Therapeutic potential and molecular mechanism of a novel, potent, non-peptide, Smac mimetic SM-164 in combination with TRAIL for cancer treatment (original) (raw)
. Author manuscript; available in PMC: 2012 May 1.
Abstract
Smac mimetics are being developed as a new class of anticancer therapies. Since the single-agent activity of Smac mimetics is very limited, rational combinations represent a viable strategy for their clinical development. The combination of Smac mimetics with TRAIL may be particularly attractive due to the low toxicity of TRAIL to normal cells and the synergistic antitumor activity observed for the combination. In the present study, we have investigated the combination synergy between TRAIL and a potent Smac mimetic, SM-164 in vitro and in vivo and the underlying molecular mechanism of action for the synergy. Our study demonstrates that SM-164 is highly synergistic with TRAIL in vitro in both TRAIL-sensitive and TRAIL-resistant cancer cell lines of breast, prostate, and colon cancer. Furthermore, the combination of SM-164 with TRAIL induces rapid tumor regression in vivo in a breast cancer xenograft model in which either agent is ineffective. Our data shows that XIAP and cIAP1, but not cIAP2, work in concert to attenuate the activity of TRAIL; SM-164 strongly enhances TRAIL activity by concurrently targeting XIAP and cIAP1. Moreover, while RIP1 plays a minimal role in TRAIL's activity as a single agent, it is required for the synergistic interaction between TRAIL and SM-164. Our present study provides a strong rationale to develop the combination of SM-164 and TRAIL as a new therapeutic strategy for the treatment of human cancer.
Keywords: Smac Mimetic, TRAIL, Synergy, Tumor Regression
Introduction
Evasion of apoptosis is a hallmark of human cancers (1, 2) and targeting key apoptosis regulators with a goal to overcome apoptosis of tumor cells is being pursued as a new cancer therapeutic strategy (3, 4).
Inhibitor of apoptosis proteins (IAPs) are a family of key apoptosis regulators, characterized by the presence of one or more Baculovirus IAP Repeat (BIR) domains (5, 6). Among them, X-linked IAP (XIAP), by binding to effector caspase-3 and -7 and an initiator caspase-9 and inhibiting the activities of these caspases, blocks both death receptor-mediated and mitochondria-mediated apoptosis, whereas cellular IAP 1 (cIAP1) and cIAP2 inhibit death receptor-mediated apoptosis by directly binding to TNF receptor-associated factor 2 (TRAF2) (5, 6). XIAP, cIAP1 and cIAP2 have been found to be overexpressed in human cancer cell lines (7, 8) and human tumor tissues (9-11) and their overexpression confers on cancer cells resistance to various anticancer drugs (12-15). IAP proteins are attractive cancer therapeutic targets (12-15).
Smac/ DIABLO (Second Mitochondria-derived Activator of Caspases or Direct IAP-Binding protein with Low PI) is a pro-apoptotic molecule (16, 17) and promotes apoptosis, at least in part, by directly antagonizing cIAP1/2 and XIAP (12-15). Smac protein interacts with XIAP and cIAP1/2 via its _N_-terminal AVPI terapeptide binding motif (18). In recent years, there have been intense research efforts in developing small-molecule Smac mimetics as a new class of anticancer drugs (19, 20) and several such compounds are now in early clinical development (21-24).
Smac mimetics can effectively induce apoptosis as single agents in certain cancer cells through a TNFα (tumor necrosis factor alpha)-dependent autocrine mechanism and activation of caspase-8 and downstream caspases (25-29) but their anticancer activity as single agents appears to be limited to approximately 10% of human cancer cell lines in vitro (27, 30). Therefore, for the successful clinical development of Smac mimetics as a new class of anticancer drugs, rational combination strategies are clearly needed.
To this end, the combination of TRAIL (TNF-related apoptosis inducing ligand) with Smac mimetics appears to be a particularly attractive strategy for several reasons. First, TRAIL is a member of the TNFα family, but unlike TNFα, TRAIL shows very low toxicity to normal cells and tissues and is well tolerated in clinical trials (31-33). Second, despite its good safety profile, TRAIL has minimal single-agent anticancer activity in clinical trials (31), and this has hampered its clinical development. Third, there is very strong synergy between TRAIL and Smac mimetics in cancer cell lines of diverse of tumor types (34-40).
