Negative cross-talk between p53 and the glucocorticoid receptor and its role in neuroblastoma cells (original) (raw)
Abstract
The tumour suppressor p53 and the glucocorticoid receptor (GR) respond to different types of stress. We found that dexamethasone-activated endogenous and exogenous GR inhibit p53-dependent functions, including transactivation, up- (Bax and p21WAF1/CIP1) and down- (Bcl2) regulation of endogenous genes, cell cycle arrest and apoptosis. GR forms a complex with p53 in vivo, resulting in cytoplasmic sequestration of both p53 and GR. In neuroblastoma (NB) cells, cytoplasmic retention and inactivation of wild-type p53 involves GR. p53 and GR form a complex that is dissociated by GR antagonists, resulting in accumulation of p53 in the nucleus, activation of p53-responsive genes, growth arrest and apoptosis. These results suggest that molecules that efficiently disrupt GR–p53 interactions would have a therapeutic potential for the treatment of neuroblastoma and perhaps other diseases in which p53 is sequestered by GR.
Keywords: apoptosis/cell cycle/cellular localization/GR antagonists/neuroblastoma
Introduction
The tumour suppressor p53 is frequently inactivated in human cancers, most commonly by mutation (Hollstein et al., 1991), but also by other mechanisms including overexpression of inhibitory proteins and sequestration in the cytoplasm (Steele et al., 1998). p53 is an ephemeral protein with a half life of 10–15 min that is stabilized and activated by diverse signals (Giaccia and Kastan, 1998). The physiological function of p53 is to mediate cell cycle arrest or apoptosis in response to various stresses, such as DNA damage (Harris, 1996; Ko and Prives, 1996; Levine, 1997). p53 is a transcription factor that regulates many different genes (Elkeles et al., 1999; Yu et al., 1999; Zhao et al., 2000), in particular, the pro-apoptotic bax (Miyashita et al., 1994b; Miyashita and Reed, 1995), the cell cycle inhibitor p21WAF1/CIP1 (Brugarolas et al., 1995; Waldman et al., 1996) and its auto-regulator mdm2 (Barak et al., 1993). Mdm2 binds to p53, inhibits its transcriptional activity (Oliner et al., 1993) and increases its degradation (Haupt et al., 1997; Kubbutat et al., 1997). p53 represses various promoters lacking p53 binding sites. Repression results from interactions with the basal transcription machinery (Ragimov et al., 1993), the corepressor mSin3A and histone deacetylases (Murphy et al., 1999), or other unidentified factors that negatively regulate the bcl2 promoter (Miyashita et al., 1994a).
p53 interacts physically with many proteins. These physical interactions modulate the activity of p53 and integrate its functions into other cellular circuits. p53 is negatively regulated by binding to viral factors, and cellular proteins such as Mdm2. p53 and Mdm2 form an auto-regulatory loop that is modulated by c-Abl (Sionov et al., 1999), pRb (Hsieh et al., 1999) and p14_Arf_ (Sherr and Weber, 2000). p53 also interacts with the Werner Syndrome protein (Blander et al., 1999), Ref-1 (Gaiddon et al., 1999), HIF-1α (An et al., 1998) and JNK (Fuchs et al., 1998). p53 cross-talks with different steroid-hormone receptors. p53 represses and interacts physically with the human thyroid receptor β-1 (Yap et al., 1996), the orphan receptor TR2 (Lin and Chang, 1996) and the oestrogen receptor (Yu et al., 1997b). Oestrogens and anti-oestrogens regulate the levels and activity of p53 and Rb (Hurd et al., 1997; Saji et al., 1999). Cofactors for steroid receptors physically and functionally interact with p53 (Lee et al., 1999). Here, we describe p53 interactions with the glucocorticoid receptor (GR).
The primary physiological function of glucocorticoids (GCs) in humans is to maintain homeostasis in response to internal or environmental changes (Tronche et al., 1998; Kellendonk et al., 1999). GCs are involved in differentiation, nervous system functions and intermediary metabolism. GCs regulate the activity of GR, a transcription factor that is ubiquitously expressed in human tissues. GR is indispensable, since targeted disruption of the gene in mice leads to death within a few hours after birth (Cole et al., 1995). GR has a modular structure, including two transactivation domains, a DNA binding domain (DBD) and a ligand binding domain (LBD). In the absence of GCs, GR remains in the cytoplasm in complex with proteins such as Hsp90. GC binding to the LBD results in GR translocation to the nucleus, where it modulates transcription. GR homodimers interact with palindromic glucocorticoid response elements (GREs) in the regulatory regions of target genes. They activate transcription through interactions with co-activators and the basic transcription machinery (Beato et al., 1995). GR represses transcription of certain genes by direct binding to negative response elements (Drouin et al., 1993) and composite elements (Diamond et al., 1990). GR cross-talk with AP-1 and NF-κB signalling leads to repression of pro-inflammatory cytokines (Barnes and Adcock, 1993). Although the mechanism of this cross-talk is still elusive, it is a major regulatory function of GR. Loss of the ability of GR to dimerize in mice blocks transactivation of canonical GREs, but does not compromise the viability or the capacity to trans-repress AP-1-regulated genes (Reichardt et al., 1998).
We have previously described tamoxifen-inducible chimeric tumour suppressors (LI-CTS), which regress tumours in a ligand-dependent manner (Sengupta et al., 2000). We found that similar dexamethasone-inducible chimeras did not have the expected properties, most probably due to negative interactions between p53 and GR. p53 and GR interact physically and functionally, and these interactions lead to inactivation of p53 by cytoplasmic sequestration in neuroblastoma cells.
Results
DITS do not transactivate and suppress cell growth in a dexamethasone-dependent manner
We constructed Dexamethasone-inducible Tumour Suppressors (DITS), which contain a mutated LBD from human GR fused to the C-terminal region of p53 or CTS1 (p53-GRD, CTS1-GRD; Figure 1A). The mutated LBD (I747T) is inducible by the synthetic GC dexamethasone (Dex), but not by natural GCs (Roux et al., 1996). CTS1 is a highly active chimeric tumour suppressor based on p53 (Conseiller et al., 1998). Transactivation by DITS, p53 and CTS1 was studied by transient transfection in p53 null cell lines, HSC-2 (Figure 1B) and Saos-2 (data not shown). p53 and CTS1 transactivated the p53-responsive reporter Bax–luc, whereas both DITS had very little effect with or without ligand (Figure 1B). As expected, GR also had little effect. Similar results were obtained with another p53-inducible reporter [WAF1–luc (el-Deiry et al., 1993) data not shown]. DITS was efficiently expressed, as shown by western blotting (Figure 1C, GFP is the internal control for transfection efficiency). We found that the activity of DITS was not induced by Dex in a variety of other assays (long-term colony formation and apoptosis) and in different cell lines (HSC-2 and Saos-2; data not shown). These results were surprising since similar molecules with an oestrogen receptor ligand-binding domain (CTS-ERT) were inducible (Sengupta et al., 2000). They raised the possibility that GR inhibits the activity of p53.


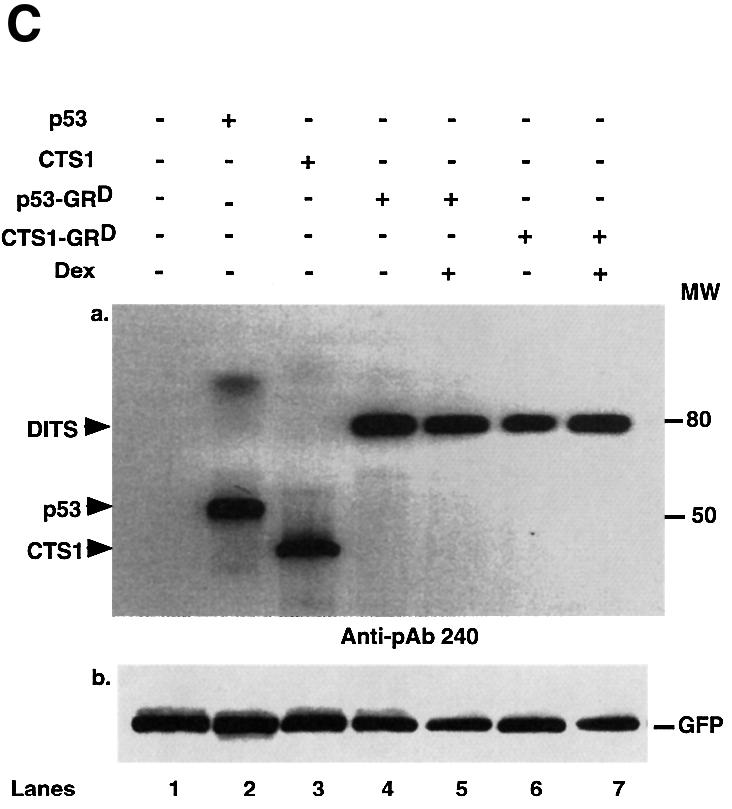
Fig. 1. DITS have attenuated transactivation and growth suppressive functions. (A) Schemes. hGRD-LBD was fused to the C-terminal end of p53 or CTS1 (Conseiller et al., 1998) to generate p53–GRD and CTS–GRD, respectively. CTS1, chimeric tumour suppressor 1; VP16, herpes simplex virus 1 VP16 transcription activation domain; DBD, DNA binding domain; LZ, leucine zipper; LBD, ligand binding domain; TET, tetramerization domain of p53; TA, transactivation domain of p53; hGR, human glucocorticoid receptor. (B) HSC-2 cells were transfected with GRD, p53, p53–GRD, CTS1 or CTS1–GRD expression vectors (250 and 500 ng/ml), the p53-responsive reporter (Bax–luc), and CMV lacZ (500 ng/ml) to measure transfection efficiencies. Activity represents the luciferase values measured 24 h after the end of transfection and corrected for transfection efficiency. The values were similar in three independent transfections. (C) Transfections were carried out as in (B) using 500 ng/ml of the expression plasmids and 500 ng/ml of pEGFP. Cell lysates (50 µg) were analysed by 10% SDS–PAGE and western blotting with (a) a monoclonal antibody that recognizes the DBD of p53 (pAb 240) or (b) anti-GFP. MW = molecular weight markers. Lane 1, mock transfection; lane 2, p53; lane 3, CTS1; lane 4, p53–GRD; lane 5, p53–GRD + Dex; lane 6, CTS1–GRD; lane 7, CTS1–GRD + Dex.
