Celluar microRNA and P-bodies modulate host-HIV-1 interactions (original) (raw)
. Author manuscript; available in PMC: 2010 Jun 26.
SUMMARY
MicroRNAs (miRNAs), ~22-nucleotide noncoding RNAs, assemble into RNA-induced silencing complexes (RISC) and localize to cytoplasmic substructures called P-bodies. Dictated by base-pair complementarity between miRNA and a target mRNA, miRNAs specifically repress posttranscriptional expression of several mRNAs. Here, we report that HIV-1 mRNA interacts with RISC proteins and that disrupting P-body structures enhances viral production and infectivity. In HIV-1-infected human T lymphocytes, we identified a highly abundant miRNA, miR-29a, which specifically targets the HIV-1 3’-UTR region. Inhibiting miR-29a enhanced HIV-1 viral production and infectivity, whereas expressing a miR-29 mimic suppressed viral replication. We also found that specific miR-29a-HIV-1 mRNA interactions enhance viral mRNA association with RISC and P-body proteins. Thus, we provide an example of a single host miRNA regulating HIV-1 production and infectivity. These studies highlight the significance of miRNAs and P-bodies in modulating host cell interactions with HIV-1 and possibly other viruses.
Keywords: HIV-1, miRNA, RNAi, RISC, P-bodies
INTRODUCTION
MicroRNAs (miRNAs) are an abundant class of small (~22-nucleotide [nt]) regulatory RNAs found in plants and animals. These noncoding RNAs play important roles in development by modulating posttranscriptional regulation of target genes (Ambros, 2004; Bartel, 2004). The human genome encodes hundreds of miRNAs with the potential to regulate protein expression by thousands of mRNAs. miRNA biogenesis begins with transcription of miRNA genes by RNA polymerase II into precursor molecules with 5’ m7G capping structures and 3’ poly-A tails. These long primary transcripts are cleaved by the complex Drosha-DGCR8 to produce ~70-nt stem-loop structured precursors (pre-miRNAs), exported to the cytoplasm, and subsequently processed by Dicer (Kim, 2005).
Dicer cleaves long dsRNAs into ~22-nt duplexes: a mature miRNA guide strand and an miRNA passenger strand with 2-nt overhangs at the 3’ termini (Kim, 2005). After Dicer processing, the miRNA guide strand is loaded into the RNA-induced silencing complex (RISC) containing Argonaute1 (Ago1), Ago2, and other RISC co-factors (Kim, 2005). Mature RISC targets specific mRNAs for cleavage or translational repression.
Ago proteins have recently been shown to be concentrated in the cytoplasm of human cells at sites known as mRNA-processing bodies (P-bodies) (reviewed in (Eulalio et al., 2007)). P-bodies contain nontranslating mRNA and proteins involved in mRNA remodeling, decapping, translational repression, and 5’ to 3’ exonuclease activity (Eulalio et al., 2007). Co-localization of miRISC and target mRNAs in P-bodies suggests that they function in miRNA-mediated gene silencing by sequestering target mRNA for storage or decay (Liu et al., 2005; Pillai et al., 2005; Sen and Blau, 2005). Several P-body components, such as GW182 and RCK/p54 (Eulalio et al., 2007; Valencia-Sanchez et al., 2006), play roles in miRNA-dependent translational repression.
miRNA-mediated gene silencing has been shown to play important roles in viral pathogenesis (Jopling et al., 2005; Lecellier et al., 2005; Triboulet et al., 2007). For example, a cellular miRNA has been shown to restrict accumulation of the retrovirus primate foamy virus type 1 (PFV-1) in human cells (Lecellier et al., 2005). In addition, a liver-specific miRNA, miR-122, can modulate replication of hepatitis C in cultured human cells (Jopling et al., 2005). Moreover, cellular miRNA expression has been shown to change upon HIV-1 infection (Triboulet et al., 2007; Yeung et al., 2005), and cellular miRNA contributes to HIV-1 latency in primary CD4+ T lymphocytes (Huang et al., 2007). Not surprisingly, HIV-1 actively suppresses expression of the polycistronic miRNA cluster, miR-17/92, an inhibition of the miRNA pathway that is required for efficient viral replication (Triboulet et al., 2007). Instead of altering host miRNA expression, viruses could use an alternative mechanism involving miRNA-mediated host-pathogen interactions. For example, viruses such as herpes simplex virus 1 and Kaposi’s sarcoma-associated herpes virus encode miRNAs that could target host or viral mRNA to regulate various stages of viral life cycles (Cullen, 2006; Umbach et al., 2008). For RNA viruses such as HIV-1, a well-defined, structured segment of viral RNA called TAR RNA (Rana and Jeang, 1999) has been reported to be processed by Dicer to release miRNAs (Klase et al., 2007; Ouellet et al., 2008) that could be involved in chromatin remodeling (Klase et al., 2007).
Since miRNAs have been shown to modulate the life cycle of human viruses and RISC proteins are localized to P-bodies, we hypothesized that HIV-1 mRNA expression is modulated by RISC and that P-bodies could be involved in innate immune mechanisms against HIV-1 propagation. This hypothesis was tested by investigating the roles of P-bodies, miRNAs, and the protein components involved in miRNA biogenesis and function in creating an innate immunity mechanism against HIV-1 or in modulating host-virus interactions. Here we demonstrate that depleting P-bodies enhances HIV-1 production and infection. In addition, we identified a human miRNA, miR-29a, which specifically targets HIV-1 mRNA and represses viral replication by accumulating viral mRNA in P-bodies.
RESULTS
Silencing Dicer and Drosha enhances HIV-1 replication
We hypothesized that HIV-1 infection alters miRNA expression in host cells, and specific sets of upregulated miRNAs could modulate HIV-1 replication. Since Dicer and Drosha are the key components of the miRNA biogenesis machinery (Chu and Rana, 2007), depleting these two enzymes lowers production of mature miRNAs. To directly determine the role of host miRNAs in HIV-1 production and infectivity, we depleted Dicer in 293T cells to downregulate production of mature miRNA (Kim, 2005) and analyzed HIV-1 production and infectivity. Dicer expression was efficiently silenced by siRNA in cells used to produce HIV-1 (producer cells; Figure 1A, top panel) and in infected (target) cells (Figure 1B, top panel). Quantitative analysis of HIV-1 production showed that depleting Dicer in producer cells increased HIV-1 production (Figure 1A, bottom panel). When HIV-1 was produced in 293T cells without Dicer depletion, depleting Dicer in target cells enhanced viral infectivity (Figure 1B, bottom panel). Similarly, depleting Drosha in producer and target cells upregulated HIV-1 production and infectivity, respectively (Figure 1 C and D). Although these results cannot rule out the possibility that knocking down Dicer and Drosha can influence HIV-1 replication by an indirect mechanism (Triboulet et al., 2007), they strongly suggest that miRNAs are involved in negatively controlling HIV-1 replication.
Figure 1. Depleting Dicer or Drosha increases virus production and infectivity.