Despite the strong synergy demonstrated between TRAIL and Smac mimetics, the precise underlying molecular mechanism of action for their synergy is not fully understood. Because Smac mimetics have been designed based upon the interaction between XIAP and Smac, previous investigations have focused on XIAP as the primary cellular target for Smac mimetics when combined with TRAIL (36-39). However, our data clearly showed that while knock-out of XIAP or efficient knock-down of XIAP by siRNA can modestly sensitize cancer cells to apoptosis induction by TRAIL, the sensitization effect is far less than that achieved by Smac mimetics. In addition, although one would expect that the underlying molecular mechanism for the synergistic interaction between TRAIL and Smac mimetics may be very similar to that between TNFα and Smac mimetics, our data showed that TNFα fails to induce apoptosis in cancer cell lines of diverse tumor types which are very sensitive to TRAIL as a single agent, and Smac mimetics can dramatically sensitize TRAIL in both TRAIL-sensitive and –resistant cancer cell lines. Finally, despite the strong synergy between TRAIL and Smac mimetics in vitro, tumor regression has not been reported in vivo for the combination when both agents are ineffective.
We have previously reported the design and evaluation of SM-164 as a potent, bivalent Smac mimetic (28, 41). SM-164 binds to XIAP, cIAP1 and cIAP2 with Ki values of 0.56 nM, 0.31 nM and 1.1 nM, respectively (28, 41). It potently antagonizes XIAP in cell-free functional assays and in cells, and induces rapid degradation of cIAP1 and cIAP2 in cancer cells at concentrations as low as 1-10 nM (28, 41). In the present study, we have employed SM-164 and evaluated its combination with recombinant TRAIL protein in a panel of 19 human breast, prostate and colon cancer cell lines in vitro and in a breast cancer xenograft model in vivo. Our study provides further insights into the molecular mechanism of action for the strong synergy between TRAIL and Smac mimetics and suggests that the combination of TRAIL with SM-164 or other Smac mimetics should be evaluated in the clinic as a new strategy for the treatment of human breast, prostate and colon cancer.
Materials and Methods
Reagents and Antibodies
SM-164 was synthesized as described previously (41) and the purity is >95% by HPLC analysis. The chemical structure of SM-164 is included in SI Fig S1. Native recombinant human TRAIL (rhTRAIL) (residues 114-281) construct without His tag was a kind gift of Dr. Arul Chinnaiyan at the Michigan Center for Translational Medicine and Department of Pathology, University of Michigan. TRAIL 114-281aa was cloned into a pHis-TEV vector in our lab. The resulting construct was transformed into Escherichia coli BL21 DE3. Cells harboring the construct were cultured at 37°C, 250 rpm in LB media with 50 μg/ml of kanamycin until the OD600 was 0.4-0.6. Protein expression was induced with 0.1 M IPTG at 30°C, 250 rpm overnight. The cells were harvested by centrifugation (7000_g_, 12 min, and 4 °C) and cell pellet was stored in -80 °C or directly used for protein purification. Cell pellet was resuspended in 40 ml lysis buffer (50mM Tris pH7.5, 200mM NaCl, 50uM ZnAc, 1mM DTT) for sonication to release soluble proteins. TRAIL 114-281aa protein was purified by Ni-NTA affinity chromatography. The Ni-NTA resin was washed with 50 ml lysis buffer and recombinant protein was eluted with lysis buffer with 80 mM imidazole. The protein was further purified with size-exclusion chromatography using an Amersham Biosciences P-920 FPLC equipped with a Superdex 200 column (Amersham Biosciences). The protein was eluted in a buffer containing 50mM Tris pH 7.5, 200 mM NaCl, 50 μM ZnAc, 10 mM DTT and 10% glycerol and then stored at -80 °C. The activity of this recombinant TRAIL was comparable with a commercial TRAIL (R&D Systems) and was found to be identical. Flag-tagged TRAIL was purchased from Alexis Biochemicals (San Diego, CA).