p53 and GR inhibit each other’s transactivation properties
We investigated whether GR affects p53 activity and the converse in HSC-2 cells (Figure 2). Dexamethasone (Dex) and cortisol (Cort) have been shown to activate the transcriptional activity of GR to similar extents (Roux et al., 1996) Vehicle alone (ethanol), Cort (a natural glucocorticoid), Dex (an agonist) and RU-486 (an antagonist) did not change the basal activity of the p53 CON reporter (Figure 2A, lanes 1–4). Increasing amounts of p53 stimulated reporter activity in a dose-dependent manner (Figure 2A, lanes 5–7). p53 was inhibited ∼3-fold by endogenous GR activated with either Cort or Dex, but was not affected by RU-486 (Figure 2A, lanes 8–16). With a constant level of transfected p53, exogenous GR decreased p53 activity to a small extent (Figure 2A, lanes 18–20), whereas GR + Dex or GR + Cort efficiently inhibited p53 (Figure 2A, lanes 21–26). In contrast, GR + RU-486 had little effect (Figure 2A, lanes 27–29). Similar results were obtained in Saos-2 and SiHa cells (data not shown). Western blots showed that the amounts of expressed protein increased as increasing amounts of expression vectors for GR and p53 were transfected (Figure 2B, lanes 1–4 and 8–11). Co-transfection of GR did not affect p53 levels (lanes 5–7) showing that the inhibition did not result from decreased expression of p53. In the converse experiments with the GR-responsive reporter GRE–tk–luc, Dex-dependent activation by exogenous GR was also inhibited by p53 (Figure 2C).





Fig. 2. The human glucocorticoid receptor and p53 inhibit each other’s transactivation properties. (A) HSC-2 cells were transfected with increasing concentrations of either p53 or GR with a constant amount of p53, a minimal p53 reporter (p53 CON) and CMV lacZ to measure transfection efficiency. Vehicle alone (ethanol, –), Cort (10–7 M), Dex (10–7 M) or RU-486 (10–7 M) were added as indicated. Lane 1, mock transfection; lane 2, + Cort; lane 3, + Dex; lane 4, + RU-486; lanes 5–7, p53 (125, 250, 500 ng/ml); lanes 8–10, p53 (125, 250, 500 ng/ml) + Cort; lanes 11–13, p53 (125, 250, 500 ng/ml) + Dex; lanes 14–16, p53 (125, 250, 500 ng/ml) + RU-486; lane 17, p53 (500 ng/ml); lanes 18–20, p53 (500 ng/ml) + GR (250, 500, 1250 ng/ml); lanes 21–23, p53 (500 ng/ml) + GR (250, 500, 1250 ng/ml) + Cort; lanes 24–26, p53 (500 ng/ml) + GR (250, 500, 1250 ng/ml) + Dex; lanes 27–29, p53 (500 ng/ml) + GR (250, 500, 1250 ng/ml) + RU-486. Luciferase activity was measured 24 h post-transfection and corrected for transfection efficiency. The values shown are from four independent experiments. p53 RE, p53-responsive element. TATA, minimal promoter element. (B) Transfections were carried out as in (A), except that 500 ng/ml of pEGFP was included to control for transfection efficiency. Cell lysates (50 µg) were analysed by 10% SDS–PAGE and western blotting with antibodies to: (a) p53 (DO-1), (b) GR (E-20 and P-20) and (c and d) GFP. Lanes 1 and 8, mock transfection with vehicle alone (ethanol); lanes 2–4, p53 (125, 250, 500 ng/ml) with vehicle alone; lanes 5–7, p53 (500 ng/ml) and GR (250, 500 and 1250 ng/ml) + Dex; lanes 9–11, GR (250, 500 and 1250 ng/ml) + Dex. (C) HSC-2 cells were transfected with increasing concentrations of the GR expression vector alone or with a constant amount of the p53 expression vector, the GR reporter (GRE–tk–luc) and CMV lacZ to measure transfection efficiency. Twenty-four hours post-transfection luciferase activities were measured and corrected for transfection efficiencies. The values were similar in three independent experiments. Lane 1, mock transfection; lane 2, mock + Dex; lanes 3–5, GR (250, 500, 1250 ng/ml); lanes 6–8, (250, 500, 1250 ng/ml) + Dex; lanes 9–11, (250, 500, 1250 ng/ml) + p53 (500 ng/ml). –, vehicle alone; +, 10–7 M Dex; GRE, glucocorticoid response element; tk, HSV tk minimal promoter. (D) (a) Transfections with expression vectors for GR or ASGR (500, 1250 ng/ml), alone or in combination, the GRE–tk–luc reporter and CMV lacZ for transfection efficiency. Luciferase was measured 24 h post-transfection and corrected for transfection efficiency. The values are from two independent experiments. Lane 1, mock transfection; lane 2, mock + Dex; lanes 3 and 4, GR (500, 1250 ng/ml); lanes 5 and 6, GR (500, 1250 ng/ml) + Dex; lanes 7 and 8, GR (1250 ng/ml) + ASGR (500, 1250 ng/ml) + Dex; lanes 9 and 10, GR (1250 ng/ml) + ASGR (500, 1250 ng/ml). (b) Cell lysates (50 µg) were prepared 48 h post-transfection and analysed by 10% SDS–PAGE and western blotting with antibodies to TBP (3G3) or GR (E-20 and P-20). Lane 1, untransfected; lane 2, ASGR; lane 3, GR. –, vehicle alone; +, 10–7 M Dex. (E) Transfections contained expression vectors for p53 (250 ng/ml), ASGR (1250 ng/ml), the p53 CON reporter and CMV lacZ to measure transfection efficiency. Luciferase activities were measured after 48 h, corrected for transfection efficiency and expressed as fold induction relative to the control (reporter in the absence of p53). The values represented are from two independent experiments. –, vehicle alone; +, 10–7 M Dex.
We investigated the mechanism of repression of p53 by GR. Antisense GR RNA (ASGR) expressed by a vector was used to down-regulate GR. We verified that ASGR inhibited transactivation of the GRE–tk–luc reporter by exogenous GR in the presence of Dex (Figure 2D, panel a), and that it reduced endogenous GR protein levels, as shown by western blotting (Figure 2D, panel b). We tested whether down-regulation of endogenous GR would stimulate p53 transactivation. Indeed, Dex inhibited p53-dependent transactivation from the p53 CON reporter and ASGR relieved this repression (Figure 2E). These results show that down-regulation of endogenous GR relieves repression of p53 by Dex.
We studied GR + Dex repression of natural p53-responsive promoters in HSC-2 cells. Dex had little effect on bax–, p21– and bcl2–luc in the absence of p53. Dex inhibited p53-mediated activation of bax– and p21–luc, and relieved repression of bcl2–luc (Figure 3A). We also studied expression of the endogenous genes by western blotting. Dex or Cort inhibited p53 induction of Bax and p21_WAF1/CIP1_, and relieved repression of Bcl2, without affecting expression of p53 (Figure 3B). These results show that activated GR regulates the expression of several p53-responsive genes in a p53-dependent manner.


Fig. 3. p53-responsive genes are modulated by Dex. (A) HSC-2 cells were transfected with an expression vector for p53 (filled bars) or the corresponding empty vector (open bars), and reporters containing the natural p53-responsive promoters bax, p21WAF1/CIP1 and bcl2 linked to luciferase, and CMV lacZ to measure transfection efficiency. Luciferase activity was assayed 24 h post-transfection and corrected for transfection efficiency. The values are from two independent experiments. (B) Transfections were carried out as in (A). Additional plates with Cort (10–7 M) were included. pEGFP (500 ng/ml) was used to measure transfection efficiency. Cell lysates (50 µg) were analysed by SDS–PAGE and western blotting with antibodies to: (a and f) Bax (N-20, polyclonal); (b and g) p21WAF1/CIP1 (1WA-IC581, monoclonal); (c) bcl2 (100, monoclonal); (d and h) p53 (DO-1, monoclonal); (e and i) GFP (monoclonal). Lanes 1 and 5, mock transfection; lane 2, mock + Dex; lanes 3 and 7, p53; lane 4, p53 + Dex; lane 6, Cort; lane 8, p53 + Cort. –, vehicle alone; +, 10–7 M Dex.