(A) Dicer knockdown enhances HIV-1 production. Producer 293T cells were transfected with siRNA mismatched to Dicer mRNA (si-control) or perfectly matched (si-Dicer) and analyzed 48 h post-transfection for virus production (pNL4-3LucR-E-). Efficient knockdown of Dicer is shown by immunoblot analysis of total cell extracts (top panel). Virus production data (lower panel) are normalized to data for cells treated with mismatched siRNA (% of si-control). (B) Dicer knockdown enhances HIV-1 infectivity. Target 293T cells were treated with siRNA mismatched to Dicer mRNA (si-control) or perfectly matched (si-Dicer), and infected for 48 h with the virus equivalent of 50 ng pNL4-3LucR-E-. Virus infectivity was analyzed by measuring luciferase activity of infected cell lysates relative to their protein content. Efficient knockdown of Dicer is shown by immunoblot analysis of total cell extracts (top panel). Virus infectivity data (bottom panel) are normalized to data for cells treated with mismatched siRNA (% of si-control). (C) Drosha knockdown enhances HIV-1 production. Producer 293T cells were transfected with siRNA mismatched to Drosha mRNA (si-control) or perfectly matched (si-Drosha) and analyzed 48 h post-transfection for virus production (bottom panel) as described in Experimental Procedures. Efficient knockdown of Drosha was confirmed by immunoblot analysis of total cell extracts (top panel). (D) Drosha knockdown enhances HIV-1 infectivity. Target 293T cells were treated with siRNA mismatched (si-control) or perfectly matched (si-Drosha) to Drosha mRNA and infected with 50 ng pNL4-3LucR-E-. Virus infectivity was analyzed by measuring luciferase activity in infected cells (bottom panel). Efficient knockdown of Drosha was confirmed by immunoblot analysis of total cell extracts (top panel). Data in all four panels are from at least 3 independent experiments. Error bars represent SD, with significance from _t_-tests shown by p< 0.05 (*) and 0.001 (***).
miR-29a represses HIV-1 replication
Which miRNAs could be involved in repressing HIV-1 mRNA expression? Target prediction analysis suggested that the HIV-1 3’-UTR can be targeted by 11 miRNAs (Table 1). To examine miRNA expression after HIV-1 infection, we infected H9 T lymphocytes with HIV-1 and allowed the infections to proceed for 5 passages. Small RNAs were isolated from HIV-1-infected H9 T lymphocytes, and miRNA expression was quantified by miRNA microarray (Table S1). Of the 11 miRNAs predicted to target HIV-1 3’-UTR (Table 1), miR-29a was most highly expressed in H9 T lymphocytes. HIV-1 infection of 293T cells also enhanced miR-29a expression (Figure 2A). Given the complementary sequences of miR-29a and HIV-1 3’-UTR (Figure 2B), we reasoned that miR-29a could repress HIV-1 gene expression. To test this possibility, we inhibited miR-29a function using 2’-_O_-methyl oligonucleotides complementary to miR-29a (anti-miR) and evaluated HIV-1 production and infectivity (see Experimental Procedures). Inhibiting miR-29a in 293T cells by anti-miR before viral infection enhanced HIV-1 production over that in cells treated with control 2’-_O_-methyl oligonucleotides (Figure 2C). Similarly, blocking miR-29a in 293T cells with anti-miR oligos increased HIV-1 infectivity (Figure 2D). To confirm the role of miR-29a in suppressing HIV-1 mRNA expression, we performed a reciprocal experiment using a miR-29a mimic and analyzed its effect on virus production. Our results show that mimicking miR-29a efficiently suppressed HIV-1 production (Figure 2E). Finally, we tested the effect of inhibiting miR-29 on HIV-1 infectivity by first transfecting H9 T lymphocytes with anti-miR and then by infecting with HIV-1. We found that HIV-1 infectivity was greater than that in lymphocytes treated with control 2’-_O_-methyl oligonucleotides (Figure 2F). HIV-1 infectivity of H9 T cells was less affected by anti-miR than that of 293T cells (Figure 2D), likely due to either lower transfection efficiency of the miR inhibitor or to higher expression of miR-29a in H9 T cells. Taken together, these results identify miR-29a as a cellular repressor of HIV-1 mRNA expression.
Table 1. Predicted targets of human miRNAs in the HIV 3’-UTRa.
The targets of human miRNAs in the HIV 3'-UTR were initially predicted with miRanda software, version 1.0 (http://www.microrna.org/miranda_new.html). Default parameters were used (Gap Open Penalty:-8.0; Gap Extend:-2.0; Score Threshold: 80; Energy Threshold: −14 kcal/Mol; Scaling Parameter: −2.0). The HIV-1 3'-UTR sequence was obtained from the NCBI database with accession number NC_001802 (86319085). miRNA sequences were downloaded from the miRBase (http://microrna.sanger.ac.uk/sequences), and 455 human miRNAs were aligned with the HIV 3'-UTR sequence. The 11 predicted targets found with perfect seed matches (http://www.targetscan.org) are summarized in this table.
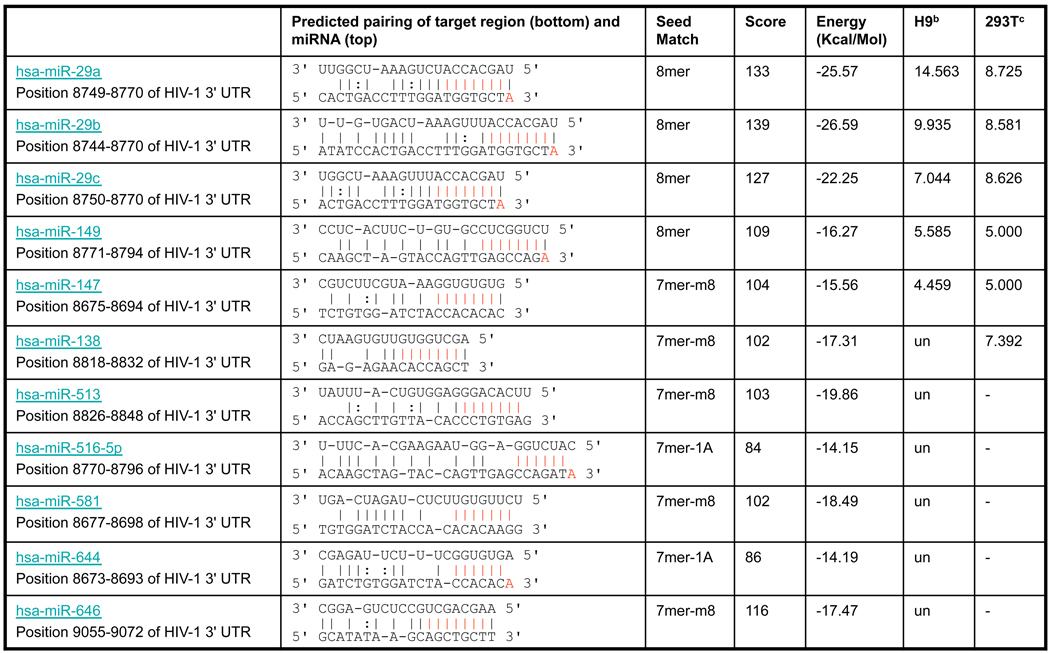
Figure 2. miR-29a modulates HIV-1 production and infectivity.

