Observations in APP Bitransgenic Mice Suggest that Diffuse and Compact Plaques Form via Independent Processes in Alzheimer's Disease (original) (raw)
Abstract
Studies of familial Alzheimer's disease suggest that misfolding and aggregation of amyloid-β (Aβ) peptides initiate the pathogenesis. The Arctic mutation of Aβ precursor protein (APP) results in AD, and Arctic Aβ is more prone to form Aβ protofibrils and extracellular deposits. Herein is demonstrated that the burden of diffuse Aβ deposits but not compact plaques is increased when tg-Swe mice are crossed with tg-ArcSwe mice synthesizing low levels of Arctic Aβ. The diffuse deposits in bitransgenic mice, which contain primarily wild-type Aβ42, accumulate in regions both with and without transgene expression. However, APP processing, when compared with tg-Swe, remains unchanged in young bitransgenic mice, whereas wild-type Aβ42 aggregation is accelerated and fibril architecture is altered in vitro and in vivo when a low level of Arctic Aβ42 is introduced. Thus, the increased number of diffuse deposits is likely due to physical interactions between Arctic Aβ and wild-type Aβ42. The selective increase of a single type of parenchymal Aβ deposit suggests that different pathways lead to formation of diffuse and compact plaques. These findings could have general implications for Alzheimer's disease pathogenesis and particular relevance to patients heterozygous for the Arctic APP mutation. Moreover, it further illustrates how Aβ neuropathologic features can be manipulated in vivo by mechanisms similar to those originally conceptualized in prion research.
The cardinal histopathologic lesions in brains affected with Alzheimer's disease (AD) are neurofibrillary tangles and extracellular compact plaques. The plaques are composed of various amyloid-β (Aβ) peptides and stain with amyloid dyes such as Congo red. Aβ is generated via endoproteolysis of the amyloid precursor protein (APP). It aggregates and gives rise to compact plaques with an amyloid core, but also parenchymal diffuse Aβ deposits and cerebrovascular amyloid angiopathy in vessel walls. Discoveries of point mutations in the APP gene that caused familial AD provided major insights into the pathogenesis.1 The Arctic mutation (E693G), located within the Aβ domain of APP, segregates with clinical AD.2,3 Because Arctic Aβ (AβArc) is more prone to form protofibrils than is wild-type Aβ (Aβwt),3,4 this mutation was used in combination with the Swedish mutation (KM670/671NL) to develop an animal model rich in soluble oligomeric Aβ assemblies.5,6 Several founder lines were generated; however, only the line with the highest APP expression (line B, Table 1) has been reported to date.5,7–9 Its neuropathologic phenotypes were compared in detail with those of a transgenic model harboring only the Swedish mutation (tg-Swe; line A, Table 1).10 In other founder lines of tg-ArcSwe and tg-Swe, the synthesis of human APP and Aβ was low, and Aβ deposition was scarce or undetectable even in very old animals. Herein we cross-bred a tg-ArcSwe founder line with very scarce Aβ pathology with a plaque-depositing tg-Swe line, in which human APP was expressed at a high level. We deemed that the experiment would be interesting for the following reasons. First, amyloid formation in human brain and APP transgenic mice is perceived to be seeded; that is, rapid fibril formation is thought to be preceded by a slow and rate-limiting reaction in which a nucleus is formed.11,12 Both in vitro and in vivo studies have clearly demonstrated that AβArc is more proto-fibrillogenic than Aβwt.4,5,13 It was hypothesized that simultaneous production of AβArc and Aβwt in bitransgenic mice would result in a prion-like aggregation mechanism14 in which the more aggregation-prone AβArc would act as a β-sheet template and accelerate Aβwt fibrillization. Second, both AβArc and Aβwt are produced in patients heterozygous for the Arctic mutation, and the pathogenic effects of such a mixture of Aβ peptides, if any, are unknown. By comparing the neuropathologic features of bitransgenic mice (tg-ArcSwe and tg-Swe) with single transgenic tg-Swe mice, whether aggregation-prone AβArc regulates Aβwt aggregation and/or formation of amyloid cores in the brain could be assessed. It was observed that the mixture of AβArc and Aβwt in bitransgenic mice led to more Aβ42-immunoreactive (ir) diffuse deposits in areas with and without transgene expression in bitransgenic mice but did not lead to more compact plaques. These findings suggest that diffuse and compact plaques develop via independent processes.
Table 1.
Primary Characteristics of Various APP-Transgenic Lines
| Model | Line | Human APP expression | + | Murine APP expression | = | APP overexpression⁎ | Age at onset of plaque deposition (months) |
|---|---|---|---|---|---|---|---|
| tg-Swe | A | 6.0 | 1 | Sevenfold APPSwe | 12 | ||
| tg-ArcSwe | B | 2.0 | 1 | Threefold APPArcSwe | 5–6 | ||
| tg-ArcSwe | D | 0.85 | 1 | Twofold APPArcSwe | 18 | ||
| tg-ArcSwe × tg-Swe | Bitransgenic | 6.85 | 1 | ∼Eightfold APP (APPArcSwe + APPSwe) | 12 | ||
| Nontransgenic | 0 | 1 |
Materials and Methods
Transgenic Mice
The expression cassette used to generate transgenic models and the procedures of genotyping have been described.5 All mice were kept at the animal facility at Uppsala University and housed under standard conditions with a 12-hour light/dark cycle and with free access to food and water. The tg-ArcSwe mice primarily used in this study were derived from a founder line with a low level of transgene APP expression. The phenotypes of this founder line differ markedly from the high-expressor tg-ArcSwe model.5,10 The various transgenic lines used in the study are given in Table 1. All mice were genotyped using PCR with primers located at 1638 to 1655 and 2299 to 2281 in Y00264, followed by digestion with MboII and separation of bands on 2% agarose gel. The PCR product from a tg-ArcSwe mouse was then cleaved into two bands (456 and 205 bp), that from a tg-Swe mouse into three bands (364, 205, and 92 bp), and that from a bitransgenic tg-ArcSwe and tg-Swe mouse harboring both transgenes into four bands (456, 364, 205, and 92 bp). All mice used in the study were genotyped at age 3 weeks (ear biopsy) and again after death (tail biopsy). The experiments were approved by an ethical committee and performed in compliance with national and local animal care and use guidelines (protocol C223/8). Mice were anesthetized using 0.4 mL tribromoethanol (Avertin), 25 mg/mL, and intracardially perfused with 0.9% (w/v) NaCl solution. Brain hemispheres were frozen on dry ice for biochemical analyses. Alternatively, they were either directly frozen in metylbutane at −25°C or immersed in 4% paraformaldehyde for 24 hours and used for immunohistochemical analyses.
Histologic and Image Analyses
Fixed brain tissue was cryoprotected via sequential immersion in 10%, 20%, and 30% (w/v) sucrose for 24 hours. Coronal sections 25-μm thick were prepared with a sledge microtome and stored at 4°C in PBS with 10 mmol/L NaN3. Two sections per individual, approximately 500 μm apart (bregma −1.0 mm to −3.0 mm), were selected for quantitative immunostaining. Fixed tissue sections were incubated in 25 mmol/L prewarmed citrate buffer (pH 7.3) for 5 minutes at 85°C, rinsed in deionized water, and immersed in 70% formic acid for 5 minutes. Endogenous peroxidase activity was quenched with 0.3% dH2O2 in Dako block (DakoCytomation, Glostrup, Denmark) for 15 minutes, and sections were permeabilized with 0.4% Triton X-100. The sections were incubated with 1 μg/mL polyclonal Aβ40- or Aβ42-specific antibodies,15 monoclonal AβArc-specific antibodies (mab27 at 1 μg/mL9), 0.5 μg/mL N-terminal Aβ antibody 6E10 (Covance, Princeton, NJ) or 82E1 (IBL International GmbH, Hamburg, Germany), 0.2 μg/mL goat anti-mouse poprotein J (AF2747; R&D Systems, Inc., Minneapolis, MN), goat anti-apolipoprotein E (ApoE) (AB947, diluted 1:5000; Chemicon International Inc., Temecula, CA), or 0.5 μg/mL monoclonal anti-human heparan sulfate (H1890, against 10E4 epitope; US Biological, Swampscott, MA) in PBS-buffered saline solution with 0.1% Tween 20 at 4°C overnight. Biotinylated goat anti-rabbit, goat anti-mouse, or rabbit anti-goat antibodies (Vector Laboratories, Inc., Burlingame, CA) were applied for 30 minutes at room temperature. Sections were then incubated with streptavidin-coupled horseradish peroxidase (Mabtech AB, Nacka Strand, Sweden) for 30 minutes and developed using the Nova Red chromogen kit (Vector Laboratories, Inc.). For double staining with the Aβ42-specific antibody and either thioflavine T (ThT) or the luminescent conjugated oligothiophene (LCO) p-FTAA, the sections were treated as described, and a fluorescent secondary antibody [Alexa Fluor 594 goat anti-rabbit (Invitrogen Corp., Carlsbad, CA)] diluted 1:250 in PBS was used to visualize the Aβ immunostaining. The sections were washed in PBS and incubated with 3 μmol/L p-FTAA16 in PBS or with 0.2% (w/v) ThT (Sigma-Aldrich Corp., St. Louis, MO) in PBS for 30 minutes with shaking at room temperature. After rinsing with PBS, the sections were washed in dH2O and mounted with Vectashield (Vector Laboratories, Inc.). Compact plaques were detected using Congo red (Sigma-Aldrich Corp.) as previously described.17 The burden of diffuse and compact deposits in the cerebral cortex was measured in two image fields per section at ×20 magnification. Images were captured using a Nikon microscope (DXM1200F; Nikon Instruments Inc., Melville, NY) equipped with a digital camera, converted to gray scale, and segmented with an autothreshold command (Image Pro-Plus; MediaCybernetics, Silver Spring, MD). Custom macros were used to measure the stained area of interest as a percentage of the total area.
p-FTAA and Fluorescence Microscopy
Frozen brain sections 10-μm thick from 18-month-old tg-ArcSwe (line B), tg-Swe (line A), and bitransgenic tg-ArcSwe (line D) and tg-Swe (line A) mice were fixed in absolute ethanol for 10 minutes and rehydrated in 50% ethanol followed by water. The sections were incubated in PBS for 10 minutes and stained with 3 μmol/L p-FTAA16 in PBS for 30 minutes at room temperature. After rinsing with PBS, the sections were mounted with Dako fluorescence mounting medium (DakoCytomation A/S). The medium was allowed to solidify for 3 hours before the slide rims were sealed with nail polish. Spectra and fluorescence images were obtained using a fluorescence microscope (Zeiss Axioplan; Carl Zeiss Ltd., Jena, Germany) equipped with a SpectraCube (optical head) module (Applied Spectral Imaging, Ltd., Migdal Ha'Emek, Israel) using bandpass filters 405/30 (LP450), 470/40 (LP515), and 546/12 (LP590). p-FTAA bound to compact plaques (10 plaques per animal, three animals from each group) was excited at 405 and 546 nm. SpectraView 3.0 EXPO software (Applied Spectral Imaging, Ltd.) was used to merge the information from the double excitation and create full visible-light emission spectra (450 to 700 nm) for p-FTAA when combining those excitation wavelengths. Fluorescent spectral unmixing, which is a function within the program, was performed to separate the emitted light intensities according to wavelength. The emission spectrum for p-FTAA bound to compact plaques in tg-ArcSwe (line B) was used to define red emission, and the emission spectrum for p-FTAA bound to compact plaques in tg-Swe (line A) was used to define green emission. By performing spectral unmixing, the fraction of each defined spectrum could be determined for each compact plaque found in tg-ArcSwe (line B), tg-Swe (line A), and bitransgenic mice, respectively.
