Loss of GluN2B-Containing NMDA Receptors in CA1 Hippocampus and Cortex Impairs Long-Term Depression, Reduces Dendritic Spine Density, and Disrupts Learning (original) (raw)
Abstract
NMDA receptors (NMDARs) are key mediators of certain forms of synaptic plasticity and learning. NMDAR complexes are heteromers composed of an obligatory GluN1 subunit and one or more GluN2 (GluN2A–GluN2D) subunits. Different subunits confer distinct physiological and molecular properties to NMDARs, but their contribution to synaptic plasticity and learning in the adult brain remains uncertain. Here, we generated mice lacking GluN2B in pyramidal neurons of cortex and CA1 subregion of hippocampus. We found that hippocampal principal neurons of adult GluN2B mutants had faster decaying NMDAR-mediated EPSCs than nonmutant controls and were insensitive to GluN2B but not NMDAR antagonism. A subsaturating form of hippocampal long-term potentiation (LTP) was impaired in the mutants, whereas a saturating form of LTP was intact. An NMDAR-dependent form of long-term depression (LTD) produced by low-frequency stimulation combined with glutamate transporter inhibition was abolished in the mutants. Additionally, mutants exhibited decreased dendritic spine density in CA1 hippocampal neurons compared with controls. On multiple assays for corticohippocampal-mediated learning and memory (hidden platform Morris water maze, T-maze spontaneous alternation, and pavlovian trace fear conditioning), mutants were impaired. These data further demonstrate the importance of GluN2B for synaptic plasticity in the adult hippocampus and suggest a particularly critical role in LTD, at least the form studied here. The finding that loss of GluN2B was sufficient to cause learning deficits illustrates the contribution of GluN2B-mediated forms of plasticity to memory formation, with implications for elucidating NMDAR-related dysfunction in disease-related cognitive impairment.
Introduction
Activation of NMDA receptors (NMDARs) initiates a cascade of molecular events underlying synaptic plasticity and learning (Malenka and Bear, 2004). Intrahippocampal administration of uncompetitive NMDAR antagonists or gene deletion of the obligatory GluN1 subunit in the rodent CA1 region of the hippocampus impairs spatial learning in the Morris water maze (MWM) and T-maze, mimicking the effects of hippocampal lesions (Morris et al., 1990; Tsien et al., 1996a; Nakazawa et al., 2004). However, NMDARs are not necessary for hippocampal-dependent forms of learning and memory under all experimental conditions (e.g., after water maze pretraining) (Bannerman et al., 2006), and the precise role of NMDARs in synaptic plasticity and memory encoding is not fully understood.
NMDARs are heteromeric assemblies composed of an obligatory GluN1 subunit and one or more GluN2 (GluN2A–GluND) subunits (Rosenmund et al., 1998). GluN2B expression decreases in favor of GluN2A during development (Hestrin, 1992). In adult cortex and hippocampus, GluN2A and GluN2B are the predominant subunits and confer distinct physiological and molecular properties to NMDARs therein. GluN2B-containing NMDARs have slower channel kinetics and lower open probabilities than those containing GluN2A (Cull-Candy et al., 2001). GluN2A/GluN2B ratio and long-term potentiation (LTP) induction thresholds are altered by synaptic activity, sensory experience, and learning (Kirkwood et al., 1996; Quinlan et al., 2004), suggesting a functional contribution to behavioral plasticity.
The contribution of these subunits, and particularly GluN2B, to NMDAR-mediated synaptic plasticity and learning remains a major issue that is not yet fully resolved. Although pharmacological studies proposed a differential role for GluN2A and GluN2B in LTP and long-term depression (LTD), respectively (Liu et al., 2004; Massey et al., 2004; Morishita et al., 2007), conclusions are limited by the nonselectivity of GluN2A antagonists and poor efficacy of GluN2B antagonists at triheteromeric receptors (Neyton and Paoletti, 2006; Kash and Winder, 2007). However, knockdown, age-related loss, or decreased tyrosine phosphorylation of GluN2B (at least partially) impaired hippocampal or cortical LTP and learning (Clayton et al., 2002; Takehara et al., 2004; Zhao et al., 2005; Nakazawa et al., 2006; Gardoni et al., 2009), whereas transgenic overexpression of GluN2B or GluN2B hypodegradation enhanced hippocampal LTP and learning (Tang et al., 1999; Hawasli et al., 2007). In addition, mice with constitutive GluN2B deletion did not show LTD when tested as neonates but did not survive to be tested in adulthood (Kutsuwada et al., 1996). Other studies have found that GluN2B antagonism did not impair hippocampal LTP (Liu et al., 2004), whereas GluN2B deletion in principal neurons throughout the forebrain disrupted various forms of hippocampal-mediated learning but only produced minor deficits in LTP (von Engelhardt et al., 2008). In contrast, GluN2B deletion in principal CA3 hippocampal neurons abolished NMDAR-dependent LTP in this region (Akashi et al., 2009).
Here we sought to clarify the role of GluN2B in synaptic plasticity and learning in the adult brain by generating mice with late-developmental deletion of GluN2B restricted to CA1 hippocampal and cortical pyramidal neurons. We assessed the consequences of this selective loss of GluN2B-containing NMDARs for hippocampal synaptic physiology and plasticity (LTP and LTD), dendritic spine density/morphology, and corticohippocampal-mediated learning.
Materials and Methods
Generation of GluN2B mutant mice.
The GluN2B gene was disrupted by inserting a loxP site downstream of the 599 bp exon 3 or exon 5 (depending on transcript) and a neomycin resistance gene cassette flanked by two loxP sites upstream of this exon (supplemental Fig. S1_A_, available at www.jneurosci.org as supplemental material). A targeting vector was constructed using a 14 kb genomic fragment isolated from a L129 phage genomic library. A 4079 bp NheI–EcoRV fragment containing the 599 bp exon was isolated from the phage clone and inserted in pBluescript. Next, a small oligonucleotide adaptor containing a loxP site was inserted at unique MfeI and SphI sites downstream of the exon. A small oligonucleotide adaptor containing SpeI and MluI sites was then inserted at a single BglII site located 500 bp upstream of the exon. A neomycin resistance gene cassette flanked by two loxP sites was introduced using the newly created SpeI and MluI sites, and the size of the left arm of recombination was increased by inserting a 5 kb EcoRI–NheI fragment. The construct was linearized using NotI. TL-1 embryonic stem (ES) cells derived from 129/SvEvTac mice were electroporated with the linearized construct and the transfected cells grown on fibroblast feeder cells in DMEM supplemented with 15% fetal bovine serum, 50 mg/ml gentamicin, 1000 U/ml leukemia inhibitory factor, 90 mm β-mercaptoethanol, and 0.2 mg/ml G418.
Three hundred fifty independent neomycin-resistant colonies were selected and grown in 96-well plates on feeder layer, expanded, and analyzed for the presence of the mutant gene by performing Southern blot analysis using genomic DNA digested with SpeI and hybridized with a 32P-labeled probe consisting of 406 bp downstream of the right arm of recombination (supplemental Fig. S1_B_, available at www.jneurosci.org as supplemental material). Two clones were independently injected into C57BL/6J blastocysts, and 11 chimeric males (>60% brown fur) were obtained with one ES cell clone and mated with C57BL/6J females. Several chimeric males produced brown fur offspring demonstrating germ-line transmission. Because these mice carry one allele containing the neomycin resistance gene cassette (three loxP), they were mated with E2a–CRE mice to eliminate the neomycin-resistance gene cassette. Progeny demonstrating two (rather than three) loxP alleles and loss of the neomycin resistance gene cassette were selected by PCR genotyping (Fig. 1C). Mice carrying one copy of the two loxP alleles were interbred to produce homozygous GluN2B-floxed mice.
Figure 1.