(A) HIV-1 infection induces miR-29a expression in 293T cells. 293T cells were infected for 48 h with the virus equivalent of 50 ng pNL4-3LucR-E-. Expression of miR-29a was quantified by RT-qPCR (see Experimental Procedures). The level of miR-29a was about 20% higher in HIV-1 infected 293T cells than in mock-infected 293T cells. (B) Predicted miR-29a target site in the HIV-1 3’-UTR (see Table 1). (C) Inhibiting miR-29a increases HIV-1 production. 293T cells were transfected with 20 nM miR inhibitor (negative control) or miR-29a inhibitor, then co-transfected with pNL4-3LucR-E- and VSVG. At 48 h post-transfection, cell supernatants were quantitated for virus production. Data are normalized to data for cells treated with anti-miR (% of control). (D) Inhibiting miR-29a increases HIV-1 infectivity. Target 293T cells were transfected with 20 nM miR inhibitor (negative control) or miR-29a inhibitor. At 24 h post-transfection, cells were infected with a 50 ng equivalent of pNL4-3LucR-E- virus. At 48 h post-infection, infectivity was measured by luciferase activity in total cell lysates relative to their protein content. Virus infectivity data are normalized to data for cells treated with anti-miR (% of control). (E) miR-29a mimic decreases HIV-1 production. 293T cells were transfected with 10 nM control miRNA or miR-29a mimic, then co-transfected with pNL4-3LucR-E- and VSVG. At 48 h post-transfection, cell supernatants were quantitated for virus production. Virus production data are normalized to data for cells treated with anti-miR (% of control). (F) Inhibiting miR-29a enhances viral infectivity in T lymphocytes. H9 T lymphocytes were transfected with 20 nM miR inhibitor (negative control) or miR-29a inhibitor. At 48 h post-transfection, H9 cells were infected by spinoculation with a virus equivalent of 1 µg pNL4-3LucR-E- virus. At 48 h post-infection, virus infectivity was measured by luciferase activity in total cell lysates relative to their protein content. Virus infectivity data are normalized to data for cells treated with anti-miR (% of control). Data in panels (A), (C), (D), (E), and (F) are from at least 3 independent experiments. Error bars represent SD, with significance from _t_-tests shown by p< 0.05 (*) and 0.001 (***).
miR-29a directly targets HIV-1 mRNA
Does miR-29a directly and specifically mediate repression of HIV-1 mRNA expression? To address these questions, we created a mutant HIV-1 reporter construct, pNL4-3-Luc-E-m29t, containing 4 mutations in the HIV-1 3’-UTR region targeted by miR-29a (Figure 3A). To match this target site in the mutant HIV-1 mRNA, we designed a mutant miRNA, miR-29a-mt (Figure 3A). The mutant HIV-1 vector, pNL4-3-Luc-E-m29t, was then transfected into 293T cells in the presence of 10 nM miR-29a or miR-29a-mt, and HIV-1 production was analyzed as described above. Viral production from the pNL4-3-Luc-E-m29t mRNA was suppressed by miR-29a-mt, but was not affected by miR-29a (Figure 3B). As a reciprocal experiment, wild-type pNL4-3-Luc-E- was transfected into 293T cells in the presence of 10 nM miR-29a or miR-29a-mt, and HIV-1 production was analyzed. Viral production from this wild-type vector mRNA was suppressed by miR-29a, but not by miR-29a-mt (Figure 3B).
Figure 3. miR-29a specifically targets HIV-1 mRNA for translation repression.

(A) Schematic representation of HIV-1 reporter constructs. HIV-1 wild-type (pNL4-3Luc wt) and miR-29a target site mutated at the seed sequence (pNL4-3Luc m29t) are shown. The miR-29a target site is indicated by a vertical line (I) and the mutant site by an x (X). Below these construct schema are base-pairing schema of the predicted miR-29a with its target site (pNL4-3Luc wt) and of the seed-mutant miR-29a-mt with its complementary target sequence (pNL4-3Luc m29t). For the mutant sequence, 4 point substitutions (in red) were introduced to disrupt base pairing to miR-29a but to retain pairing to miR-29a-mt. (B) miR-29a specifically represses HIV-1 virus production. 293T cells were co-transfected with HIV-1 construct (pNL4-3Luc wt or pNL4-3Luc m29t) and either miR-29a mimic or seed-mutant mimic (miR-29a-mt). At 48 h post-transfection, virus production in cell supernatants was determined by p24 ELISA. Virus production data are normalized to data for cells treated with miRNA control (% of control). (C) Virus production increases from miR-29a mutant viral constructs and is insensitive to repression by miR-29a. 293 T cells were first transfected with 10 nM miR-29a or control, then co-transfected with pNL4-3LucR E (wt), HIV-1 construct lacking the miR-29a target site pNL4-3LucR E(Δ29t), or pNL4-3LucRE(m29t) and VSVG. At 48 h post-transfection, virus production from cell supernatants was determined by p24 ELISA. Virus production data are normalized to data for cells treated with pNL4-3LucR E (wt) miR (% relative virus production). (D) Mutant viruses pNL4-3LucR E(Δ29t) and pNL4-3LucRE(m29t) are more infectious than pNL4-3LucR E (wt). Target cells were transfected with 10 nM miR-29a or control and infected after 18 h with the virus equivalent of 10 ng pNL4-3LucR E (wt), pNL4-3LucRE(Δ29t), or pNL4-3LucRE(m29t) virus. At 72 h post-infection, virus infectivity was analyzed by measuring luciferase activity in infected cells relative to their protein contents. Virus infectivity data are normalized to data for cells treated with pNL4-3LucR E (wt) (% relative virus infectivity). (E) Mutant virus pNL4-3LucR E(m29t) is significantly more infectious in T lymphocytes than pNL4-3LucR E (wt). H9 T lymphocytes were infected by spinoculation with a virus equivalent of 0.3 µg pNL4-3LucR-E- virus. At 72 h post-infection, virus infectivity was measured by luciferase activity in total cell lysates relative to their protein content. Virus infectivity data are normalized to data for cells treated with pNL4-3LucR E (wt) (% relative virus infectivity). (F) Inhibiting miR-29a increases production of wild-type HIV-1 but not of HIV-1 lacking the miR-29a target site (Δ29t) or HIV-1 containing the mutant miR-29a target site (m29t). 293T cells were first transfected with 20 nM control or miR-29a inhibitor, then co-transfected with pNL4-3LucR-E- (wt), pNL4-3LucR-E-(Δ29t), or pNL4-3LucR-E-(m29t) and VSVG. At 48 h post-transfection, cell supernatants were quantitated for virus production. Virus production data are normalized to data for cells treated with anti-miR (% of control). (G) miR-29a decreases HIV-1 production in Dicer-depleted cells. 293T cells were transfected with 10 nM miRNA control or miR-29a plus mismatched Dicer siRNA (si-control) or perfectly matched Dicer siRNA (si-Dicer). For the second round of transfections, cells were co-transfected with miRNAs, si-RNAs (Figure 1 above), pNL4-3LucR-E-, VSVG, and analyzed 48 h later for virus production. Efficient knockdown of Dicer is indicated by immunoblot analysis of total cell extracts for both miRNA control- and miR-29a-treated cells (top panel). Virus production data (lower panel) are normalized to data for cells treated with miRNA control and si-control (% control). Data in panels (B), (C), (D), (E), (F), and (G) are from at least 3 independent experiments. Error bars represent SD, with significance from _t_-tests shown by p<0.05 (*), 0.01 (**) and 0.001 (***).