The following primary antibodies were used in this study: cleaved-caspase-8, XIAP, PAPR, TRAF2 (Cell Signaling Technology, Beverly, MA); caspase-3, caspase-9, FADD (Stressgen Biotechnologies, Victoria, Canada), caspase 8 clone 6E (Epitomics, Inc., Burlingame, CA); DR4 and DR5 (Prosci, Flint Place Poway, CA), cIAP1 (J. Silke, La Trobe University) and cIAP2 (R&D Systems, Minneapolis, MN).
Cell lines
Human breast cancer MDA-MB-436, SK-BR-3, MDA-MB-453, MDA-MB-468, SUM159, SUM52, T47D cell lines, human colon cancer HCC116, SW620, SW480, RKO, HT29, DLD1 cell lines and human prostate cancer PC-3, DU-145, CL-1, 22RV1, LNCaP cell line are purchased from ATCC. Human breast cancer 2LMP cell line was a subclone of the MDA-MB-231 cell line and was a kind gift of Dr. Dajun Yang, Department of Internal Medicine, University of Michigan. All 19 cell lines were passaged fewer than 6 months in our laboratory either after receipt or resuscitation. Isogenic XIAP+/+ and XIAP−/− HCT116 colon cancer cell lines were a kind gift from Dr. Fred Bunz (Johns Hopkins University, Baltimore, MD).
Cell Viability, Cell Death, and Apoptosis
Cell viability was evaluated by a lactate dehydrogenase (LDH) based WST-8 assay (Dojindo Molecular Technologies, Rockville, MD) as described previously (23). Cell death was quantitated by microscopic examination in a trypan blue excluson assay. Apoptosis analysis was done using an Annexin V/propidium iodide apoptosis detection kit (Roche Applied Science, Indianapolis, IN) by flow cytometry according to the manufacturer's instructions. Total Annexin V (+) cells plus Annexin V (-)/PI (+) were counted as apoptotic cells.
Western Blot Analysis
Cells or xenograft tumor tissues were lysed using radioimmunoprecipitation assay lysis buffer (PBS containing 1% NP40, 0.5% Na-deoxycholate, and 0.1% SDS) supplemented with 1 μmol/L phenylmethylsulfonyl fluoride and 1 protease inhibitor cocktail tablet per 10 mL on ice for 20 min, and lysates were then cleared by centrifugation before determination of protein concentration using the Bio-Rad protein assay kit according to the manufacturer's instructions. Proteins were electrophoresed onto 4-20% SDS-PAGE gels (Invitrogen, Carlsbad, CA) and transferred onto polyvinylidene difluoride membranes. Following blocking in 5% milk, membranes were incubated with a specific primary antibody, washed, and incubated with horseradish peroxidase–linked secondary antibody (GE Healthcare, Piscataway, NJ). Signals were visualized with chemiluminescent horseradish peroxidase antibody detection reagent (Denville Scientific, Metuchen, NJ). Where indicated, the blots were stripped and reprobed with a different antibody.
Co-immunoprecipitation
TRAIL-receptor complex was immunoprecipitated with Flag-tagged TRAIL, based on the reported protocol (28). A total of 5×107 cells of each sample in 10 ml culture medium were treated with the mixture of 100 ng/ml of Flag-tagged TRAIL and anti-Flag M2 IgG (3 μg/ml for each sample) at 37°C and then lysed for 30 min on ice with the lysis buffer. The soluble fraction was pulled down with sepharose 4B beads overnight at 4°C and subjected to Western blot analysis. For the analysis of the TRAIL-dependent secondary signaling complex, after immunoprecipitation of the DISC, DISC-depleted cell lysates were subjected to a second round of immunoprecipitation using anti-RIP1, followed by western blotting analysis of co-immunoprecipitated procaspase-8 and cIAP1.
RNA interference
RNA interference was performed as described previously (23). Briefly, siRNA was used to knock down XIAP, cIAP1, cIAP2, and caspase-8, -9, and -3 (Dhamarcon Research, Inc.). Non-targeting control siRNA was purchased from Ambion. Transfections were performed using Lipofectamine RNAiMAX (Invitrogen) in the reverse manner according to the manufacturer's instructions. Between 5 and 10 pmol siRNA and 5 μL Lipofectamine RNAiMAX were mixed in each well of six-well plates for 20 min, followed by culturing 3 × 105 cells in the siRNA mix for 24 to 48 h; knockdown efficacy was assessed by Western blotting.