Loss of corticohippocampal GluN2B mRNA expression and protein levels. GluN2B mRNA quantification by in situ hybridization in parasagittal section showed extensive loss of GluN2B mRNA in neocortex (Cx) and dorsal CA1 region of the hippocampus (HP) in mutants (B, G, arrowheads) relative to controls (A, E). No obvious GluN2B mRNA loss in ventral CA1, CA3, or dentate gyrus (DG) in a horizontal section of mutants (D) relative to controls (C). No differences in GluN2B mRNA in striatum (ST) (A, B), olfactory bulbs (C, D), or lateral (LA), basolateral (BLA), or central (Ce) amygdala nuclei (F, H). I, Loss of GluN2B protein levels in CA1 hippocampal tissue of mutants relative to controls (n = 4–12 per genotype). J, Loss of GluN2B and GluN1 but not GluN2A, CaMKIIα, or CaMKIIβ protein levels in cortical tissue of mutants relative to controls (n = 4–6 per genotype). Data are mean ± SEM normalized to levels in control genotype. *p < 0.05 versus control. Cb, Cerebellum; BS, brainstem; Con, control; Mut, mutant.
We took advantage of the fact that CaMKII-driven Cre/LoxP recombination occurs late in postnatal development to generate mice lacking GluN2B in principal neurons of the cortex and CA1 subregion of the hippocampus by crossing floxed mutants with transgenic mice expressing Cre recombinase driven by the CaMKII promoter (T29-1 line) (Tsien et al., 1996b). Double mutants were interbred to produce wild-type, Cre-positive, homozygous GluN2B-floxed, and mutant mice (i.e., Cre-positive homozygous GluN2B-floxed mice). Additional mutants were produced by interbreeding Cre-positive homozygous GluN2B-floxed and Cre-negative homozygous GluN2B-floxed mice. Floxed/Cre-negative, non-floxed Cre-positive, and wild-type mice served as controls.
Both the GluN2B-floxed and Cre transgenic lines were backcrossed onto the C57BL/6J strain; analysis of 150 single nucleotide polymorphism markers at ∼15–20 Mb intervals estimated the genetic background of the mutant cross to be ∼95% C57BL/6J (JRS Allele Typing Services, The Jackson Laboratory). Male and female mice used for experimentation were bred at The Jackson Laboratory, shipped at 7 weeks of age, and allowed at least 1 week of acclimation before experimentation. Mice were housed with a same-sex littermate in a temperature- and humidity-controlled vivarium under a 12 h light/dark cycle (lights on 6:00 A.M.). Experimenters were blind to genotype during all experiments except those examining NMDAR–EPSC amplitude and pharmacology. Procedures were approved by National Institute on Alcohol Abuse and Alcoholism Animal Care and Use Committee and strictly followed the National Institutes of Health guidelines Using Animals in Intramural Research.
In situ hybridization. Fresh-frozen brain sections (14 μm in thickness) from experimentally naive 12-week-old mice were prepared in the parasagittal or horizontal plane with a cryostat and mounted onto silane-coated glass slides. Sections were postfixed with 4% paraformaldehyde in PBS for 15 min, followed by 0.2 m HCl for 10 min. After rinsing, sections were further incubated in 0.25% acetic anhydride and 0.1 m triethanolamine for 10 min to avoid nonspecific binding of the probe. After dehydration with ethanol, hybridization was performed at 55°C for 18 h in a hybridization buffer containing 50% formamide. For detection of GluN2B mRNA, a cRNA probe, derived from the whole exon 2 sequence (599 bp) of mouse GluN2B genome, was labeled with [33P]UTP (5 × 105 cpm) and added to the hybridization buffer. For detection of GluN1 mRNA, a cRNA probe, derived from the AvrII–SphI 0.4 kb antisense DNA fragment of rat GluN1 cDNA containing exon 13 to exon 16, was radiolabeled as described previously (Iwasato et al., 1997). Brain sections were serially washed at 55°C with a set of SSC buffers of decreasing strength, the final strength being 0.2× and then treated with RNase A (12.5 μg/ml) at 37°C for 30 min. The sections were exposed to x-ray film (Kodak BioMax MR) for 2 d and were dipped in nuclear emulsion (Kodak NTB) for exposure for 3–4 weeks. Images were collected with a digital camera attached to a microscope.
Western blot analysis.
To confirm loss of GluN2B protein levels in the CA1 hippocampal subregion of the mutants, tissue was taken from sections obtained from the same mice as used for the electrophysiological experiments below. Dorsal and ventral CA1 regions were rapidly dissected from 500 μg coronal vibratome slices and frozen immediately on dry ice. Tissue was homogenized by sonication in protease and phosphatase inhibitors (Sigma protease inhibitor cocktail and phosphatase inhibitor cocktails 1 and 2, 10 μm NaF, 1% Triton X-100, and 25 mm Tris, pH 6.8) by sonication, and protein concentration was determined by the BCA method. Protein was diluted to 1.25 μg/ml and then mixed with 4× sample buffer (Laemmli, 1970) to yield a final concentration of 1 μg/ml. β-Mercaptoethanol was added to a final concentration of 5% (v/v), and samples were boiled for 5 min. Thirty micrograms of protein was subjected to discontinuous pH 7.5% SDS-PAGE, pH 8.3, with a 4% stacking gel, pH 6.8 (Laemmli, 1970), using a triple-wide electrophoresis apparatus (CBS Scientific). Proteins were transferred to polyvinylidene difluoride membranes for 3 h at 400 mA as described for nitrocellulose membranes (Towbin et al., 1979). Blots were washed in TTBS (150 mm NaCl, 25 mm Tris, and 0.05% Tween 20, pH 7.3) and blocked for 1 h in 5% nonfat powdered milk. A GluN2B-specific antibody (AB1557P; Millipore Corporation) was used at 1:1000. Blots were incubated overnight in primary antibody, washed three times in TTBS, and then incubated in HRP-conjugated goat anti-rabbit (Pierce) for 1 h. After three washes, immunoreactivity was detected using SuperSignal West Dura chemiluminescence detection reagent and collected using a Kodak Image Station 1000. Net intensity values were determined using the Kodak 1D software and were normalized to total tubulin (1:1000; Cell Signaling Technology) immunodetected as described above. Data were normalized to control genotype levels and compared across genotypes by t tests.
To perform a more complete analysis of protein levels of NMDAR subunits and their CaMKII isoform binding partners, we took punches from ventromedial prefrontal cortex from experimentally naive 8- to 14-week-old mice and froze them immediately on dry ice. Samples were sonicated in 400 μl of 2% SDS containing 10 μg/ml leupeptin and 1 μg/ml pepstatin. Homogenates were diluted to a final concentration 1 mg/ml, as determined using a Pierce BCA protein assay, and then heated at 60°C for 10 min. Samples were separated by PAGE and then transferred to nitrocellulose membranes in 10 mm _N_-cyclohexyl-3-aminopropanesulfonic acid and 10% MeOH, pH 11.0. Membranes were stained for total protein in each lane using Ponceau-S, digitally scanned, and then washed and blocked in 5% milk in TTBS [50 mm Tris-HCl, pH 7.4, 200 mm NaCl, and 0.1% (v/v) Tween 20] at 4°C overnight. Membranes were then incubated at 4°C overnight with primary antibody diluted in 5% milk in TTBS as follows: mouse anti-GluN2B (1:250; catalog #610417; BD Transduction Laboratory); rabbit anti-GluN2A (1:1000; catalog #AB1555P; Millipore Corporation); mouse anti-GluN1 (1:1000; catalog #556308; BD Pharmingen); mouse anti-CaMKIIα (1:4000; catalog #MA1-048; Thermo Fisher Scientific); and mouse anti-CaMKIIβ (1:4000; catalog #13-9800; Zymed). After washing four times for 10 min in TTBS, membranes were incubated for 1.5 h with secondary antibodies diluted 1:2000–1:5000 in 5% milk in TTBS: donkey anti-mouse HRP (catalog #SC-2096; Santa Cruz Biotechnologies); donkey anti-rabbit HRP (catalog #SC-2077; Santa Cruz Biotechnologies); and goat anti-mouse IgG1–HRP (catalog #SC-2969; Santa Cruz Biotechnologies). After washing four times for 10 min in TTBS, signals were detected using Western Lighting Enhanced Luminol Reagent-Plus (GE Healthcare Plus-ECL). x-ray films exposed in the linear range were digitally scanned. Densitometry of the digital scans of Ponceau-S-stained membranes and x-ray films was performed using NIH ImageJ software. Western blot signals were normalized to the Ponceau-S protein stain, essentially as described previously (Brown et al., 2005). Data were normalized to control genotype levels and compared across genotypes using t tests.
Hippocampal electrophysiology.