To further analyze the specific effects of miR-29a on HIV-1 mRNA expression, we made another mutant HIV-1 reporter construct, pNL4-3-Luc-E-Δ29t, containing a 20-nt deletion at the miR-29a target site in the HIV-1 3’-UTR region. This mutant construct or a control HIV-1 construct, pNL4-3-Luc-E, was transfected into 293T cells in the presence of 10 nM miR-29a or an miR control, and HIV-1 production was analyzed. Similar to the results in Figure 3B, viral production was suppressed by miR-29a only when its complete target site was in the HIV-1 sequence, but not when the target site contained a deletion mutation (data not shown).
We next determined the effect of these miR-29a target-site mutations on viral fitness. First, we analyzed the viral production from various mutant viral constructs. We transfected 293 T cells with 10 nM miR-29a or control, then co-transfected with pNL4-3LucR−E− (wt), the HIV-1 construct lacking the miR-29a target site pNL4-3LucR−E−(Δ29t), or pNL4-3LucR−E−(m29t) and vesicular stomatitis virus glycoprotein (VSVG). At 48 h post-transfection with the miR-29a mutant viral constructs, viral production in cell supernatants (see Experimental Procedures) increased and was insensitive to repression by miR-29a (Figure 3C). Second, we determined the effect of miR-29 target-site mutations on viral infectivity. Target cells were transfected with 10 nM miR-29a or control and infected 18 h later with the virus equivalent of 10 ng pNL4-3LucR−E−(wt), pNL4-3LucR−E−(Δ29t), or pNL4-3LucR−E−(m29t). At 72 h post-infection, viral infectivity was analyzed as described above. As shown in Figure 3D, mutant viruses, pNL4-3LucR−E−(Δ29t) and pNL4-3LucR−E−(m29t), were more infectious than the pNL4-3LucR −E− (wt). Third, we investigated the infectivity of mutant and wild-type viruses in T lymphocytes. H9 T lymphocytes were infected by spinoculation with a virus equivalent of 0.3 µg pNL4-3LucR-E- virus. At 72 h post-infection, luciferase activity was measured in total cell lysates relative to their protein content and virus infectivity data were analyzed (Figure 3E). We found that mutant virus pNL4-3LucR−E−(m29t) was significantly more infectious in T lymphocytes than pNL4-3LucR−E− (wt). Collectively, these results show that miR-29a target-site mutations enhance viral fitness and lead to loss of viral sensitivity to repression by miR-29a.
Our observation that mutant virus pNL4-3LucR−E−(m29t) was significantly more infectious than pNL4-3LucR−E− (wt) in T lymphocytes (Figure 3E) suggests that miR-29a, which is highly expressed in T lymphocytes (Table S1), would suppress wild-type virus more potently than the mutant viruses. These results, taken with those shown in Figure 3B, indicate that miR-29a specifically targets the predicted site in the HIV-1 3’-UTR (Figure 3A) and directly mediates miRNA suppression of HIV-1 mRNA expression.
As an alternative approach to determining the specificity of the interaction between miR-29a and its target, we analyzed the ability of miR-29a inhibitors to relieve HIV-1 mRNA repression as described above (Figure 2B). Inhibiting miR-29a de-repressed only HIV-1 expression of the wild-type pNL4-3-Luc-E construct, but not of the pNL4-3-Luc-E-m29t or pNL4-3-Luc-E-Δ29t reporters (Figure 3F). As an additional control, we determined the effect of miR-29a on HIV-1 production in Dicer-depleted cells (Figure 3G). Dicer depletion enhanced HIV-1 production as shown in Figure 1, and miR-29a reversed this effect by suppressing HIV-1 production to that in normal cells (Figure 3G). Together, these results indicate that miR-29a directly targets a specific region in the HIV-1 3’-UTR.
miR-29a specifically enhances HIV-1 mRNA interactions with RISC
Since miR-29a directly targets HIV-1 mRNA (Figure 3), we next addressed whether HIV-1 mRNA interacts with RISC where specificity is dictated by miR-29a interactions with viral mRNA. To that end, we analyzed the effect of wt and mutant miR-29a on the association of wt and mutant HIV-1 mRNA with the immunopurified Myc-tagged RISC component, Ago2. Cells were transfected sequentially with miR-29a or miR-mt29a and with pNL4-3LucR−E−(wt) or pNL4-3LucR−E−m29t, VSVG, and Myc-Ago2. Cell extracts were immunopurified for Ago2 (Figure 4A, top panel). miRNA expression was monitored (bottom panel) to confirm the presence of corresponding miRs in cells. The RISC and P-body protein-binding mRNA, connexin 43 (Beitzinger et al., 2007), was used as a control. We found that in the presence of miR-29a, wild-type HIV-1 gag mRNA associated with the Ago2 immunoprecipitate, but HIV-1 mRNA containing miR-29a target-site mutations did not efficiently associate with immunopurified Ago2. In a reciprocal experiment, mutant miR-29a enhanced the association of mutant HIV-1 with Ago2 immunoprecipitates (Figure 4A, middle panel). Quantification of gag mRNA associated with Ago2 revealed that miRNAs specifically enhanced ~3-fold accumulation of the target mRNAs with Ago2 (Figure 4A, bar graph).
Figure 4. miR-29a directly targets HIV-1 mRNA for its association with RISC and P-body RNP assemblies.

(A) miR-29a specifically enhances the association of HIV-1 mRNA with RISC RNP complexes. 293T cells were first transfected with miR-29a or miR-mt29a, then with pNL4-3LucRE(wt) or pNL4-3LucREm29t, VSVG, and Myc-Ago2. At 48 h post-transfection, total cell extracts were immunoprecipitated with antibodies against Myc tag and immunoblotted for Myc-Ago2 (top panel). The mRNA was amplified by RT-PCR for HIV-1 gag and connexin 43 mRNA (middle panels). miRNA expression was monitored (bottom panel) by a commercially available kit (see Experimental Procedures). The mRNA associated with Ago2 was quantified by RT-qPCR (bar graph). Data in the bar graph represent % gag mRNA in the presence of wt and mutant miRNAs normalized to data for transfection with pNL4-3LucR-E- (wt) in the presence of miR-29a and for transfection with pNL4-3LucR-E-m29t in the presence of miR-29a-mt, respectively. (B) miR-29 specifically enhances HIV-1 mRNA co-purification with the immunopurified, endogenous P-body protein, RCK/p54. 293T cells were first transfected with miR-29a or miR-mt29a, then with pNL4-3LucR E(wt) or pNL4-3LucREm29t, and VSVG. At 48 h post-transfection, total cell extracts were immunoprecipitated with antibodies against RCK/p54 and immunoblotted (top panel). The mRNA was amplified by RT-PCR for HIV-1 gag and connexin 43 mRNA (middle panels). miRNA expression was monitored (bottom panel) as in (A). The mRNA associated with RCK/p54 was quantified by RT-qPCR (Figure 4B, bar graph; as described in A). Data in the bar graph represent % gag mRNA in the presence of wt and mutant miRNAs normalized to data for pNL4-3LucR E (wt) with miR-29a and for pNL4-3LucR Em29t with miR-29a-mt, respectively.