In vivo studies
Nude athymic mice bearing s.c. 2LMP tumor xenografts were used. For determination of PARP cleavage, caspase activation and cIAP1 degradation in vivo, tissues were harvested, and tissue lysates were analyzed by Western blotting as described in the Supplementary Information (SI). For apoptosis, TUNEL and histopathology analysis of tissues, paraffin-embedded tissues were examined as described in the SI. For the efficacy experiment, mice bearing 2LMP xenograft tumors (8-12 mice per group) were treated with 5 mg/kg of SM-164, i.v. 10 mg/kg of TRAIL i.p. or their combination daily, 5 times a week for 2 weeks. Tumor volume was measured 3 times per week. Data are represented as mean tumor volumes ± SEM. All animal experiments were performed under the guidelines of the University of Michigan Committee for Use and Care of Animals.
Combination index (CI) calculation
Synergy was quantified by Combination Index (CI) analysis. CI value was calculated by equation as described previously (42, 43): CI = (CA, x/ICx,A) + (CB, x/ICx,B). CA,x and CB,x are the concentrations of drug A and drug B used in combination to achieve x% drug effect. ICx,A and ICx,B are the concentrations for single agents to achieve the same effect. We used IC50 values (x% = 50%) to calculate the combination index in the present study. A CI of less than 0.3 indicates very strong synergy; 0.3-0.7 strong synergy; 0.7-1 modest synergy, and more than 1 synergy, antagonism, respectively.
Statistical Analysis
Statistical analyses were performed by two-way ANOVA and unpaired two-tailed t test, using Prism (version 4.0, GraphPad, La Jolla, CA). P < 0.05 was considered statistically significant.
Results
SM-164 greatly enhances the anticancer activity of TRAIL in both TRAIL-sensitive and TRAIL–resistant cancer cell lines
We tested the combination of SM-164 with TRAIL in a panel of 8 breast, 6 colon and 5 prostate cancer cell lines using the cell viability inhibition assay. SM-164 has no or minimal single-agent activity at concentrations up to 100 nM in these cancer cell lines but shows the strong synergistic activity in combination with TRAIL in 15 cancer cell lines (Fig. 1 and SI Fig. S2-S4). At concentrations of 100 nM, SM-164 reduces the IC50 values of TRAIL by one to three orders of magnitude in 12 of these 19 cancer cell lines. SM-164 greatly enhances the anticancer activity of TRAIL not only in TRAIL-sensitive but also in TRAIL-resistant cancer cell lines in the cell viability assay. The interaction between SM-164 and TRAIL in these 12 cancer cell lines is highly synergistic based upon the calculated combination index (CI).
Figure 1.

SM-164 potentiates cell viability inhibition by TRAIL in both TRAIL-sensitive and TRAIL-resistant cancer cell lines of three tumor types. A panel of breast cancer (2LMP, MDA-MB-436, SK-BR-3, MDA-MB-453), prostate cancer (PC-3 and DU-145) and colon cancer (SW620 and SW480) cell lines were treated with TRAIL (T) alone, SM-164 (SM) (100 nM) alone and their combination for 4 days. Cell viability inhibition was determined using a WST-8 assay.
SM-164 enhances TRAIL-induced apoptosis in cancer cells through amplification of the caspase-8-mediated extrinsic apoptosis pathway
To gain insights into the underlying mechanism of action for the strong synergy between TRAIL and SM-164, we selected 2LMP, a TRAIL-sensitive breast cancer cell line and MDA-MB-453, a TRAIL-resistant breast cancer cell line for further investigations.
SM-164 at 10 nM effectively induces rapid degradation of both cIAP1 and cIAP2 in 2LMP and MDA-MB-453 cancer cell lines (Fig. 2A and SI Fig. S5A). While either TRAIL or SM-164 (10 and 100 nM) has minimal effect in induction of apoptosis in both cell lines, the combination is very effective (Fig. 2B and SI Fig. S5B). Although either TRAIL or SM-164 at indicated concentrations has little or no effect on caspase-8, caspase-3 and PARP cleavage, their combination effectively induces robust processing of these proteins at 8- and 16-h time-points in both cell lines (Fig. 2C and SI Fig. S5C). Interestingly, the combination has minimal effect on caspase-9 processing in both cell lines. These data show that SM-164 can greatly enhance apoptosis induction by TRAIL in both TRAIL-sensitive and -resistant cancer cell lines.