GR inhibits p53-induced cell cycle arrest and apoptosis
We tested whether GR + Dex reduced p53-mediated cell cycle arrest, apoptosis and inhibition of colony formation. Apoptosis was measured by PARP cleavage (Figure 4A), DNA fragmentation (Figure 4B) and sub-G1 cells in flow cytometry (Figure 4C). CD20 co-expression was used to select transfected cells. PARP cleavage, which is an early event in apoptosis, showed that apoptosis induced by p53 expression was partially inhibited by exogenous GR, and strongly inhibited by exogenous GR with Dex (Figure 4A). DNA fragmentation, which is a later event, gave similar results (Figure 4B). p53-induced cell cycle arrest, as measured by the increase in G0–G1 cells by flow cytometry, was inhibited partially by GR and totally by GR + Dex (Figure 4C). Activation of endogenous GR by Dex also rescued the cells from p53-mediated apoptosis (Figure 4C). p53-mediated inhibition of colony formation was relieved by Dex (Figure 4D). These results show that endogenous and exogenous GR inhibit p53-mediated functions. The degree of inhibition depends on the amount of GR and is enhanced when GR is activated by ligands.
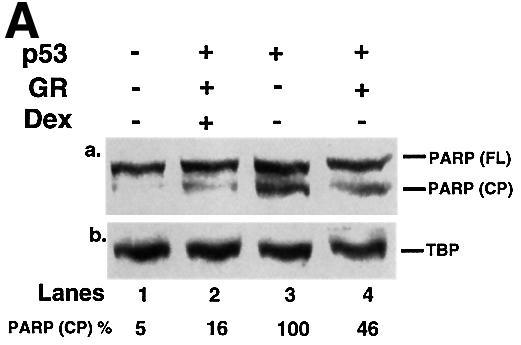
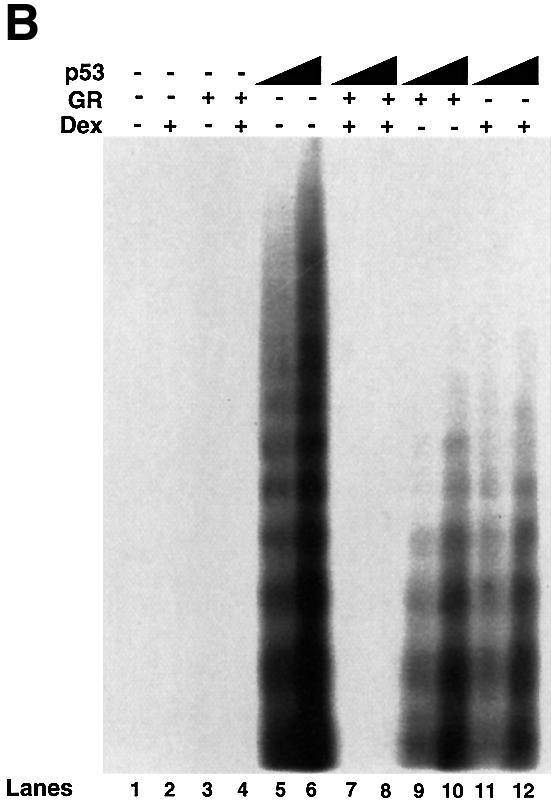

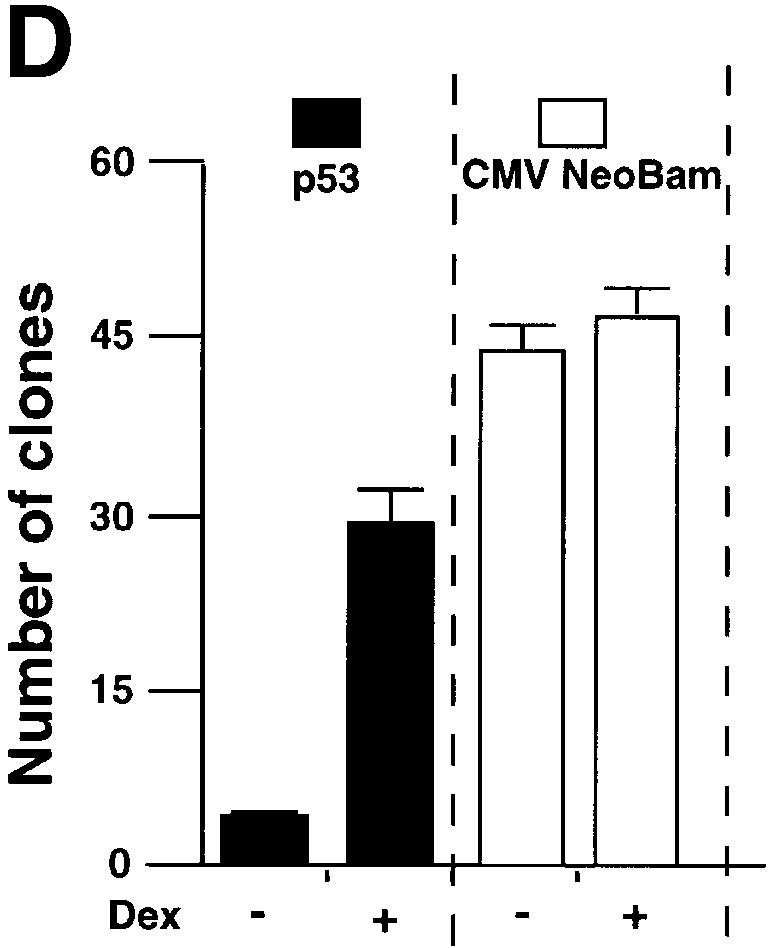
Fig. 4. GR inhibits p53-mediated apoptosis and growth suppression. (A) Cells were transfected with expression vectors for p53 (250 ng/ml) and/or GR (1250 ng/ml) in the presence of either vehicle (ethanol) or 10–7 Dex. Cell lysates (50 ng) prepared 16 h post-transfection were analysed by SDS–PAGE and western blotting with antibodies to (a) PARP (polyclonal) and (b) TBP (3G3, monoclonal). Lane 1, mock transfection; lane 2, p53 + GR + Dex; lane 3, p53; lane 4, p53 + GR. –, vehicle; +, 10–7 M Dex. The PARP cleavage product (CP) was scanned and the values are the amount of CP relative to p53 alone (lane 3). (B) Cells were transfected with p53 (250 and 500 ng/ml) and/or GR (1250 ng/ml) expression plasmids in the presence of either vehicle (ethanol) or 10–7 M Dex. Low molecular weight DNA was extracted 24 h post-transfection, radiolabelled, run on 1.5% TBE agarose gels, transferred to nylon membranes and visualized by autoradiography. Lane 1, mock transfection with vehicle alone (–); lane 2, mock + Dex; lane 3, GR; lane 4, GR + Dex; lanes 5 and 6, p53 (250, 500 ng/ml); lanes 7 and 8; p53 (250, 500 ng/ml) + GR + Dex; lanes 9 and 10, p53 (250, 500 ng/ml) + GR; lanes 11 and 12, p53 (250, 500 ng/ml) + Dex. (C) Transfections as in (A) with expression plasmids for p53 (500 ng/ml), GR (1250 ng/ml), CD20 (4 µg/ml, to select transfected cells), and vehicle (ethanol) or Dex. Twenty-four hours post-transfection, the cells were harvested and analysed by flow cytometry. Transfections were duplicated and the whole experiment was repeated. The values (with standard deviation) are from four experimental points. The percentage of transfected cells in each stage of the cell cycle was substracted from the control transfected with empty vector (control). Hence the control (with no change) is not visible. The cell cycle distribution of the control was 10% sub-G0, 36% G0–G1, 44% S, 10% G2–M. (D) Clonogenic assays with an expression vector for p53 and the corresponding empty vector (CMV NeoBam) with vehicle (–) or 10–7 M Dex (+). Triplicate independent transfections were carried out for each point. The values are from two independent experiments.
GR forms a complex with p53 in the cytoplasm
p53 and GR have been reported to interact physically in vitro (Yu et al., 1997a). We investigated whether they interact in vivo. Immunoprecipitation assays were performed with extracts of p53 null HSC-2 cells co-transfected with expression plasmids encoding p53 and GR, in the presence of Dex. Complexes were immunoprecipitated with p53- or GR-specific antibodies and probed by western blotting (Figure 5A). GR was present in p53 antibody immunoprecipitates, whether or not GR was co-expressed with p53 (Figure 5A, lanes 3 and 4, panel a), showing that both exogenous and endogenous GR bind to p53. There was no GR in the p53 antibody immunoprecipitates when p53 was not expressed (Figure 5A, lane 2, panel a), showing that the immunoprecipitations were specific. As expected, p53 was only detected when it was transfected (Figure 5A, lanes 1–4, panel b). In the converse experiments, p53 was detected in the GR antibody immunoprecipitates only when p53 was expressed (Figure 5A, lanes 6–8, panel c). GR was detected in all the immunoprecipitates (Figure 5A, panel d), even when GR was not co-transfected, as expected from the presence of endogenous GR.