To determine the effect of miR-29a on HIV-1 gag mRNA association with endogenous RISC proteins, we transfected 293T cells with miR-29a or miR-mt29a followed by pNL4-3LucR−E−(wt) or pNL4-3LucR−E−m29t, and VSVG. Cell extracts were immunopurified for the P-body protein, RCK/p54 (Figure 4B, top panel). miRNA expression in cells was monitored (bottom panel) to confirm the presence of corresponding miRs. The RISC and P-body protein-binding mRNA, connexin 43 (Beitzinger et al., 2007), was used as a control. We found that in the presence of miR-29a, wild-type HIV-1 gag mRNA eluted with immunopurified RCK/p54, and HIV-1 mRNA containing miR-29a target-site mutations did not efficiently associate with immunopurified RCK/p54. In a reciprocal experiment, mutant miR-29a enhanced the association of mutant HIV-1 with RCK/p54 immunoprecipitates. Quantification of gag mRNA associated with RCK/p54 showed that miRNAs specifically increased ~3-fold association of the target mRNAs with RCK/p54 (Figure 4B, bar graph). The specificity of these interactions was verified by two experiments: (a) RCK/p54 immunoprecipitation experiments were repeated with wt-HIV-1 in the absence of miRNA, (b) HA antibodies were used to immunoprecipitate RNA in the presence of miR-29a. Our results showed that miR-29a specifically enhanced association of HIV-1 mRNA with endogenous P-body proteins, and the HA-immunopurified complexes showed no interaction between HIV-1 mRNA and miR-29a (Figure S1). Altogether, these results indicate that miR-29a specifically targets HIV-1 mRNA and enhances its association with ribonucleoprotein (RNP) complexes containing RISC proteins.
HIV-1 mRNA interacts with P-body proteins, and depleting P-bodies enhances HIV-1 replication
One mechanism proposed for miRNA-mediated gene silencing suggests that the miRNA in RISC provides the sequence specificity for target mRNA interactions, and RISC effector proteins shuttle the target mRNA toward the fate of storage or processing in P-bodies (Chu and Rana, 2006; Rana, 2007). We found that miR-29a specifically enhances HIV-1 mRNA interactions with RISC (Figure 4). Therefore, we reasoned that these HIV-1 mRNA-RISC interactions lead to the accumulation of viral mRNA at P-bodies for translational suppression.
We asked whether HIV-1 mRNA assembles into large RNP complexes and associates with P-body proteins. To address this question, we first immunopurified a P-body marker from HIV-1-infected cells and analyzed the resulting RNP complex for HIV-1 mRNA. As a P-body marker, we used a DNA-editing enzyme, APOBEC3G (Sheehy et al., 2002), which has been shown to form RNP complexes with P-body proteins that have established roles in cap-dependent translation (eIF4E and eIF4E-T), translation suppression (RCK/p54), RNAi-mediated post-transcriptional gene silencing (Ago2), and de-capping of mRNA (DCP2) (Wichroski et al., 2006). In immunoprecipitation experiments with HIV-1-infected cells, we used Vif-deleted HIV-1 vectors because HIV-1 Vif protein degrades APOBEC3G (Sheehy et al., 2002). HIV-1 gag mRNA was found only in HA-APOBEC3G immunoprecipitates from cells expressing both HA-APOBEC3G and HIV-1 (Figure S2A). In similar experiments, we found that HIV-1 gag mRNA immunopurified with other P-body proteins such as Ago2 and endogenous RCK/p54 (Figure 4 and Figure S2B–D). No HIV-1 gag mRNA bands were observed in the absence of APOBEC3G or Ago2. Control experiments showed that HIV-1 mRNA did not co-purify with HA- or PTEN-HA (HA-tagged phosphatase and tensin homolog deleted on chromosome 10) proteins that did not localize to P-bodies (Figure S2E). These results indicate that HIV-1 mRNA interacts with RNP complexes containing P-body proteins.
To determine the functional significance of HIV-1 mRNA association with P-bodies, we disrupted P-body structures in host cells by depleting various P-body proteins, infected cells with HIV-1, and analyzed HIV-1 production and infectivity. Three P-body proteins, RCK/p54, Lsm1, and Ago2, were depleted in 293T cells by siRNA-mediated RNAi in the presence of APOBEC3G. Specific knockdown was confirmed 24 h after siRNA transfection by immunoblot analysis showing that targeted protein levels decreased significantly without affecting the expression of other proteins (Figure S3). Next, cell structure and function were monitored after disrupting P-bodies, as described (Chu and Rana, 2006). To that end, we first depleted P-body proteins by transfecting cells with siRNAs and examined the localization of YFP-tagged APOBEC3G. As shown in Figure 5A, depleting RCK/p54 disrupted cellular P-body structures. In these P-body-deficient cells, APOBEC3G proteins were diffused throughout the cytoplasm, no longer accumulating at specific foci (Figure 5A, panels d-f). Similarly, depleting Lsm1 and Ago2 resulted in a diffuse cytoplasmic distribution of APOBEC3G (Figure 5B, 5C, panels d-f). Together, these results show that knockdown of RCK/p54, Lsm1, or Ago2 disrupts P-body structures and disperses APOBEC3G throughout the cytoplasm.
Figure 5. Depleting P-body proteins disrupts P-body formation and increases HIV-1 production and infectivity.