SDS-PAGE and Western Blot Analysis
For analysis of Aβ, multiphasic urea/SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis were performed. This gel system enables differentiation of Aβ of various lengths and also Aβwt and AβArc. Samples were subjected to electrophoresis using a 15% T, 5% C bicine-Tris separation gel [T, total concentration of acrylamide; C, bis-acrylamide cross-linker content (acrylamide/bis-acrylamide, 40%; Sigma-Aldrich Corp.)] containing 8 mol/L urea. They were run at 24 mA, 30 V. Gels and buffers have been described previously,18,19 with the distinction that in the present study, 8 mol/L urea was included in the stacking gel. For analysis of total brain Aβ, tissues at bregma −1.0 mm to −3.0 mm were used. Tissue sections (30 μm; bregma 1.0 mm to −0.1 mm) were dissected and analyzed for Aβ levels in striatum and cerebral cortex. Brain tissue was homogenized in a final concentration of 70% formic acid at a ratio of 1:10 (w/v) using a Dounce homogenizer, sonicated for 30 seconds, and centrifuged at 100,000_g_ at 4°C for 60 minutes. The supernatants were stored at −80°C before they were analyzed with enzyme-linked immunosorbent assay (ELISA) and SDS-PAGE Western blot analysis. For analysis of APP levels, brain tissues (bregma −1.0 mm to −3.0 mm) from 2-month-old mice were homogenized in Tris-buffered saline solution [20 mmol/L Tris and 137 mmol/L NaCl (pH 7.6)] supplemented with 2% (w/v) SDS and complete protease inhibitor (Hoffman-La Roche AG, Basel, Switzerland), sonicated for 30 seconds, and centrifuged at 100,000_g_ at 4°C for 60 minutes. Proteins in the supernatant were separated on 4% to 20% Tris-tricine gels (Invitrogen Corp.). Protein transfer and Western blot analysis were performed as previously described.5
Aβ Enzyme-Linked Immunosorbent Assay
Ninety-six–well plates were coated for 12 hours at 4°C with antibodies specific for Aβ40 or Aβ42 (200 ng per well) and used for measuring Aβ1-40 or Aβ1-42 levels. For the total Aβ assay, plates were coated with 100 ng per well of the N-terminal Aβ-specific antibody 82E1 (IBL International GmbH) in PBS [130 mmol/L NaCl, 7 mmol/L NaH2PO4, and 3 mmol/L Na2HPO4 (pH 7.4)] and blocked with 1% bovine serum albumin in phosphate-buffered NaCl, 0.15% Kathon (Rohm and Haas Co., Philadelphia, PA). Formic acid extracts of mouse brain were neutralized in 1 mol/L Tris (pH 10) and diluted in ELISA incubation buffer (PBS with 0.1% bovine serum albumin, 0.05% Tween 20). Samples or standards (recombinant wt/Arctic Aβ1-40 or 1-42) were added to the plates in triplicate and incubated for 2 hours at room temperature. Biotinylated mAb27, 0.5 μg/mL, with an epitope in the mid-domain of Arctic Aβ,9 was used to measure total AβArc. Biotinylated 4G8, 0.5 μg/mL (Covance, Inc.), was used to detect total Aβ, and biotinylated mAb1C3, 0.5 μg/mL,7 was used to quantify Aβ1-40 or Aβ1-42. Both biotinylated detection antibodies and streptavidin-coupled horseradish peroxidase (Mabtech AB), diluted 1:2000, were incubated for 1 hour in successive steps. K-blue aqueous (ANL-Produkter AB, älvsjö, Sweden) was used as horseradish peroxidase substrate, the reaction was stopped using 1 mol/L H2SO4, and the optical density was measured at 450 nm with a spectrophotometer (SpectraMax 190; Molecular Devices, Inc., Sunnyvale, CA). Wells were washed three times between each step in ELISA washing buffer (phosphate-buffered NaCl with 0.1% Tween 20 and 0.15% Kathon).
mAb158 Protofibril/Oligomer ELISA
The mAb158 ELISA7 detects oligomers larger than 100 kDa, which is consistent with the original description of oligomeric assemblies of Aβ.20 In brief, each well in 96-well plates was coated with 200 ng mAb158 in PBS overnight at 4°C and blocked with 1% bovine serum albumin. The samples were centrifuged at 17,900 × g for 5 minutes before incubation to ensure that they were devoid of any insoluble Aβ aggregates. Samples were added to plates in duplicate and incubated for 2 hours at room temperature. Biotinylated mAb158, 0.5 μg/mL, was added and incubated for 1 hour at room temperature, followed by incubation with streptaviden-coupled horseradish peroxidase for 1 hour at room temperature. K-blue aqueous (ANL-Produkter AB) horseradish peroxidase substrate was used, and the reaction was stopped using 1 mol/L H2SO4. After the blocking step, plates were washed three times between each step. All samples and antibodies were diluted in ELISA incubation buffer (PBS with 0.1% bovine serum albumin and 0.02% Tween 20). The standard was generated by incubating Aβ1-42wt (American Peptide Co., Inc., Sunnyvale, CA) in 50 μmol/L PBS for 30 minutes at 37°C, followed by centrifugation at 16,000 × g for 5 minutes. The supernatant was loaded on a Superdex 75 10/300 GL column, resulting in a single high-molecular-weight peak, which was collected.
Kinetic Measurement of Aβ Fibrillation
Recombinant Aβ1-42wt and Aβ1-42Arc (American Peptide Co., Inc.) were treated with 100% hexafluoroisopropanol (Sigma-Aldrich Corp.), reaching a final concentration of 1 mmol/L. Hexafluoroisopropanol was allowed to evaporate in a chemical fume hood, and Aβ was then solubilized as a 400-μmol/L stock solution in 10 mmol/L NaOH and stored at −80°C. Immediately before use, Aβ1-42wt and Aβ1-42Arc were diluted in PBS [50 mmol/L phosphate buffer and 100 mmol/L NaCl (pH 7.4)] to 20 μmol/L and added to wells of a nonbinding 96-well microtiter plate (Greiner Bio-One GmbH, Frickenhausen, Germany). ThT (Sigma Corp.) and p-FTAA16 were prepared as 15-μmol/L stock solutions in PBS (pH 7.4) and added to the wells at a final concentration of 1.2 μmol/L. Plates were incubated at room temperature for up to 22 hours under agitation. Aβ aggregation was recorded with a fluorometer (Wallac Victor 2 multilabel counter; Perkin Elmer Inc., Waltham, MA) as described for ThT21 and p-FTAA.16 The wavelengths of excitation and emission were 440 and 485 nm, respectively. Experiments were performed in triplicate, and fluorescence from free ThT or p-FTAA, incubated without Aβ in parallel, was subtracted from the readings.
Electron Microscopy
The ultrastructure of Aβ aggregates was examined by collecting samples of Aβ-solutions, 20 μmol/L, from the kinetic experiment. Solutions of Aβ were diluted to 1 μmol/L in mH2O and spotted on grids coated with carbon-formvar. Excess fluid was removed after 30 seconds, and 2.5% uranyl acetate, 5 to 10 μL, was added to the grids, and again excess fluid was quickly removed. The negatively stained specimen were examined and photographed using an electron microscope (JEOL 1230; JEOL Ltd., Tokyo, Japan) at 100 kV.
Statistical Analyses
Data were analyzed using the Student's _t_-test or with one- or two-way analysis of variance followed by Tukey's multiple comparison post hoc test (GraphPad Software, Inc., San Diego, CA). Results were presented as values representing the group mean and standard error of the mean (SEM). The relationship between Aβ40-ir and Aβ42-ir burden was investigated using linear regression, and slopes were compared using the F-test. P < 0.05, P < 0.01, and P < 0.001 were considered significant.
Results
Transgene Expression in Founder Lines of Tg-Swe and Tg-ArcSwe Mice
Intraneuronal Aβ accumulates in young tg-ArcSwe mice, and extracellular deposition of compact plaques begins at age 5 to 6 months, whereas the frequency of diffuse Aβ deposits remains extremely low.5 This high-expressing founder line, with threefold APP expression compared with the level of murine APP, is hereafter referred to as tg-ArcSwe line B. In tg-Swe line A, hereafter referred to as tg-Swe, APP expression is sevenfold higher than in nontransgenic mice, and extracellular Aβ deposition emerges at approximately 12 months of age.10 Several additional founder lines of tg-ArcSwe with lower transgene expression were generated. It is not known whether a small amount of highly aggregation-prone peptides such as AβArc can influence the comparably slower aggregation of Aβwt. To investigate this, tg-Swe mice were crossed with tg-ArcSwe (line D), which express human APPArcSwe at a low level (total APP is less than twofold compared with murine APP) (Figure 1A; Table 1). The total level of APP protein, human plus murine, in 2-month-old mice was analyzed using Western blot analysis. Human APP was sevenfold more abundant in young tg-Swe than in age-matched tg-ArcSwe (line D) as measured at densitometry. For comparison, a tg-ArcSwe mouse (line B)5 with threefold APP expression and a nontransgenic mouse were included in the analyses (Figure 1B; Table 1). In tg-Swe, with sevenfold APP expression, an abundance of human APP was observed in the hippocampal pyramidal cell layers CA1 and CA3, in the lower layers of cerebral cortex, and to some extent in the upper cortical lamina (Figure 1, C and F). The anatomical distribution was similar in tg-ArcSwe (line D), although the laminar expression pattern in the cerebral cortex was attenuated (Figure 1, D and G). As expected, in a nontransgenic mouse, only a faint nonspecific background signal was observed (Figure 1, E and H).
Figure 1.
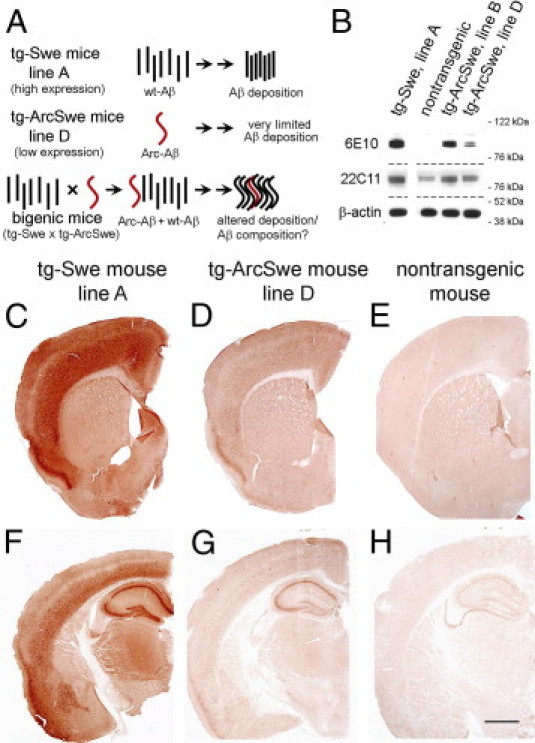
High-level APP-expressing tg-Swe mice (line A) produced seven times more human APP than did tg-ArcSwe mice (line D). A: Experimental design of the study is schematically illustrated. B: APP protein synthesis of transgenic models, tg-Swe (line A), and nontransgenic mice, tg-ArcSwe (lines B and D), was analyzed using Western blot analysis and antibodies 6E10 (human APP), 22C11 (human plus endogenous APP), and β-actin. Anatomical distribution of human APP detected with 6E10-immunostaining in 2-month-old tg-Swe, line A (C and F), low-expressing founder line of tg-ArcSwe, line D (D and G), and a nontransgenic mouse (E and H). Scale bars = 1 mm.