For electrophysiological experiments, experimentally naive mice were anesthetized by halothane or isoflurane inhalation at 14–22 weeks of age, and the brain was rapidly removed and placed in ice-cold cutting solution (in mm): 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 glucose, pH 7.3 (equilibrated with 95% O2 and 5% CO2, mOsm 320). Hippocampal coronal slices were cut with a Vibratome 1000 series or an Integraslice 7550 (Campden Instruments) and incubated in artificial CSF (aCSF) (in mm): 124 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, 10 glucose, and 2 CaCl2, pH 7.3 (equilibrated with 95% O2 and 5% CO2 at 34°C, for 30 min and then at room temperature for at least 30 min before starting experiments).
NMDAR-mediated synaptic currents.
NMDAR-mediated evoked EPSC (eEPSC) amplitude and decay time constant were measured via whole-cell patch-clamp electrophysiological recordings from CA1 pyramidal neurons in brain slices. Slices were transferred onto a nylon net in a recording chamber and superfused with aCSF at a rate of 2 ml/min and at a temperature of 34°C. Whole-cell patch-clamp electrophysiological recordings from CA1 pyramidal neurons were performed with an Axopatch 200-B amplifier (Molecular Devices). Patch microelectrodes were pulled from borosilicate capillaries with filament (1.5 mm outer diameter; WPI) with a horizontal Flaming-Brown puller (model P-97; Sutter Instruments) using a four-step program. Electrode resistances were 3–5 MΩ when filled with a solution containing the following (in mm): 120 CsMeSO3, 5 NaCl, 10 tetraethylammonium-Cl, 10 HEPES, 5 QX-314 [2(triethylamino)-_N_-(2,6-dimethylphenyl) acetamine], 1.1 EGTA, 0.3 Na-GTP, and 4 Mg-ATP, pH 7.2 (adjusted with CsOH, 300 mOsm). NMDAR-mediated eEPSCs were recorded from CA1 hippocampal pyramidal neurons under conditions in which GABAA and AMPA receptors were inhibited by inclusion of picrotoxin (50 μm) and 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[_f_]quinoxaline (NBQX) (5 μm) in the aCSF (GABAB receptor-activated K+ currents are blocked by the Cs-based internal solution). Picrotoxin and NBQX were obtained from Sigma-Aldrich. The neuronal membrane potential was clamped at −30 mV. Access resistance varied between 10 and 25 MΩ, and, if it changed by ≥20% during an experiment, the recording was discarded. eEPSCs were evoked at 10 s intervals by stimulation of Shaffer collaterals with a concentric bipolar stainless steel and platinum–iridium electrode (core electrode diameter of 25 μm; FHC) placed in the stratum radiatum. Current was filtered at 2 kHz and digitized at 5.5 kHz with pClamp 9.2 software (Molecular Devices).
NMDAR-mediated eEPSC amplitude and decay time constant were analyzed using Clampfit 9.2 software. Amplitude was measured as the difference between measurements with cursors placed in the prestimulus baseline period and the eEPSC peak. Decay time constant was estimated from single-exponential fits to the decay phase of the current starting just after the current peak. Genotype effects on eEPSC decay times were analyzed using t test.
For pharmacological studies, we evaluated the effect of application of the GluN2B-selective antagonist Ro 25-6981 maleate [_R_-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol] (dissolved in DMSO to a final concentration of 0.01%, 0.1 μl/ml) on NMDAR-mediated eEPSCs recorded from CA1 pyramidal neurons after applying 1 μm of the compound and measuring the effects of the drug on peak amplitude and decay kinetics. This concentration was chosen to provide effective inhibition while retaining specificity for GluN2B over GluN2A, based on previous studies (Chazot et al., 2002; Berberich et al., 2007). The subunit-nonspecific NMDAR antagonist dl-AP-5 (50 μm, from a 50 mm stock in dH2O) was applied to determine whether eEPSCs were truly NMDAR mediated. Drugs were obtained from Tocris Bioscience. Drug effects on eEPSC amplitude were compared with baseline using Bonferroni's corrected paired t test.
Subsaturating hippocampal LTP.
Synaptic transmission and plasticity in the CA1 hippocampal subregion was evaluated via field potential recordings. Brain slices were placed onto a nylon mesh in a Teflon/Plexiglas recording chamber (Warner Instruments) and superfused with aCSF at a rate of 2–3 ml/min. Recording temperature was 26–28°C, stable within 1°C during a recording. Schaffer collateral afferents were activated by a twisted bipolar electrode fabricated from Teflon-coated tungsten wire (0.003 inch diameter; A-M Systems). Field EPSPs (fEPSPs) were recorded using a patch pipette (resistance of 2–3 MΩ) filled with 0.9% saline and placed in stratum radiatum. For input–output and paired-pulse experiments, as well as baseline and post-high-frequency stimulation (HFS) recordings in LTP experiments, stimulation was delivered at 0.1 Hz, at a stimulus intensity that evoked the maximal amplitude fEPSP that was uncontaminated by a population spike. Input–output curves were constructed by applying stimuli of increasing intensity with responses to three stimuli recorded at each intensity.
To evoke synaptic plasticity, a 10 min period of baseline stimulation at 0.1 Hz was followed by two trains of 100 Hz stimulation delivered once every 10 s at a stimulus intensity marginally suprathreshold for the population spike. Stimulation at 0.1 Hz was maintained in the interval between trains and throughout the poststimulation period. The amplitude of the fiber volley was measured as the difference between cursors placed in the prestimulus baseline period and the peak of the fiber volley. The fEPSP slope was measured as the linear slope of the rising phase of the response between time points at which the response amplitude was 20 and 50% of the maximal amplitude for each fEPSP in the input–output and paired-pulse experiments and 20–50% of the post-HFS response in LTP experiments. Time effects on fEPSP slope were analyzed for this and the other LTP and LTD experiments using repeated-measures ANOVA.
Saturating hippocampal LTP.
The procedure was the same as above with two exceptions. First, the stimulation frequency during the baseline, interstimulation and poststimulation periods was 0.033 Hz. Second, we applied three, rather than just one, sets of the two trains of 100 Hz stimulation and delivered these 20 min apart. The rationale for these changes was that in some, but not all, previous studies, differences in baseline stimulation (Fonseca et al., 2006) and train number (Kiyama et al., 1998) have been associated with differences in the strength of LTP.
Hippocampal LTD.
We assessed hippocampal LTD at an adult age corresponding to our other physiological, morphological, and behavioral phenotypic measures. Brain slices were prepared as above for the LTP experiments. Baseline stimulation was delivered 160 s in aCSF using a stimulus intensity that evoked the maximal amplitude fEPSP that was uncontaminated by a population spike. Given the difficulty of producing hippocampal LTD in adults by low-frequency stimulation (LFS) alone, we induced LTD by combining 1 Hz, 900 s stimulation with application of the glutamate transporter inhibitor _L_-trans-pyrrolidine-2,_4_-dicarboxylic acid (tPDC) (Massey et al., 2004; Duffy et al., 2008). After applying tPDC in aCSF (300 μm) and allowing it to equilibrate for 10 min at baseline frequency, we delivered 15 min of 1 Hz stimulation (i.e., LFS) at baseline stimulus intensity in the continued presence of tPDC. After an additional 5 min at baseline stimulation frequency, tPDC was washed out with aCSF, and recording continued for 60 min.
In a separate experiment to confirm that LTD was NMDAR dependent, the same procedure was used with the exception that the NMDAR antagonist dl-AP-5 was coapplied with tPDC before LFS. To confirm that LTD could be induced in these brain slices, both drugs were washed out, and then tPDC was reapplied alone, before a second round of LFS.
Dendritic spine density and morphology.
Dendritic spine density and morphology in CA1 neurons was examined by quantifying spines in neurons ballistically labeled with fluorescent dye 1-1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate (DiI) (Grutzendler et al., 2003) in 14- to 22-week-old mice. Coronal brain slices (200 μm) were cut on a vibratome (Leica VT 1200S) in a modified aCSF, in which NaCl was replaced by sucrose (as for the electrophysiological experiments) given evidence that this cold cutting solution prevents unwanted morphological changes seen with cold aCSF (Kirov and Harris, 1999). Slices were then immediately fixed in 4% paraformaldehyde/4% sucrose solution for 30 min at room temperature and washed thoroughly with PBS. Particle-mediated ballistic delivery of fluorescent dyes was performed based on methods described previously (Grutzendler et al., 2003; Lee et al., 2006).