Figure 2.
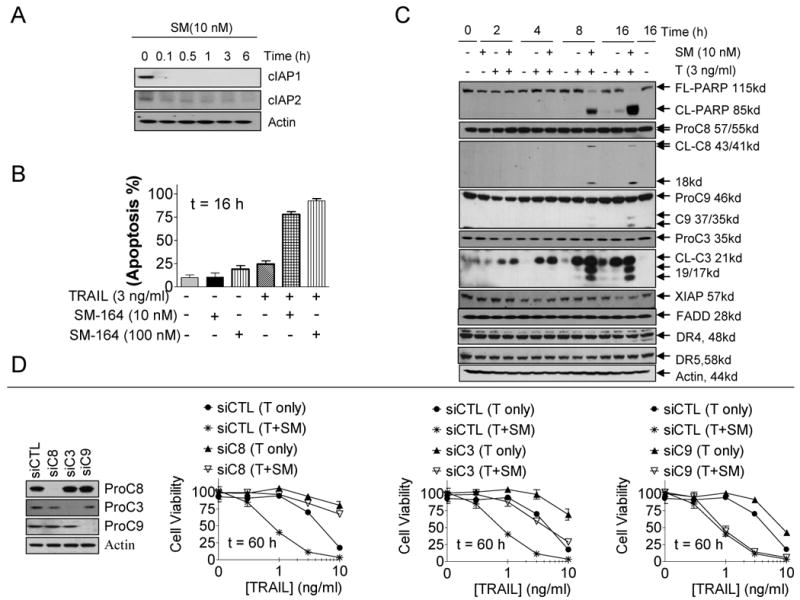
SM-164 enhances TRAIL-induced apoptosis through amplification of caspase-8 mediated apoptotic signals. (A) 2LMP cells were treated with SM-164 at 10 nM at indicated time-points, degradation of cIAP1 and cIAP2 was assessed by Western blotting. (B) 2LMP cells were treated as indicated, and apoptosis was determined using Annexin-V/PI double staining and flow cytometry assay. (C) 2LMP cells were treated as indicated, PARP cleavage and activation of caspases were determined by Western blotting. (D) 2LMP cells were transfected with control siRNA, siRNA against caspase-8, -9 or -3. Cells were treated as indicated and cell viability inhibitory activity was determined by a WST-8 assay.
We next examined if the anticancer activity of TRAIL and of the combination depends upon caspase activity. We found that knock-down of either caspase-8 or caspase-3 effectively attenuates the cell viability inhibition by TRAIL alone in the 2LMP cell line or by the combination in both cell lines (Fig. 2D and SI Fig. S5D). Knock-down of caspase-9 by siRNA has minimal effect on the cell viability inhibition by TRAIL alone or by the combination in both cancer cell lines; although the same siRNA effectively attenuates cell viability inhibition by ABT-737, a potent Bcl-2/Bcl-xL inhibitor, whose activity is known to be dependent upon caspase-9 (Fig. 2D and SI Fig. S5D-E) (44).
To investigate if the combination synergy depends upon the TRAIL receptors DR4 and/or DR5, DR4 or DR5 was knocked down individually or concurrently by siRNA. While knock-down of either DR4 or DR5 can attenuate the activity of TRAIL or the combination in the 2LMP cell line, knocking-down both receptors is more effective (SI Fig. S6A). In the MDA-MB-453 cell line resistant to TRAIL as a single agent, knock-down of DR4 clearly reduces the activity of the combination, but knock-down of DR5 alone or of both receptors blocks the combination activity (SI Fig. S6B). No increase of DR4 or DR5 protein was observed with TRAIL, SM-164 or their combination in both 2LMP and MDA-MB-453 cell lines (Fig. 2C and SI Fig. S5C). These data indicate that the activity of SM-164 in combination with TRAIL depends upon the caspase-8-mediated extrinsic pathway.