Burden of Compact Plaques in Tg-Swe Is Not Altered by a Minute Amount of AβArc
Whether AβArc, at a level that seemed insufficient on its own to generate extracellular Aβ deposition, could instigate or modify the neuropathologic conditions produced by an excess of Aβwt was investigated. Therefore, tg-ArcSwe (line D) and tg-Swe were crossed, and the neuropathologic features of bitransgenic mice were compared with those of singly tg-Swe mice. The neuropathologic conditions of singly tg-ArcSwe (line D) mice were reassessed in two aged individuals generated from the cross-breeding experiment at the study end point (ie, 18-month-old mice). Six tissue sections distributed along the rostrocaudal axis of the brain were stained with different Aβ-antibodies (6E10, 82E1, and Aβ42-specific). In one of the 18-month-old mice, a single plaque was observed, and in the other mouse, three Aβ-immunopositive plaques were observed in six tissue sections altogether. Thus, Aβ deposition and intraneuronal Aβ immunostaining in aged tg-ArcSwe (line D) was limited. Biochemical analyses confirmed that age-dependent Aβ accumulation was modest. The mean (SEM) level of Aβ1-40 was 9.9 (1.2) pmol/g (n = 2) at 2 months, and 19.5 (1.7) pmol/g at 18 months (n = 2) in tg-ArcSwe (line D). This was in contrast to findings in 11-month-old tg-ArcSwe (line B, n = 2) with a cortical Aβ-burden of 2.5% (0.3%), total Aβ1-40 levels of 8982 (770) pmol/g, and strong intraneuronal Aβ immunostaining (see Supplemental Figure S1 at http://ajp.amjpathol.org). These results were not surprising because it is well known that Aβ deposition will occur only if a sufficiently high level of Aβ synthesis is attained and that age at onset depends on the level of transgene expression.22 If the effect of transgenes on pathologic features were simply additive, bitransgenic mice, tg-ArcSwe (line D) and tg-Swe, and singly tg-Swe would be expected to exhibit essentially the same type and quantity of Aβ deposits.
Brains from bitransgenic and tg-Swe mice were first examined with Congo red and an Aβ40-specific antibody. Both of these markers are associated with compact plaques in tg-Swe. At age 12 months, Aβ40 burden was not higher in bitransgenic mice [mean (SEM), 1.2% (0.3%); n = 17] than in tg-Swe [1.0% (0.2%); _n_ = 16; _P_ = 0.55; Figure 2, A–C]. Likewise, Congo red burden was not increased in bitransgenic mice [0.25% (0.09%); n = 17] compared with singly tg-Swe [0.19% (0.07%); _n_ = 16; _P_ = 0.57; see Supplemental Figure S2, A–C, at http://ajp.amjpathol.org]. At age 18 months, again Aβ40-ir burden was not higher in bitransgenic compared with tg-Swe mice [3.6% (0.6%); _n_ = 7, and 2.8% (0.6%); _n_ = 8; _P_ = 0.43; Figure 2C]. Likewise, Congo red burden was not elevated in bitransgenic mice [1.3% (0.6%); n = 7] compared with age-matched tg-Swe [1.0% (0.3%); _n_ = 8; _P_ = 0.61; see Supplemental Figure S2C at http://ajp.amjpathol.org]. Sections were then stained using an LCO, p-FTAA,16 a conformational-dependent probe with a flexible structure that can serve as an optical tool to reveal heterogeneity among Aβ and prion deposits in a manner similar to luminescent conjugated polythiophenes.10,23,24 As expected from previous imaging experiments using Drosophila models,25 the p-FTAA emission spectrum of compact plaques in tg-ArcSwe (line B; Figure 2D) and tg-Swe mice (Figure 2E) differed. The p-FTAA fluorescence of compact plaques in bitransgenic mice (Figure 2F) resembled that in tg-Swe; however, small areas within the deposits were more similar to compact plaques in tg-ArcSwe (line B). Spectral analyses of emitted fluorescence from multiple individual compact plaques confirmed that signals in bitransgenic mice were intermediate to those of compact plaques in tg-ArcSwe and tg-Swe (Figure 2, G and H). Thus, a minute amount of AβArc in bitransgenic mice did not influence burden of compact plaques, but did result in a different LCO spectral profile in individual compact plaques.
Figure 2.
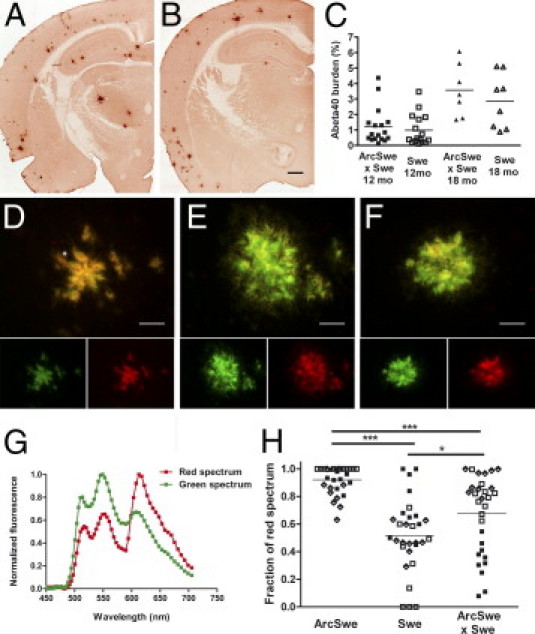
AβArc did not change Aβ40 burden but influenced p-FTAA fluorescence of individual compact plaques in bitransgenic mice. Representative 12-month-old bitransgenic (A) and singly tg-Swe (B) mice. C: There was no statistically significant difference in cortical Aβ40-burden between 12- and 18-month-old bitransgenic and singly tg-Swe mice. Fluorescence images of p-FTAA bound to compact plaques and excited at 470 and 546 nm. Images illustrate a variation in p-FTAA emission spectrum between compact plaques found in tg-ArcSwe, line B (D), singly tg-Swe (E), and bitransgenic mice (F), respectively. The resulting emission when the excitation wavelengths (470 and 546 nm) are separated into two channels is shown below. Spectral unmixing of p-FTAA emission when bound to compact plaques in bitransgenic mice revealed a shift toward that of compact plaques in tg-ArcSwe compared with singly tg-Swe. G and H, Fraction of red spectrum of 10 individual plaques from three animals in each group (filled and open boxes and diamonds). Scale bars: 500 μm (B); 30 μm (D–F).
Increased Accumulation of Diffuse Aβ42-Immunoreactive Deposits in Bitransgenic Mice
Tissue sections were immunostained with an Aβ42-specific antibody, which in brains affected with AD typically detects both compact plaques and diffuse Aβ deposits, although diffuse Aβ deposits are rarely observed in tg-Swe. At age 12 months, the mean (SEM) burden of Aβ42-ir plaques was more than fourfold higher in bitransgenic mice [4.1% (0.9%); n = 17] than in tg-Swe [0.9% (0.3%); _n_ = 16; _P_ < 0.01 Figure 3, A, D, and G]. In 18-month-old mice, Aβ42 burden was still increased more than threefold in bitransgenic mice [9.9% (1.3%); n = 7] compared with tg-Swe mice [3.1% (0.8%); _n_ = 8; _P_ < 0.001; Figure 3, B, E, and G]. Groups of bitransgenic (r = 0.86; P < 0.0001) and tg-Swe mice (r = 0.90; P < 0.0001) fitted well into separate linear regression models when Aβ40 burden was plotted against Aβ42 burden for each animal. The slope of linear regression for bitransgenic mice [2.3 (0.3)] and tg-Swe mice differed [1.1 (0.1); F1,44 = 14.8; P < 0.001]. Thus, introduction of a small amount of AβArc, produced by low expression of the tg-ArcSwe transgene, strongly and selectively increased the burden of diffuse Aβ42-immunopositive deposits (Figure 3H). Next, consecutive sections were stained to investigate which Aβ peptides were located in diffuse and compact plaques. Aβ42, Aβ40, AβArc, and Congo red staining co-localized in compact plaques in bitransgenic mice. There was an additional population of diffuse Aβ42-ir deposits in the cerebral cortex in these mice, but also in the striatum, where there is essentially no transgene expression (Figure 4, A–D and I–L). Neither using a protocol for silver intensification of Ni-DAB product,26 nor using unfixed tissue section and a 10-fold higher concentration of mAb279 resulted in staining of the diffuse deposits with the AβArc antibody (data not shown). In tg-Swe, Aβ40, Aβ42, and Congo red staining were most often co-localized, and the compact plaques, as expected, did not stain with an AβArc antibody (Figure 4, E–H and M–P). It is important to note that the selectivity of the AβArc-specific antibody, mAb27, depends on a unique conformation around glycine 22 and not on the linear amino acid sequence. Thus, mAb27 is selective for AβArc at ELISA analysis but not, for example, at denaturing Western blot analysis.9 The selectivity of mAb27 immunostaining was determined using tissue sections from plaque-bearing tg-ArcSwe, line B (positive control) and tg-Swe (negative control) mice that were prepared and stained in parallel to those from bitransgenic mice.
Figure 3.
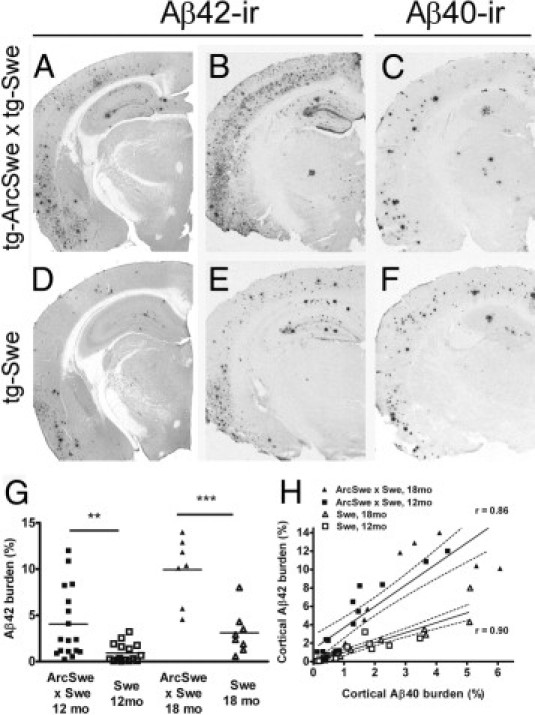
AβArc favored an increased Aβ42-burden in bitransgenic mice. Aβ burden in the cerebral cortex of 12-month-old (A and D) and 18-month-old (B, C, E, and F) bitransgenic (A–C) and singly tg-Swe (D–F) mice. Aβ42 burden in representative 12-month-old (A) and 18-month-old (B) bitransgenic mice and in 12-month-old (D) and 18-month-old (E) tg-Swe. Aβ40 burden is shown from semiadjacent sections of 18-month-old bitransgenic (C) and tg-Swe (F) mice. G: Aβ42 burden was investigated in all animals using quantitative image analysis. H: There was a linear correlation between Aβ40 burden and Aβ42 burden among individuals within the experimental groups, but a substantial difference between bitransgenic and singly tg-Swe mice. Scale bar = 900 μm.
