protease (type XXIV, Sigma, St. Louis, Missouri) and 0.62 mg/ml collagenase (type-A, Boehringer-Mannheim, Indianapolis, Indiana) in preparation for recording. Electrophysiological recordings were made in the whole-cell voltage clamp configuration (holding potential, −60 mV) at room temperature (21°C) using an EPC-9 patch-clamp amplifier (HEKA, Lambrecht, Germany) and digitized at 2.9 kHz via an ITC-16 interface (Instrutech, Great Neck, New York). A stimulating electrode positioned close to the ventral nerve cord was used to evoke release using a 2-ms depolarizing current controlled by a Grass S48 stimulator (Warwick, Rhode Island). Data were acquired by Pulse software (HEKA) run on a Power Mac 6500/225. The bath solution contained 140 mM NaCl, 5 mM KCl, 3 mM CaCl2, 5 mM MgCl2, 11 mM glucose and 5 mM HEPES, pH 7.2, ~330 mOsm. For the zero calcium bath solution, which was applied by gravity flow, CaCl2 was omitted and 0.5 mM EGTA was added. For the ratioed Ca2+ evoked responses, two consecutive evoked response was obtained during bath perfusion with 0.4 mM Ca2+ followed by 5 mM Ca2+ saline. The pipette solution contained 120 mM KCl, 20 mM KOH, 4 mM MgCl2, 5 mM (N-tris[Hydroxymethyl]methyl-2-aminoethane-sulfonic acid), 0.25 mM CaCl2, 4 mM NaATP, 36 mM sucrose, 5 mM EGTA, pH 7.2, ~315 mOsm. Subsequent analysis and graphing were performed using Pulsefit (HEKA), Mini Analysis (Synaptosoft, Decatur, Georgia) and Igor Pro (Wavemetrics, Lake Oswego, Oregon). All statistically derived values are given as mean ± s.e.m. (Instat, Statistical Services Center, Univ. of Reading, Reading, UK).
Acknowledgments
We thank G. Philips for cutting serial sections, L. Wei for technical assistance, R. Kohn, J. Deurr and J. Rand for providing UNC-13 antisera and unc-10 alleles, and N. Brose and T. Südhof for communicating unpublished data. This work was funded by a grant from the U.S. PHS.
Footnotes
GenBank accession number. The GenBank accession number for UNC-10 is AF257062.
References
- 1.Burns M, Augustine G. Synaptic structure and function: dynamic organization yields architectural precision. Cell. 1995;83:187–194. doi: 10.1016/0092-8674(95)90160-4. [DOI] [PubMed] [Google Scholar]
- 2.Heuser JE, Reese TS. In: Handbook of Physiology I: The Nervous System. Kandel ER, editor. American Physiological Society: Baltimore; 1973. pp. 261–294. [Google Scholar]
- 3.Landis DM, Hall AK, Weinstein LA, Reese TS. The organization of cytoplasm at the presynaptic active zone of a central nervous system synapse. Neuron. 1988;1:201–209. doi: 10.1016/0896-6273(88)90140-7. [DOI] [PubMed] [Google Scholar]
- 4.Garner CC, Kindler S, Gundelfinger ED. Molecular determinants of presynaptic active zones. Curr Opin Neurobiol. 2000;10:321–327. doi: 10.1016/s0959-4388(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Okamoto M, Schmitz F, Hofmann K, Südhof TC. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 1997;388:593–598. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- 6.Ozaki N, et al. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805–811. doi: 10.1038/35041046. [DOI] [PubMed] [Google Scholar]
- 7.Betz A, et al. Functional interaction of the active zone proteins munc13-1 and rim1 in synaptic vesicle priming. Neuron. 2001;30:183–196. doi: 10.1016/s0896-6273(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 8.Fischer von Mollard G, et al. rab3 is a small GTP-binding protein exclusively localized to synaptic vesicles. Proc Natl Acad Sci USA. 1990;87:1988–1992. doi: 10.1073/pnas.87.5.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geppert M, et al. The role of Rab3A in neurotransmitter release. Nature. 1994;369:493–497. doi: 10.1038/369493a0. [DOI] [PubMed] [Google Scholar]
- 10.Geppert M, Südhof TC. RAB3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annu Rev Neurosci. 1998;21:75–95. doi: 10.1146/annurev.neuro.21.1.75. [DOI] [PubMed] [Google Scholar]
- 11.Nonet ML, et al. C. elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci. 1997;17:8021–8073. doi: 10.1523/JNEUROSCI.17-21-08061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christoforidis S, McBride HM, Burgoyne RD, Zerial M. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 1999;397:621–625. doi: 10.1038/17618. [DOI] [PubMed] [Google Scholar]
- 13.Dixon D, Atwood HL. Adenylate cyclase system is essential for long-term facilitation at the crayfish neuromuscular junction. J Neurosci. 1989;9:4246–4252. doi: 10.1523/JNEUROSCI.09-12-04246.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong Y, Wu CF. Altered synaptic plasticity in Drosophila memory mutants with a defective cyclic AMP cascade. Science. 1991;251:198–201. doi: 10.1126/science.1670967. [DOI] [PubMed] [Google Scholar]
