Genome-wide association study identifies novel breast cancer susceptibility loci (original) (raw)
. Author manuscript; available in PMC: 2009 Jul 24.
Published in final edited form as: Nature. 2007 Jun 28;447(7148):1087–1093. doi: 10.1038/nature05887
Abstract
Breast cancer exhibits familial aggregation, consistent with variation in genetic susceptibility to the disease. Known susceptibility genes account for less than 25% of the familial risk of breast cancer, and the residual genetic variance is likely to be due to variants conferring more moderate risks. To identify further susceptibility alleles, we conducted a two-stage genome-wide association study in 4,398 breast cancer cases and 4,316 controls, followed by a third stage in which 30 single nucleotide polymorphisms (SNPs) were tested for confirmation in 21,860 cases and 22,578 controls from 22 studies. We used 227,876 SNPs that were estimated to correlate with 77% of known common SNPs in Europeans at _r_2>0.5. SNPs in five novel independent loci exhibited strong and consistent evidence of association with breast cancer (P<10−7). Four of these contain plausible causative genes (FGFR2, TNRC9, MAP3K1 and LSP1). At the second stage, 1,792 SNPs were significant at the P<0.05 level compared with an estimated 1,343 that would be expected by chance, indicating that many additional common susceptibility alleles may be identifiable by this approach.
Breast cancer is about twice as common in the first-degree relatives of women with the disease as in the general population, consistent with variation in genetic susceptibility to the disease1. In the 1990s, two major susceptibility genes for breast cancer, BRCA1 and BRCA2, were identified2,3. Inherited mutations in these genes lead to a high risk of breast and other cancers4. However, the majority of multiple case breast cancer families do not segregate mutations in these genes. Subsequent genetic linkage studies have failed to identify further major breast cancer genes5. These observations have led to the proposal that breast cancer susceptibility is largely ‘polygenic’: that is, susceptibility is conferred by a large number of loci, each with a small effect on breast cancer risk6. This model is consistent with the observed patterns of familial aggregation of breast cancer7. However, progress in identifying the relevant loci has been slow. As linkage studies lack power to detect alleles with moderate effects on risk, large case-control association studies are required. Such studies have identified variants in the DNA repair genes _CHEK2_, _ATM_, BRIP1 and PALB2 that confer an approximately twofold risk of breast cancer, but these variants are rare in the population8-14. A recent study has shown that a common coding variant in CASP8 is associated with a moderate reduction in breast cancer risk15. After accounting for all the known breast cancer loci, more than 75% of the familial risk of the disease remains unexplained16.
Recent technological advances have provided platforms that allow hundreds of thousands of SNPs to be analysed in association studies, thus providing a basis for identifying moderate risk alleles without prior knowledge of position or function. It has been estimated that there are 7 million common SNPs in the human genome (with minor allele frequency, m.a.f., >5%)17. However, because recombination tends to occur at distinct ‘hot-spots’, neighbouring polymorphisms are often strongly correlated (in ‘linkage disequilibrium’, LD) with each other. The majority of common genetic variants can therefore be evaluated for association using a few hundred thousand SNPs as tags for all the other variants18. We aimed to identify further breast cancer susceptibility loci in a three-stage association study19. In the first stage, we used a panel of 266,722 SNPs, selected to tag known common variants across the entire genome18. These SNPs were genotyped in 408 breast cancer cases and 400 controls from the UK; data were analysed for 390 cases and 364 controls genotyped for ≥80% of the SNPs. The cases were selected to have a strong family history of breast cancer, equivalent to at least two affected female first-degree relatives, because such cases are more likely to carry susceptibility alleles20. Initally, we analysed 227,876 SNPs (85%) with genotypes on at least 80% of the subjects. We estimate that these SNPs are correlated with 58% of common SNPs in the HapMap CEPH/CEU (Utah residents with ancestry from northern and western Europe) samples at _r_2>0.8, and 77% at _r_2>0.5 (mean _r_2=0.75; see Supplementary Fig. 1) (http://www.hapmap.org/)21. As expected, coverage was strongly related to m.a.f.: 70% of SNPs with m.a.f.>10% were tagged at _r_2>0.8, compared with 23% of SNPs with m.a.f. 5–10%. The main analyses were restricted to 205,586 SNPs that had a call rate of 90% and whose genotype distributions did not differ from Hardy–Weinberg equilibrium in controls (at P<10−5).
For the second stage we selected 12,711 SNPs, approximately 5% of those typed in stage 1, on the basis of the significance of the difference in genotype frequency between cases and controls. These SNPs were then genotyped in a further 3,990 invasive breast cancer cases and 3,916 controls from the SEARCH study, using a custom-designed oligonucleotide array. In the main analyses, we considered 10,405 SNPs with call rate of >95% that did not deviate from Hardy–Weinberg equilibrium in controls.
Comparison of the observed and expected distribution of test statistics showed some evidence for an inflation of the test statistics in both stage 1 (inflation factor _λ_=1.03, 95% confidence interval (CI) 1.02–1.04) and stage 2 (_λ_=1.06, 95% CI 1.04–1.12), based on the 90% least significant SNPs (Fig. 1). Possible explanations for this inflation include population stratification, cryptic relatedness among subjects, and differential genotype calling between cases and controls. There was evidence for an excess of low call rate SNPs among the most significant SNPs (P<0.01) in stage 1, but not in stage 2, suggesting that some of this effect is a genotyping artefact (Supplementary Table 1). However, the inflation was still present among SNPs with call rate >99% in both cases and controls, possibly reflecting population sub-structure. We computed 1 degree of freedom (d.f.) association tests for each SNP, combining stages 1 and 2. After adjustment for this inflation by the genomic control method22, we observed more associations than would have been expected by chance at P<0.05 (Table 1). One SNP (dbSNP rs2981582) was significant at the P<10−7 level that has been proposed as appropriate for genome-wide studies23.
Figure 1. Quantile–quantile plots for the test statistics (Cochran-Armitage 1 d.f. _χ_2 trend tests) for stages 1 and 2.