Figure 4.
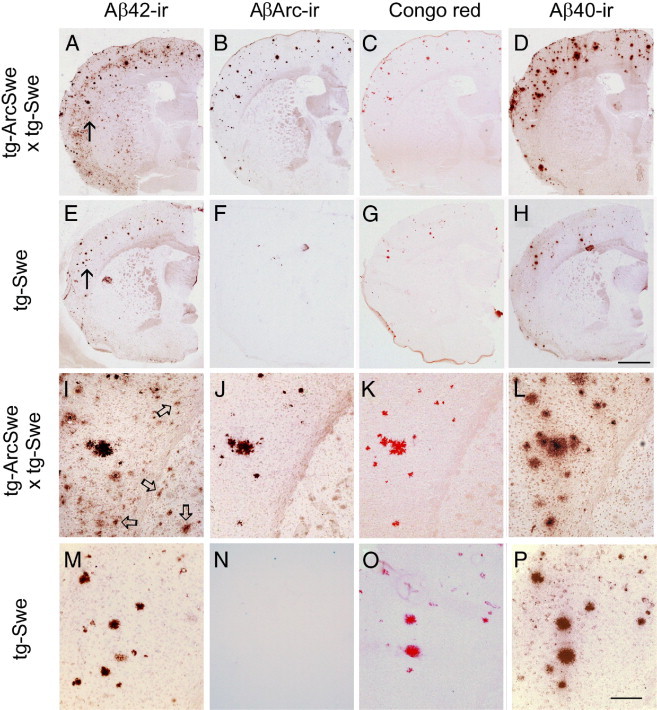
Increased Aβ42 burden in bitransgenic mice was due to diffuse deposits that were not recognized by an antibody specific for AβArc. Aβ-deposition in 18-month-old bitransgenic (A–D and I–L) and singly tg-Swe mice (E–H and M–P). Adjacent sections (10 μm) were immunostained with Aβ42-specific antibody (A, E, I, and M), AβArc-specific antibody (B, F, J, and N), Congo red (C, G, K, and O), and Aβ40-specific antibody (D, H, L, and P). Arrows in A and E indicate areas shown at higher magnification (I and M, respectively), and arrows in I indicate some diffuse Aβ42-immunoreactive deposits. Scale bars: 1 mm (A–H); 200 μm (I–P). The spots in (F) are unspecific staining that are due to slight damage to parts of this tissue section.
Factors were then searched that could further explain how diffuse and compact plaques could form via independent processes. ApoE resides in extracellular deposits and regulates Aβ deposition in vivo.27 Other plaque-associated factors such as apolipoprotein J and heparan sulfate selectively affect burden of compact plaques.28,29 Therefore, we searched for selective markers of diffuse deposits and compact plaques. Among those, only heparan sulfate was specifically associated with compact plaques, whereas ApoE and apolipoprotein J were found in both diffuse and compact plaques (see Supplemental Figure S3 at http://ajp.amjpathol.org).
AβArc Facilitates Striatal Aβ1-42wt Accumulation and Formation of Diffuse Deposits
Aβ deposition in bitransgenic and tg-Swe mice was biochemically analyzed using urea/SDS-PAGE and Western blot analysis. Extracts of 70% formic acid (total Aβ) from bitransgenic and tg-Swe mouse brain with early- and late-stage plaque (ages 12 and 18 months) were investigated. The system was calibrated with synthetic Aβ peptides (lanes 1 to 4, Figure 5A), and a 6-month-old tg-ArcSwe (line B) with high level of APP expression was used as reference material (lane 7).10 In 18-month-old bitransgenic mice, Aβ1-38wt, Aβ1-40wt, and Aβ1-42wt were predominant, with low levels of AβArc (Aβ1-40Arc and/or Aβ1-42Arc) (lane 5, Figure 5A). These Aβ species were present at 12 months in bitransgenic mice (lane 8), although the levels of all Aβ species were much lower and Aβ1-40wt was the predominant species. As expected, high levels of Aβ1-40wt were observed in 18-month-old tg-Swe, and Aβ1-38wt and Aβ1-42wt were also detected (lane 6). Brain tissue from a tg-ArcSwe line B mouse with plaque deposition contained AβArc peptides, primarily Aβ1-40Arc and Aβ1-42Arc and presumably Aβ1-38Arc (lane 7), as previously described.10 An immunohistochemical and a biochemical technique were combined to further determine the identity of Aβ in the diffuse deposits in bitransgenic mice. In thin tissue sections, the striatum, which contained only diffuse Aβ deposits, was separated from the cerebral cortex, in which both diffuse and compact deposits were observed (Figure 5B). Extracts of 70% formic acid in striatal and cerebral cortical tissue (layers I to VI) from a representative 18-month-old bitransgenic mouse were analyzed using urea/SDS-PAGE. There was a marked region-specific difference, with Aβ1-42wt as the predominant Aβ peptide in the striatum, whereas Aβ1-42wt and Aβ1-40wt with some Aβ1-38wt were present in the cerebral cortex (Figure 5C). Because the sensitivity of SDS-PAGE and Western blot analysis is somewhat limited, ELISA was used to measure AβArc levels (82E1/mAb27 sandwich) and total Aβ levels (82E1/4G8 sandwich) in the cerebral cortex and striatum. The mean (SEM) levels of AβArc and total Aβ in cerebral cortex were 160 (24) pmol/g and 2300 (190) pmol/g (n = 4), respectively; that is, approximately 7% of total Aβ was AβArc. In the striatum, the levels of AβArc and total Aβ were 16 (4) pmol/g and 770 (50) pmol/g, respectively (n = 4); that is, 2% of total Aβ was AβArc (Figure 5D). These findings suggest that AβArc/APP facilitates formation of diffuse Aβ deposits, which consist primarily of Aβ1-42wt and possibly small amounts of AβArc. In old (age 24 months) tg-Swe mice, diffuse Aβ42-immunoreactive deposits were also observed in areas both with and without transgene expression, for example, in the striatum (see Supplemental Figure S4 at http://ajp.amjpathol.org). Thus, AβArc strongly facilitated a process whereby diffuse deposits formed. It did not create a unique process of Aβ aggregation that does not normally occur in tg-Swe mice.
Figure 5.
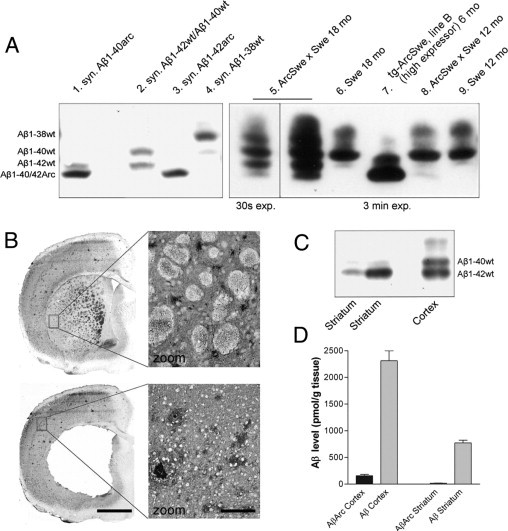
AβArc led to increased accumulation of Aβ42wt and more diffuse deposits in the striatum in bitransgenic mice. Urea/SDS-PAGE and Western blot analysis (with 6E10 antibody) of extracts of 70% formic acid (total Aβ) of plaque-bearing bitransgenic and tg-Swe mice at ages 12 and 18 months. A: For comparison, tissue from a 6-month-old tg-ArcSwe (line B; high-expressor line) with plaques and synthetic Aβ peptides were included in the experiments. B: Immunostaining of a brain section from an 18-month-old bitransgenic mouse with an Aβ42-specific antibody before and after the striatum (caudate putamen) had been dissected. Extracts of 70% formic acid from dissected striatum (caudate putamen) and cerebral cortex layers I to VI in an 18-month-old bitransgenic mouse at the level of the frontal cortex (bregma 1.0 mm to −0.1 mm). C: Localization of Aβ peptides differed markedly, and Aβ1-42wt was the predominant Aβ peptide in the striatum, whereas both Aβ1-40wt and Aβ1-42wt were abundant in the cerebral cortex. D: Further analyses of the same formic acid extracts of dissected tissues at ELISA (pmol/g tissue) demonstrated that AβArc was present but represented only a small fraction of total Aβ in both cerebral cortex and striatum (n = 4). Scale bars: 1 mm (B); 100 μm in enlarged images.
APPArcSwe Does Not Alter APPSwe Processing in Bitransgenic Mice
APPArcSwe could possibly interact with APPSwe and, thereby, quantitatively or qualitatively influence its endoproteolysis, for example, through APP dimerization.30 This could explain different Aβ neuropathologic features in aged bitransgenic and tg-Swe mice. If so, alterations in the steady-state level of Aβ would be expected in young bitransgenic mice without extracellular Aβ deposition. The total Aβ level tended to be higher in formic acid extracts from brains of 2-month-old bitransgenic mice [mean (SEM), 8.0 (0.5) pmol/g Aβ1-40 and 6.7 (0.3) pmol/g Aβ1-42; n = 5] compared with age-matched tg-Swe [7.0 (0.6) pmol/g Aβ1-40; P = 0.28, and 6.0 (0.3) pmol/g Aβ1-42; n = 6; P = 0.14] (see Supplemental Figure S5A at http://ajp.amjpathol.org); however, the Aβ42/Aβ40 ratio in the bitransgenic mice [0.84 (0.04)] and tg-Swe [0.85 (0.04)] did not differ (P = 0.75) (see Supplemental Figure S5B at http://ajp.amjpathol.org). When a total AβArc ELISA (Aβ1-xArc) was used, the level of AβArc in bitransgenic mice was 1.60 (0.20) pmol/g (n = 5), and, as expected, was not detectable in tg-Swe (n = 5) (see Supplemental Figure S5C at http://ajp.amjpathol.org). Thus, total Aβ levels were higher in young bitransgenic mice than in age-matched tg-Swe mice; however, this increase was not more than what would be expected by the increased transgene expression (ie, the additional approximately one-eighth processing of APPArcSwe and AβArc synthesis in bitransgenic mice). Thus, it was concluded that APPArcSwe did not selectively increase Aβ1-42wt synthesis from the APPSwe transgene in bitransgenic mice.