Briefly, neurons were labeled by shooting the slices with tungsten beads (1.7 μm in diameter; Bio-Rad) coated with DiI (Invitrogen) using a biolistic Helios gene gun (Bio-Rad) (180 psi helium gas pressure) through a membrane filter with a 3 μm pore size (Millipore Corporation). Slices were rinsed with PBS and mounted on slides using ProLong Antifade Gold (Invitrogen). A confocal microscope (Carl Zeiss LSM 5 Pascal) was used to acquire a stack of images (z spacing of 0.8 μm) of the apical dendrites from isolated CA1 pyramidal neurons using a 63× oil objective (numerical aperture 1.4) at 1.0 μm optical section. Regions with dense DiI staining in which individual neurons could not be distinguished were avoided. Three to five dendrites per cell were imaged (512 × 512 pixels; x–y scaling, 0.0952 μm/pixel) and at least three cells per mouse collected. All dendrites were secondary branches from the apical dendritic tree and of a similar distance from the cell body across genotypes.
Dendritic spine morphology was analyzed using ImageViewer (Steiner et al., 2008), a custom software for spine analysis written in Matlab (MathWorks). Spine density was determined by manually identifying spines and using the three-dimensional images to measure dendrite length. The effect of genotype on spine density was measured at the level of individual spines as well as cell averages. Spine length was measured from the dendritic shaft junction to the spine tip. To measure spine head width, a line was drawn across the thickest part of the spine head, parallel to the dendrite, and the fluorescence distribution along this line was determined. Width was defined as width at which fluorescent intensity fell to 30% of maximum.
Spine length and width were plotted by displaying the mean ± SEM of the average distributions for each dendritic field image in control or mutant. Given that images varied in the total number of spines present, it is not possible to directly average the distribution plots. Each of the cumulative distributions was resampled every 2.2% to make a distribution of 45 points in all cases, and the mean and SEM of all control or mutant distributions was calculated by averaging the 45 points. The effect of genotype on spine head width and spine length was measured using the Kolmogorov–Smirnov (K–S) test to statistically capture genotype differences in distribution and (because parametric assumptions for normalcy and homogeneity were violated) Mann–Whitney U test.
Behavioral phenotyping.
Mice were phenotyped for multiple forms of learning and memory at 8–14 weeks of age.
Functional observation battery.
To assess gross behavioral or neurological abnormalities that might confound testing for learning and memory, mice were placed in a bare empty home cage and observed for the presence of freezing, trembling, wild running, grooming, sniffing, licking, rearing, jumping, spontaneous seizure, defecation, urination, head bobbing, circling, abnormal gait, and retropulsion over 60 s (Boyce-Rustay and Holmes, 2006). Basic physical health was evaluated by examining for missing whiskers, bald patches, exophthalmus, straub tail, kinked tail, kyphosis, lordosis, body weight, and core body temperature. Simple sensory reflexes were measured via orienting responses to an approaching hand and to physical touch and via palpebral closure on touch of the eye, twitch of the pinna on touch, and an orienting response to tail pinch. Basic motor and neurological function was assessed by observing for splayed limbs, forepaw clutch, and hindlimb clutch when mice were suspended upside down by the tail. To measure grip strength, the mouse was placed on a grid surface made of 2-mm-diameter metal rods running lengthwise at 10 mm intervals, which was then slowly rotated, and the latency of the mouse to fall was manually recorded (60 s maximum).
Morris water maze.
The MWM tested for corticohippocampal-mediated spatial learning based on methods described previously (Holmes et al., 2002; von Engelhardt et al., 2008). The apparatus was a 164-cm-diameter circular pool filled with 24 ± 1°C water made opaque with white Crayola play paint in which a 15.5-cm-diameter circular escape platform was submerged 0.5 cm below the water at one of four positions equidistant (30 cm) from the edge of the pool. Extramaze cues were positioned around the pool. The Ethovision video-tracking system was used to measure the latency to find the platform, average swimming speed, time spent within 10 cm of the perimeter (thigmotaxis), and percentage time spent in the trained platform-quadrant during the probe trial.
For the hidden platform task, mice were placed at one of four pseudorandomly chosen start positions and given up to 60 s to find the platform that was hidden in a fixed location for a given mouse (counterbalanced across genotypes). Mice that failed to find the platform were manually guided there. Once on the platform, mice remained there for 15 s. There were four trials per day (30 min intertrial interval) over seven sessions. Twenty-four hours after hidden platform task sessions 5 and 7 mice were placed in the pool in the opposite quadrant to the trained platform location with the platform absent for 60 s.
For the visible platform task, the procedure was the same as for hidden platform task with the exception that platform was made visible by a protruding cue and its location was moved on each trial in a pseudorandom order between each of four possible quadrant locations. There were four trials per day (30 min intertrial interval) over three sessions.
Genotype × session effects on latencies, thigmotaxis, and swim speed in the hidden and visible tasks were analyzed using two-factor ANOVA with repeated measures for session. Paired t tests were used to compare time spent in the target quadrant versus (averaged) nontarget quadrants in the probe trials.
Discrete trial T-maze spontaneous alternation.
Corticohippocampal-mediated spatial working memory was tested using a discrete-trial spontaneous alternation T-maze task based on methods described previously (von Engelhardt et al., 2008). The apparatus was an enclosed T-shaped maze constructed of black Plexiglas. The start arm was 8.5 × 10.5 × 33.0 cm, and the two choice arms were each 8.5 × 10.5 × 30.0 cm. Black Plexiglas dividers were inserted into the start arm to create a start box and placed at the entrance of a choice arm to sequester mice in the arm after a choice was made. Each trial commenced with mice placed in the start box for 5 s. The insert was then removed to allow mice to run the start arm and select a choice arm (choice was all four paws in an arm), in which they were confined for 30 s (sample phase). Mice were returned to the start box for 5 s and then allowed to make another choice (choice phase). There were 10 trials (i.e., sample + choice phases), with a 15 min intertrial interval. Testing was conducted under normal room lighting. Spontaneous alternation was compared across genotypes and compared with chance (50%) using t tests.
Trace and delay pavlovian fear conditioning.
Corticohippocampal-mediated trace pavlovian cued fear conditioning was assessed in one group of mice, and non-corticohippocampal-dependent delay cued fear conditioning was tested in another. Conditioning was conducted as described previously (Kim et al., 1995; Huerta et al., 2000; Misane et al., 2005; Feyder et al., 2007; Yang et al., 2008) in a 27 × 27 × 11 cm chamber with white walls and a metal rod floor. To provide a distinctive olfactory environment, the chamber was cleaned between subjects with a 79.5% water/19.5% ethanol/1% vanilla extract solution. After an initial 180 s acclimation period, the mouse received three pairings (60–120 s interval after each pairing) between the conditioned stimulus (CS) (30 s, 80 dB, 3 kHz white noise auditory tone) and the unconditioned stimulus (US) (2 s, 0.6 mA scrambled footshock). For the trace paradigm, the US was presented 30 s after the end of the CS. For the delay paradigm, the US was presented during the last 2 s of the CS. The presentation of stimuli was controlled by the Med Associates Video Freeze Monitor system.
For both conditioning paradigms, memory for the CS was tested in a novel context in a different room from training 24 h after conditioning. The novel context was a 27 × 27 × 11 cm chamber with black and white checkered walls and Plexiglas black and white checkered floor. This chamber was cleaned between subjects with a 50% ethanol/50% water solution. After an initial 180 s acclimation period, the CS was continuously presented for 180 s. Freezing was defined as absence of any visible movement except that required for respiration and was scored at 5 s intervals by an observer who was blind to genotype. The number of observations scored as freezing were converted to a percentage [(number of freezing observations/total number of observations) × 100] for analysis. Freezing was compared across genotypes using t tests.