a, Stage 1; b, stage 2. Black dots are the uncorrected test statistics. Red dots are the statistics corrected by the genomic control method (_λ_=1.03 for stage 1, _λ_=1.06 for stage 2). Under the null hypothesis of no association at any locus, the points would be expected to follow the black line.
Table 1. Number of significant associations after stage 2.
| Level of significance | Observed | Observed adjusted* | Expected | Ratio |
|---|---|---|---|---|
| 0.01–0.05 | 1,239 | 1,162 | 934.3 | 1.24 |
| 0.001–0.01 | 574 | 517 | 347.6 | 1.49 |
| 0.0001–0.001 | 112 | 88 | 53.3 | 1.65 |
| 0.00001–0.0001 | 16 | 12 | 7.0 | 1.71 |
| <0.00001 | 15 | 13 | 0.96 | 13.5 |
| All P<0.05 | 1,956 | 1,792 | 1,343.2 | 1.33 |
In the third stage, to establish whether any SNPs were definitely associated with risk, we tested 30 of the most significant SNPs in 22 additional case-control studies, comprising 21,860 cases of invasive breast cancer, 988 cases of carcinoma in situ (CIS) and 22,578 controls (Supplementary Table 2). Six SNPs showed associations in stage 3 that were significant at _P_≤10−5 with effects in the same direction as in stages 1 and 2 (Table 2, Supplementary Table 3, and Fig. 2). All these SNPs reached a combined significance level of P<10−7 (ranging from 2×10−76 to 3×10−9). Of these six SNPs, five were within genes or LD blocks containing genes. SNP rs2981582 lies in intron 2 of FGFR2 (also known as CEK3), which encodes the fibroblast growth factor receptor 2. SNPs rs12443621 and rs8051542 are both located in an LD block containing the 5′ end of TNRC9 (also known as TOX3), a gene of uncertain function containing a tri-nucleotide repeat motif, as well as the hypothetical gene, LOC643714. SNP rs889312 lies in an LD block of approximately 280 kb that contains MAP3K1 (also known as MEKK), which encodes the signalling protein mitogen-activated protein kinase kinase kinase 1, in addition to two other genes: MGC33648 and MIER3. SNP rs3817198 lies in intron 10 of LSP1 (also known as WP43), encoding lymphocyte-specific protein 1, an F-actin bundling cytoskeletal protein expressed in haematopoietic and endothelial cells. A further SNP, rs2107425, located just 110 kilobases (kb) from rs3817198, was also identified (overall _P_=0.00002). rs2107425 is within the H19 gene, an imprinted maternally expressed untranslated messenger RNA closely involved in regulation of the insulin growth factor gene, IGF2. In stage 3, however, rs2107425 was only weakly significant after adjustment for rs3817198 by logistic regression (_P_=0.06). This suggests that the association with breast cancer risk may be driven by variants in LSP1 rather than in H19. The sixth SNP reaching a combined P<10−7 was rs13281615, which lies on 8q. It is correlated with SNPs in a 110 kb LD block that contains no known genes. The basis of this association therefore remains obscure. This SNP is approximately 130 kb proximal to rs1447295, 60 kb proximal to rs6983267 and 230 kb distal to rs16901979, recently shown to be associated with prostate cancer24-26.
Table 2. Summary of results for eleven SNPs selected for stage 3 that showed evidence of an association with breast cancer.
| rs Number | Gene | Position* | m.a.f.† | Per allele OR (95% CI) | HetOR (95% CI) | HomOR (95% CI) | _P_-trend | ||
|---|---|---|---|---|---|---|---|---|---|
| Stages 1 and 2 | Stage3 | Combined | |||||||
| rs2981582 | FGFR2 | 10q 123342307 | 0.38 (0.30) | 1.26 (1.23–1.30) | 1.23 (1.18–1.28) | 1.63 (1.53–1.72) | 4×10−16 | 5×10−62 | 2×10−76 |
| rs12443621 | TNRC9/ LOC643714 | 16q 51105538 | 0.46 (0.60) | 1.11 (1.08–1.14) | 1.14 (1.09–1.20) | 1.23 (1.17–1.30) | 10−7 | 9×10−14 | 2×10−19 |
| rs8051542 | TNRC9/ LOC643714 | 16q 51091668 | 0.44 (0.20) | 1.09 (1.06–1.13) | 1.10 (1.05–1.16) | 1.19 (1.12–1.27) | 4×10−6 | 4×10−8 | 10−12 |
| rs889312 | MAP3K1 | 5q 56067641 | 0.28 (0.54) | 1.13 (1.10–1.16) | 1.13 (1.09–1.18) | 1.27 (1.19–1.36) | 4×10−6 | 3×10−15 | 7×10−20 |
| rs3817198 | LSP1 | 11p 1865582 | 0.30 (0.14) | 1.07 (1.04–1.11) | 1.06 (1.02–1.11) | 1.17 (1.08–1.25) | 8×10−6 | 10−5 | 3×10−9 |
| rs2107425 | H19 | 11p 1977651 | 0.31 (0.44) | 0.96 (0.93–0.99) | 0.94 (0.90–0.98) | 0.95 (0.89–1.01) | 7×10−6 | 0.01 | 2×10−5 |
| rs13281615 | 8q 128424800 | 0.40 (0.56) | 1.08 (1.05–1.11) | 1.06 (1.01–1.11) | 1.18 (1.10–1.25) | 2×10−7 | 6×10−7 | 5×10−12 | |
| rs981782 | 5p 45321475 | 0.47 (0.37) | 0.96 (0.93–0.99) | 0.96 (0.92–1.01) | 0.92 (0.87–0.97) | 8×10−5 | 0.003 | 9×10−6 | |
| rs30099 | 5q 52454339 | 0.08 (0.39) | 1.05 (1.01–1.10) | 1.06 (1.00–1.11) | 1.09 (0.96–1.24) | 0.003 | 0.02 | 0.001 | |
| rs4666451 | 2p 19150424 | 0.41 (0.04) | 0.97 (0.94–1.00) | 0.98 (0.93–1.02) | 0.93 (0.87–0.99) | 5×10−6 | 0.04 | 6×10−5 | |
| rs3803662‡ | TNRC9/ LOC643714 | 16q 51143842 | 0.25 (0.60) | 1.20 (1.16–1.24) | 1.23 (1.18–1.29) | 1.39 (1.26–1.45) | 3×10−12 | 10−26 | 10−36 |
Figure 2. Forest plots of the per-allele odds ratios for each of the five SNPs reaching genome-wide significance.