- 15.Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 17.Aravamudan B, Fergestad T, Davis WS, Rodesch CK, Broadie K. Drosophila UNC-13 is essential for synaptic transmission. Nat Neurosci. 1999;2:965–971. doi: 10.1038/14764. [DOI] [PubMed] [Google Scholar]
- 18.Augustin I, Rosenmund C, Südhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- 19.Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci. 1999;2:959–964. doi: 10.1038/14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richmond JE, Weimer RM, Jorgensen EM. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature. 2001;412:338–341. doi: 10.1038/35085583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen M, Alfonso A, Johnson CD, Rand JB. Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics. 1995;140:527–535. doi: 10.1093/genetics/140.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller KG, et al. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci USA. 1996;93:12593–12598. doi: 10.1073/pnas.93.22.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rand JB, Nonet ML. Synaptic transmission. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C elegans II. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1997. pp. 611–644. [PubMed] [Google Scholar]
- 25.Kohn RE, et al. Expression of multiple UNC-13 proteins in the caenorhabditis elegans nervous system. Mol Biol Cell. 2000;11:3441–3452. doi: 10.1091/mbc.11.10.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nonet ML, Saifee O, Zhao H, Rand JB, Wei L. Synaptic transmission deficits in C. elegans synaptobrevin mutants. J Neurosci. 1998;18:70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saifee O, Wei LP, Nonet ML. The C. elegans unc-64 gene encodes a syntaxin which interacts genetically with synaptobrevin. Mol Biol Cell. 1998;9:1235–1252. doi: 10.1091/mbc.9.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwasaki K, Staunton J, Saifee O, Nonet ML, Thomas J. aex-3 encodes a novel regulator of presynaptic activity in C. elegans. Neuron. 1997;18:613–622. doi: 10.1016/s0896-6273(00)80302-5. [DOI] [PubMed] [Google Scholar]
- 29.Shirataki H, et al. Rabphilin-3A, a putative target protein for smg p25A/rab3A p25 small GTP-binding protein related to synaptotagmin. Mol Cell Biol. 1993;13:2061–2068. doi: 10.1128/mcb.13.4.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Staunton J, Ganetzky B, Nonet ML. Rabphilin potentiates SNARE function independently of rab3. J Neurosci. doi: 10.1523/JNEUROSCI.21-23-09255.2001. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Sugita S, Sudhof TC. The RIM/NIM family of neuronal C2 domain proteins. Interactions with Rab3 and a new class of Src homology 3 domain proteins. J Biol Chem. 2000;275:20033–20044. doi: 10.1074/jbc.M909008199. [DOI] [PubMed] [Google Scholar]
- 32.Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- 33.Nonet ML, Grundahl K, Meyer BJ, Rand JB. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 1993;73:1291–1305. doi: 10.1016/0092-8674(93)90357-v. [DOI] [PubMed] [Google Scholar]
- 34.Richmond JE, Jorgensen EM. One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci. 1999;2:791–797. doi: 10.1038/12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klenchin VA, Martin TF. Priming in exocytosis: attaining fusion-competence after vesicle docking. Biochimie. 2000;82:399–407. doi: 10.1016/s0300-9084(00)00208-x. [DOI] [PubMed] [Google Scholar]
- 36.Rizo J, Südhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem. 1998;273:15879–15882. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- 37.Chen YA, Scales SJ, Scheller RH. Sequential SNARE assembly underlies priming and triggering of exocytosis. Neuron. 2001;30:161–170. doi: 10.1016/s0896-6273(01)00270-7. [DOI] [PubMed] [Google Scholar]
- 38.Dulubova I, et al. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J. 1999;18:4372–4382. doi: 10.1093/emboj/18.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris TW, Hartwieg E, Horvitz HR, Jorgensen EM. Mutations in synaptojanin disrupt synaptic vesicle recycling. J Cell Biol. 2000;150:589–600. doi: 10.1083/jcb.150.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nonet ML, et al. UNC-11, a C. elegans AP180 homolog, regulates the size and protein composition of synaptic vesicles. Mol Biol Cell. 1999;10:2343–2360. doi: 10.1091/mbc.10.7.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jorgensen EM, et al. Defective recycling of synaptic vesicles in synaptotagmin mutants of Caenorhabditis elegans. Nature. 1995;378:196–199. doi: 10.1038/378196a0. [DOI] [PubMed] [Google Scholar]
- 42.Sulston J, Hodgkin J. In: The Nematode Caenorhabditis elegans. Wood WB, editor. Cold Spring Harbor Laboratory; Cold Spring Harbor, New York: 1988. pp. 587–606. [Google Scholar]
- 43.Schultz J, Milpetz F, Bork P, Ponting CP. SMART a simple modular architecture research tool identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]