Results
Conditional corticohippocampal GluN2B deletion
In situ hybridization on 12-week-old mice confirmed GluN2B mRNA loss in the dorsal CA1 hippocampal subregion and parahippocampal regions including entorhinal cortex, as well as the entire neocortex (Fig. 1A–E,G). Residual expression in these areas was likely attributable to GluN2B mRNA in interneurons. mRNA distribution in dorsal CA3/dentate gyrus, and ventral hippocampus and other forebrain regions, including striatum and amygdala, was normal in mutants (Fig. 1A,B,F,H). The distribution of GluN1 mRNA was normal in mutants, as measured by in situ hybridization (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Western blot showed significantly reduced protein levels of GluN2B in homogenate tissue taken from the dorsal (but not ventral) CA1 hippocampal subregion of mutants compared with controls (t = 2.31, df = 14, p < 0.05) (Fig. 1I). Western blot of cortical homogenate tissue also showed significantly reduced protein levels of GluN2B in mutants, as well as reduced GluN1 but no change in GluN2A, CaMKIIα, or CaMKIIβ (Fig. 1J) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Faster decaying NMDAR-mediated eEPSCs and insensitivity to GluN2B antagonist
NMDAR–eEPSC decay time constants were almost twice as fast in mutants as controls (t = 2.17, df = 22, p < 0.05) (Fig. 2A), whereas amplitude was slightly reduced in the mutants (Fig. 2B) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material).
Figure 2.

Faster decaying NMDAR-mediated eEPSCs at CA1 hippocampus synapses. A, Decay time constant data showing decreased NMDAR-mediated EPSC tau for mutants compared with controls (n = 8–14 per genotype) (blue lines represent a single-exponential fit to the decay phase of the eEPSCs) (*p < 0.05 vs control). B, Peak amplitude of eEPSCs was not significantly different between genotypes (n = 8–14 per genotype). C, The selective GluN2B antagonist Ro 25-6981 (1 μm) reduced NMDAR-mediated eEPSCs in controls (Con), not mutants (Mut). The subunit-nonselective NMDAR antagonist dl-AP-5 (50 μm) reduced NMDAR-mediated eEPSCs equally in controls and mutants (n = 8–14 per genotype) (*p < 0.05 vs baseline/same genotype). Data are means ± SEM.
dl-AP-5 inhibited eEPSCs to a similar extent in mutants (t = 10.03, df = 4, p < 0.01) and controls (t = 14.91, df = 7, p < 0.01) (Fig. 2C). Ro 25-6981 produced a modest but significant inhibition in controls (t = 4.00, df = 13, p < 0.01) as a result of sensitivity in a subpopulation of the cells (supplemental Fig. S4, available at www.jneurosci.org as supplemental material) but was without effect in mutants (Fig. 2C).
Partial deficiency of hippocampal LTP
NMDAR-mediated synaptic transmission in the CA1 hippocampal subregion was evaluated via field potential recordings (fEPSPs) evoked by stimulation of Schaffer collateral/commissural fibers in 14- to 22-week-old mice.
A protocol designed to evoke modest, subsaturating LTP produced weak but significant lasting increase in field EPSP slope in controls (time effect, F(34,170) = 2.45, p < 0.01) (Fig. 2A) but was without effect in mutants (time effect, NS) (Fig. 3A). Genotypes did not differ in paired-pulse ratio or input–output relationship (supplemental Fig. S5, available at www.jneurosci.org as supplemental material).
Figure 3.

Partially impaired hippocampal LTP and hippocampal LTD. A, HFS (2 100-Hz/s trains) produced subsaturating LTP in controls but not mutants (n = 5–6 per genotype) (arrow denotes stimulation). B, Representative traces and data showing three trains of HFS (2 100-Hz/s trains) produced saturating LTP in mutants and controls (n = 5–6 per genotype). C, Representative traces and data showing LFS (1 Hz/s) combined with the glutamate transporter blocker tPDC application produced LTD in controls, not mutants (n = 6–8 per genotype). D, LTD produced by LFS plus tPDC was blocked by application of AP-5 (n = 6–7 per genotype). Arrows denote LFS. Thin line denotes application of tPDC alone. Thick line denotes application of tPDC plus dl-AP-5. Time in C and D is from last stimulation. Data are means ± SEM.
Using a protocol designed to produce saturating LTP, the first set of trains produced a significant lasting increase in fEPSP slope that was similar between genotypes, although there was a decrease in fEPSP during the first time point after stimulation in controls but not mutants (genotype × time interaction, F(31,279) = 2.00, p < 0.01) (Fig. 3B). A significant lasting increase in fEPSP slope was also produced by the second (time effect, F(31,279) = 5.35, p < 0.01) (Fig. 3B) and third (time effect, F(31,248) = 3.24, p < 0.01) (Fig. 3B) set of trains, and again this did not differ between genotypes.
Loss of NMDAR-dependent hippocampal LTD
Our LTD protocol produced a significant lasting decrease in fEPSP slope in controls (time effect, F(29,203) = 4.64, p < 0.01) but not mutants (Fig. 3C). To determine whether this form of LTD was NMDAR mediated, we repeated the protocol with dl-AP-5 coapplied with tPDC. After stimulation, there was a transient decrease in fEPSP slope in both genotypes. Controls failed to show a lasting decrease in slope, whereas mutants showed a significant lasting increase (genotype × time effect, F(27,270) = 1.90, p < 0.01) (Fig. 3D). After AP-5 washout and another round of stimulation, there was a significant lasting decrease in fEPSP slope in controls (time effect, F(23,115) = 5.61, p < 0.01) but not mutants (Fig. 3D), replicating our previous observation. In a separate experiment, application of tPDC alone was sufficient produce LTD in controls but again not in mutants (supplemental Fig. S6, available at www.jneurosci.org as supplemental material).
Decreased dendritic spine density
Spine density of dendrites in CA1 pyramidal neurons was significantly lower in mutants than controls (control, 1.15 ± 0.04; mutant, 0.96 ± 0.03, Mann–Whitney U = 40.00, p < 0.05). This was apparent when spine density was averaged by either dendrite (K–S test, D = 0.395, p < 0.01, n = 34–62) (Fig. 4C) or neuron (K–S test, D = 0.567, p < 0.05, n = 10–16) (Fig. 4D). Average spine head width did not differ between genotypes (Fig. 4E), although there was a nonsignificant trend for spines to be longer in the mutants (Fig. 4F).
Figure 4.
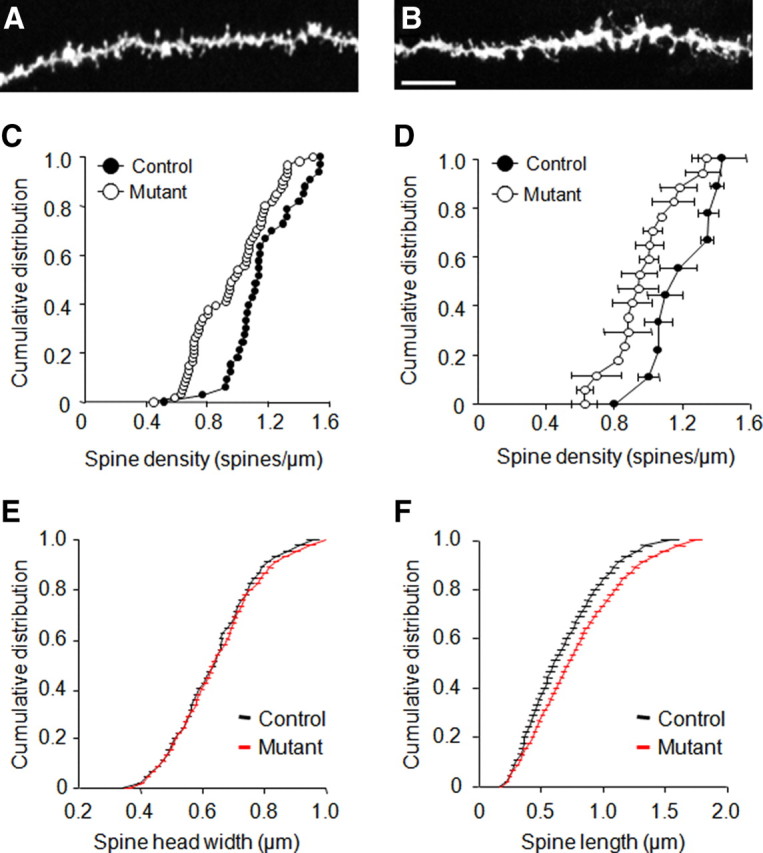
A, B, Decreased hippocampal neuron dendritic spine density. Example of labeled spines along apical dendrites ballistically labeled with fluorescent dye DiI in controls (A) and mutants (B). Scale bars, 7 μm. C, D, Mutants showed decreased spine density relative to controls as depicted at the level of individual dendrites (C) and neuron averages (D). E, F, Neither spine head width (E) nor spine length (F) distributions significantly differed between genotypes. n = 4–5 per genotype (10–18 neurons per genotype, 34–62 dendrites per genotype). Data are means ± SEM protrusions.