a, rs2981582; b, rs3803662; c, rs889312; d, rs13281615; and e, rs3817198. The _x_-axis gives the per-allele odds ratio. Each row represents one study (see Supplementary Table 2), with summary odds ratios for all European and all Asian studies, and all studies combined. The area of the square for each study is proportional to the inverse of the variance of the estimate. Horizontal lines represent 95% confidence intervals. Diamonds represent the summary odds ratios, with 95% confidence intervals, based on the stage 3 studies only.
In addition to the seven SNPs described above, there was evidence of association among the remaining 23 SNPs (global _P_=0.001 in stage 3). In particular, three SNPs showed some evidence of association in stage 3 (P<0.05, in each case in the same direction as in stages 1 and 2; Table 2). SNPs rs981782 and rs30099 both lie in the centromeric region of chromosome 5. rs4666451 lies on 2p, a region for which some evidence of linkage to breast cancer in families has been reported5. The 20 other SNPs showed no evidence of association in stage 3 (global _P_=0.11), suggesting that most of these associations from stages 1 and 2 were false positives.
FGFR2
The most significantly associated SNP, rs2981582, lies within a 25 kb LD block almost entirely within intron 2 of FGFR2. We found no evidence of association with SNPs elsewhere in the gene (Fig. 3a). In an attempt to identify a causal variant, we first identified the 19 common variants (m.a.f.>0.05) in this block from HapMap CEU data. These were tagged (_r_2>0.8) by 7 SNPs including rs2981582. The additional tag SNPs were genotyped in the SEARCH study cases and controls. Multiple logistic regression analysis of these variants found no additional evidence for association after adjusting for rs2981582. Haplotype analysis of these 7 SNPs indicated that multiple haplotypes carrying the minor (a) allele of rs2981582 were associated with an increased risk of breast cancer, implying that the association was being driven by rs2981582 itself or a variant strongly correlated with it (Supplementary Table 4).
Figure 3. The FGFR2 locus.

a, Map of the whole FGFR2 gene, viewed relative to common SNPs on HapMap. The gene is 126 kb long and in reverse 3′–5′ orientation on chromosome 10. Exon positions are illustrated with respect to the 67 SNPs with m.a.f.>5% in HapMap CEU (therefore the map is not to physical scale). Numbered SNPs are those tested in the genomewide study. SNPs in black were not significant in stage 1. Those in red were significant at P<0.0001 after stage 2. rs10510097 (orange) was significant in stage 1, but failed quality control in stage 2 owing to deviation from Hardy–Weinberg equilibrium. Squares indicate pairwise _r_2 on a greyscale (black=1, white=0). Red circle indicates rs2981582. **b**, Resequenced 32 kb region, shown relative to SNPs in CEU with m.a.f.>5%, showing pairwise LD for SNPs in HapMap CEU (left panel) and JPT/CHB (right panel). Red circle indicates rs2981582, shown in bold black. c, Sequence conservation of 32 kb region in five species, relative to human sequence (http://pipeline.lbl.gov/methods.shtml)35. Red circle indicates rs2981582. SNPs in grey are those used in the initial tagging of known common HapMap SNPs within the block. SNPs in black are correlated with rs2981582 with _r_2>0.6 in European samples. Six SNPs in red were those consistent with being the causative variant on the basis of the genetic data (not excluded at odds of 100:1 relative to the SNP with the strongest association, rs7895676).
Resequencing of this region in 45 subjects of European origin identified 29 variants that were strongly correlated with rs2981582 (_r_2>0.6) (http://cgwb.nci.nih.gov; Fig. 3b and Supplementary Tables 5–8). A subset of 14 variants tagged 27 of these in European (_r_2>0.95) and Asian (Korean) samples (_r_2>0.86). Two variants could not be genotyped reliably. This new tagging set was then genotyped in SEARCH and 3 studies from Asian populations; the Asian studies were included because the LD is weaker, providing greater power to resolve the causal variant (Fig. 3b, left panel). The strongest association was found with rs7895676. On the assumption that there is a single disease-causing allele, we calculated a likelihood for each variant. 21 SNPs (including rs2981582) had a likelihood ratio of <1/100 relative to rs7895676, indicating that none of these are likely to be the causal variant (Supplementary Table 8). Six variants were too strongly correlated for their individual effects to be separated using a genetic epidemiological approach. Functional assays will be required to determine which is causally related to breast cancer risk.
Intron 2 of FGFR2 shows a high degree of conservation in mammals, and contains several putative transcription-factor binding sites (http://genomequebec.mcgill.ca/PReMod)27, some of which lie in close proximity to the relevant SNPs. We therefore speculate that the association with breast cancer risk is mediated through regulation of FGFR2 expression. Of possible relevance is that only three of these variants (rs10736303, rs2981578 and rs35054928) are within sequences conserved across all placental mammals (Fig. 3c and Supplementary Table 8). Of these, the disease associated allele of rs10736303 generates a putative oestrogen receptor (ER) binding site. rs35054928 lies immediately adjacent to a perfect POU domain protein octamer (Oct) binding site. However, multiple splice variants have been reported in FGFR2, and differential splicing might provide an alternative mechanism for the association. FGFR2 is a receptor tyrosine kinase that is amplified and overexpressed in 5–10% of breast tumours28-30. Somatic missense mutations of FGFR2 that are likely to be implicated in cancer development have also been demonstrated in primary tumours and cell lines of multiple tumour types (http://www.sanger.ac.uk/genetics/CGP/cosmic/)30,31.