SM-164 enhances TRAIL activity by concurrently targeting XIAP and cIAP1
Previous studies have focused on XIAP as the primary molecular target for Smac mimetics in combination with TRAIL (36-39). However, since Smac mimetics also effectively induce degradation of cIAP1/2 (25, 26, 28, 29), all these IAP proteins may play a role in the strong synergy of the combination of Smac mimetics and TRAIL. We have thus examined the roles of XIAP and cIAP1/2 using 2LMP and MDA-MB-436, two TRAIL-sensitive cell lines and MDA-MB-453, a TRAIL-resistant cell line.
2LMP, MDA-MB-436 and MDA-MB-453 cells were treated with smartpool siRNA against XIAP, cIAP1 and cIAP2, individually or in combinations. Western blotting showed that each of these siRNA or their combinations knocked down their intended target genes efficiently (Fig. 3A and SI Fig. S7) and individual knockdowns, or any combination of knockdowns, had little or no effect on cell viability. Knockdown of cIAP1 in all these three cell lines induces robust upregulation of cIAP2 protein, presumably due to the loss of cIAP1-mediated degradation of cIAP2 (45). Knockdown of cIAP1 or XIAP alone has a modest effect in sensitizing TRAIL in these cell lines, whereas knockdown of cIAP2 alone has little or no effect compared to the control siRNA (Fig. 3A, SI Fig. 7A-B).
Figure 3.
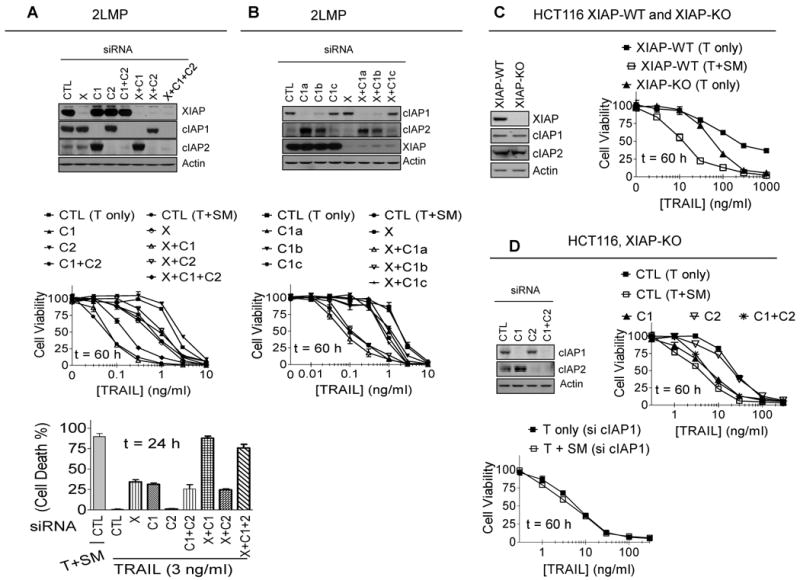
SM-164 enhances TRAIL-induced anticancer activity by targeting cIAP1 and XIAP, but not by targeting cIAP2. (A) 2LMP cells were transfected with control (CTL) siRNA, or siRNA against XIAP (X), cIAP1 (C1), or cIAP2 (C2) individually or in combination for 48 h, siRNA transfection efficiency was examined by western blotting (upper panel); cells were treated as indicated, cell death induction was determined with trypan blue exclusion assay (middle panel); cell viability inhibition was determined by a WST-8 assay (lower panel). (B) 2LMP cells were transfected with control (CTL) siRNA, and siRNA against XIAP (X), or cIAP1 (C1), individually or in combination for 48 h, siRNA transfection efficiency was examined by western blotting (upper panel); cells were treated for another 60 h, cell viability inhibition was determined by a WST-8 assay (lower panel). (C) HCT116 XIAP wild type (XIAP-WT) and knockout (XIAP-KO) human colon cancer cells were treated for 60 h, cell viability inhibitory activity was determined by a WST-8 assay. (D) HCT116 XIAP knockout (XIAP-KO) cells were transfected with control siRNA or siRNA against cIAP1 (C1) or cIAP2 (C2) individually or in combination for 48 h, siRNA transfection efficiency was examined by western blotting (upper left panel); cells were treated for a further 60 h, cell viability inhibition was determined by a WST-8 assay (upper right panel). HCT116 XIAP knockout (XIAP-KO) cells were transfected with cIAP1 siRNA, cells were treated for a further 60 h, cell viability inhibitory activity was determined by a WST-8 assay (lower panel).