Impaired corticohippocampal-mediated learning and memory
During hidden platform training, mutants had significantly longer latencies to find a hidden platform than controls (genotype effect, F(1,14) = 18.13, p < 0.01; session effect, F(6,84) = 13.78, p < 0.01; genotype × session, NS) (Fig. 5A). During probe tests, controls spent significantly more time in the platform quadrant location than non-platform quadrants on the second (t = 3.30, df = 7, p < 0.05) but not first probe, whereas mutants showed no preference on either probe (Fig. 5B). Swim speed was significantly lower (genotype effect, F(1,14) = 9.63, p < 0.01; session effect, F(6,84) = 7.43, p < 0.01; genotype × session, NS) (supplemental Fig. S7_A_, available at www.jneurosci.org as supplemental material) and thigmotaxis was higher (genotype, F(1,14) = 4.75, p < 0.05; session, F(6,84) = 16.21, p < 0.01; genotype × session, NS) (supplemental Fig. S7_B_, available at www.jneurosci.org as supplemental material) in mutants than controls across training sessions.
Figure 5.

Cognitive deficits. A, Mutants showed longer latencies to find a hidden platform in the Morris water maze than controls (n = 8 per genotype). B, Controls (Con) showed significant preference of the target quadrant over nontrained quadrants during a second, but not first, probe trial transfer test, whereas mutants (Mut) showed no preference on either (*p < 0.05 vs nontarget). C, Latencies to find a visible platform in the Morris water maze was not different between genotypes (n = 6 per genotype). D, Controls, but not mutants, showed significant spontaneous alternation (relative to chance) in a discrete-trial T-maze task (#p < 0.05 vs 50%/chance) (n = 6 per genotype). E, Mutants showed significantly less conditioned (tone) freezing after trace fear conditioning (*p < 0.05 vs control) (n = 32–33 per genotype). F, Genotypes did not differ in conditioned (tone) freezing after delay fear conditioning (n = 9 per genotype). Data are means ± SEM.
During visible platform training, mutants learned to find the platform as well as controls (genotype effect, NS; session effect, F(2,14) = 37.26, p < 0.01; genotype × session, NS) (Fig. 5C). Neither swim speed nor thigmotaxis differed between genotypes (supplemental Fig. S7_C_,D, available at www.jneurosci.org as supplemental material), indicating that differences in these behaviors on the hidden platform task were likely a consequence of the failure of the mutants to acquire the task rather than a more generalized behavioral impairment. Mutants showed no obvious physical, neurological, or gross behavioral abnormalities (supplemental Table S1, available at www.jneurosci.org as supplemental material).
On the T-maze, controls spontaneously alternated significantly above chance (t = 4.25, df = 7, p < 0.01 vs 50%), whereas mutants did not (Fig. 5D).
Mutants froze significantly less than controls during retrieval of a trace-conditioned fear memory (t = 10.69, df = 63, p < 0.01) (Fig. 5E) but not delay-conditioned fear memory (Fig. 5F). Genotypes did not differ in freezing during conditioning or before tone presentation in either task.
Discussion
We successfully generated viable mice with postdevelopmental GluN2B deletion in principal neurons of the dorsal CA1 subregion of the hippocampus and the entire neocortex, providing an unique tool for addressing the contribution of GluN2B in these cells to synaptic and behavioral plasticity in the adult brain.
These mutants retained normal NMDAR-mediated eEPSC amplitude at CA1 hippocampal synapses, but decay time constants were almost half that of controls. Furthermore, although the NMDAR antagonist dl-AP-5 markedly inhibited hippocampal eEPSCs in the mutants, the GluN2B-selective antagonist Ro 25-6981 was without affect. Similar changes were recently reported in mice with forebrain-wide GluN2B deletion (von Engelhardt et al., 2008) and together affirm the contribution of GluN2B to NMDAR kinetics but not overall NMDAR-mediated synaptic function (Kash and Winder, 2007). Although we found no compensatory increases in GluN2A protein levels and no loss of GluN1 mRNA by in situ hybridization, the total levels of GluN1 protein in tissue homogenates were significantly reduced in the mutants. Together with our electrophysiological data indicating no change in synaptic NMDAR function, this could suggest that deletion of GluN2B causes a loss of nonsynaptic GluN1. Indeed, previous work has shown that the synaptic pool of GluN1 subunits is more stable than an intracellular pool of GluN1 that does not appear to be associated with GluN2 subunits (Huh and Wenthold, 1999). Thus, absent the normal association with GluN2B, the “free” intracellular pool of GluN1 subject to more rapid degradation may be greater. Similar interactions involving subunit stability have been observed after deletion of specific subunits of other hetero-multimeric protein complexes (e.g., protein phosphatase 2A) (Silverstein et al., 2002). However, additional studies will be needed to more fully understand these mechanisms in GluN complexes.
GluN2B antagonism impaired hippocampal or cortical LTP in some previous studies (Köhr et al., 2003; Berberich et al., 2005; Fox et al., 2006; Berberich et al., 2007) but had partial (Köhr et al., 2003; Zhao et al., 2005) or no effect (Liu et al., 2004; Massey et al., 2004) in others. However, these data were sometimes obtained in young mice, when GluN2B-to-GluN2A expression is higher than in adulthood. A recent study found that GluN2B deletion in adult CA3 hippocampal principal neurons abolished LTP in these neurons (Akashi et al., 2009). However, these mice also showed significant loss of GluN1 and NMDAR-mediated EPSCs. Forebrain-wide GluN2B deletion produced only modest diminution of adult CA1 hippocampal LTP induced by a single HFS protocol (von Engelhardt et al., 2008). Consistent with the latter finding, our mutants showed loss of potentiation in a procedure that produced subsaturating LTP in control mice but normal LTP with a saturating induction protocol. These protocols differed only in baseline stimulation frequency, and, although previous evidence has suggested that this can affect LTP (Fonseca et al., 2006), it is not fully clear why this is so or why loss of GluN2B would selectively affect the “subsaturating” form. Our data are, however, generally consistent with an increase in LTP threshold in the mutants. This could be a consequence of decreased calcium entry from shortened NMDAR synaptic currents. It could also reflect disruption of the anchoring of CaMKII to the NMDAR complex, an interaction that has been shown to modulate NMDAR activity in heterologous cells (Sessoms-Sikes et al., 2005) and LTP in cultured hippocampal slices (Barria and Malinow, 2005). However, we did not find reduced total levels of either CaMKIIα or CaMKIIβ in the mutants.
There has been considerable debate about the necessity of GluN2B for LTD. Previous studies supporting such a role have been based on pharmacological or neonatal data that have important limitations (Kutsuwada et al., 1996; Zhao et al., 2005; Fox et al., 2006; Morishita et al., 2007; Duffy et al., 2008), namely inefficacy at triheteromeric NMDARs and high GluN2B expression, respectively. The difficulty of inducing LTD in the adult hippocampus has confounded efforts to tackle this question. By combining LFS with application of a glutamate transporter (tPDC) to increase synaptic glutamate (possibly causing spillover onto extrasynaptic GluN2B-containing NMDARs) (Massey et al., 2004; Duffy et al., 2008), we produced significant lasting synaptic depression. Interestingly, we also found that tPDC alone may be sufficient to produce LTD in controls and that this effect was absent in mutants, possibly further reflecting a consequence of loss of extrasynaptic GluN2B for this form of LTD. Another noteworthy observation was that the NMDAR antagonist AP-5, which blocked LTD in control mice, appeared to unmask a degree of LTP in the mutants (hinting at GluN2B modulation of an NMDAR-independent form of LTP in this preparation).