TNRC9/LOC643714 locus
As two SNPs in the TNRC9/LOC643714 locus, rs12443621 and rs8051542, both showed convincing evidence of association, we further evaluated this region by genotyping, in the SEARCH set, an additional 19 SNPs tagging 101 common variants within the entire TNRC9 and LOC643714 genes, based on the HapMap CEU data. SNPs tagging the coding region of TNRC9 showed no evidence of association. The strongest association was observed with rs3803662, a synonymous coding SNP of LOC643714 that lies 8 kb upstream of TNRC9. This SNP was therefore genotyped in the stage 3 set (Table 2). Logistic regression analysis indicated that rs3803662 exhibited a stronger association with disease than other SNPs, and the associations with other SNPs were non-significant after adjustment for rs3803662. These results suggest that the causal variant is closely correlated with rs3803662. Four SNPs in the HapMap CEU data (rs17271951, rs1362548, rs3095604 and rs4784227) that span LOC643714 and the 5′ regulatory regions of TNRC9 are strongly correlated with rs3803662, and it therefore remains unclear in which gene the causative variant lies. TNRC9 contains a putative HMG (high mobility group) box motif, suggesting that it might act as a transcription factor.
Pattern of risks
We assessed in more detail, in the stage 3 data, the pattern of the risks associated with the five independent SNPs that reached an overall P<10−7: rs2981582 (FGFR2), rs3803662 (TNRC9/LOC643714), rs889312 (MAP3K1), rs13281615 (8q) and rs3817198 (LSP1). For each of these five SNPs, the minor allele in Europeans was associated with an increased risk of breast cancer in a dose-dependent manner, with a higher risk of breast cancer in homozygous than in heterozygous carriers. Simple dominant and recessive models could be rejected for each SNP (all _P_=0.02 or less). There was a marked difference in allele frequencies between populations, with the risk-associated alleles of rs8051542, rs889312 and rs13281615 being the major allele in Asian populations. The per allele odds ratio associated with rs2981582 was significantly smaller, though still elevated, in the Asian versus European populations (_P_=0.04 for difference in odds ratio). This difference is consistent with the hypothesis that rs2981582 is not the functional variant at the FGFR2 locus, and was not seen for SNPs exhibiting stronger evidence in the fine-scale mapping. No other evidence for heterogeneity in the per-allele odds ratio among studies was observed (Fig. 2).
Three of the SNPs (rs2981582, rs3803662 and rs889312) also showed evidence of association with breast CIS (Supplementary Table 9). For rs2981582 and rs3803662, the estimated odds ratios were greater for a diagnosis of breast cancer before age 40 years, but the trends by age were not statistically significant (Supplementary Table 10). There was evidence of an association with family history of breast cancer for three SNPs: for rs2981582 (_P_=0.02), rs3803662 (_P_=0.03) and rs13281615 (_P_=0.05), the susceptibility allele was commoner in women with a first-degree relative with the disease than in those without (Supplementary Table 11). rs2981582 was also associated with bilaterality (_P_=0.02). The associations with family history and bilaterality are to be expected for susceptibility loci, and are similar to previous observations for alleles in CHEK2 and ATM (refs 10, 12, 14).
Discussion
This study has identified five novel breast cancer susceptibility loci, and demonstrated conclusively that some of the variation in breast cancer risk is due to common alleles. None of the loci we identified had been previously reported in association studies. Most previously identified breast cancer susceptibility genes are involved in DNA repair, and many association studies in breast cancer have concentrated on genes in DNA repair and sex hormone synthesis and metabolism pathways. None of the associations reported here appear to relate to genes in these pathways. It is notable that three of the five loci contain genes related to control of cell growth or to cell signalling, but only one (FGFR2) had a clear prior relevance to breast cancer. These results should, therefore, open up new avenues for basic research.
Our results emphasize the critical importance of study size in genetic association studies. It is notable that none of the confirmed associations reached genome-wide significance after stage 1 and only one reached this level after stage 2. As most common cancers have similar familial relative risks to breast cancer, it is likely that similarly large studies will be required to identify common alleles for other cancers. The fine-scale mapping of the FGFR2 locus demonstrates that, even with a clear association, identification of the causative variant can be extremely problematic. However, the use of studies from multiple populations with different patterns of LD can substantially reduce the number of variants that need to be subjected to functional analysis.
As these susceptibility alleles are very common, a high proportion of the general population are carriers of at-risk genotypes. For example, approximately 14% of the UK population and 19% of UK breast cancer cases are homozygous for the rare allele at rs2981582. On the other hand, the increased risks associated with these alleles are relatively small—on the basis of UK population rates, the estimated breast cancer risk by age 70 years for rare homozygotes at rs2981582 is 10.5%, compared to 6.7% in heterozygotes and 5.5% in common homozygotes. At this stage, it is unlikely that these SNPs will be appropriate for predictive genetic testing, either alone or in combination with each other. However, as further susceptibility alleles are identified, a combination of such alleles together with other breast cancer risk factors may become sufficiently predictive to be important clinically.
On the basis of the relative risk estimates from stage 3, and assuming that the five most significant loci interact multiplicatively on disease risk, these loci explain an estimated 3.6% of the excess familial risk of breast cancer. On the basis of our staged design and the estimated distribution of linkage disequilibrium between the typed SNPs and those in HapMap, we estimate that the power to identify the five most significant associations at P<10−7 (rs2981582, rs3803662, rs889312, rs13281615 and rs3817198) was 93%, 71%, 25%, 3% and 1% respectively. These estimates are uncertain, notably because the true coverage of HapMap SNPs is unknown. Nevertheless, these calculations indicate that the power to detect the two strongest associations was high, and suggest that there are likely to be few other common variants with a similar effect on variation in breast cancer risk to rs2981582. In contrast, the low power to detect rs13281615 and rs3817198 suggests that these variants may represent a much larger class of loci, each explaining of the order of 0.1% of the familial risk of breast cancer. An example of such a locus is provided by CASP8 D302H, which showed strong evidence of association in a previous large study15. This SNP was tested in stage 1, but the association was missed because it did not reach the threshold for testing in stage 2. The excess of associations after stage 2 is also consistent with the existence of many such loci. In addition, because the coverage for SNPs with m.a.f.<10% was low, many low frequency alleles may have been missed. The detection of further susceptibility loci will require genome-wide studies with more complete coverage and using larger numbers of cases and controls, together with the combination of results across multiple studies. The present study demonstrates that common susceptibility loci can be reliably identified, and that they may together explain an appreciable fraction of the genetic variance in breast cancer risk.