In contrast to the modest effect of knock-down of cIAP1 or XIAP alone in TRAIL sensitization, knockdown of both XIAP and cIAP1 greatly sensitizes TRAIL in all these three cell lines, closely mimicking the strong synergy achieved by SM-164. However, knockdown of cIAP2, in addition to knockdown of XIAP and/or cIAP1, fails to further sensitize TRAIL as compared to the respective knockdown in all of the three cell lines (Fig. 3A, SI Fig. S7A-B).
To further test the role of cIAP1 and cIAP2, we have employed siRNA oligos with different knockdown efficacy. While the combination of efficient knock-down of XIAP and cIAP1 by two different siRNA oligos results in robust sensitization of TRAIL in 2LMP cells, closely mimicking the effect achieved by SM-164, inefficient knockdown of cIAP1 by a third oligo does not further enhance TRAIL-activity as compared with XIAP knock-down alone (Fig. 3B).
Three siRNA oligos that target different segments of the cIAP2 mRNA were also employed to further test the role of cIAP2 in TRAIL sensitization. All three oligos efficiently down-regulate cIAP2 in the 2LMP (SI Fig. S8) cell line but none of them significantly enhances cell-viability inhibition by TRAIL as compared to the non-target control siRNA. Furthermore, as compared to the XIAP siRNA alone, none of these three cIAP2 siRNA oligos in combination with XIAP siRNA further enhances cell viability inhibition by TRAIL. Taken together, our data show that cIAP2 has a minimal role in blocking TRAIL activity in these cancer cell lines.
To complement these siRNA experiments, we employed HCT116 XIAP+/+ and XIAP-/- isogenic cell lines (46). Consistent with the original study (46), knockout of XIAP makes the HCT116 cells more sensitive than its wild-type counterpart to TRAIL-induced cell viability inhibition, but SM-164 is much more effective than the XIAP gene knockout in sensitizing TRAIL in HCT116 XIAP-/- cells (Fig. 3C). Furthermore, in the XIAP knock-out HCT116 cells, SM-164 still greatly enhances the activity of TRAIL based upon cell viability inhibition assay and the activation of caspase-3 and -8 and cleavage of PARP (Fig. 3D and SI Fig. S9).
To examine if degradation of cIAP1 or cIAP2 by SM-164 accounts for the further sensitization of TRAIL in the HCT116 XIAP knockout cells, cIAP1 or cIAP2 were knocked down individually or concurrently. While knockdown of cIAP1 in the HCT116 XIAP-/- cells further sensitizes the cells to TRAIL and closely mimics the effect achieved by SM-164, knockdown of cIAP2 has a minimal effect (Fig. 3D). Interestingly, although knockdown of cIAP1 again greatly increases the levels of cIAP2 protein, simultaneous knockdown of cIAP1 and cIAP2 in the HCT116 XIAP-/- cells does not further sensitize the cells to TRAIL as compared to knock-down of cIAP1 alone (Fig. 3D). In addition, when cIAP1 is knocked down in the HCT116 XIAP-/- cells, addition of SM-164 fails to further sensitize the cells to TRAIL (Fig. 3D).
Collectively, our data provide strong evidence that XIAP and cIAP1 are two non-redundant blockades of the activity of TRAIL in both TRAIL-sensitive and TRAIL-resistant cancer cell lines, while cIAP2 plays a minimal role. Furthermore, XIAP and cIAP1 work in concert in inhibition of the activity of TRAIL and SM-164 enhances the activity of TRAIL by concurrently targeting cIAP1 and XIAP.
Ablation of cIAP1 markedly enhances the TRAIL-DISC formation
Previous studies have established that cIAP1 plays a critical role in TNFα-mediated apoptosis induced by Smac mimetics (25-28). In our hands, TNFα as a single agent is ineffective in induction of apoptosis in cancer cell lines such as 2LMP and MDA-MB-436 (data not shown), that are sensitive to TRAIL as a single agent, suggesting certain key differences between TRAIL-mediated and TNFα-mediated apoptosis pathways. We have therefore investigated the role of cIAP1 in regulation of apoptosis induction by TRAIL alone or in combination with SM-164 in both TRAIL-sensitive and TRAIL-resistant cancer cell lines.