Fig. 5. p53 and GR interact physically and co-localize in the cell. (A) HSC-2 cells were transfected with p53 (500 ng/ml) and/or GR (1250 ng/ml) expression plasmids, as indicated, in the presence of 10–7 M Dex. Lysates prepared 24 h post-transfection were immunoprecipitated (IP) with either p53 (DO-1, monoclonal) or GR (E-20 and P-20, polyclonal) antibodies (Ab). After the washes, the immunoprecipitates were analysed by 10% SDS–PAGE and western blotting with antibodies against either GR (p53 IP, panel a) or p53 (GR IP, panel c). The efficiency of immunoprecipitation was checked by western blotting with self antibodies (panels b and d). Lane 1, transfected GR, no IP (20% of p53 input); lane 2, mock transfection, IP with p53 Ab; lane 3, transfected GR and p53, IP with p53 Ab; lane 4, transfected p53, IP with p53 Ab; lane 5, transfected p53, no IP (20% of input for GR IP); lane 6, transfected p53 and GR, IP with GR Ab; lane 7, mock transfection, IP with GR Ab; lane 8, transfected GR, IP with GR Ab. (B) HSC-2 (a) or Saos-2 (b) cells were transfected with p53 (500 ng/ml) and/or GR (1250 ng/ml) expression plasmids in the presence of either vehicle (ethanol) or 10–7 M Dex. Twenty-four hours post-transfection the cells were fixed, incubated with antibodies against GR (E-20 and P-20, polyclonal) or p53 (DO-1, monoclonal) and analysed by confocal microscopy. (C) Quantitation of the transfections in (B). At least 500 cells were counted for each transfection. The percentage of cells with the protein in the nucleus (N), the cytoplasm (C) and both (C + N) is shown. (a) GR; (b) GR + Dex; (c) p53; (d) p53 + GR; (e) p53 + GR + Dex; (f) p53 + Dex; (g) p53 + ASGR + Dex. Open bars, GR; filled bars, p53.
GR is a cytoplasmic protein that translocates to the nucleus upon ligand binding. We investigated how GR–p53 interactions affect their localization by immunocytochemistry (IHC) in HSC-2 (Figure 5B, panel a) and Saos-2 (panel b) cells. Typical cells are shown in Figure 5B, and the quantification in Figure 5C. In HSC-2 cells expressing low amounts of exogenous GR (determined in titration experiments), the receptor is almost exclusively cytoplasmic (96%) and predominantly nuclear in the presence of Dex. p53 is predominantly nuclear (60%) but in many cells it is both cytoplasmic and nuclear (C+N, 38%). When the two proteins are co-transfected in the absence of ligand, GR remains cytoplasmic whereas the number of cells with p53 in both the cytoplasm and nucleus, or the cytoplasm alone, increases slightly. Strikingly, in the presence of Dex, p53 and GR co-localize in the cytoplasm in >90% of the transfected cells. Similar cytoplasmic co-localization was observed in Saos-2 (Figure 5B, panel b) and PAP (data not shown) cells, indicating the generality of the phenomenon. Endogenous GR can also affect the localization of p53. The addition of Dex alone decreased the proportion of cells with exclusively nuclear p53, and increased the proportion with both cytoplasmic and nuclear or just cytoplasmic p53 (Figure 5C, compare c and f). Down-regulation of endogenous GR with ASGR in the presence of Dex markedly increased the number of cells with exclusively nuclear p53 (Figure 5C, compare g with f). These results show that the interaction between p53 and GR in the presence of ligand results in efficient mutual sequestration in the cytoplasm.
p53 cytoplasmic sequestration in neuroblastoma cells involves interactions with GR
The novel subcellular re-localization of p53 in the presence of GR suggested that, in cells with constitutive cytoplasmic p53, p53 might be sequestered by GR. p53 is cytoplasmic in many primary neuroblastomas (NBs) (Moll et al., 1995) and the corresponding cell lines (Moll et al., 1996). We tested whether p53 was complexed with GR in IMR 32 NB cells. Immunoprecipitates formed with an antibody against GR contained p53, as detected by western blotting (Figure 6A, lane 3). An immunoprecipitate with a control antibody (anti-HA) did not contain p53 or GR (Figure 6A, lane 5, panels a and b), showing that it was specific. In the converse experiment, the p53 antibody immunoprecipitates contained GR (Figure 6A, lane 8). We tested whether GR antagonists would weaken the complex. Indeed, when the cells were treated with the antagonist RU-38,486 (RU-486), the GR antibody immunoprecipitates contained much less p53 (Figure 6A, lane 4, ∼20% remained) and conversely, the p53 antibody immunoprecipitates had less GR (lane 9, ∼5% remained). This was not a consequence of decreased expression of either p53 or GR in the presence of RU486 (Figure 6A, lanes 1 and 2, panels a and b; lanes 6 and 7, panels c and d).



Fig. 6. The GR antagonist RU-486 disrupts the GR–p53 complex and increases p53 localization in the nucleus in neuroblastoma cells. (A) IMR 32 cells were grown for 24 h in the presence of vehicle (ethanol) or 10–7 M RU-486. Immunoprecipitates (IP) formed with Ab against p53 (DO-1, monoclonal), HA (12CA5, monoclonal) or GR (E-20 and P-20, polyclonal) were washed, subjected to 10% SDS–PAGE and western blotted with Abs against p53 (GR and HA IP, panel a) or GR (p53 IP, panel c). The efficiency of IP was checked by western blotting with self antibodies (panels b and d). Lane 1, IMR 32 cells with vehicle (–, 5% of the lysate used for GR/HA IP); lane 2, IMR 32 cells with RU-486 (+, 5% of input for GR/HA IP); lane 3, vehicle (–), IP with GR Ab; lane 4, RU-486 (+), IP with GR Ab; lane 5, vehicle (–), IP with HA Ab; lane 6, vehicle (–, 20% of input for p53 IP); lane 7, RU-486 (+, 20% of input for p53 IP); lane 8, vehicle (–), IP with p53 Ab; lane 9, RU-486 (+), IP with p53 Ab. (B) IMR 32 cells were cultured with vehicle (control), RU-486 or Dex, and after 24 h they were fixed, incubated with antibodies against GR (E-20 and p20, polyclonal) or p53 (DO-1, monoclonal) and examined by confocal microscopy. (C) Quantification of (B). For each group ∼1000 cells were counted. (a) control; (b) RU-486; (c) Dex. N, nuclear; C, cytoplasmic; C + N, both cytoplasmic and nuclear.
Dissociation of the GR–p53 complex by RU-486 was predicted to result in nuclear accumulation of wild-type p53. GR is known to be translocated to the nucleus in the presence of RU-486 (van Steensel et al., 1995; Savory et al., 1999). IMR 32 cells were grown for 24 h in the presence of either the vehicle (control) or RU-486 or Dex, and analysed by IHC. In the control, as expected, p53 and GR were cytoplasmic in virtually all the cells (Figure 6B and C). Addition of Dex had very little effect on the subcellular localization of p53 and a small effect on GR (<10% of the cells had exclusively nuclear GR). In the presence of RU-486, a dramatic redistribution of both proteins was apparent, with >80% of the cells containing p53 and GR in the nucleus as well as in the cytoplasm. RU-486 can also function as a weak agonist for GR (Baulieu, 1997) and an antagonist for the progesterone receptor (PR) (Meyer et al., 1990). Another GR-specific antagonist, RU-43,044 (RU-044) (Bocquel et al., 1993), also induced nuclear localization of p53 (data not shown), suggesting that nuclear localization was due to GR antagonism rather than other side-effects of a particular antagonist. Nuclear accumulation was detected with different p53 antibodies (DO1, 421 and 1801; data not shown). Two other neuroblastoma cell lines, SK-SY5Y and SK-N-SH, gave a similar redistribution of p53 and GR in the presence of RU-486 and RU-044 (data not shown). These results show that GR antagonists dissociate the GR–p53 complex and induce nuclear accumulation of p53.
p53 is activated by GR antagonists in neuroblastoma cells
We tested whether nuclear accumulation of p53 in neuroblastoma cells treated with GR antagonists increased p53 activity. When IMR 32 cells were plated in the presence of RU-486 there was a reproducible inhibition of cell growth (Figure 7A). Twenty-four hours of treatment with RU-486 and RU-044 induced expression of the p53 downstream genes, p21_WAF1/CIP1_ and bax, as detected by western blotting (Figure 7B, panels b and c). There was no change in the level of p53 (Figure 7B, panel a) suggesting that p53 stabilization is not involved. Flow cytometry showed that both antagonists affect cell cycle progression (Figure 7C). They increased the proportion of cells in the sub-G0–G1 (∼5%) and G0–G1 (∼14%) phases, and decreased S phase (∼15%), and G2–M (∼4%). These results are consistent with the model that GR antagonists release p53, which translocates to the nucleus and induces downstream effects, including gene expression, cell cycle arrest and apoptosis.