This form of NMDAR-dependent LTD was absent in the GluN2B mutants, providing some of the clearest evidence to date that loss of GluN2B impairs LTD in the adult hippocampus and helping to resolve the issue of the contribution of GluN2B, versus GluN2A, to LTD. Interestingly, mutants arguably showed signs of weak LTP under these LTD-inducing conditions. Indeed, a similar phenomenon was seen under the same conditions after GluN2B antagonist application (Massey et al., 2004; Duffy et al., 2008). One interpretation of these data is that this protocol can produce some LTP but that this is normally masked by the much larger NR2B-dependent LTD. Such LTP could be driven by non-NMDAR mechanisms (e.g., dopaminergic) (Shen et al., 2008). This could account for the trend we observed for LTP (under LTD-inducing conditions) in both mutants and controls in the presence of the NMDAR antagonist AP-5. Nonetheless, the main conclusion of the current work is that this form of LTD is absent in mice lacking GluN2B.
Most excitatory synaptic transmission occurs at dendritic spines, and, in those, spine number and morphology are a readout of the functional state of glutamatergic synapses (Alvarez and Sabatini, 2007). GluN2B mutants had reduced spine density in apical dendrites of CA1 neurons and a trend for increased length in extant spines. This finding is consistent with the reduced spine density found in cultured hippocampal neurons after GluN2B RNA interference (Kim et al., 2005) and in apical spine density in CA3 hippocampal neurons, ex vivo, after GluN2B deletion restricted to this subregion (Akashi et al., 2009). Collectively, these findings point to a significant contribution of GluN2B-containing NMDARs to either the formation and/or long-term stability of dendritic spines. The precise nature of this role is not fully clear, but it may reflect functional interactions between GluN2B and actin cytoskeleton proteins that regulate the motility and maintenance of spines. Because spines can be rapidly upregulated after acute slicing (Kirov and Harris, 1999), a caveat we cannot fully exclude is that the genotype differences are attributable to differential responses to this procedural trauma. However, in vivo spine differences would relate to our physiological data, given evidence that LTD is associated with shrinkage of spines (Zhou et al., 2004). One possibility is that GluN2B deletion produces a state of tonic synaptic depression that leads to loss of spines. This could also account for the occlusion of additional LTD and the increased threshold for LTP we observed in the mutants.
Morphological and plasticity abnormalities caused by GluN2B loss were associated with selective learning deficits. Mutants were impaired on multiple learning assays that tax spatial reference (Morris water maze) or working memory (T-maze) or the associative bridging of discrete events over an intervening delay period (trace fear), which are known to require intact hippocampal, and to some extent cortical (Kesner and Rogers, 2004), function (Morris et al., 1990; Tsien et al., 1996a; Huerta et al., 2000; Nakazawa et al., 2004; Quinn et al., 2005; Zhao et al., 2005). In contrast, striatal- and amygdala-mediated learning was normal. These data show that GluN2B in principal neurons of the CA1 and the cortex is sufficient to impair spatial reference and working memory, without residual effects in non-corticohippocampal-mediated learning. It is particularly notable that this pattern of learning deficits is striking in the context of impaired LTD but only marginally deficient LTP, given that both forms of plasticity are posited to be key molecular correlates of learning. However, in lieu of additional studies (e.g., examining cortical plasticity), it would be premature to claim that this demonstrates a causative relationship between loss of hippocampal LTD and these cognitive deficits in the mutants.
Notwithstanding, the cognitive deficits we found in GluN2B phenocopy the effects of hippocampal lesions or intrahippocampal administration of NMDAR antagonists and GluN1 gene deletion in CA1 principal cells (Morris et al., 1990; Tsien et al., 1996a; Nakazawa et al., 2004). GluN2A deletion also impairs performance on certain of these and other tasks (Bannerman et al., 2008; Brigman et al., 2008). Recent work has also shown that forebrain-wide GluN2B deletion impaired performance on both hippocampal and extrahippocampal tasks, whereas GluN2B deletion in CA1 principal cells and dentate gyrus granule cells impaired some (spatial reversal and working memory) but not all (spatial reference) forms of hippocampal learning tested (von Engelhardt et al., 2008). These findings may be reconciled by a model in which either the combined loss of GluN2B loss in CA1 and cortex (and just not CA1) or the combined loss of both GluN2B and GluN2A (or GluN1 per se) in CA1 is necessary to produce spatial reference memory deficits.
In conclusion, the current study describes a novel mouse model of postdevelopment corticohippocampal GluN2B loss. We found that GluN2B shortened NMDAR-mediated synaptic currents, caused modest impairment in hippocampal LTP and significant impairment of a form of hippocampal LTD, decreased CA1 hippocampal neuronal dendritic spine density, and produced a profile of selective cognitive deficits. The findings suggest a major role for GluN2B in the LTD-mediated component of corticohippocampal memory formation and help elucidate the contribution of GluN2B to different forms of synaptic plasticity and learning.
Footnotes
This work was supported by the National Institute on Alcohol Abuse and Alcoholism and National Institute of Mental Health Intramural Research Programs, National Institute of Mental Health Grant RO1-MH063232 (R.J.C.), National Institute of Neurological Disorders and Stroke Grant F31-NS061537 (R.M.G.), and Integrative Neuroscience Initiative on Alcoholism Stress Consortium Grant U01 AA013514 (E.D.). We are grateful to Bai Lu and Yuan Lu for advice on the LTD protocol, Guoxiang Luo for genotyping, and Yoshihiro Kashiwaya for use of the confocal microscope.
References
- Akashi K, Kakizaki T, Kamiya H, Fukaya M, Yamasaki M, Abe M, Natsume R, Watanabe M, Sakimura K. NMDA receptor GluN2B (GluRε2/NR2B) subunit is crucial for channel function, postsynaptic macromolecular organization, and actin cytoskeleton at hippocampal CA3 synapses. J Neurosci. 2009;29:10869–10882. doi: 10.1523/JNEUROSCI.5531-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Rawlins JN, Good MA. The drugs don't work-or do they? Pharmacological and transgenic studies of the contribution of NMDA and GluR-A-containing AMPA receptors to hippocampal-dependent memory. Psychopharmacology (Berl) 2006;188:552–566. doi: 10.1007/s00213-006-0403-6. [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Niewoehner B, Lyon L, Romberg C, Schmitt WB, Taylor A, Sanderson DJ, Cottam J, Sprengel R, Seeburg PH, Köhr G, Rawlins JN. NMDA receptor subunit NR2A is required for rapidly acquired spatial working memory but not incremental spatial reference memory. J Neurosci. 2008;28:3623–3630. doi: 10.1523/JNEUROSCI.3639-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby Ø, Köhr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberich S, Jensen V, Hvalby Ø, Seeburg PH, Köhr G. The role of NMDAR subtypes and charge transfer during hippocampal LTP induction. Neuropharmacology. 2007;52:77–86. doi: 10.1016/j.neuropharm.2006.07.016. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Holmes A. Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology. 2006;31:2405–2414. doi: 10.1038/sj.npp.1301039. [DOI] [PubMed] [Google Scholar]
- Brigman JL, Feyder M, Saksida LM, Bussey TJ, Mishina M, Holmes A. Impaired discrimination learning in mice lacking the NMDA receptor NR2A subunit. Learn Mem. 2008;15:50–54. doi: 10.1101/lm.777308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Deutch AY, Colbran RJ. Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. Eur J Neurosci. 2005;22:247–256. doi: 10.1111/j.1460-9568.2005.04190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazot PL, Lawrence S, Thompson CL. Studies on the subtype selectivity of CP-101,606: evidence for two classes of NR2B-selective NMDA receptor antagonists. Neuropharmacology. 2002;42:319–324. doi: 10.1016/s0028-3908(01)00191-5. [DOI] [PubMed] [Google Scholar]