(A) Depleting RCK/p54 disrupts P-bodies. To silence expression of RCK/p54, 293T cells were co-transfected with APOBEC3G–YFP (APO-YFP) and with siRNA against RCK/p54 mRNA (si-RCK/p54) or with mismatched siRNA (si-control). At 24 h post-transfection, cells were fixed and analyzed by confocal microscopy. APO-YFP was detected by direct YFP fluorescence (panels a, d), whereas endogenous RCK/p54 was detected by immunofluorescence with anti-RCK/p54 (panels b, e). Cells were stained to visualize nuclei and images were digitally merged (panels c, f). (B) Depleting Lsm1 disrupts P-bodies. Lsm1 expression was silenced and cells were treated with siRNA against Lsm1 or si-control as in (A). (C) Depleting Ago2 disrupts P-bodies. Ago2 expression was silenced and cells were treated with siRNA against Ago2 or si-control as in (A). (D) Knockdown of RCK/p54, Ago2, or Lsm1 increases HIV-1 production. Producer 293T cells were first transfected with siRNAs mismatched (mm) or perfectly matched (pm) to target P-body protein mRNAs, then co-transfected with pNL4-3LucR-E- and VSVG. Protein knockdown was monitored by immunoblotting (Figure S3). At 48 h post-transfection, viral production was determined by measuring HIV-1 p24 antigen in culture supernatants. Data are normalized to data from mm siRNA-treated cells (si-control). Error bars represent SD, with significance from _t_-tests shown by p< 0.01 (**) and 0.001 (***). (E) Depleting RCK/p54, Ago2, or Lsm1 increases HIV-1 infectivity. Target 293T cells were transfected with mm or pm siRNA. Protein knockdown was confirmed by immunoblotting (data not shown). Approximately 4 h after the second siRNA transfection, cells were infected for 48 h with the virus equivalent of 50 ng pNL4-3LucR-E. Virus infectivity was determined in total cell lysates from infected cells by measuring luciferase activity determined relative to lysate protein content. Virus infectivity data are normalized to data from mm siRNA-treated cells (% control). Error bars represent SD, with significance from _t_-tests shown by p< 0.05 (*) and 0.01 (**). (F) Endogenous miRNAs and exogenous HIV-1 mRNAs localize to P-bodies in 293T cells. To detect the localization of miR-18a, 293T cells were transfected with APOBEC3G–YFP (APO-YFP), fixed at 24 h post-transfection, immunostained for APO-YPF or RCK/p54, hybridized in situ for miR-18a with fluorescein-labeled LNA probe, and analyzed by confocal microscopy. APO-YFP and endogenous RCK/p54 were detected by immunofluorescence with anti-YFP and anti-RCK/p54, respectively (panels b, e). Cells were stained to visualize nuclei and images were digitally merged (panels c, f). Arrowheads indicate co-localization of miR-18a and APOBEC3G or RCK/p54 in P-bodies (G) HIV-1 mRNAs co-localize with APOBEC3G. 293T cells were transfected with APO-YFP and pNL4-3, fixed at 36 h post-transfection, immunostained, and hybridized in situ for HIV-1 mRNA as described above. LNA probes complementary to HIV-1 Nef mRNA were labeled with Cy3 and used for FISH.
To examine the effect of P-body disruption on HIV-1 production, we depleted 293T cells of RCK/p54, Lsm1, or Ago2 before exposure to HIV-1 and VSVG constructs. This treatment enhanced HIV-1 production over that in cells pretreated with mismatched siRNA (si-control, Figure 5D). RCK/p54 knockdown increased viral production more than Ago2 or Lsm1 knockdown because RCK/p54 is a general translational suppressor. Therefore, its knockdown could have a dual or additive effect on viral production, i.e., release translational suppression plus disrupt P-bodies. Next, we produced HIV-1 in 293T cells without first depleting P-body proteins and infected target 293T cells in which these three P-body proteins had been depleted. In single-round infections, depleting RCK/p54, Lsm1, or Ago2 increased viral infectivity (Figure 5E). We observed that Lsm1 knockdown increased viral infectivity less than it increased viral production. These results show that depleting RCK/p54, Lsm1, and Ago2 disrupts P-body structures and enhances HIV-1 production and infectivity.
To visualize HIV-1 mRNA in P-bodies, we analyzed co-localization of a highly abundant miRNA in 293T cells, miR-18a, with various P-body proteins and HIV-1 mRNA. 293T cells were transfected with APOBEC3G-YFP (APO-YFP) vectors, fixed at 24 h post-transfection, immunostained for APO-YPF or RCK/p54, hybridized in situ for miR-18a with fluorescein-labeled locked nucleic acid (LNA) probe, and analyzed by confocal microscopy. APO-YFP and endogenous RCK/p54 were detected by immunofluorescence with anti-YFP and anti-RCK/p54, respectively (panels b, e). Cells were stained to visualize nuclei and images were digitally merged (panels c, f). As expected, miRNA-18a localized at P-bodies with APOBEC3G or RCK/p54 (arrowheads, Figure 5F). To detect HIV-1 mRNA with APOBEC3G, 293T cells were transfected with APO-YFP and pNL4-3 constructs, fixed at 36 h post-transfection, immunostained, and hybridized in situ for HIV-1 mRNA as described above. LNA probes complementary to HIV-1 Nef mRNA were labeled with Cy3 and used for FISH. We found that HIV-1 mRNA co-localized with APO-YFP (Figure 5G). These results show that HIV-1 mRNA can be detected in P-bodies with endogenous miRNAs.
RCK/p54 depletion increases viral protein synthesis without affecting its mRNA levels
Based on the above findings, we hypothesized that the repression of viral protein translation is modulated by miRNAs that enhance the association between viral mRNAs and P-bodies. To test our hypothesis, we analyzed expression of HIV-1 protein and GFP (as a control) by quantifying p24 levels in producer cells depleted of RCK/p54 by RNAi (Figure S4A). For control experiments, cells were treated with a mismatched siRNA duplex. Total cellular protein was extracted from equal numbers of cells and measured by ELISA or GFP fluorescence (Figure S4B and C). GFP protein levels were measured as a control. HIV-1 protein expression increased more robustly ~ 3-fold in RCK/p54-depleted cells than in control cells treated with mismatched siRNA (Figure S4B). HIV-1 mRNA levels did not differ in producer cells after treatment with RCK/p54 or control siRNA (Figure S4D). Since RCK/p54 is a general translational repressor, its depletion led to a moderate increase in GFP protein with no significant change in GFP mRNA levels (Figure S4C and E). Remarkably, RCK/p54 depletion released ~3-fold translation suppression by viral mRNA (Figure S4), and miR-29 enhanced ~3-fold target viral mRNA association with P-bodies (Figure 4). Taken together, these results strongly suggest that miR-29a facilitates suppression of HIV-1 translation by enhancing the association of viral mRNA with P-bodies and that disrupting P-bodies by RCK/p54 depletion releases translation suppression and enhances production of viral proteins.
A highly conserved miR-29 family modulates HIV-1 production
miR-29a is a member of an miRNA family whose mature miRNAs contain the same seed region, which is highly conserved across humans, mice, rats, dogs, and chickens (http://www.targetscan.org/). Besides miR-29a, miR-29b and miR-29c were also expressed in H9 T cells although at much lower levels (Table 1). To determine whether these 3 miR family members all modulate HIV-1 mRNA expression, we analyzed the effects of miR-29a, b, and c inhibitors on viral production. Inhibiting miR-29a, b, or c increased HIV-1 production, but inhibiting miR-29a resulted in the highest viral production (Figure 6A). Similarly, using miRNA mimics showed that all 3 miR family members decreased viral production, with 29-a again showing the strongest effect (Figure 6B). These findings strongly suggest that that miR-29 family members influence virus replication. Further, our analysis of the miR-29a target site in various HIV-1 subtypes revealed that the sequence targeted by the seed region of miR 29a family is highly conserved (Figure 6C; see Discussion).
Figure 6. A highly conserved miR-29 family modulates HIV-1 production and infectivity.