AβArc Facilitates Formation of Prefibrillar and Fibrillar Assemblies of Aβwt in Vitro
The increased levels of diffuse deposits in bitransgenic mice could perhaps be explained by the ability of AβArc to facilitate refolding and aggregation of Aβwt. To investigate this, an in vitro system was designed to evaluate the influence of Aβ1-42Arc on Aβ1-42wt aggregation. LCOs enable staining and analysis of prefibrillar Aβ assemblies.16 Thus, the fluorescence of an LCO, p-FTAA, was used as a readout of prefibrillar Aβ aggregates, and that of ThT was used as a measure of amyloid fibril formation. Recombinant Aβ1-42wt and Aβ1-42Arc were treated with hexafluoroisopropanol to dissociate possible preformed Aβ aggregates that could seed fibril formation. The monomeric Aβ peptides were allowed to form fibrils, and the kinetics was monitored with ThT (Figure 6A) and p-FTAA (Figure 6B). Aβ1-42wt and Aβ1-42Arc were incubated either alone or together in ratios of 7:1 or 1:1 (Aβwt/AβArc). The total concentration of Aβ (ie, Aβ1-42wt and/or Aβ1-42Arc either alone or in combination) was always 20 μmol/L. A lag phase was followed by an exponential growth phase and then a plateau phase. With both ThT and p-FTAA, the lag phase was shorter when Aβ1-42wt and Aβ1-42Arc were mixed before polymerization, at 7:1 or 1:1. The effect seemed to depend on the presence of AβArc but not on the percentages of AβArc and Aβwt. When mixing Aβ1-42wt and Aβ1-42Arc, the intensity of ThT fluorescence was noticeably lower in the plateau phase; however, this was not observed when using p-FTAA. The ultrastructure of in vitro preparations was then investigated. The starting material for all samples contained only small aggregates but no fibrils (Figure 6, C–E). An almost evenly distributed carpet of proteins was present at time 0 when Aβ1-42wt and Aβ1-42Arc were mixed (Figure 6D). At 100 minutes, when p-FTAA-fluorescence was increased, small clusters of thin fibrils were observed in samples consisting only of Aβ1-42Arc (Figure 6F), whereas a few fibrillar aggregates with varying morphologic appearance were present at 100 minutes when Aβ1-42wt and Aβ1-42Arc were mixed (7:1; Figure 6G). In contrast, in samples that contained only Aβ1-42wt peptide, p-FTAA fluorescence intensity was low (Figure 6B), and both fibrils and low-molecular-weight aggregates were rare at this time point (Figure 6H). At 1200 minutes, incubation of Aβ1-42Arc alone gave rise to an abundance of tightly aggregated thin fibrils (Figure 6I). In contrast, mixtures of Aβ1-42wt and Aβ1-42Arc contained large numbers of massive fibrillar aggregates. These mixtures of peptides formed thin fibrils that seemed to be composed of globular subunits (Figure 6J). Their appearance was different from the straighter fibrils observed with Aβ1-42wt (Figure 6K).
Figure 6.
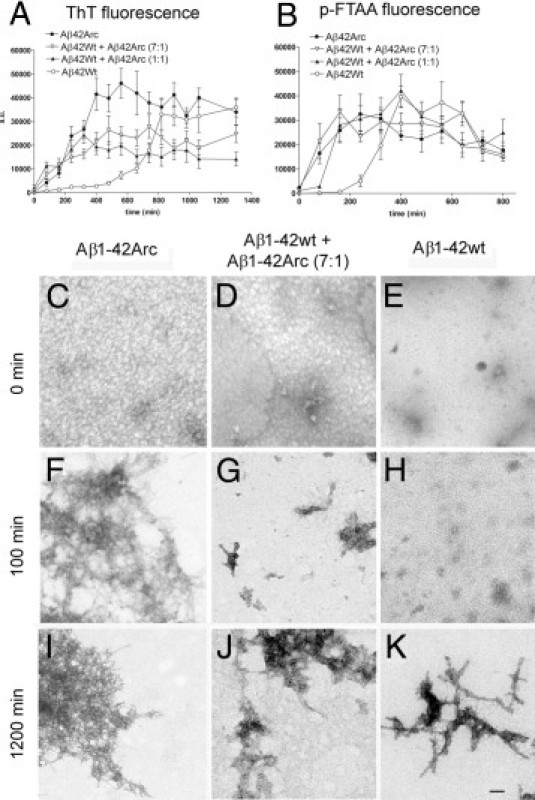
A mixture of AβArc and Aβwt accelerated prefibrillar Aβ aggregate formation in vitro. Aggregation of recombinant Aβ1–42 was monitored with ThT (A) and p-FTAA (B). Fibrillation kinetics of Aβ1-42wt (open circles; 20 μmol/L), Aβ1-42wt and Aβ1-42Arc in a ratio of 7:1 (open triangles; 20 μmol/L total Aβ), Aβ1-42wt and Aβ1-42Arc in a ratio of 1:1 (solid triangles; 20 μmol/L total Aβ), and Aβ1-42Arc (solid circles; 20 μmol/L). p-FTAA detects early events in the aggregation process.16 With both probes, the lag phase was shorter when AβArc was present. Each symbol represents the mean (SEM) of two or three experiments. Ultrastructural analysis of Aβ aggregates at electron microscopy. Samples of Aβ1-42Arc (20 μmol/L; C, F, and I), Aβ1-42wt and Aβ1-42Arc at a ratio of 7:1 (20 μmol/L total Aβ; D, G, and J), and Aβ1-42wt (20 μmol/L; E, H, and K) were analyzed at 0 minutes (C–E), 100 minutes (F–H), and 1200 minutes (I–K). Scale bars: 100 nm (C–K).
Analyses of Aβ Protofibrils, p-FTAA Staining, and Aβ42-Immunoreactivity in Bitransgenic Mice
Staining with ThT or p-FTAA was combined with Aβ IHC on paraformaldehyde-fixed sections. Many but not all diffuse Aβ42-immunoreactive deposits in the cerebral cortex and striatum were also stained using p-FTAA in an 18-month-old bitransgenic mouse (Figure 7, A, C–H). In contrast, there was no overlap between ThT staining and Aβ42 immunoreactivity in the striatum of bitransgenic mice (see Supplemental Figure S6 at http://ajp.amjpathol.org). The same results were observed when thioflavine S was used instead of ThT (data not shown). Moreover, Aβ protofibrils, which are large soluble Aβ oligomers, were more abundant in Tris-buffered saline solution extracts in 12-month-old bitransgenic tg-ArcSwe and tg-Swe [mean (SEM), 163 (20) pmol/L; n = 11] than in age-matched tg-Swe [83 (13) pmol/L; n = 9; P < 0.01].
Figure 7.
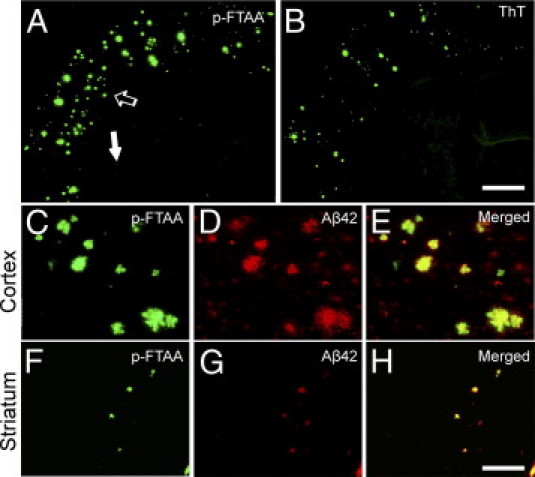
Diffuse Aβ deposits in bitransgenic mice were stained using p-FTAA. Tissue sections 25 μm thick from a bitransgenic mouse were fixed in paraformaldehyde and stained with an Aβ42-specific antibody together with either p-FTAA (A) or ThT (B). The area at the tip of the open arrow in A is magnified (C–E), and the area at the tip of the solid arrow in B is enlarged (F–H). Both dyes enabled visualization of compact plaques, and p-FTAA also stained numerous diffuse Aβ deposits (F–H). Scale bars: 1 mm (A and B); 100 μm (C–H).
Discussion
Enhanced deposition of small and diffuse Aβ42-immunopositive deposits were observed in regions with and without transgene expression in bitransgenic mice producing both Aβwt and AβArc. In contrast, the burden of compact plaques was similar in bitransgenic and singly tg-Swe mice. Transgenic mice express only mutant human APP, for example, with the Arctic and/or Swedish mutations,5,31 whereas patients are heterozygous for the Arctic mutation or any other APP mutation and, therefore, produce a mixture of both Aβwt and Aβmutant peptides. The Arctic mutation results in a neuropathologic condition consisting of cerebral amyloid angiopathy and an abundance of parenchymal Aβ deposits lacking amyloid cores, and the symptoms are those of typical AD.2 In the present experiments, a mixture of Aβwt and AβArc in bitransgenic mice facilitated diffuse Aβ deposition. Recently, in a similar experimental approach, transgenic models APPDutch and APP23 were cross-bred. The APPDutch model mimics hereditary cerebral hemorrhage with amyloidosis–Dutch type,32,33 with cerebral amyloid angiopathy and diffuse Aβ deposits. APP23/APPDutch mice produced a mixture of Aβwt and AβDutch, and because Aβ levels were substantially higher in young APP23 mice than in age-matched APPDutch mice, the conditions were comparable to those of the present study. Parenchymal Aβ deposition was reduced in APP23/APPDutch, and vascular load with microhemorrhages was increased, which suggests that AβDutch interacted with Aβwt and altered the pathogenic phenotypes.34 Thus, direct physical interactions between AβArc/Aβwt and AβDutch/Aβwt may be relevant pathogenic mechanisms in patients heterozygous for either of the mutations, both of which are located in APP at the same amino acid position. The conditions in human brain affected with AD unquestionably differ from those of transgenic mouse brain because N- and C-terminal truncated Aβ that have undergone posttranslational modifications are far more prevalent in human brain.35,36 In the transgenic models used, the population of Aβ species was more homogeneous and dominated by a few types of Aβ peptides.10 If it is assumed that AβArc and Aβwt are produced in equimolar amounts in Arctic human brain, an interesting future experiment would be to cross-breed mice with equal amounts of AβArc and Aβwt. The effect, if any, of murine Aβ on extracellular Aβ deposition is likely small because Aβ pathologic features in APP23 mice with or without the murine APP gene were indistinguishable at the light and ultrastructural levels.37 Even overexpression of murine APP did not alter the extent of parenchymal Aβ deposition in APPSwe and PS1dE9 mice (line 85), but did increase the solubility of Aβwt42 and led to more cerebral amyloid angiopathy.38 Nonetheless, ideally, bitransgenic mice should be further bred on an APP-knockout background. In general, considering the differences to human brain, experiments in transgenic models must be interpreted cautiously with respect to patients with AD, for example, those with the Arctic mutation.