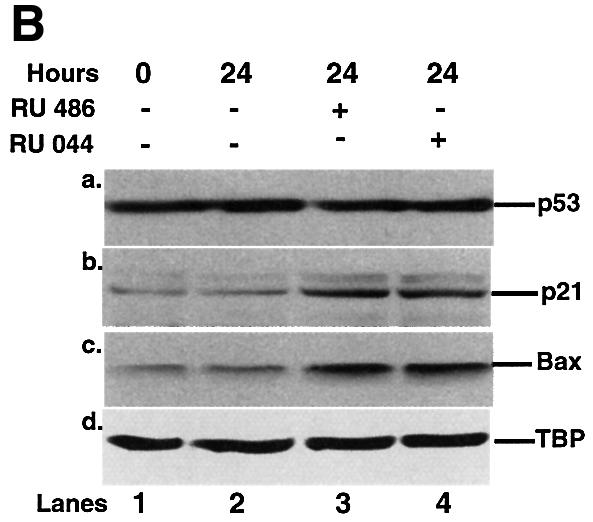

Fig. 7. Effect of GR antagonists on the growth, expression of p53-responsive genes and the cell cycle of IMR 32 neuroblastoma cells. (A) Equal numbers (1.7 × 10–6) of IMR 32 cells were plated in the presence of vehicle (open symbols) or 10–7 M RU-486 (closed symbols). At different times the number of viable cells was counted. The values are from three independent experiments. (B) IMR 32 cells were cultured with vehicle, 10–7 M RU-486 or 10–7 M RU-044 for 24 h. Lysates (100 µg) were analysed by SDS–PAGE and western blotting with antibodies to (a) p53 (DO-1, monoclonal), (b) p21_WAF1/CIP1_ (1WA-IC581, monoclonal), (c) Bax, (N-20, polyclonal), (d) TBP (3G3, monoclonal). Lane 1, cells before ligand addition; lane 2, vehicle (–); lane 3, RU-486 (+); lane 4, RU-044 (+). (C) IMR 32 cells were cultured as in (B) and additionally with mitomycin C (1 µM) as indicated. After 24 h of treatment, the cells were harvested and analysed by flow cytometry. Experimental points were in duplicate and the whole experiment was repeated. The values (with standard deviation) are from four experimental points. The percentage of cells in each stage of the cell cycle was subtracted from the control (2% sub-G0, 39% G0–G1, 48% S, 11% G2–M). The control, with zero change, is not visible.
Nuclear localization alone may not be sufficient to fully activate p53. We tested whether the DNA damaging agent mitomycin C enhances the effects of GR antagonists in IMR 32 cells. Mitomycin C alone increased the proportion of cells in the sub-G0 and G0–G1 phases by 4 and 9%, respectively (Figure 7C). GR antagonists together with mitomycin C had a much greater effect, with an increase of ∼9% in sub-G0 and 26% in G0–G1 compared with the control. These results show that GR antagonists can considerably increase the effects of DNA damaging agents on neuroblastoma cells.
Discussion
Our attempts to make glucocorticoid-inducible p53 chimeras that resembled the successful tamoxifen- inducible chimeras (Sengupta et al., 2000) apparently failed because of the mutually antagonistic activities of p53 and GR. We found that endogenous and exogenous GR decreased the transcriptional activity of p53 in a dexamethasone-dependent manner. Similar observations have been made in another study (Maiyar et al., 1997). We have extended these observations, by showing that antisense GR RNA blocks endogenous GR + Dex inhibition of p53. Furthermore, we showed that GR + Dex or Cort antagonize p53’s positive and negative effects on the natural downstream targets, p21WAF1/CIP1, bax and bcl2, at the level of both their promoters and expression of the endogenous genes. Finally, we found that GR + Dex antagonize p53-mediated functions, including cell cycle arrest and apoptosis.
GR has been shown to inhibit apoptosis and stimulate cell cycle progression in many contexts, but has the opposite effects in others. Dex induces apoptosis in thymocytes (Cohen et al., 1992), lymphocytes (D’Adamio et al., 1997) and multiple myeloma cells (Chauhan et al., 1997). Dex has been shown to arrest the cell cycle by direct regulation of p21_WAF1/CIP1_ (Cha et al., 1998). In contrast, glucocorticoids repress apoptosis and increase proliferation in cancer cell lines (Chang et al., 1997), neutrophils (Daffern et al., 1999), retinal pigment cells (He et al., 1994), fibroblasts (Li et al., 1998), rat hepatoma (Yamamoto et al., 1998) and serum-depleted T lymphocytes that express Bcl2 (Huang and Cidlowski, 1999). Interestingly, Dex inhibits apoptosis induced by various chemotherapeutic agents more efficiently in cell lines with wild-type as opposed to mutant p53 (Weller et al., 1997). Steroids are known to decrease the efficacy of chemotherapy for some tumours (Green et al., 1983). Our study raises the possibility that GR inhibition of p53 may be involved in these effects.
In vitro studies have shown that GST–p53 interacts with GR (Yu et al., 1997a). We extended these observations by showing that GR and p53 interact in vivo. Furthermore, in the presence of Dex, GR and p53 co-localize in the cytoplasm, which could account for their mutually antagonistic activities. This observation raised the possibility that pathological and natural situations in which p53 is cytoplasmic could result from sequestration by GR. This could be the case in various situations. Wild-type p53 is cytoplasmic in inflammatory breast carcinoma (Moll et al., 1992), undifferentiated neuroblastomas (Moll et al., 1995), colon adenocarcinomas (Bosari et al., 1995), differentiated rat oligodendrocytes, neurons and PC12 pheochromocytoma cells (Eizenberg et al., 1996), neuroblastoma cell lines (Moll et al., 1996), Balb/c 3T3 cells in certain stages of the cell cycle (Shaulsky et al., 1990) and normal mouse embryonic stem cells (Aladjem et al., 1998).
In neuroblastoma cell lines we found that GR antagonists dissociate the GR–p53 complex, leading to nuclear accumulation of both p53 and GR, whereas a GR agonist did not have the same effect. The nuclear accumulation of p53 is associated with augmented expression of downstream genes, cell cycle arrest and apoptosis. The antagonist RU-486 has been observed to decrease proliferation of neuroblastoma cells in another report (Maggi et al., 1998), but the mechanism was not studied. Various attempts have been made to understand the mechanism of cytoplasmic accumulation of p53 in NB cell lines. A C-terminal peptide was shown to induce p53 re-localization to the nucleus (Ostermeyer et al., 1996), due to the unmasking of a nuclear export signal (NES) in the tetramerization domain (Stommel et al., 1999). Inhibi tion of nuclear export by leptomycin B also results in nuclear accumulation of p53 (Stommel et al., 1999). p53 is sequestered in large protein complexes in the cytoplasm (Moll et al., 1996), which was proposed to account for its resistance to Mdm-2-mediated degradation (Zaika et al., 1999). A model taking into account these observations and our results is that p53 is sequestered in the cytoplasm in a complex with GR, which is in equilibrium with molecules free to cycle in and out of the nucleus. In the complex, the nuclear localization signal of either molecule may be masked. The inactive DITS chimeras minimally contain the p53 DNA binding domain and the GR LBD, suggesting that these domains of the two proteins interact. GR antagonists, upon binding to the LBD, presumably alter the conformation and thereby weaken the interaction with p53. Release of p53 would disturb the equilibrium and thereby increase the pool of p53 that can move into the nucleus. Increasing the rate of entry or decreasing the rate of exit from the nucleus of molecules free to cycle would also lead to nuclear accumulation.
The mechanisms that regulate ligand-independent formation of GR–p53 complexes and control the amount of p53 that is in the cytoplasm remain to be established. An interesting possibility is that the extent of p53–GR interaction in the absence of ligand is set at different levels, which are low in most cell types and high in neuroblastoma. In several cell lines other than neuroblastoma we observed that GR inhibits p53 functions to a low extent in the absence of ligand, suggesting that there could be a low level of ligand-independent interaction. The amount of endogenous glucocorticoids in the serum most likely is not enough to activate the receptor. Furthermore, in preliminary experiments, we found that GR inhibits p53 in stripped medium (data not shown). The mechanisms that regulate ligand-independent interactions could involve post-translational modifications and interactions with other proteins. Interestingly, a protein kinase inhibitor, 1-(5-isoquinolinesulfonyl)-2-methylpiperazine (H-7), has been demonstrated to induce apoptosis in NB cell lines in a p53-dependent manner (Ronca et al., 1997), raising the possibility that phosphorylation may regulate the GR–p53 interaction. It would also be interesting to determine whether down-regulation of Hsp90, which is known to bind to and inactivate GR, affects p53 function.
The normal physiological function of cross-talk between GR and p53 is an interesting field for further study. Glucocorticoids and p53 have antagonistic effects under various conditions that could be imagined to require cross-talk. One of these is that glucocorticoids increase glucose metabolism in response to psychological stress. Hypoxic stress leads to p53 activation, increased apoptosis and decreased glucose metabolism (Riva et al., 1998). Perhaps the choice between adaptation (increased glucose metabolism) and death in response to hypoxic stress involves cross-talk between GR and p53. Interestingly, the type II hexokinase gene, whose overexpression is associated with increased glucose catabolism in cancer cells, is apparently regulated in opposing manners by p53 and GR (Mathupala et al., 1997).
In summary, we have demonstrated that p53 and GR repress each other’s functions. Mutual repression apparently results from their physical interaction in vivo and co-localization in the cytoplasm. However, we cannot exclude that other mechanisms also contribute to p53 inactivation by GR (effects on DNA binding or others). Strikingly, p53 is able to prevent ligand-mediated transport of GR to the nucleus. In a pathological situation, represented by neuroblastoma cells, endogenous GR and p53 also interact physically. Disruption of this complex results in translocation of wild-type p53 from the cytoplasm into the nucleus, activation of p53-dependent endogenous genes, growth arrest and apoptosis. Thus, this report provides the first evidence for the inactivation of p53 by a steroid hormone-dependent signalling pathway in a human pathological condition.
Materials and methods
Recombinants
_p53–GR_D. PCR amplification of the coding region of p53 was used to remove the stop codon and introduce _Eco_RI and _Xho_I sites at 5′ and 3′ ends, respectively. p53–GRD was generated by co-ligating the 1.2 kb _Eco_RI–_Xho_I digested PCR product, the 5.4 kb _Eco_RI–_Xho_I fragment from pcDNA3 and the 1 kb _Xho_I–_Sal_I fragment from pCre-GRD containing GRD-LBD.