(A) Inhibiting miR-29a, 29b, or 29c increases HIV-1 production. 293T cells were first transfected with 10 nM miR inhibitor (negative control), miR-29a, b, or c inhibitor, then co-transfected with pNL4-3LucR-E- and VSVG. At 48 h post-transfection, cell supernatants were quantitated for virus production. Data are normalized to data for treatment with anti-miR control (% of control). Error bars represent SD, with significance from _t_-tests shown by p<0.001 (***). (B) miR-29a, −29b, or −29c decreases HIV-1 production. 293T cells were first transfected with 20 nM control, miR-29a, b, or c mimic, then co-transfected with pNL4-3LucR-E- and VSVG. At 48 h post-transfection, cell supernatants were quantitated for virus production. Data are normalized to data for treatment with miRNA control (% of control). Error bars represent SD, with significance from _t_-tests shown by p<0.001 (***). (C) Sequence conservation of the miR-29a target nef/LTR region in various HIV-1 subtypes. Sequences of HIV-1 subtypes were downloaded from HIV sequence database (http://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html). Sequences for nef or 3’LTR were aligned using CLC Sequence Viewer 4 software.
DISCUSSION
Our results indicate that miRNA-mediated gene silencing limits HIV-1 replication, and this antiviral response is substantially modulated by a single cellular miRNA. Not surprisingly, HIV-1 has developed counter-defense mechanisms for suppressing cellular gene silencing pathways (Triboulet et al., 2007). miRNAs could modulate host-viral interactions by at least four possible mechanisms. (1) Viruses can encode miRNAs that target viral mRNAs to regulate various stages of the viral life cycle. This mechanism is supported by recent evidence that herpes viruses express miRNAs that regulate viral gene expression, implicating their role in establishing and maintaining viral latency (Murphy et al., 2008; Umbach et al., 2008). (2) Virus-encoded miRNAs can suppress the expression of a specific set of host genes to benefit viral survival and propagation (Cullen, 2006). (3) Viral infections induce expression of host miRNAs that inhibit expression of various cellular genes (Triboulet et al., 2007). (4) In response to viral infections, host cells mount an antiviral defense mechanism that includes expressing specific miRNAs that suppress viral mRNA expression. Our findings provide an example of a single miRNA influencing HIV-1 production and infectivity, thus targeting expression of foreign RNA. These findings support the hypothesis that cellular miRNAs play important immune functions during viral infections and modulate host-pathogen interactions. This concept is further supported by recent evidence that expression of cellular miRNAs was rapidly enhanced by interferon beta (IFNβ) in hepatitis C-infected cells, resulting in limited viral replication by host miRNAs targeting viral genomic RNA (Pedersen et al., 2007). In this case, mammalian organisms may have evolved a miRNA-mediated antiviral mechanism using IFNβ (Pedersen et al., 2007).
Our findings also suggest that cytoplasmic P-body structures are involved in modulating host cell interactions with HIV-1 and possibly other viruses. We found that HIV-1 mRNA associated with P-body and RISC proteins, and this interaction was significantly enhanced by miR-29a. In addition, disrupting P-bodies by RNAi released translation suppression and enhanced HIV-1 production and infectivity. P-bodies could play a role in modulating viral replication by several mechanisms. First, viral messages could be translationally suppressed by miRNAs and shuttled to P-bodies, a notion supported by our results. Second, viral mRNA transported to P-bodies for suppression of viral protein synthesis could be released by certain stimuli, e.g., host or environmental cues, to activate viral replication (Figure 7). This possibility is supported by cellular proteins such as APOBEC3G being localized to P-bodies and assembling into RNP complexes for packaging into virus particles (Wichroski et al., 2006). Since miR-29 is a conserved miRNA that emerged many millions of years before HIV-1, we hypothesize that HIV-1 has evolved a mechanism utilizing miR-29a to modulate its own life cycle. Thus, miRNA-mediated control of HIV-1 replication could provide a checkpoint in the cycle of viral latency to activation. Such mechanisms would allow viruses to evade the immune system in various tissues or to create viral reservoirs as a hiding place during chemotherapy.
Figure 7. Model for cellular miRNAs modulating host-virus interactions.

HIV-1 infects human cells and integrates its DNA into the host genome. HIV-1 infection induces expression of specific sets of cellular miRNAs, including miR-29a. HIV-1 mRNA is transcribed, exported from the nucleus, and translated into viral proteins. Cellular RISC containing specific miRNAs, such as miR-29a, targets HIV-1 mRNA and sequesters the ribonucleoprotein complex in P-bodies. Depending upon cellular stimuli or viral pathogenesis cues, HIV-1 mRNA could be stored in P-bodies and released for subsequent translation of viral proteins. Alternatively, viral mRNA could be degraded in P-bodies.
To test this hypothesis, we analyzed miR-29a target-site sequence conservation in various HIV-1 subtypes and found that the miR-29a target site complementary to the RNA seed region was highly conserved (Figure 6C). Interestingly, HIV-1 group O contained non-conserved nucleotides in the 5’-end of the miRNA seed region, making it less likely the mRNA of this HIV-1 group would be targeted by miR-29a. HIV-1 group O is endemic only in west and central Africa and is typically 100-fold less infectious than the main HIV-1 group responsible for the global AIDS epidemic, HIV-1 group M (Arien et al., 2005). Similarly, HIV-1 with deletions in the nef gene and U3 region of the LTR, which also contains an miR-29a target-site deletion, did not cause disease in humans 10 to 14 years after infection (Deacon et al., 1995). Therefore, miR-29a interactions with viral mRNA can be plausibly linked to HIV-1 infectious capabilities that are lost in group O and in _nef_-deleted HIV-1 because this mRNA cannot be targeted by miR-29a for transport to P-bodies. However, the possibility cannot be ruled out that loss of infection efficiency in group O and _nef_-deleted HIV-1 involved deletion of other regions. Since our experiments using miR-29 target-site mutant virus were single-round infections and were not carried out in humans under physiological conditions, we could not detect loss of infectious behavior in these virions. Based on these findings, it is feasible that miRNAs played an important role during evolution in defining the pathogenicity of animal viruses.
EXPERIMENTAL PROCEDURES
Expression vectors
HIV-1 luciferase reporter constructs pNL4-3LucR-E- and pNL4-3ΔVif LucR-E were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, made available by Dr. Nathaniel Landau’s laboratory. APOBEC3G-YFP and APOBEC3G-HA were described previously (Wichroski et al., 2005) and pFlag/HA-Ago2 plasmid was a generous gift from Dr. Tom Tuschl. Myc-Ago2 and HA-PTEN vectors were kind gifts from Drs. Hannon and Ross, respectively. The plasmid pEYFP-C1 and plasmids YFP-RCK/p54, YFP-Ago2, and YFP-Lsm1 were described previously (Chu and Rana, 2006).
Immunoprecipitation and immunoblotting
293T cells were transfected with 50 fmoles of pNL4-3ΔVif LucR-E alone or with either 60 fmoles APOBEC3G-HA or 120 fmoles pFlag/HA-Ago2. As a specificity control, 293T cells were transfected with 60 fmoles APOBEC3G-HA or 120 fmoles pFlag/HA-Ago2. Preparation of total cell extracts, immunoaffinity purification, and immunoblotting were performed as described (Chu and Rana, 2006; Wichroski et al., 2005; Wichroski et al., 2006).