Parenchymal Aβ deposits in human brain are classified according to morphologic features, amyloid tinctorial characteristics, and neuritic component of the corona. Whether one plaque type converts to another and, if so, whether plaque evolution reflects disease progression have been long-standing questions.39 Diffuse Aβ deposits are observed in patients without dementia symptoms at death and in primates and dogs. They are commonly assumed to be less pathogenic or even inert, and little is known about their genesis. Few animal models are rich in diffuse Aβ deposits. PDAPP mice with a high metabolic Aβ42 level are an exception.40 It has been suggested that aggregated Aβ in diffuse plaques form an amyloid core that becomes surrounded by dystrophic neurites (ie, emergence of a mature plaque), which are then removed via phagocytosis, leading to a “burnt out” plaque. The hypothesis has been challenged in cross-sectional studies of brains postmortem using IHC and various silver staining protocols.41 However, the conclusions rest on the assumption of AD as a homogeneous pathogenic entity. The strongest experimental evidence for the “plaque evolution theory” comes from investigations of patients with Down's syndrome, who invariably develop dementia and neuropathologic features of AD due to trisomy of chromosome 21. Postmortem brains of different ages have, therefore, been investigated to understand disease progression. Extracellular diffuse deposits of Aβ ending at amino acid 42 (Aβx-42) were observed in 12-year-old children, whereas compact plaques with Aβ ending at amino acid 40 (Aβx-40) were not evident until age 30 years.42,43 Because diffuse Aβ deposits predate compact plaques in brains in patients with Down's syndrome, it has been thought that with age they transform into compact plaques. In essence, formation of an amyloid core is a slow process involving extracellular Aβ accumulation in tissues, followed by structural modification, isomerization, and truncation of assembled Aβ peptides. In the present study, accelerated accumulation of diffuse deposits was observed that occurred independent of compact plaque formation. It was interpreted that diffuse and compact plaques can form via distinct processes, at least in transgenic mice. Consistent with this hypothesis, rapid formation of compact plaques has been demonstrated in transgenic models, which are known to develop few diffuse Aβ deposits.44,45
Aβ begins to accumulate inside neurons.46 The composition of intraneuronal Aβ aggregates could depend on the subcellular location, facilitators (eg, certain lipids or amyloid-associated factors), and origin (eg, Aβ metabolism in a particular neuronal subtype). Stable Aβ aggregates or Aβ monomers, which have been released from neurons, would then guide downstream events, leading to a specific type of extracellular Aβ deposit. The existence of prion strains illustrates that conformational differences can generate distinct pathologic phenotypes.47 Axonal transport and synaptic release are clearly involved in extracellular plaque formation.48,49 Thus, diffuse deposits in the striatum of bitransgenic mice were likely due to corticostriatal connections, resulting in axonal transport of APP/Aβ aggregates and/or extracellular aggregation after synaptic release of Aβ monomers. No compact plaques were observed in the striatum, at least at 18 months of age, again arguing that diffuse and compact plaques form via different mechanisms.
An important question is whether AβArc co-aggregate with Aβwt in diffuse deposits or whether they behave more like enzymes and facilitate a conformational change in Aβwt without becoming a part of the final Aβwt aggregates. The latter idea is reminiscent to that of pathologic chaperones, heterologous proteins such as ApoE and α1-antichymotrypsin, both of which regulate formation of Aβ deposits26,27 and are thought to induce or stabilize the β-sheet conformation.50,51 Although both ApoE and antichymotrypsin stain Aβ deposits, they do not necessarily co-polymerize with Aβ in the fibrils.52 Data from the present study suggest that AβArc behaved as a pathologic chaperone; that is, diffuse deposits were not AβArc-immunoreactive. However, because small amounts of AβArc were detected in striatal tissue extracts at ELISA analysis, that AβArc to some extent became building blocks of the diffuse deposits cannot be ruled out. Heparan sulfate was found only in compact plaques, possibly suggesting that they might selectively favor formation of an amyloid core,28 whereas ApoE and antichymotrypsin facilitate both diffuse and compact extracellular Aβ deposition.26,27 Selective association of heparan sulfate with compact plaques is consistent with previous observations in brains affected with AD 53 and with the dependence of heparin-binding properties of Aβ on its aggregation state.54 Cross-breeding of bitransgenic mice with animals in which the biologic characteristics of heparan sulfate proteoglycans have been manipulated28 would be needed to further challenge the hypothesis of different pathways leading to either diffuse Aβ deposits or compact plaques.
Mixtures of AβArc/Aβwt seemed to inhibit fibrillar but not prefibrillar growth in vitro compared with AβArc or Aβwt alone, possibly suggesting that the interaction occurred at an early stage. Interpreting differences in ThT signal in the plateau phase is not straightforward. The signal should reflect amyloid fibril concentration; however, it also depends on the number of available binding sites and the affinity of ThT for those sites in a heterogeneous sample, and on quantum yield.55 The percentage of AβArc of total Aβ in aged bitransgenic mice (<10%; Figure 5D) was lower than metabolic AβArc levels in young mice (>10% (Figure S5), although AβArc alone is much more prone to aggregate and form amyloid fibrils than is Aβwt, both in vitro and in vivo.4,5,13 Apparently, end products consisting of mixtures of Aβwt and AβArc did not easily form, perhaps because fibrils have an ordered structure. As demonstrated by others, Aβ40wt interacted with Aβ42wt and inhibited fibril formation both in vitro and in vivo.56,57 Moreover, the efficiency of seeded elongation of Aβ40wt by Aβwt40 fibrils was superior to that of Aβ40wt by Aβ40Arc fibrils in vitro.58 Consistent with this, it was observed that co-incubation of AβArc with Aβwt affected fibril structure in vitro and in vivo, as assessed at p-FTAA spectral analysis and electron microscopy. Others have reported that interactions between monomeric and protofibrillar Aβ interfered with the assembly and elongation of amyloid fibrils and stabilized Aβ protofibrils,59 and the present study detected higher Aβ protofibrillar levels in bitransgenic mice. Heterologous interactions between Aβ variants selectively increased diffuse Aβ deposits. That is, AβArc behaved as a facilitator; however, in principle, it illustrates that Aβ deposition is tractable to Aβ fragments or other binders serving as decelerators.
Acknowledgments
We thank the Uppsala University Transgenic Facility for assistance in developing APP transgenic models and Paul O′Callaghan, B.Sc., for English corrections.
Footnotes
Supported by grants from Uppsala University, Landstinget i Uppsala län, the Swedish Brain Fund, Alzheimerfonden, Demensfonden, Gamla Tjänarinnor, Gun och Bertil Stohnes Stiftelse, Magnus Bergvall, Åhlénsstiftelsen, Lars Hierta, Lundströms Minne, Frimurarstiftelsen, Svenska Läkaresällskapet, the Swedish Research Council (No. 2009-4389 to L.N.G.N.; No. 2008-4169 to P.H.; No. 2009-5343 to G.W.), the Swedish Foundation for Strategic Research and the Knut and Alice Wallenberg Foundation (P.H. and K.P.R.N.), Linköping University (P.H. and K.P.R.N.), the Swedish Foundation for Strategic Research (P.H. and K.P.R.N.), The European Union FP7 HEALTH (Project LUPAS) (K.P.R.N. and P.H.), the European Research Council (Project MUMID) (K.P.R.N.), and Astrid and Georg Olsson (P.H. and K.P.R.N.).
A.L. and O.P. contributed equally to the study.
Supplementary data
Supplemental Figure S1
Biochemical and immunohistologic Aβ analyses of young and aged tg-ArcSwe mice, line D, with a low level of transgenic APP expression and a tg-ArcSwe mouse (line B) with amyloid plaques. Tissue sections 25μm thick along the rostrocaudal axis from an 11-month-old tg-ArcSwe, line B (A and C), and an 18-month-old tg-ArcSwe, line D (B and D), immunostained with Aβ-antibody 82E1. Eleven-month-old tg-ArcSwe mice, line B (n = 2; positive control), and 2-month-old nontransgenic mice (n = 2; negative control) were simultaneously prepared and immunostained. As expected, high-expressing 11-month-old tg-ArcSwe mice, line B, were rich in extracellular Aβ deposits [mean (SEM) cortical Aβ burden 2.5% (0.3%); n = 2] and intracellular Aβ aggregates [A and C (location of C is marked by an asterisk in A)]. B, In contrast, in six sections from an 18-month-old tg-ArcSwe, line D, mouse, only a single plaque was found. Weak granular intracellular Aβ immunostaining was occasionally observed, as illustrated in CA3 pyramidal neurons in the hippocampus [D (location of D is marked with an asterisk in B)]. Scale bars: 470 μm (A and B); 48 μm (C and D). E and F, Frozen brain tissue from tg-ArcSwe, lines B and D, and nontransgenic mice were directly extracted in 70% formic acid, neutralized, and analyzed using Western blot analysis with 6E10 antibody (E) and Aβ1-40 ELISA (F). Total Aβ40 content was 9.9 (1.2) pmol/g at 2 months and 19.5 (1.7) pmol/g at 18 months in tg-ArcSwe mice, line D; that is, it increased modestly with age, whereas it was 8982 (770) pmol/g in 11-month-old tg-ArcSwe mice, line B.
Supplemental Figure S2
AβArc did not change Congo red burden in bitransgenic mice. There was no statistically significant difference in Congo red burden in the cerebral cortex at 12 and 18 months of age between bitransgenic mice and singly tg-Swe. Representative 12-month-old bitransgenic (A) and tg-Swe (B) mice are depicted. C: Graph summarizes the results of image analysis. Scale bar = 500 μm.
Supplemental Figure S3
Heparan sulfate, but neither ApoE nor ApoJ, was selectively associated with compact plaques in Aβ-depositing bitransgenic mice. Each row includes sets of three 10-μm consecutive sections from an 18-month-old bitransgenic mouse. They were fixed in 4% paraformaldehyde and immunostained with Congo red (A, D, and G), ApoE (B), Aβ42 (C, F, I, and L), ApoJ (E), Aβ40 (J), and heparan sulfate–specific (H and K) antibodies. A small area (arrow) is depicted in greater detail below each image to better visualize co-localization or lack thereof. Immunostaining with ApoE- and ApoJ-specific antibodies essentially overlaps with Aβ42 immunostaining, although immunostaining of ApoJ is faint in the diffuse Aβ deposits. Thus, ApoE and ApoJ are present in both diffuse plaques and compact plaques (A–F). In contrast, heparan sulfate immunostaining is observed only in compact plaques, possibly suggesting that it could selectively regulate their formation (G–L). Scale bars: 825 μm (A–L); 200 μm (detailed images).
Supplemental Figure S4
Diffuse Aβ42-immunoreactive deposits in areas with and without transgene expression in a very old tg-Swe mouse. Three consecutive 14-μm coronal sections from a 24-month-old tg-Swe, line A, mouse are shown in each row. Overview at different anatomic levels (A–C and J–L) and enlargements [D–F (star in A–C) and G–I (arrow in A–C)]. Many diffuse Aβ42-immunoreactive deposits were present in the striatum (B, H, and K) and also in the cerebral cortex (E). Scale bars: 670 μm (A–C); 130 μm (D–I); 340 μm (J–L).