_CTS1–GR_D. Initially, modified CTS1 without the stop codon was produced by ligating the 6.4 kb _Xho_I–_Pfl_MI fragment from pcDNA3CTS1 (Conseiller et al., 1998) with the _Pfl_MI–_Xho_I 100 bp fragment from a PCR product that was amplified with a primer before the _Pfl_MI site (within CTS1) and a reverse primer excluding the stop codon but incorporating a _Xho_I site. The 6.5 kb _Xho_I-digested modified-CTS1 plasmid was ligated to the 1 kb _Xho_I–_Sal_I fragment from pCre-GRD containing the GRD-LBD, to generate CTS1–GRD.
Antisense GR (ASGR). The 2.7 kb _Eco_RI fragment from HG1 (Kumar et al., 1987) was ligated to _Eco_RI-linearized pcDNA3 to generate pcDNA3ASGR or pcDNA3GR.
Reporters and expression vectors. p53CON, a gift from J.Shay (Funk et al., 1992); p21WAF1/CIP1–luc, a gift from B.Vogelstein (el-Deiry et al., 1993); bax–luc, a gift from J.Reed (Miyashita and Reed, 1995); bcl2–luc, a gift from L.Boxer (Budhram-Mahadeo et al., 1999); GRE–tk–luc, a gift grom H.Gronemeyer (Roux et al., 1996); CTS1, a gift from E.Conseiller (Conseiller et al., 1998); pC53-SN3, a gift from A.J.Levine (Hinds et al., 1990); pCre-GRD, a gift from D.Metzger (Brocard et al., 1998); HG1, a gift from P.Chambon (Kumar et al., 1987); CMV-CD20, a gift from L.Zhu (Zhu et al., 1993); CMV-LacZ, IGBMC core facility.
Cells
HSC-2 (head and neck carcinoma), a gift from N.Tsuchida (Momose et al., 1989); PAP (head and neck carcinoma), IGBMC cell culture facility; Saos-2 (osteosarcoma), SiHa (cervical carcinoma), IMR-32 (neuroblastoma), SK-SY5Y (neuroblastoma) and SK-N-SH (neuroblastoma), ATCC.
Transfections
Cell lines were maintained in Dulbecco’s modified Eagle’s medium + 10% serum + antibiotics at 37°C with 5% CO2. They were transfected using calcium phosphate (Chen and Okayama, 1987) with 4 µg of plasmid, in 36 mm plates (6-well cluster, Costar 3516) in the presence of either the vehicle (ethanol) or the ligands (Dex or RU-486, Sigma; RU-044, a gift from H.Gronemeyer). After 16 h the cells were washed with medium and antibiotics and re-fed with full growth medium ± ligands. Post-transfection is defined as after removal of the precipitate.
Colony formation assays
Forty-eight hours post-transfection the cells were washed and cultured in 1 mg/ml G418 for 3 weeks, with medium changes every 5–6 days. Three independent transfections were performed for each experimental point and the whole experiment was repeated at least twice. Clones were stained with Giemsa and counted.
Transcriptional activity and DNA fragmentation
Cell lysates were analysed for luciferase and β-galactosidase activities, and DNA fragmentation, as described previously (Sengupta et al., 2000).
Immunoprecipitations
Standard procedures were used. The cells were lysed on ice in NP-40 buffer (150 mM NaCl, 1.0% NP-40, 50 mM Tris–HCl pH 8.0) and the cleared supernatant used for immunoprecipitation. The antigen–antibody complex, formed on ice for 1 h, was immobilized on protein G beads, washed three times, boiled in Laemmli buffer and analysed by SDS–PAGE.
Western blots
The protein content of RIPA buffer lysates was estimated by the Bio-Rad assay. Equal quantities were fractionated by SDS–PAGE, transferred to nitrocellulose membranes (Schleicher and Schuell) and processed for western blotting. Primary and secondary antibodies: Bax, N20 (Santa Cruz Biotechnology); p21_WAF1/CIP1_, 1WA-IC581 (L.Andera and the IGBMC core facility); TBP, 3G3 (Brou et al., 1993); p53, DO-1 (Vojtesek et al., 1992); p53, pAb 240 (Gannon et al., 1990); GFP (Clontech); GR, E-20 and P-20 (Santa Cruz Biotechnology); Bcl2, 100 (Santa Cruz Biotechnology); PARP (Kaufmann et al., 1993); HA, 12CA5 (Boehringer Mannheim); anti-CD20 (Pharmingen); peroxidase-coupled anti-rabbit or mouse antibody (Jackson Laboratories). The blots were revealed by chemiluminescence (Pierce Super Signal).
Flow cytometry
HSC-2 cells were co-transfected with the appropriate vectors and CMV-CD20 in 10 cm dishes (20 µg total plasmid). Twenty-four hours post-transfection the cells were processed as described previously (Wasylyk et al., 1999). About 15 000 CD-20 positive cells for HSC-2 and 50 000 cells for IMR 32 were analysed.
Immunofluorescence
The cells were fixed with acetone:methanol (1:1), blocked with 10% normal goat serum, incubated with primary antibodies (mouse for p53 and rabbit for GR) for 2 h, followed by appropriate secondary antibodies linked either to Cy3 or fluorescein isothiocyanate (Moll et al., 1996), and visualized by confocal microscopy (Leica TCS 4D). Hoechst dye was used to stain nuclei.
Acknowledgments
Acknowledgements
We would like to thank H.Gronemeyer and P.Anglard for critical reading of the manuscript, P.Chambon, J.Shay, B.Vogelstein, J.Reed, L.Boxer, H.Gronemeyer, E.Conseiller, A.J.Levine, D.Metzger and L.Zhu for the gifts of recombinants, N.Tsuchida for HSC-2 cells, L.Andera and G.de Murcia for the gift of antibodies, the IGBMC core facilities for help and support, CEFIPRA for their financial and scientific contribution to the early stages of this work, BioAvenir (Rhone-Poulenc, now Aventis), the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, the Hôpital Universitaire de Strasbourg, the Association pour la Recherche sur le Cancer, the Fondation pour la Recherche Médicale, the Ligue Nationale Française contre le Cancer, the Ligue Régionale (Haut-Rhin) contre le Cancer and the Ligue Régionale (Bas-Rhin) contre le Cancer for financial help.
References
- Aladjem M.I., Spike,B.T., Rodewald,L.W., Hope,T.J., Klemm,M., Jaenisch,R. and Wahl,G.M. (1998) ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr. Biol., 8, 145–155. [DOI] [PubMed] [Google Scholar]
- An W.G., Kanekal,M., Simon,M.C., Maltepe,E., Blagosklonny,M.V. and Neckers,L.M. (1998) Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature, 392, 405–408. [DOI] [PubMed] [Google Scholar]
- Barak Y., Juven,T., Haffner,R. and Oren,M. (1993) mdm2 expression is induced by wild-type p53 activity. EMBO J., 12, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes P.J. and Adcock,I. (1993) Anti-inflammatory actions of steroids: molecular mechanisms. Trends Pharmacol. Sci., 14, 436–441. [DOI] [PubMed] [Google Scholar]
- Baulieu E.E. (1997) RU 486 (mifepristone). A short overview of its mechanisms of action and clinical uses at the end of 1996. Ann. NY Acad. Sci., 828, 47–58. [DOI] [PubMed] [Google Scholar]
- Beato M., Herrlich,P. and Schutz,G. (1995) Steroid hormone receptors: many actors in search of a plot. Cell, 83, 851–857. [DOI] [PubMed] [Google Scholar]
- Blander G., Kipnis,J., Leal,J.F., Yu,C.E., Schellenberg,G.D. and Oren,M. (1999) Physical and functional interaction between p53 and the Werner’s syndrome protein. J. Biol. Chem., 274, 29463–29469. [DOI] [PubMed] [Google Scholar]
- Bocquel M.T., Ji,J., Ylikomi,T., Benhamou,B., Vergezac,A., Chambon,P. and Gronemeyer,H. (1993) Type II antagonists impair the DNA binding of steroid hormone receptors without affecting dimerization. J. Steroid Biochem. Mol. Biol., 45, 205–215. [DOI] [PubMed] [Google Scholar]
- Bosari S., Viale,G., Roncalli,M., Graziani,D., Borsani,G., Lee,A.K. and Coggi,G. (1995) p53 gene mutations, p53 protein accumulation and compartmentalization in colorectal adenocarcinoma. Am. J. Pathol., 147, 790–798. [PMC free article] [PubMed] [Google Scholar]
- Brocard J., Feil,R., Chambon,P. and Metzger,D. (1998) A chimeric Cre recombinase inducible by synthetic,but not by natural ligands of the glucocorticoid receptor. Nucleic Acids Res., 26, 4086–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brou C., Chaudhary,S., Davidson,I., Lutz,Y., Wu,J., Egly,J.M., Tora,L. and Chambon,P. (1993) Distinct TFIID complexes mediate the effect of different transcriptional activators. EMBO J., 12, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J., Chandrasekaran,C., Gordon,J.I., Beach,D., Jacks,T. and Hannon,G.J. (1995) Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature, 377, 552–557. [DOI] [PubMed] [Google Scholar]
- Budhram-Mahadeo V., Morris,P.J., Smith,M.D., Midgley,C.A., Boxer,L.M. and Latchman,D.S. (1999) p53 suppresses the activation of the Bcl-2 promoter by the Brn-3a POU family transcription factor. J. Biol. Chem., 274, 15237–15244. [DOI] [PubMed] [Google Scholar]