METHODS SUMMARY
Cases for stage 1 were identified through clinical genetics centres in the UK and a national study of bilateral breast cancer. Cases in stage 2 were drawn from a population-based study of breast cancer (SEARCH)32. Controls for stages 2 and 3 were drawn from EPIC-Norfolk, a population-based study of diet and cancer33.
Cases and controls for stage 3 were identified through case-control studies in Europe, North America, South-East Asia and Australia participating in the Breast Cancer Association Consortium (Supplementary Table 2)34.
Genotyping for stages 1 and 2 was conducted using high-density oligonucleotide microarrays. For the main analyses, we excluded samples called on ≤80% of SNPs in either stage. We also excluded SNPs that achieved a call rate of ≤90% in stage 1 and ≤95% in stage 2, and SNPs whose frequency deviated from Hardy–Weinberg equilibrium in controls at P<0.00001. Genotyping for stage 3, and for the fine-scale mapping of the FGFR2 locus, was conducted using either a 5′ nuclease assay (Taqman, Applied Biosystems) or MALDI-TOF mass spectrometry using the Sequenom iPLEX system. For each centre, we excluded any sample called on ≤80% of SNPs, and any SNP with a call rate of ≤95% or a deviation from Hardy–Weinberg equilibrium in controls at P<0.00001. Tests of association were 1 d.f. Cochran-Armitage tests, stratified for stage, centre and ethnic group (European or Asian). Odds ratios for each SNP were estimated using stratified logistic regression, using the stage 3 data only.
METHODS
Subjects
Cases in stage 1 were identified through clinical genetics centres in Cambridge (n = 91), Manchester (96) and Southampton (136), and a national study of bilateral breast cancer (85). Cases were women diagnosed with invasive breast cancer under the age of 60 years who had a family history score of at least 2, where the score was computed as the total number of first-degree relatives plus half the number of second-degree relatives affected with breast cancer. The score for women with bilateral breast cancer was increased by 1, so that women were eligible if they were diagnosed with bilateral breast cancer and had one affected first-degree relative. Cases known to carry a BRCA1 or BRCA2 mutation were excluded. Controls were selected from the EPIC-Norfolk study, a population-based cohort study of diet and cancer based in Norfolk, East Anglia, UK33. Controls were chosen to be women aged over 50 years and free of cancer at the time of entry. Genotyping was attempted on 408 cases, plus 32 duplicate case samples, and 400 controls. For the analysis in Table 1, 54 samples with genotype call rates <80% were excluded, so the final analyses were based on 390 cases and 364 controls. The minimum genotype call rate for the remaining samples was 89%. The overall genotype discordance rate between duplicate samples in stage 1 was 0.01%.
For stage 2, invasive breast cancer cases were drawn from SEARCH, a population-based study of cancer in East Anglia32. Controls were women selected from the EPIC-Norfolk study, as previously described33. Eighty-eight subjects who were also genotyped in stage 1, and 35 controls who subsequently developed breast cancer and were also in the case series, were excluded from the analysis, leaving 3,990 breast cancer cases and 3,916 controls, plus five duplicates. The overall rate of discordance of genotypes between duplicate samples in stage 2 was 0.008%.
Twenty-one additional studies were included in stage 3 (see Supplementary Table 2). These studies participated through the Breast Cancer Association Consortium, an ongoing collaboration among investigators conducting case-control association studies in breast cancer15,33. All studies provided information on disease status (invasive breast cancer, carcinoma in situ or control), age at diagnosis/observation, ethnic group, first-degree family history of breast cancer and bilaterality of breast cancer. One further study (Breast Cancer Study of Taiwan) was included in the fine-scale mapping of the FGFR2 locus.
Genotyping
For stage 1, genotyping was performed on 200 ng DNA that was first subjected to whole genome amplification using Multiple Displacement Amplification (MDA)36. Samples were then genotyped for a set of 266,732 SNPs using high-density oligonucleotide, photolithographic microarrays at Perlegen Sciences. For stage 2, genotyping was performed using 2.5 μg genomic DNA. These samples were genotyped for a set of 13,023 SNPs selected on the basis of the stage 1 results, using a custom designed oligonucleotide array. For both stages, each SNP was interrogated by 24 25-mer oligonucleotide probes synthesized by photolithography on a glass substrate. The 24 features comprise 4 sets of 6 features interrogating the neighbourhoods of SNP reference and alternative alleles on forward and reference strands. Each allele and strand is represented by five offsets: −2, −1, 0, 1 and 2 indicating the position of the SNP within the 25-mer, with zero being at the thirteenth base. At offset 0 a quartet was tiled, which included the perfect match to reference and alternative SNP alleles, and the two remaining nucleotides as mismatch probes. When possible, the mismatch features were selected as a purine nucleotide substitution for a purine perfect match nucleotide and a pyrimidine nucleotide substitution for a pyrimidine perfect match nucleotide. Thus, each strand and allele tiling consisted of 6 features comprising five perfect match probes and one mismatch.
Individual genotypes were determined by clustering all SNP scans in the two-dimensional space defined by reference and alternative trimmed mean intensities, corrected for background. Allele frequencies were approximated using the intensities collected from the high-density oligonucleotide arrays. An SNP's allele frequency, p, was estimated as the ratio of the relative amount of the DNA with reference allele to the total amount of DNA. The 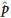 value was computed from the trimmed mean intensities of perfect match features, after subtracting a measure of background computed from trimmed means of intensities of mismatch features. The trimmed mean disregarded the highest and the lowest intensity from the five perfect match intensities before computing the arithmetic mean. For the mismatch features, the trimmed mean is the individual intensity of the specified mismatch feature.
value was computed from the trimmed mean intensities of perfect match features, after subtracting a measure of background computed from trimmed means of intensities of mismatch features. The trimmed mean disregarded the highest and the lowest intensity from the five perfect match intensities before computing the arithmetic mean. For the mismatch features, the trimmed mean is the individual intensity of the specified mismatch feature.