Supplemental Figure S5
APP-ArcSwe neither alters processing of APP-Swe nor ratio of Aβ1-42wt/Aβ1-40wt in young tg-Swe mice. A: Steady-state levels of Aβ1-40 and Aβ1-42 in total brain (formic acid extracts) in 2-month-old bitransgenic (n = 5) and tg-Swe mice (n = 6). Levels of Aβx-40 (P = 0.28) and Aβx-42 (P = 0.14) tended to be increased in bitransgenic mice compared with age-matched tg-Swe; however, the effect was not significant and not greater then would be expected by increased transgene APP expression. B: The Aβ1-42/Aβ1-40 ratio in bitransgenic and tg-Swe mice did not differ (P = 0.75). C: AβArc could be detected only in bitransgenic mice using the total AβArc ELISA (AβArc1-x).
Supplemental Figure S6
ThT, a conventional amyloid dye, stained compact plaques but not diffuse Aβ deposits in bitransgenic mice. Tissue sections 25 μm thick from an 18-month-old bitransgenic mouse were fixed in paraformaldehyde and stained with ThT (A and D) and an Aβ42-specific antibody (B and E). Merged images illustrate the extent of overlap between ThT and Aβ42 (C and F). Primarily, large Aβ42-immunoreactive deposits were stained by ThT in cerebral cortex (A–C). In contrast, Aβ42 immunopositive deposits, especially in the striatum (D–F), did not stain with ThT. The faint ThT signal (D) was due to autofluorescence, which did not overlap with the Aβ42 immunoreactivity. Scale bars = 100 μm.
References
- 1.Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 2.Basun H., Bogdanovic N., Ingelsson M., Almkvist O., Näslund J., Axelman K., Bird T.D., Nochlin D., Schellenberg G.D., Wahlund L.O., Lannfelt L. Clinical and neuropathological features of the Arctic APP gene mutation causing early-onset Alzheimer disease. Arch Neurol. 2008;65:499–505. doi: 10.1001/archneur.65.4.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nilsberth C., Westlind-Danielsson A., Eckman C.B., Condron M.M., Axelman K., Forsell C., Stenh C., Luthman J., Teplow D.B., Younkin S.G., Näslund J., Lannfelt L. The “Arctic” APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 4.Johansson A.S., Berglind-Dehlin F., Karlsson G., Edwards K., Gellerfors P., Lannfelt L. Physiochemical characterization of the Alzheimer's disease–related peptides A beta 1-42Arctic and A beta 1-42wt. FEBS J. 2006;273:2618–2630. doi: 10.1111/j.1742-4658.2006.05263.x. [DOI] [PubMed] [Google Scholar]
- 5.Lord A., Kalimo H., Eckman C., Zhang X.Q., Lannfelt L., Nilsson L.N. The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006;27:67–77. doi: 10.1016/j.neurobiolaging.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Stenh C., Englund H., Lord A., Johansson A.S., Almeida C.G., Gellerfors P., Greengard P., Gouras G.K., Lannfelt L., Nilsson L.N. Amyloid-beta oligomers are inefficiently measured by enzyme-linked immunosorbent assay. Ann Neurol. 2005;58:147–150. doi: 10.1002/ana.20524. [DOI] [PubMed] [Google Scholar]
- 7.Englund H., Sehlin D., Johansson A.S., Nilsson L.N., Gellerfors P., Paulie S., Lannfelt L., Pettersson F.E. Sensitive ELISA detection of amyloid-beta protofibrils in biological samples. J Neurochem. 2007;103:334–345. doi: 10.1111/j.1471-4159.2007.04759.x. [DOI] [PubMed] [Google Scholar]
- 8.Sahlin C., Lord A., Magnusson K., Englund H., Almeida C.G., Greengard P., Nyberg F., Gouras G.K., Lannfelt L., Nilsson L.N. The Arctic Alzheimer mutation favors intracellular amyloid-beta production by making amyloid precursor protein less available to alpha-secretase. J Neurochem. 2007;101:854–862. doi: 10.1111/j.1471-4159.2006.04443.x. [DOI] [PubMed] [Google Scholar]
- 9.Lord A., Englund H., Söderberg L., Tucker S., Clausen F., Hillered L., Gordon M., Morgan D., Lannfelt L., Pettersson F.E., Nilsson L.N. Amyloid-beta protofibril levels correlate with spatial learning in Arctic Alzheimer's disease transgenic mice. FEBS J. 2009;276:995–1006. doi: 10.1111/j.1742-4658.2008.06836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Philipson O., HammarströM P., Nilsson K.P., Portelius E., Olofsson T., Ingelsson M., Hyman B.T., Blennow K., Lannfelt L., Kalimo H., Nilsson L.N. A highly insoluble state of Abeta similar to that of Alzheimer's disease brain is found in Arctic APP transgenic mice. Neurobiol Aging. 2009;30:1393–1405. doi: 10.1016/j.neurobiolaging.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 11.Jarrett J.T., Lansbury P.T., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 12.Gajdusek D.C. Transmissible and nontransmissible dementias: distinction between primary cause and pathogenetic mechanisms in Alzheimer's disease and aging. Mt Sinai J Med. 1988;55:3–5. [PubMed] [Google Scholar]
- 13.Cheng I.H., Palop J.J., Esposito L.A., Bien-Ly N., Yan F., Mucke L. Aggressive amyloidosis in mice expressing human amyloid peptides with the Arctic mutation. Nat Med. 2004;10:1190–1192. doi: 10.1038/nm1123. [DOI] [PubMed] [Google Scholar]
- 14.Prusiner S.B. Molecular biology of prion diseases. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 15.Näslund J., Haroutunian V., Mohs R., Davis K.L., Davies P., Greengard P., Buxbaum J.D. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 16.Åslund A., Sigurdson C.J., Klingstedt T., Grathwohl S., Bolmont T., Dickstein D.L., Glimsdal E., Prokop S., Lindgren M., Konradsson P., Holtzman D.M., Hof P.R., Heppner F.L., Gandy S., Jucker M., Aguzzi A., Hammarström P., Nilsson K.P. Novel pentameric thiophene derivatives for in vitro and in vivo optical imaging of a plethora of protein aggregates in cerebral amyloidoses. ACS Chem Biol. 2009;4:673–684. doi: 10.1021/cb900112v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nilsson L.N., Bales K.R., DiCarlo G., Gordon M.N., Morgan D., Paul S.M., Potter H. Alpha-1-antichymotrypsin promotes beta-sheet amyloid plaque deposition in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:1444–1451. doi: 10.1523/JNEUROSCI.21-05-01444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiltfang J., Arold N., Neuhoff V. A new multiphasic buffer system for sodium dodecyl sulfate–polyacrylamide gel electrophoresis of proteins and peptides with molecular masses 100,000–1000, and their detection with picomolar sensitivity. Electrophoresis. 1991;12:352–366. doi: 10.1002/elps.1150120507. [DOI] [PubMed] [Google Scholar]
- 19.Klafki H.W., Wiltfang J., Staufenbiel M. Electrophoretic separation of betaA4 peptides (1–40) and (1–42) Anal Biochem. 1996;237:24–29. doi: 10.1006/abio.1996.0195. [DOI] [PubMed] [Google Scholar]
- 20.Walsh D.M., Lomakin A., Benedek G.B., Condron M.M., Teplow D.B. Amyloid beta-protein fibrillogenesis: detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 21.LeVine H., III Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mucke L., Masliah E., Yu G.Q., Mallory M., Rockenstein E.M., Tatsuno G., Hu K., Kholodenko D., Johnson-Wood K., McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sigurdson C.J., Nilsson K.P., Hornemann S., Manco G., Polymenidou M., Schwarz P., Leclerc M., HammarströM P., Wuthrich K., Aguzzi A. Prion strain discrimination using luminescent conjugated polymers. Nat Methods. 2007;4:1023–1030. doi: 10.1038/nmeth1131. [DOI] [PubMed] [Google Scholar]
- 24.Nilsson K.P., Åslund A., Berg I., Nystrom S., Konradsson P., Herland A., Inganäs O., Stabo-Eeg F., Lindgren M., Westermark G.T., Lannfelt L., Nilsson L.N., HammarströM P. Imaging distinct conformational states of amyloid-beta fibrils in Alzheimer's disease using novel luminescent probes. ACS Chem Biol. 2007;2:553–560. doi: 10.1021/cb700116u. [DOI] [PubMed] [Google Scholar]
- 25.Berg I., Nilsson K.P., Thor S., Hammarström P. Efficient imaging of amyloid deposits in Drosophila models of human amyloidoses. Nat Protoc. 2010;5:935–944. doi: 10.1038/nprot.2010.41. [DOI] [PubMed] [Google Scholar]
- 26.Nilsson L.N., Arendash G.W., Leighty R.E., Costa D.A., Low M.A., Garcia M.F., Cracciolo J.R., Rojiani A., Wu X., Bales K.R., Paul S.M., Potter H. Cognitive impairment in PDAPP mice depends on ApoE and ACT-catalyzed amyloid formation. Neurobiol Aging. 2004;25:1153–1167. doi: 10.1016/j.neurobiolaging.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 27.Bales K.R., Verina T., Dodel R.C., Du Y., Altstiel L., Bender M., Hyslop P., Johnstone E.M., Little S.P., Cummins D.J., Piccardo P., Ghetti B., Paul S.M. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 28.Li J.P., Galvis M.L., Gong F., Zhang X., Zcharia E., Metzger S., Vlodavsky I., Kisilevsky R., Lindahl U. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc Natl Acad Sci USA. 2005;102:6473–6477. doi: 10.1073/pnas.0502287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeMattos R.B., O'Dell M.A., Parsadanian M., Taylor J.W., Harmony J.A., Bales K.R., Paul S.M., Aronow B.J., Holtzman D.M. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2002;99:10843–10848. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheuermann S., Hambsch B., Hesse L., Stumm J., Schmidt C., Beher D., Bayer T.A., Beyreuther K., Multhaup G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease. J Biol Chem. 2001;276:33923–33929. doi: 10.1074/jbc.M105410200. [DOI] [PubMed] [Google Scholar]
- 31.Rönnbäck A., Zhu S., Dillner K., Aoki M., Lilius L., Näslund J., Winblad B., Graff C. Progressive neuropathology and cognitive decline in a single Arctic APP transgenic mouse model. Neurobiol Aging. 2011;32:280–292. doi: 10.1016/j.neurobiolaging.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 32.Levy E., Carman M.D., Fernandez-Madrid I.J., Power M.D., Lieberburg I., van Duinen S.G., Bots G.T., Luyendijk W., Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 33.Herzig M.C., Winkler D.T., Burgermeister P., Pfeifer M., Kohler E., Schmidt S.D., Danner S., Abramowski D., Sturchler-Pierrat C., Burki K., van Duinen S.G., Maat-Schieman M.L., Staufenbiel M., Mathews P.M., Jucker M. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 34.Herzig M.C., Eisele Y.S., Staufenbiel M., Jucker M. E22Q-mutant Abeta peptide (AbetaDutch) increases vascular but reduces parenchymal Abeta deposition. Am J Pathol. 2009;174:722–726. doi: 10.2353/ajpath.2009.080790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mori H., Takio K., Ogawara M., Selkoe D.J. Mass spectrometry of purified amyloid beta protein in Alzheimer's disease. J Biol Chem. 1992;267:17082–17086. [PubMed] [Google Scholar]