- Cha H.H., Cram,E.J., Wang,E.C., Huang,A.J., Kasler,H.G. and Firestone,G.L. (1998) Glucocorticoids stimulate p21 gene expression by targeting multiple transcriptional elements within a steroid responsive region of the p21waf1/cip1 promoter in rat hepatoma cells. J. Biol. Chem., 273, 1998–2007. [DOI] [PubMed] [Google Scholar]
- Chang T.C., Hung,M.W., Jiang,S.Y., Chu,J.T., Chu,L.L. and Tsai,L.C. (1997) Dexamethasone suppresses apoptosis in a human gastric cancer cell line through modulation of bcl-x gene expression. FEBS Lett., 415, 11–15. [DOI] [PubMed] [Google Scholar]
- Chauhan D., Pandey,P., Ogata,A., Teoh,G., Treon,S., Urashima,M., Kharbanda,S. and Anderson,K.C. (1997) Dexamethasone induces apoptosis of multiple myeloma cells in a JNK/SAP kinase independent mechanism. Oncogene, 15, 837–843. [DOI] [PubMed] [Google Scholar]
- Chen C. and Okayama,H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol., 7, 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J.J., Duke,R.C., Fadok,V.A. and Sellins,K.S. (1992) Apoptosis and programmed cell death in immunity. Annu. Rev. Immunol., 10, 267–293. [DOI] [PubMed] [Google Scholar]
- Cole T.J. et al. (1995) Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev., 9, 1608–1621. [DOI] [PubMed] [Google Scholar]
- Conseiller E., Debussche,L., Landais,D., Venot,C., Maratrat,M., Sierra,V., Tocque,B. and Bracco,L. (1998) CTS1: a p53-derived chimeric tumor suppressor gene with enhanced in vitro apoptotic properties. J. Clin. Invest., 101, 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Adamio F., Zollo,O., Moraca,R., Ayroldi,E., Bruscoli,S., Bartoli,A., Cannarile,L., Migliorati,G. and Riccardi,C. (1997) A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity, 7, 803–812. [DOI] [PubMed] [Google Scholar]
- Daffern P.J., Jagels,M.A. and Hugli,T.E. (1999) Multiple epithelial cell-derived factors enhance neutrophil survival. Regulation by glucocorticoids and tumor necrosis factor-α. Am. J. Respir. Cell Mol. Biol., 21, 259–267. [DOI] [PubMed] [Google Scholar]
- Diamond M.I., Miner,J.N., Yoshinaga,S.K. and Yamamoto,K.R. (1990) Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science, 249, 1266–1272. [DOI] [PubMed] [Google Scholar]
- Drouin J., Sun,Y.L., Chamberland,M., Gauthier,Y., De Lean,A., Nemer,M. and Schmidt,T.J. (1993) Novel glucocorticoid receptor complex with DNA element of the hormone-repressed POMC gene. EMBO J., 12, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizenberg O., Faber-Elman,A., Gottlieb,E., Oren,M., Rotter,V. and Schwartz,M. (1996) p53 plays a regulatory role in differentiation and apoptosis of central nervous system-associated cells. Mol. Cell. Biol., 16, 5178–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry W.S. et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell, 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Elkeles A., Juven-Gershon,T., Israeli,D., Wilder,S., Zalcenstein,A. and Oren,M. (1999) The c-fos proto-oncogene is a target for transactivation by the p53 tumor suppressor. Mol. Cell. Biol., 19, 2594–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S.Y., Adler,V., Buschmann,T., Yin,Z., Wu,X., Jones,S.N. and Ronai,Z. (1998) JNK targets p53 ubiquitination and degradation in non-stressed cells. Genes Dev., 12, 2658–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk W.D., Pak,D.T., Karas,R.H., Wright,W.E. and Shay,J.W. (1992) A transcriptionally active DNA-binding site for human p53 protein complexes. Mol. Cell. Biol., 12, 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiddon C., Moorthy,N.C. and Prives,C. (1999) Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J., 18, 5609–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon J.V., Greaves,R., Iggo,R. and Lane,D.P. (1990) Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J., 9, 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccia A.J. and Kastan,M.B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Green S.B. et al. (1983) Comparisons of carmustine, procarbazine and high-dose methylprednisolone as additions to surgery and radiotherapy for the treatment of malignant glioma. Cancer Treat. Rep., 67, 121–132. [PubMed] [Google Scholar]
- Harris C.C. (1996) Structure and function of the p53 tumor suppressor gene: clues for rational cancer therapeutic strategies. J. Natl Cancer Inst., 88, 1442–1455. [DOI] [PubMed] [Google Scholar]
- Haupt Y., Maya,R., Kazaz,A. and Oren,M. (1997) Mdm2 promotes the rapid degradation of p53. Nature, 387, 296–299. [DOI] [PubMed] [Google Scholar]
- He S., Wang,H.M., Ye,J., Ogden,T.E., Ryan,S.J. and Hinton,D.R. (1994) Dexamethasone induced proliferation of cultured retinal pigment epithelial cells. Curr. Eye Res., 13, 257–261. [DOI] [PubMed] [Google Scholar]
- Hinds P.W., Finlay,C.A., Quartin,R.S., Baker,S.J., Fearon,E.R., Vogelstein,B. and Levine,A.J. (1990) Mutant p53 DNA clones from human colon carcinomas cooperate with ras in transforming primary rat cells: a comparison of the ‘hot spot’ mutant phenotypes. Cell Growth Differ., 1, 571–580. [PubMed] [Google Scholar]
- Hollstein M., Sidransky,D., Vogelstein,B. and Harris,C.C. (1991) p53 mutations in human cancers. Science, 253, 49–53. [DOI] [PubMed] [Google Scholar]
- Hsieh J.K., Chan,F.S., O’Connor,D.J., Mittnacht,S., Zhong,S. and Lu,X. (1999) RB regulates the stability and the apoptotic function of p53 via MDM2. Mol. Cell, 3, 181–193. [DOI] [PubMed] [Google Scholar]
- Huang S.T. and Cidlowski,J.A. (1999) Glucocorticoids inhibit serum depletion-induced apoptosis in T lymphocytes expressing Bcl-2. FASEB J., 13, 467–476. [DOI] [PubMed] [Google Scholar]
- Hurd C., Khattree,N., Dinda,S., Alban,P. and Moudgil,V.K. (1997) Regulation of tumor suppressor proteins, p53 and retinoblastoma, by estrogen and antiestrogens in breast cancer cells. Oncogene, 15, 991–995. [DOI] [PubMed] [Google Scholar]
- Kaufmann S.H., Desnoyers,S., Ottaviano,Y., Davidson,N.E. and Poirier,G.G. (1993) Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res., 53, 3976–3985. [PubMed] [Google Scholar]
- Kellendonk C., Tronche,F., Reichardt,H.M. and Schutz,G. (1999) Mutagenesis of the glucocorticoid receptor in mice. J. Steroid Biochem. Mol. Biol., 69, 253–259. [DOI] [PubMed] [Google Scholar]
- Ko L.J. and Prives,C. (1996) p53: puzzle and paradigm. Genes Dev., 10, 1054–1072. [DOI] [PubMed] [Google Scholar]
- Kubbutat M.H., Jones,S.N. and Vousden,K.H. (1997) Regulation of p53 stability by Mdm2. Nature, 387, 299–303. [DOI] [PubMed] [Google Scholar]
- Kumar V., Green,S., Stack,G., Berry,M., Jin,J.R. and Chambon,P. (1987) Functional domains of the human estrogen receptor. Cell, 51, 941–951. [DOI] [PubMed] [Google Scholar]
- Lee S.K., Kim,H.J., Kim,J.W. and Lee,J.W. (1999) Steroid receptor coactivator-1 and its family members differentially regulate transactivation by the tumor suppressor protein p53. Mol. Endocrinol., 13, 1924–1933. [DOI] [PubMed] [Google Scholar]
- Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Li S., Mawal-Dewan,M., Cristofalo,V.J. and Sell,C. (1998) Enhanced proliferation of human fibroblasts, in the presence of dexamethasone, is accompanied by changes in p21Waf1/Cip1/Sdi1 and the insulin-like growth factor type 1 receptor. J. Cell. Physiol., 177, 396–401. [DOI] [PubMed] [Google Scholar]
- Lin D.L. and Chang,C. (1996) p53 is a mediator for radiation-repressed human TR2 orphan receptor expression in MCF-7 cells, a new pathway from tumor suppressor to member of the steroid receptor superfamily. J. Biol. Chem., 271, 14649–14652. [DOI] [PubMed] [Google Scholar]
- Maggi R., Poletti,A., Casulari,L.A., Pimpinelli,F., Piva,F., Zanisi,M.R. and Martini,L. (1998) Effects and metabolism of steroid hormones in human neuroblastoma cells. Steroids, 63, 257–262. [DOI] [PubMed] [Google Scholar]
- Maiyar A.C., Phu,P.T., Huang,A.J. and Firestone,G.L. (1997) Repression of glucocorticoid receptor transactivation and DNA binding of a glucocorticoid response element within the serum/glucocorticoid-inducible protein kinase (sgk) gene promoter by the p53 tumor suppressor protein. Mol. Endocrinol., 11, 312–329. [DOI] [PubMed] [Google Scholar]
- Mathupala S.P., Heese,C. and Pedersen,P.L. (1997) Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J. Biol. Chem., 272, 22776–22780. [DOI] [PubMed] [Google Scholar]