- Clayton DA, Mesches MH, Alvarez E, Bickford PC, Browning MD. A hippocampal NR2B deficit can mimic age-related changes in long-term potentiation and spatial learning in the Fischer 344 rat. J Neurosci. 2002;22:3628–3637. doi: 10.1523/JNEUROSCI.22-09-03628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- Duffy S, Labrie V, Roder JC. d-Serine augments NMDA-NR2B receptor-dependent hippocampal long-term depression and spatial reversal learning. Neuropsychopharmacology. 2008;33:1004–1018. doi: 10.1038/sj.npp.1301486. [DOI] [PubMed] [Google Scholar]
- Feyder M, Wiedholz L, Sprengel R, Holmes A. Impaired associative fear learning in mice with complete loss or haploinsufficiency of AMPA GluR1 receptors. Front Behav Neurosci. 2007;1:4. doi: 10.3389/neuro.08.004.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca R, Nägerl UV, Bonhoeffer T. Neuronal activity determines the protein synthesis dependence of long-term potentiation. Nat Neurosci. 2006;9:478–480. doi: 10.1038/nn1667. [DOI] [PubMed] [Google Scholar]
- Fox CJ, Russell KI, Wang YT, Christie BR. Contribution of NR2A and NR2B NMDA subunits to bidirectional synaptic plasticity in the hippocampus in vivo. Hippocampus. 2006;16:907–915. doi: 10.1002/hipo.20230. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Mauceri D, Malinverno M, Polli F, Costa C, Tozzi A, Siliquini S, Picconi B, Cattabeni F, Calabresi P, Di Luca M. Decreased NR2B subunit synaptic levels cause impaired long-term potentiation but not long-term depression. J Neurosci. 2009;29:669–677. doi: 10.1523/JNEUROSCI.3921-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Tsai J, Gan WB. Rapid labeling of neuronal populations by ballistic delivery of fluorescent dyes. Methods. 2003;30:79–85. doi: 10.1016/s1046-2023(03)00009-4. [DOI] [PubMed] [Google Scholar]
- Hawasli AH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, Greengard P, Powell CM, Cooper DC, Bibb JA. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci. 2007;10:880–886. doi: 10.1038/nn1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestrin S. Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature. 1992;357:686–689. doi: 10.1038/357686a0. [DOI] [PubMed] [Google Scholar]
- Holmes A, Wrenn CC, Harris AP, Thayer KE, Crawley JN. Behavioral profiles of inbred strains on novel olfactory, spatial and emotional tests for reference memory in mice. Genes Brain Behav. 2002;1:55–69. doi: 10.1046/j.1601-1848.2001.00005.x. [DOI] [PubMed] [Google Scholar]
- Huerta PT, Sun LD, Wilson MA, Tonegawa S. Formation of temporal memory requires NMDA receptors within CA1 pyramidal neurons. Neuron. 2000;25:473–480. doi: 10.1016/s0896-6273(00)80909-5. [DOI] [PubMed] [Google Scholar]
- Huh KH, Wenthold RJ. Turnover analysis of glutamate receptors identifies a rapidly degraded pool of the N-methyl-d-aspartate receptor subunit, NR1, in cultured cerebellar granule cells. J Biol Chem. 1999;274:151–157. doi: 10.1074/jbc.274.1.151. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Erzurumlu RS, Huerta PT, Chen DF, Sasaoka T, Ulupinar E, Tonegawa S. NMDA receptor-dependent refinement of somatotopic maps. Neuron. 1997;19:1201–1210. doi: 10.1016/s0896-6273(00)80412-2. [DOI] [PubMed] [Google Scholar]
- Kash T, Winder DG. NMDAR LTP and LTD induction: 2B or Not 2B. Is that the question? Debates Neurosci. 2007;1:79–84. [Google Scholar]
- Kesner RP, Rogers J. An analysis of independence and interactions of brain substrates that subserve multiple attributes, memory systems, and underlying processes. Neurobiol Learn Mem. 2004;82:199–215. doi: 10.1016/j.nlm.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Clark RE, Thompson RF. Hippocampectomy impairs the memory of recently, but not remotely, acquired trace eyeblink conditioned responses. Behav Neurosci. 1995;109:195–203. doi: 10.1037//0735-7044.109.2.195. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Rioult MC, Bear MF. Experience-dependent modification of synaptic plasticity in visual cortex. Nature. 1996;381:526–528. doi: 10.1038/381526a0. [DOI] [PubMed] [Google Scholar]
- Kirov SA, Harris KM. Dendrites are more spiny on mature hippocampal neurons when synapses are inactivated. Nat Neurosci. 1999;2:878–883. doi: 10.1038/13178. [DOI] [PubMed] [Google Scholar]
- Kiyama Y, Manabe T, Sakimura K, Kawakami F, Mori H, Mishina M. Increased thresholds for long-term potentiation and contextual learning in mice lacking the NMDA-type glutamate receptor epsilon1 subunit. J Neurosci. 1998;18:6704–6712. doi: 10.1523/JNEUROSCI.18-17-06704.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhr G, Jensen V, Koester HJ, Mihaljevic AL, Utvik JK, Kvello A, Ottersen OP, Seeburg PH, Sprengel R, Hvalby Ø. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. J Neurosci. 2003;23:10791–10799. doi: 10.1523/JNEUROSCI.23-34-10791.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron. 1996;16:333–344. doi: 10.1016/s0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lee KW, Kim Y, Kim AM, Helmin K, Nairn AC, Greengard P. Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc Natl Acad Sci U S A. 2006;103:3399–3404. doi: 10.1073/pnas.0511244103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misane I, Tovote P, Meyer M, Spiess J, Ogren SO, Stiedl O. Time-dependent involvement of the dorsal hippocampus in trace fear conditioning in mice. Hippocampus. 2005;15:418–426. doi: 10.1002/hipo.20067. [DOI] [PubMed] [Google Scholar]
- Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology. 2007;52:71–76. doi: 10.1016/j.neuropharm.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Morris RG, Davis S, Butcher SP. Hippocampal synaptic plasticity and NMDA receptors: a role in information storage? Philos Trans R Soc Lond B Biol Sci. 1990;329:187–204. doi: 10.1098/rstb.1990.0164. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci. 2004;5:361–372. doi: 10.1038/nrn1385. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Watabe AM, Kiyama Y, Fukaya M, Arima-Yoshida F, Horai R, Sudo K, Ebine K, Delawary M, Goto J, Umemori H, Tezuka T, Iwakura Y, Watanabe M, Yamamoto T, Manabe T. NR2B tyrosine phosphorylation modulates fear learning as well as amygdaloid synaptic plasticity. EMBO J. 2006;25:2867–2877. doi: 10.1038/sj.emboj.7601156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J Neurosci. 2006;26:1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Lebel D, Brosh I, Barkai E. A molecular mechanism for stabilization of learning-induced synaptic modifications. Neuron. 2004;41:185–192. doi: 10.1016/s0896-6273(03)00874-2. [DOI] [PubMed] [Google Scholar]
- Quinn JJ, Loya F, Ma QD, Fanselow MS. Dorsal hippocampus NMDA receptors differentially mediate trace and contextual fear conditioning. Hippocampus. 2005;15:665–674. doi: 10.1002/hipo.20088. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596–1599. doi: 10.1126/science.280.5369.1596. [DOI] [PubMed] [Google Scholar]
- Sessoms-Sikes S, Honse Y, Lovinger DM, Colbran RJ. CaMKIIalpha enhances the desensitization of NR2B-containing NMDA receptors by an autophosphorylation-dependent mechanism. Mol Cell Neurosci. 2005;29:139–147. doi: 10.1016/j.mcn.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein AM, Barrow CA, Davis AJ, Mumby MC. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci U S A. 2002;99:4221–4226. doi: 10.1073/pnas.072071699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takehara K, Kawahara S, Munemoto Y, Kuriyama H, Mori H, Mishina M, Kirino Y. The N-methyl-d-aspartate (NMDA)-type glutamate receptor GluRepsilon2 is important for delay and trace eyeblink conditioning in mice. Neurosci Lett. 2004;364:43–47. doi: 10.1016/j.neulet.2004.04.024. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996a;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996b;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- von Engelhardt J, Doganci B, Jensen V, Hvalby Ø, Göngrich C, Taylor A, Barkus C, Sanderson DJ, Rawlins JN, Seeburg PH, Bannerman DM, Monyer H. Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron. 2008;60:846–860. doi: 10.1016/j.neuron.2008.09.039. [DOI] [PubMed] [Google Scholar]
- Yang RJ, Mozhui K, Karlsson RM, Cameron HA, Williams RW, Holmes A. Variation in mouse basolateral amygdala volume is associated with differences in stress reactivity and fear learning. Neuropsychopharmacology. 2008;33:2595–2604. doi: 10.1038/sj.npp.1301665. [DOI] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]