- 36.Kalback W., Watson M.D., Kokjohn T.A., Kuo Y.M., Weiss N., Luehrs D.C., Lopez J., Brune D., Sisodia S.S., Staufenbiel M., Emmerling M., Roher A.E. APP transgenic mice Tg2576 accumulate Abeta peptides that are distinct from the chemically modified and insoluble peptides deposited in Alzheimer's disease senile plaques. Biochemistry. 2002;41:922–928. doi: 10.1021/bi015685+. [DOI] [PubMed] [Google Scholar]
- 37.Calhoun M.E., Burgermeister P., Phinney A.L., Stalder M., Tolnay M., Wiederhold K.H., Abramowski D., Sturchler-Pierrat C., Sommer B., Staufenbiel M., Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jankowsky J.L., Younkin L.H., Gonzales V., Fadale D.J., Slunt H.H., Lester H.A., Younkin S.G., Borchelt D.R. Rodent A beta modulates the solubility and distribution of amyloid deposits in transgenic mice. J Biol Chem. 2007;282:22707–22720. doi: 10.1074/jbc.M611050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armstrong R.A. Beta-amyloid plaques: stages in life history or independent origin? Dement Geriatr Cogn Disord. 1998;9:227–238. doi: 10.1159/000017051. [DOI] [PubMed] [Google Scholar]
- 40.Johnson-Wood K., Lee M., Motter R., Hu K., Gordon G., Barbour R., Khan K., Gordon M., Tan H., Games D., Lieberburg I., Schenk D., Seubert P., McConlogue L. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibson P.H. Relationship between numbers of cortical argentophilic and congophilic senile plaques in the brains of elderly people with and without senile dementia of Alzheimer type. Gerontology. 1985;31:321–324. doi: 10.1159/000212716. [DOI] [PubMed] [Google Scholar]
- 42.Lemere C.A., Blusztajn J.K., Yamaguchi H., Wisniewski T., Saido T.C., Selkoe D.J. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis. 1996;3:16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 43.Iwatsubo T., Mann D.M., Odaka A., Suzuki N., Ihara Y. Amyloid beta protein (A beta) deposition: a beta 42(43) precedes A beta 40 in Down syndrome. Ann Neurol. 1995;37:294–299. doi: 10.1002/ana.410370305. [DOI] [PubMed] [Google Scholar]
- 44.Meyer-Luehmann M., Spires-Jones T.L., Prada C., Garcia-Alloza M., de Calignon A., Rozkalne A., Koenigsknecht-Talboo J., Holtzman D.M., Bacskai B.J., Hyman B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan P., Bero A.W., Cirrito J.R., Xiao Q., Hu X., Wang Y., Gonzales E., Holtzman D.M., Lee J.M. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci. 2009;29:10706–10714. doi: 10.1523/JNEUROSCI.2637-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Philipson O., Lannfelt L., Nilsson L.N. Genetic and pharmacological evidence of intraneuronal Abeta accumulation in APP transgenic mice. FEBS Lett. 2009;583:3021–3026. doi: 10.1016/j.febslet.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 47.Prusiner S.B. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lazarov O., Lee M., Peterson D.A., Sisodia S.S. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheng J.G., Price D.L., Koliatsos V.E. Disruption of corticocortical connections ameliorates amyloid burden in terminal fields in a transgenic model of Abeta amyloidosis. J Neurosci. 2002;22:9794–9799. doi: 10.1523/JNEUROSCI.22-22-09794.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wisniewski T., Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992;135:235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 51.Potter H., Wefes I.M., Nilsson L.N. The inflammation-induced pathological chaperones ACT and Apo-E are necessary catalysts of Alzheimer amyloid formation. Neurobiol Aging. 2001;22:923–930. doi: 10.1016/s0197-4580(01)00308-6. [DOI] [PubMed] [Google Scholar]
- 52.Söderberg L., Bogdanovic N., Axelsson B., Winblad B., Naslund J., Tjernberg L.O. Analysis of single Alzheimer solid plaque cores by laser capture microscopy and nanoelectrospray/tandem mass spectrometry. Biochemistry. 2006;45:9849–9856. doi: 10.1021/bi060331+. [DOI] [PubMed] [Google Scholar]
- 53.O'Callaghan P., Sandwall E., Li J.P., Yu H., Ravid R., Guan Z.Z., van Kuppevelt T.H., Nilsson L.N., Ingelsson M., Hyman B.T., Kalimo H., Lindahl U., Lannfelt L., Zhang X. Heparan sulfate accumulation with Abeta deposits in Alzheimer's disease and Tg2576 mice is contributed by glial cells. Brain Pathol. 2008;18:548–561. doi: 10.1111/j.1750-3639.2008.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watson D.J., Lander A.D., Selkoe D.J. Heparin-binding properties of the amyloidogenic peptides Abeta and amylin: dependence on aggregation state and inhibition by Congo red. J Biol Chem. 1997;272:31617–31624. doi: 10.1074/jbc.272.50.31617. [DOI] [PubMed] [Google Scholar]
- 55.LeVine H., III Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 56.Hasegawa K., Yamaguchi I., Omata S., Gejyo F., Naiki H. Interaction between A beta(1–42) and A beta(1–40) in Alzheimer's beta-amyloid fibril formation in vitro. Biochemistry. 1999;38:15514–15521. doi: 10.1021/bi991161m. [DOI] [PubMed] [Google Scholar]
- 57.Kim J., Onstead L., Randle S., Price R., Smithson L., Zwizinski C., Dickson D.W., Golde T., McGowan E. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O'Nuallain B., Williams A.D., Westermark P., Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem. 2004;279:17490–17499. doi: 10.1074/jbc.M311300200. [DOI] [PubMed] [Google Scholar]
- 59.Lashuel H.A., Hartley D.M., Petre B.M., Wall J.S., Simon M.N., Walz T., Lansbury P.T., Jr Mixtures of wild-type and a pathogenic (E22G) form of Abeta40 in vitro accumulate protofibrils, including amyloid pores. J Mol Biol. 2003;332:795–808. doi: 10.1016/s0022-2836(03)00927-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1
Biochemical and immunohistologic Aβ analyses of young and aged tg-ArcSwe mice, line D, with a low level of transgenic APP expression and a tg-ArcSwe mouse (line B) with amyloid plaques. Tissue sections 25μm thick along the rostrocaudal axis from an 11-month-old tg-ArcSwe, line B (A and C), and an 18-month-old tg-ArcSwe, line D (B and D), immunostained with Aβ-antibody 82E1. Eleven-month-old tg-ArcSwe mice, line B (n = 2; positive control), and 2-month-old nontransgenic mice (n = 2; negative control) were simultaneously prepared and immunostained. As expected, high-expressing 11-month-old tg-ArcSwe mice, line B, were rich in extracellular Aβ deposits [mean (SEM) cortical Aβ burden 2.5% (0.3%); n = 2] and intracellular Aβ aggregates [A and C (location of C is marked by an asterisk in A)]. B, In contrast, in six sections from an 18-month-old tg-ArcSwe, line D, mouse, only a single plaque was found. Weak granular intracellular Aβ immunostaining was occasionally observed, as illustrated in CA3 pyramidal neurons in the hippocampus [D (location of D is marked with an asterisk in B)]. Scale bars: 470 μm (A and B); 48 μm (C and D). E and F, Frozen brain tissue from tg-ArcSwe, lines B and D, and nontransgenic mice were directly extracted in 70% formic acid, neutralized, and analyzed using Western blot analysis with 6E10 antibody (E) and Aβ1-40 ELISA (F). Total Aβ40 content was 9.9 (1.2) pmol/g at 2 months and 19.5 (1.7) pmol/g at 18 months in tg-ArcSwe mice, line D; that is, it increased modestly with age, whereas it was 8982 (770) pmol/g in 11-month-old tg-ArcSwe mice, line B.
Supplemental Figure S2
AβArc did not change Congo red burden in bitransgenic mice. There was no statistically significant difference in Congo red burden in the cerebral cortex at 12 and 18 months of age between bitransgenic mice and singly tg-Swe. Representative 12-month-old bitransgenic (A) and tg-Swe (B) mice are depicted. C: Graph summarizes the results of image analysis. Scale bar = 500 μm.
Supplemental Figure S3
Heparan sulfate, but neither ApoE nor ApoJ, was selectively associated with compact plaques in Aβ-depositing bitransgenic mice. Each row includes sets of three 10-μm consecutive sections from an 18-month-old bitransgenic mouse. They were fixed in 4% paraformaldehyde and immunostained with Congo red (A, D, and G), ApoE (B), Aβ42 (C, F, I, and L), ApoJ (E), Aβ40 (J), and heparan sulfate–specific (H and K) antibodies. A small area (arrow) is depicted in greater detail below each image to better visualize co-localization or lack thereof. Immunostaining with ApoE- and ApoJ-specific antibodies essentially overlaps with Aβ42 immunostaining, although immunostaining of ApoJ is faint in the diffuse Aβ deposits. Thus, ApoE and ApoJ are present in both diffuse plaques and compact plaques (A–F). In contrast, heparan sulfate immunostaining is observed only in compact plaques, possibly suggesting that it could selectively regulate their formation (G–L). Scale bars: 825 μm (A–L); 200 μm (detailed images).
Supplemental Figure S4
Diffuse Aβ42-immunoreactive deposits in areas with and without transgene expression in a very old tg-Swe mouse. Three consecutive 14-μm coronal sections from a 24-month-old tg-Swe, line A, mouse are shown in each row. Overview at different anatomic levels (A–C and J–L) and enlargements [D–F (star in A–C) and G–I (arrow in A–C)]. Many diffuse Aβ42-immunoreactive deposits were present in the striatum (B, H, and K) and also in the cerebral cortex (E). Scale bars: 670 μm (A–C); 130 μm (D–I); 340 μm (J–L).
Supplemental Figure S5
APP-ArcSwe neither alters processing of APP-Swe nor ratio of Aβ1-42wt/Aβ1-40wt in young tg-Swe mice. A: Steady-state levels of Aβ1-40 and Aβ1-42 in total brain (formic acid extracts) in 2-month-old bitransgenic (n = 5) and tg-Swe mice (n = 6). Levels of Aβx-40 (P = 0.28) and Aβx-42 (P = 0.14) tended to be increased in bitransgenic mice compared with age-matched tg-Swe; however, the effect was not significant and not greater then would be expected by increased transgene APP expression. B: The Aβ1-42/Aβ1-40 ratio in bitransgenic and tg-Swe mice did not differ (P = 0.75). C: AβArc could be detected only in bitransgenic mice using the total AβArc ELISA (AβArc1-x).
Supplemental Figure S6
ThT, a conventional amyloid dye, stained compact plaques but not diffuse Aβ deposits in bitransgenic mice. Tissue sections 25 μm thick from an 18-month-old bitransgenic mouse were fixed in paraformaldehyde and stained with ThT (A and D) and an Aβ42-specific antibody (B and E). Merged images illustrate the extent of overlap between ThT and Aβ42 (C and F). Primarily, large Aβ42-immunoreactive deposits were stained by ThT in cerebral cortex (A–C). In contrast, Aβ42 immunopositive deposits, especially in the striatum (D–F), did not stain with ThT. The faint ThT signal (D) was due to autofluorescence, which did not overlap with the Aβ42 immunoreactivity. Scale bars = 100 μm.