The genotype clustering procedure was an iterative algorithm developed as a combination of K-means and constrained multiple linear regressions. The K-means at each step re-evaluated the cluster membership representing distinct diploid genotypes. The multiple linear regressions minimized the variance in within each cluster while optimizing the regression lines' common intersect. The common intersect defined a measure of common background that was used to adjust the allele frequencies for the next step of K-means. The K-means and multiple linear regression steps were iterated until the cluster membership and background estimates converged. The best number of clusters was selected by maximizing the total likelihood over the possible cluster counts of 1, 2 and 3 (representing the combinations of the three possible diploid genotypes). The total likelihood was composed of data likelihood and model likelihood. The data likelihood was determined using a normal mixture model for the distribution of around the cluster means. The model likelihood was calculated using a prior distribution of expected cluster positions, resulting in optimal positions of 0.8 for the homozygous reference cluster, 0.5 for the heterozygous cluster and 0.2 for the homozygous alternative cluster.
A genotyping quality metric was compiled for each genotype from 15 input metrics that described the quality of the SNP and the genotype. The genotyping quality metric correlated with a probability of having a discordant call between the Perlegen platform and outside genotyping platforms (that is, non-Perlegen HapMap project genotypes). A system of 10 bootstrap aggregated regression trees was trained using an independent data set of concordance data between Perlegen genotypes and HapMap project genotypes. The trained predictor was then used to predict the genotyping quality for each of the genotypes in this data set. Genotypes with quality scores of less than 7 were discarded. Data were analysed for 227,876 SNPs in stage 1 and 12,026 (of 13,023 selected) in stage 2, for which the call rate was >80%.
The 12,711 SNPs for stage 2 were primarily selected on the basis of a 1 d.f. Cochran-Armitage trend test (11,809, all with P<0.052). We also included 826 SNPs with P<0.01 testing for the difference in frequency of either homozygote between cases and controls (that is, assuming either a dominant or recessive model) and 76 SNPs that achieved P<0.01 on a Cochran-Armitage test, weighting individuals by their family history score as above.
For the main analyses, we discarded SNPs with a call rate <90% in stage 1 and 95% in stage 2, and SNPs with a deviation from Hardy–Weinberg equilibrium significant at P<0.00001 in either stage, leaving 205,586 SNPs in stage 1 and 10,621 SNPs in stage 2.
The 30 SNPs included in the stage 3 analyses were initially selected on the basis of a combined analysis of stage 1 and stage 2. We included all SNPs achieving a combined P<0.00002 (based on either the Cochran-Armitage or 2 d.f. test, see below). Following re-evaluation of the stage 2 genotyping by 5′ nuclease assay (Taqman, Applied Biosystems) using the ABI PRISM 7900HT (Applied Biosystems), and exclusion of some samples, 16 of these SNPs were significant at P<0.00002 and 24 at P<0.0002 (Supplementary Table 3). One additional SNP, rs3803662, was added as a result of fine-scale mapping of the TNRC9/LOC643714 locus.
The 31 stage 3 SNPs were genotyped in 22 studies (including cases and controls from SEARCH not used in stage 2, together with 21 other studies). For 18 of the studies, genotyping was performed by 5′ nuclease assay (Taqman) using the ABI PRISM 7900HT or 7500 Sequence Detection Systems according to manufacturer's instructions. Primers and probes were supplied directly by Applied Biosystems (http://www.appliedbiosystems.com/) as Assays-by-Design. All assays were carried out in 384-well or 96-well format, with each plate including negative controls (with no DNA). Duplicate genotypes were provided for at least 2% of samples in each study. For three studies, SNPs were genotyped using matrix assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) for the determination of allele-specific primer extension products using Sequenom's MassARRAY system and iPLEX technology. The design of oligonucleotides was carried out according to the guidelines of Sequenom and performed using MassARRAY Assay Design software (version 1.0). Multiplex PCR amplification of amplicons containing SNPs of interest was performed using Qiagen HotStart Taq Polymerase on a Perkin Elmer GeneAmp 2400 thermal cycler (MJ Research) with 5 ng genomic DNA. Primer extension reactions were carried out according to manufacturer's instructions for iPLEX chemistry. Assay data were analysed using Sequenom TYPER software (version 3.0). One study used both the Taqman and MALDI-TOF MS approaches. The SNPs genotyped in stage 3 were also regenotyped in the stage 2 samples using Taqman; these genotype calls were used in the overall analyses (Table 2, Supplementary Table 3, and Fig. 2).
We eliminated any sample that could not be scored on 20% of the SNPs attempted. We also removed data for any centre/SNP combination for which the call rate was less than 90%. In any instances where the call rate was 90–95%, the clustering of genotype calls was re-evaluated by an independent observer to determine whether the clustering was sufficiently clear for inclusion. We also eliminated all the data for a given SNP/centre where the reproducibility in duplicate samples was <97%, or where there was marked deviation from Hardy–Weinberg equilibrium in the controls (P<0.00001).
Fine-scale mapping of FGFR2
Initial tagging of the associated region was done by identifying all SNPs with an m.a.f.>5% in the HapMap CEPH/CEU set (Utah residents with ancestry from northern and western Europe). We then selected 7 SNPs (in addition to rs2981582) that tagged these variants with a pairwise _r_2>0.8, using the program Tagger (http://www.broad.mit.edu/mpg/tagger/)37. To identify additional common variants within the 32.5 kb region of linkage around the associated SNP, we resequenced 45 lymphocyte DNA samples from a subset of European subjects also genotyped by HapMap and other publicly available data sets. Seventy overlapping PCR amplicons were designed from positions 123317613 to 123348192 of chromosome 10 (average amplicon size 650 bp, 160 bp overlap). M13-tagged PCR products were bidirectionally sequenced using Big Dye 3.0 (Applied Biosystems) and processed using automated trace analysis through the Cancer Genome Workbench (cgwb.nci.nih.gov). Eighty-six per cent of the nucleotides across the region could be scored for polymorphisms in at least 80% of subjects. This set gave a >97% probability of detecting a variant with an m.a.f.>5%. One hundred and seventeen variants were identified, including 27 present in dbSNP but without individual genotype information in European subjects, and an additional 46 not in dbSNP. Individual genotype information was then compared and merged with publicly available genotypes from Caucasian subjects (HapMap release 21 for 60 CEU parents, 22 European subjects from the Environmental Genome Project (EGP) resequencing effort (http://egp.gs.washington.edu/data/fgfr2/), and 24 European subjects from Perlegen (retrieved through http://gvs.gs.washington.edu/GVS)). There were 2 discrepancies among 389 genotype calls among subjects in common between our resequencing effort and EGP or Perlegen data, and 10 out of 926 compared to HapMap genotypes.