Live cell imaging and immunofluorescence
293T cells were cultured in 35 mm dishes with glass cover slip bottoms (MatTek Corporation, Ashland, MA). Cells were transfected with expression vectors for YFP-tagged proteins using Lipofectamine 2000 according to the manufacturer’s protocol. 24 h later, cells were fixed and monitored for YFP signals of transiently expressed proteins. Signals were detected by a Leica confocal imaging spectrophotometer system (TCS-SP2) attached to a Leica DMIRE inverted fluorescence microscope equipped with an argon laser, two HeNe lasers, an acousto-optic tunable filter (AOTF) to attenuate individual visible laser lines, and a tunable acousto-optical beam splitter (AOBS). A 63x, 1.32 NA oil-immersion objective was employed.
For immunofluorescence studies, cells transfected with APO-YFP and siRNA were fixed with 4% paraformaldehyde in PBS at room temperature for 20 min and permeabilized for 5 min with 0.25% (v/v) Triton X-100. Samples were washed 3 times with PBST (0.1% [v/v] Triton X-100 in PBS) and blocked for 30 min in PBST containing 2% (w/v) BSA. Primary and secondary antibodies were diluted in blocking solution during incubation. Antibodies used in these experiments included anti-RCK/p54, anti-Lsm1, anti-Ago2 as previously described (Wichroski et al., 2006). Secondary antibodies against rabbit and mouse IgG were directly conjugated to Alexa Fluor dyes (Molecular Probes, Inc., Eugene, OR). After the final wash, samples were counterstained with Hoechst 33258 to visualize nuclei with a Leica confocal imaging system as described above.
Virus production and knockdown of P-body proteins
293T cells were transfected twice by appropriate siRNAs prior as described (Robb and Rana, 2007). On the first day, cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. On the second day, cells were re-seeded and transfected 4 h later with the following constructs: siRNA, pNL4-3LucR-E-, vesicular stomatitis virus glycoprotein (VSVG), or empty vector. At 48 h post-transfection, virus supernatants were harvested, clarified by low-speed centrifugation (500 × g, 10 min), and filtered using 0.22 µM syringe filters (Millipore Corporation, Bedford, MA). Virus production was quantified using a HIV-1 p24 ELISA kit (ZeptoMetrix, Buffalo, NY) according to the manufacturer’s instructions. Data for virus production represent the average of at least 4 independent experiments, presented as % of control transfection with mismatched siRNA (si-control), and analyzed by Prism Software.
To verify knockdown of P-body proteins, total cell lysates (20 µg) were prepared as described (Chu and Rana, 2006), treated with SDS-PAGE sample loading buffer, heated for 5 min at 100 °C, and separated by SDS-PAGE. Proteins were separated and detected by immunoblot analysis with various antibodies as described (Wichroski et al., 2006).
Virus production and treatment with miR-29a or miR-29a inhibitor
293T cells were transfected as described above for knockdown of P-body proteins, but siRNA transfection was replaced by transfection with 20 nM miR inhibitor 29a or 10 nM miR-29a. Negative controls for miR inhibitor 29a and miR-29a were negative control miR inhibitor 1# and GFP mm siRNA, respectively. Virus production was monitored as described above. Data for virus production represent the average of at least 4 independent experiments, normalized to the anti-miR control or miRNA control, and analyzed by Prism Software.
Supplementary Material
01
02
Acknowledgments
We thank David Bartel, Nikolaus Rajewsky, and Mihaela Zavolan for advice on miRNA target prediction; and Drs. Hannon, Landau, Ross, and Tuschl for reagents. We also thank Victor Ambrose, David Baltimore, Greg Hannon, Craig Mello, and Rana laboratory members for helpful discussions and support, and the University of Massachusetts Center for AIDS Research for virology support. This work was supported in part by an NIH grant to TMR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Arien KK, Abraha A, Quinones-Mateu ME, Kestens L, Vanham G, Arts EJ. The replicative fitness of primary human immunodeficiency virus type 1 (HIV-1) group M, HIV-1 group O, and HIV-2 isolates. J Virol. 2005;79:8979–8990. doi: 10.1128/JVI.79.14.8979-8990.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Beitzinger M, Peters L, Zhu JY, Kremmer E, Meister G. Identification of human microRNA targets from isolated argonaute protein complexes. RNA Biol. 2007;4:76–84. doi: 10.4161/rna.4.2.4640. [DOI] [PubMed] [Google Scholar]
- Chu CY, Rana TM. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006;4:e210. doi: 10.1371/journal.pbio.0040210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CY, Rana TM. Small RNAs: regulators and guardians of the genome. J Cell Physiol. 2007;213:412–419. doi: 10.1002/jcp.21230. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Viruses and microRNAs. Nat Genet. 2006;38 Suppl:S25–S30. doi: 10.1038/ng1793. [DOI] [PubMed] [Google Scholar]
- Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, et al. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270:988–991. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol. 2007;8:9–22. doi: 10.1038/nrm2080. http://www.targetscan.org/ Targetscan. [DOI] [PubMed] [Google Scholar]
- Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007;13:1241–1247. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- Klase Z, Kale P, Winograd R, Gupta MV, Heydarian M, Berro R, McCaffrey T, Kashanchi F. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol Biol. 2007;8:63. doi: 10.1186/1471-2199-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, Saib A, Voinnet O. A cellular microRNA mediates antiviral defense in human cells. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol. 2005;7:719–723. doi: 10.1038/ncb1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc Natl Acad Sci U S A. 2008;105:5453–5458. doi: 10.1073/pnas.0711910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet DL, Plante I, Landry P, Barat C, Janelle ME, Flamand L, Tremblay MJ, Provost P. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucleic Acids Res. 2008;36:2353–2365. doi: 10.1093/nar/gkn076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- Rana TM, Jeang KT. Biochemical and functional interactions between HIV-1 Tat protein and TAR RNA. Arch Biochem Biophys. 1999;365:175–185. doi: 10.1006/abbi.1999.1206. [DOI] [PubMed] [Google Scholar]
- Robb GB, Rana TM. RNA Helicase A Interacts with RISC in Human Cells and Functions in RISC Loading. Mol Cell. 2007;26:523–537. doi: 10.1016/j.molcel.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Sen GL, Blau HM. Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat Cell Biol. 2005;7:633–636. doi: 10.1038/ncb1265. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Triboulet R, Mari B, Lin YL, Chable-Bessia C, Bennasser Y, Lebrigand K, Cardinaud B, Maurin T, Barbry P, Baillat V, et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science. 2007;315:1579–1582. doi: 10.1126/science.1136319. [DOI] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008 doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- Wichroski MJ, Ichiyama K, Rana TM. Analysis of HIV-1 viral infectivity factor-mediated proteasome-dependent depletion of APOBEC3G: correlating function and subcellular localization. J Biol Chem. 2005;280:8387–8396. doi: 10.1074/jbc.M408048200. [DOI] [PubMed] [Google Scholar]
- Wichroski MJ, Robb GB, Rana TM. Human Retroviral Host Restriction Factors APOBEC3G and APOBEC3F Localize to mRNA Processing Bodies. PLoS Pathog. 2006;2:e41. doi: 10.1371/journal.ppat.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung ML, Bennasser Y, Myers TG, Jiang G, Benkirane M, Jeang KT. Changes in microRNA expression profiles in HIV-1-transfected human cells. Retrovirology. 2005;2:81. doi: 10.1186/1742-4690-2-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
02