- Meyer M.E., Pornon,A., Ji,J.W., Bocquel,M.T., Chambon,P. and Gronemeyer,H. (1990) Agonistic and antagonistic activities of RU486 on the functions of the human progesterone receptor. EMBO J., 9, 3923–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita T. and Reed,J.C. (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell, 80, 293–299. [DOI] [PubMed] [Google Scholar]
- Miyashita T., Harigai,M., Hanada,M. and Reed,J.C. (1994a) Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res., 54, 3131–3135. [PubMed] [Google Scholar]
- Miyashita T., Krajewski,S., Krajewska,M., Wang,H.G., Lin,H.K., Liebermann,D.A., Hoffman,B. and Reed,J.C. (1994b) Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene, 9, 1799–1805. [PubMed] [Google Scholar]
- Moll U.M., Riou,G. and Levine,A.J. (1992) Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc. Natl Acad. Sci. USA, 89, 7262–7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll U.M., LaQuaglia,M., Benard,J. and Riou,G. (1995) Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc. Natl Acad. Sci. USA, 92, 4407–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll U.M., Ostermeyer,A.G., Haladay,R., Winkfield,B., Frazier,M. and Zambetti,G. (1996) Cytoplasmic sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. Mol. Cell. Biol., 16, 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momose F., Araida,T., Negishi,A., Ichijo,H., Shioda,S. and Sasaki,S. (1989) Variant sublines with different metastatic potentials selected in nude mice from human oral squamous cell carcinomas. J. Oral Pathol. Med., 18, 391–395. [DOI] [PubMed] [Google Scholar]
- Murphy M., Ahn,J., Walker,K.K., Hoffman,W.H., Evans,R.M., Levine,A.J. and George,D.L. (1999) Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev., 13, 2490–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner J.D., Pietenpol,J.A., Thiagalingam,S., Gyuris,J., Kinzler,K.W. and Vogelstein,B. (1993) Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature, 362, 857–860. [DOI] [PubMed] [Google Scholar]
- Ostermeyer A.G., Runko,E., Winkfield,B., Ahn,B. and Moll,U.M. (1996) Cytoplasmically sequestered wild-type p53 protein in neuroblastoma is relocated to the nucleus by a C-terminal peptide. Proc. Natl Acad. Sci. USA, 93, 15190–15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragimov N., Krauskopf,A., Navot,N., Rotter,V., Oren,M. and Aloni,Y. (1993) Wild-type but not mutant p53 can repress transcription initiation in vitro by interfering with the binding of basal transcription factors to the TATA motif. Oncogene, 8, 1183–1193. [PubMed] [Google Scholar]
- Reichardt H.M. et al. (1998) DNA binding of the glucocorticoid receptor is not essential for survival. Cell, 93, 531–541. [DOI] [PubMed] [Google Scholar]
- Riva C., Chauvin,C., Pison,C. and Leverve,X. (1998) Cellular physiology and molecular events in hypoxia-induced apoptosis. Anticancer Res., 18, 4729–4736. [PubMed] [Google Scholar]
- Ronca F., Chan,S.L. and Yu,V.C. (1997) 1-(5-Isoquinolinesulfonyl)-2-methylpiperazine induces apoptosis in human neuroblastoma cells, SH-SY5Y, through a p53-dependent pathway. J. Biol. Chem., 272, 4252–4260. [DOI] [PubMed] [Google Scholar]
- Roux S., Terouanne,B., Balaguer,P., Jausons-Loffreda,N., Pons,M., Chambon,P., Gronemeyer,H. and Nicolas,J.C. (1996) Mutation of isoleucine 747 by a threonine alters the ligand responsiveness of the human glucocorticoid receptor. Mol. Endocrinol., 10, 1214–1226. [DOI] [PubMed] [Google Scholar]
- Saji S., Nakashima,S., Hayashi,S., Toi,M. and Nozawa,Y. (1999) Overexpression of MDM2 in MCF-7 promotes both growth advantage and p53 accumulation in response to estradiol. Jpn. J. Cancer Res., 90, 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savory J.G., Hsu,B., Laquian,I.R., Giffin,W., Reich,T., Hache,R.J. and Lefebvre,Y.A. (1999) Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol. Cell. Biol., 19, 1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S., Ralhan,R. and Wasylyk,B. (2000) Tumour regression in a ligand inducible manner mediated by a chimeric tumour suppressor derived from p53. Oncogene, 19, 337–350. [DOI] [PubMed] [Google Scholar]
- Shaulsky G., Ben-Ze′ev,A. and Rotter,V. (1990) Subcellular distribution of the p53 protein during the cell cycle of Balb/c 3T3 cells. Oncogene, 5, 1707–1711. [PubMed] [Google Scholar]
- Sherr C.J. and Weber,J.D. (2000) The ARF/p53 pathway. Curr. Opin. Genet. Dev., 10, 94–99. [DOI] [PubMed] [Google Scholar]
- Sionov R.V., Moallem,E., Berger,M., Kazaz,A., Gerlitz,O., Ben-Neriah,Y., Oren,M. and Haupt,Y. (1999) c-Abl neutralizes the inhibitory effect of Mdm2 on p53. J. Biol. Chem., 274, 8371–8374. [DOI] [PubMed] [Google Scholar]
- Steele R.J., Thompson,A.M., Hall,P.A. and Lane,D.P. (1998) The p53 tumour suppressor gene. Br. J. Surg., 85, 1460–1467. [DOI] [PubMed] [Google Scholar]
- Stommel J.M., Marchenko,N.D., Jimenez,G.S., Moll,U.M., Hope,T.J. and Wahl,G.M. (1999) A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J., 18, 1660–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F., Kellendonk,C., Reichardt,H.M. and Schutz,G. (1998) Genetic dissection of glucocorticoid receptor function in mice. Curr. Opin. Genet. Dev., 8, 532–538. [DOI] [PubMed] [Google Scholar]
- van Steensel B., Brink,M., van der Meulen,K., van Binnendijk,E.P., Wansink,D.G., de Jong,L., de Kloet,E.R. and van Driel,R. (1995) Localization of the glucocorticoid receptor in discrete clusters in the cell nucleus. J. Cell Sci., 108, 3003–3011. [DOI] [PubMed] [Google Scholar]
- Vojtesek B., Bartek,J., Midgley,C.A. and Lane,D.P. (1992) An immunochemical analysis of the human nuclear phosphoprotein p53. New monoclonal antibodies and epitope mapping using recombinant p53. J. Immunol. Methods, 151, 237–244. [DOI] [PubMed] [Google Scholar]
- Waldman T., Lengauer,C., Kinzler,K.W. and Vogelstein,B. (1996) Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature, 381, 713–716. [DOI] [PubMed] [Google Scholar]
- Wasylyk C., Salvi,R., Argentini,M., Dureuil,C., Delumeau,I., Abecassis,J., Debussche,L. and Wasylyk,B. (1999) p53 mediated death of cells overexpressing MDM2 by an inhibitor of MDM2 interaction with p53. Oncogene, 18, 1921–1934. [DOI] [PubMed] [Google Scholar]
- Weller M., Schmidt,C., Roth,W. and Dichgans,J. (1997) Chemotherapy of human malignant glioma: prevention of efficacy by dexamethasone? Neurology, 48, 1704–1709. [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Fukuda,K., Miura,N., Suzuki,R., Kido,T. and Komatsu,Y. (1998) Inhibition by dexamethasone of transforming growth factor β1-induced apoptosis in rat hepatoma cells: a possible association with Bcl-xL induction. Hepatology, 27, 959–966. [DOI] [PubMed] [Google Scholar]
- Yap N., Yu,C.L. and Cheng,S.Y. (1996) Modulation of the transcriptional activity of thyroid hormone receptors by the tumor suppressor p53. Proc. Natl Acad. Sci. USA, 93, 4273–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C., Yap,N., Chen,D. and Cheng,S. (1997a) Modulation of hormone-dependent transcriptional activity of the glucocorticoid receptor by the tumor suppressor p53. Cancer Lett., 116, 191–196. [DOI] [PubMed] [Google Scholar]
- Yu C.L., Driggers,P., Barrera-Hernandez,G., Nunez,S.B., Segars,J.H. and Cheng,S. (1997b) The tumor suppressor p53 is a negative regulator of estrogen receptor signaling pathways. Biochem. Biophys. Res. Commun., 239, 617–620. [DOI] [PubMed] [Google Scholar]
- Yu J., Zhang,L., Hwang,P.M., Rago,C., Kinzler,K.W. and Vogelstein,B. (1999) Identification and classification of p53-regulated genes. Proc. Natl Acad. Sci. USA, 96, 14517–14522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaika A., Marchenko,N. and Moll,U.M. (1999) Cytoplasmically “sequestered” wild type p53 protein is resistant to Mdm2-mediated degradation [published erratum appears in J. Biol. Chem., 275, 11538]. J. Biol. Chem., 274, 27474–27480. [DOI] [PubMed] [Google Scholar]
- Zhao R., Gish,K., Murphy,M., Yin,Y., Notterman,D., Hoffman,W.H., Tom,E., Mack,D.H. and Levine,A.J. (2000) Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev., 14, 981–993. [PMC free article] [PubMed] [Google Scholar]
- Zhu L., van den Heuvel,S., Helin,K., Fattaey,A., Ewen,M., Livingston,D., Dyson,N. and Harlow,E. (1993) Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes Dev., 7, 1111–1125. [DOI] [PubMed] [Google Scholar]