On the basis of these data, we identified 28 SNPs correlated with rs2981582 with _r_2>0.6. We then attempted to genotype these 28 SNPs, plus rs2981582, in a subset of 80 controls from SEARCH and 84 controls from the Seoul Breast Cancer Study. Twenty-two of the variants were genotyped using Taqman. Four further variants (rs34032268, rs2912778, rs2912781 and rs7895676), which were not amenable to Taqman, were genotyped by Pyrosequencing (Biotage; http://www.biotagebio.com/). Assays were designed using Pyrosequencing Assay Design Software 1.0. The remaining 2 SNPs (rs35393331 and rs33971856) could not be genotyped using either technology and were excluded from further analyses. We cannot therefore comment on their likelihood of being the causal variant. Using these data, we selected tagging sets of 11 SNPs for UK subjects and 14 SNPs for Korean subjects (including rs2981582), such that each of the remaining variants was correlated with a tagging SNP with _r_2>0.95 in the UK study or _r_2>0.86 in the Korean study. After genotyping the 11 tag SNPs in SEARCH, two of these SNPs (rs4752569 and rs35012336) showed strong evidence against being the causative variant and were not considered further. The remaining 12 tag SNPs from the Korean subset were then genotyped in the samples from the IARC-Thai Breast Cancer Study, the Breast Cancer Study in Taiwan and the Multi-Ethnic Cohort (MEC), by Taqman.
Statistical methods
The primary test used for each SNP was a Cochran-Armitage 1 d.f. score test for association between disease status and allele dose. In the combined analysis, we performed a stratified Cochran-Armitage test. Stage 1 was given a weight of 4 in this analysis (corresponding to a weight of 2 in the score statistic), to allow for the expected greater effect size given the inclusion of cases with a family history. In the stage 3 analyses, each study was treated as a separate stratum, except for the MEC, in which the European American and Japanese American subgroups were treated as separate strata. For all studies except the MEC, individuals from a minor ethnic group for that study were excluded. Per-allele and genotype-specific odds ratios, and confidence intervals, were estimated using logistic regression, adjusting for the same strata. The summary odds ratios in Fig. 2 are based on the data from the stage 3 studies only, to avoid the bias inherent in estimates from the stage 1 and 2 data for SNPs exhibiting an association (the so called ‘winner's curse’). The effects of genotype on family history of breast cancer (first degree yes/no) and bilaterality were examined by treating these variables as outcomes in a stratified Cochran-Armitage test.
To assess the global significance of the SNPs in stage 3, we computed the sum of the χ2 trend statistics (excluding the 6 SNPs reaching genome-wide significance, plus rs2107425 as it was in LD with rs3817198) over those SNPs (17 of 23) for which the estimated odds ratios in stage 3 were in the same direction as the combined stage 1/stage 238. Under the null hypothesis of no association, the asymptotic distribution of this statistic is χ2 with n degrees of freedom, where n has a binomial distribution with parameters 23 and 1/2. The significance of this statistic was then assessed by computing a weighted sum of the tails of the relevant χ2 distributions.
For the fine-scale mapping of the FGFR2 locus, we first derived haplotype frequencies using the haplo.stats package in S-plus39, separately for the European and Asian populations, using data from the case-control studies on whom the tag SNPs were typed plus the 164 control individuals on whom all SNPs were typed. These were used to impute genotype probabilities for each identified SNP in each individual. We then used an EM algorithm to fit a logistic regression model assuming that each SNP in turn was the causal variant, allowing for uncertainty in the genotypes of untyped SNPs, and hence to determine the likelihood that each SNP was the causal variant.
Coverage of the stage 1 tagging set was estimated using HapMap phase II as a reference. We based estimates on 2,116,183 SNPs with an m.a.f. of >5% in the CEU population. Of the SNPs successfully genotyped in stage 1, 187,663 were also on HapMap. For those SNPs not on HapMap, we identified ‘surrogate’ SNPs that were in perfect LD based on genotyping of 24 Caucasians by Perlegen Sciences (269,203 SNPs)18. To estimate coverage, we determined the best pairwise _r_2 for each HapMap SNP and each tag SNP or a surrogate SNP, using the HapMap CEU data. This coverage was summarized in terms of the distribution of _r_2 by allele frequency in 10 categories.
To estimate the power to detect each of the associations found, we computed the non-centrality parameter for the test statistic at each stage, based on the perallele relative risk, allele frequency and _r_2. This was used to estimate the power for a given _r_2, based on a simulated trivariate normal distribution for the score statistics after each stage to allow for the correlations in the test statistics. We assumed a cut-off of P<0.05 for stage 1, P<0.00002 for stage 2 and P<10−7 for stage 3 (the first is slightly conservative, as more SNPs than this were actually taken forward). The overall power was obtained by averaging the power estimates for each _r_2 over the distribution of _r_2 obtained from the HapMap data, applicable to a SNP of that frequency.
The expected number of significant associations after stage 2 (Table 1) was calculated using a bivariate normal distribution for the joint distribution of the (weighted) Cochran-Armitage score statistics after stage 1 and after both stages, using a correlation of 0.525 between the two statistics (reflecting the weighted sizes of the two studies). These calculations were based on the 205,586 SNPs reaching the required quality control in stage 1. Of these, 11,313 reached a P<0.05, of which 7,405 (65.5%) were successfully genotyped to the required quality control in stage 2. Thus the expected number reaching a given significance level with good quality control was calculated from the total number expected to reach this level × 65.5%. We adjusted the variances of the test statistics, separately for stages 1 and 2, using the genomic control method22. The adjustment factor, λ, was estimated from the median of the smallest 90% of the test statistics for SNPs typed in that stage, divided by the predicted median for the smallest 90% of a sample of χ21 distributions (that is, the 45% percentile of a χ21 distribution, 0.375).
Supplementary Material
suppl 1
suppl 2
suppl 3
Acknowledgements
The authors thank the women who took part in this research, and all the funders and support staff who made this study possible. The principal funding for this study was provided by Cancer Research UK. Detailed acknowledgements are provided in Supplementary Information.
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
The authors declare no competing financial interests.
References
- 1.Collaborative Group on Hormonal Factors in Breast Cancer Familial breast cancer: Collaborative reanalysis of individual data from 52 epidemiological studies including 58 209 women with breast cancer and 101 986 women without the disease. Lancet. 2001;358:1389–1399. doi: 10.1016/S0140-6736(01)06524-2. [DOI] [PubMed] [Google Scholar]
- 2.Miki Y, et al. A strong candidate for the breast and ovarian-cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 3.Wooster R, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 4.Antoniou A, et al. Average risks of breast and ovarian cancer associated with mutations in BRCA1 or BRCA2 detected in case series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003;72:1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith P, et al. A genome wide linkage search for breast cancer susceptibility genes. Genes Chromosom. Cancer. 2006;45:646–655. doi: 10.1002/gcc.20330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pharoah PDP, et al. Polygenic susceptibility to breast cancer and implications for prevention. Nature Genet. 2002;31:33–36. doi: 10.1038/ng853. [DOI] [PubMed] [Google Scholar]
- 7.Antoniou AC, Pharoah PDP, Smith P, Easton DF. The BOADICEA model of genetic susceptibility to breast and ovarian cancer. Br. J. Cancer. 2004;91:1580–1590. doi: 10.1038/sj.bjc.6602175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahman N, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nature Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson D, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J. Natl Cancer Inst. 2005;97:813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- 10.Meijers-Heijboer H, et al. Low-penetrance susceptibility to breast cancer due to CHEK2*1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nature Genet. 2002;31:55–59. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 11.Erkko H, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–319. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 12.Renwick A, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nature Genet. 2006;38:873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 13.Seal S, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are lowpenetrance breast cancer susceptibility alleles. Nature Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 14.The CHEK2 Breast Cancer Case-Control Consortium CHEK2*1100delC and susceptibility to breast cancer: A collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from ten studies. Am. J. Hum. Genet. 2004;74:1175–1182. doi: 10.1086/421251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox A, et al. A common coding variant in CASP8 is associated with breast cancer risk. Nature Genetics. 2007;39:352–358. doi: 10.1038/ng1981. corrigendum 39, 688 (2007) [DOI] [PubMed] [Google Scholar]
- 16.Easton DF. How many more breast cancer predisposition genes are there? Breast Cancer Res. 1999;1:1–4. doi: 10.1186/bcr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kruglyak L, Nickerson DA. Variation is the spice of life. Nature Genet. 2001;27:234–236. doi: 10.1038/85776. [DOI] [PubMed] [Google Scholar]
- 18.Hinds DA, et al. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 19.Satagopan JM, Verbel DA, Venkatraman ES, Offit KE, Begg CB. Two-stage designs for gene-disease association studies. Biometrics. 2002;58:163–170. doi: 10.1111/j.0006-341x.2002.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antoniou AC, Easton DF. Polygenic inheritance of breast cancer: Implications for design of association studies. Genet. Epidemiol. 2003;25:190–202. doi: 10.1002/gepi.10261. [DOI] [PubMed] [Google Scholar]
- 21.Altshuler D, et al. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 23.Thomas DC, Haile RW, Duggan D. Recent developments in genomewide association scans: A workshop summary and review. Am. J. Hum. Genet. 2005;77:337–345. doi: 10.1086/432962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amundadottir LT, et al. A common variant associated with prostate cancer in European and African populations. Nature Genet. 2006;38:652–658. doi: 10.1038/ng1808. [DOI] [PubMed] [Google Scholar]
- 25.Yeager M, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nature Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 26.Gudmundsson J, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nature Genet. 2007;39:631–637. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 27.Ferretti V, et al. PReMod: a database of genome-wide mammalian cis-regulatory module predictions. Nucleic Acids Res. 2007;35:D122–D126. doi: 10.1093/nar/gkl879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moffa AB, Tannheimer SL, Ethier SP. Transforming potential of alternatively spliced variants of fibroblast growth factor receptor 2 in human mammary epithelial cells. Mol. Cancer Res. 2004;2:643–652. [PubMed] [Google Scholar]
- 29.Adnane J, et al. Bek and Flg, 2 receptors to members of the Fgf family, are amplified in subsets of human breast cancers. Oncogene. 1991;6:659–663. [PubMed] [Google Scholar]
- 30.Jang JH, Shin KH, Park JG. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res. 2001;61:3541–3543. [PubMed] [Google Scholar]
- 31.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lesueur F, et al. Allelic association of the human homologue of the mouse modifier Ptprj with breast cancer. Hum. Mol. Genet. 2005;14:2349–2356. doi: 10.1093/hmg/ddi237. [DOI] [PubMed] [Google Scholar]
- 33.Day N, et al. EPIC-Norfolk: Study design and characteristics of the cohort. Br. J. Cancer. 1999;80:95–103. [PubMed] [Google Scholar]
- 34.Breast Cancer Association Consortium Commonly studied SNPs and breast cancer: Negative results from 12,000 – 32,000 cases and controls from the Breast Cancer Association Consortium. J. Natl Cancer Inst. 2006;98:1382–1396. doi: 10.1093/jnci/djj374. [DOI] [PubMed] [Google Scholar]
- 35.Hubbard T, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dean FB, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl Acad. Sci. USA. 2002;99:5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Bakker PIW, et al. Efficiency and power in genetic association studies. Nature Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 38.Tyrer J, Pharoah PDP, Easton DF. The admixture maximum likelihood test: A novel experiment-wise test of association between disease and multiple SNPs. Genet. Epidemiol. 2006;30:636–643. doi: 10.1002/gepi.20175. [DOI] [PubMed] [Google Scholar]
- 39.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am. J. Hum. Genet. 2002;70:425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
suppl 1
suppl 2
suppl 3