To investigate early events in TRAIL-mediated apoptosis signaling, we treated 2LMP cancer cells for 5, 10 and 30 min with a Flag-tagged recombinant TRAIL protein, with or without pretreatment with SM-164. TRAIL-receptor death-inducing signaling complex (TRAIL-DISC) was then immunoprecipitated with anti-Flag antibody from the lysates of the treated cells, and the key components in the complex were analyzed by Western blotting (Fig. 4 and SI Fig. S10). Our data showed that TRAIL treatment not only time-dependently induces FADD and procaspase-8 to form DISC, but it also rapidly recruits TRAF2, cIAP1 and RIP1 to the complex (Fig. 4A and SI Fig. S10). While similar levels of DR4 and DR5 are found in the TRAIL-DISC complex with or without treatment of SM-164, the recruitments of procaspase-8 to the TRAIL-DISC are greatly enhanced at very early time-points with SM-164 (Fig. 4 and SI Fig. S10).
Figure 4.
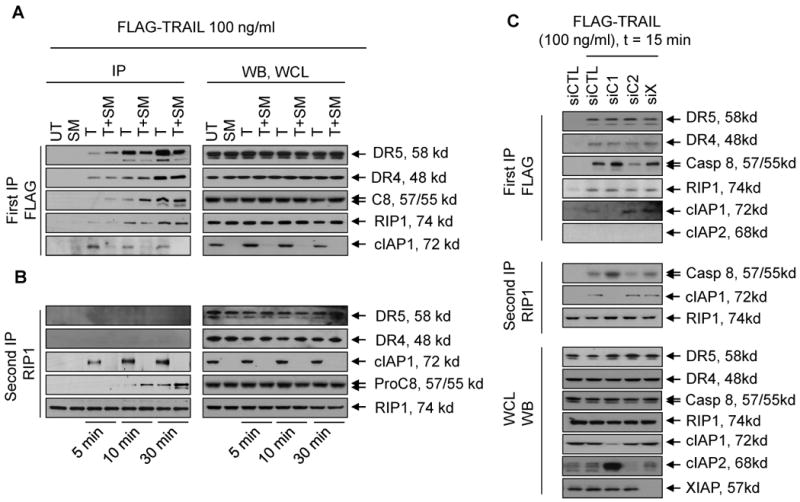
Ablation of cIAP1 markedly enhances TRAIL-DISC formation, and facilitates the intracellular interaction of RIP1 and caspase-8. (A) 2LMP cells were treated with a mixture of Flag-tagged TRAIL and anti-Flag M2 IgG with or without pretreatment of 100 nM of SM-164 for 5, 10 and 30 min. Cells were lysed and the associated proteins in the lysates were pulled down with sepharose 4B beads, and subjected to Western blotting as indicated (left panel); Expression of DR4, DR5, procaspase-8, RIP1 and cIAP1 in the whole cell lysates was examined by Western blotting (right panel). (B). Secondary signaling complexes were immunoprecipitated with an RIP1 antibody from DISC-depleted lysates, and analyzed by western blotting assay. (C) 2LMP cells transfected with siRNA against IAPs were treated with a mixture of Flag-tagged TRAIL and anti-Flag M2 IgG for 15 min. Cells were lysed and the associated proteins in the lysates were pulled down with sepharose 4B beads, and subjected to western blotting as indicated (upper panel); Secondary signaling complexes were immunoprecipitated with an RIP1 antibody from DISC-depleted lysates, and analyzed by Western blotting (middle panel); expression of DR4, DR5, procaspase-8, RIP1 and cIAP1 in the cell lysates were examined by Western blotting (lower panel).
To test directly whether SM-164 promotes the TRAIL-DISC formation through induction of cIAP1 degradation, the TRAIL-receptor complex was analyzed in 2LMP cells transfected with siRNA against cIAP1, cIAP2 or XIAP. Knockdown of cIAP1 strongly enhances procaspase-8 recruitment following TRAIL treatment (Fig. 4B) as compared with non-targeting siRNA, whereas recruitment of procaspase-8 was essentially unchanged in cells transfected with XIAP siRNA and non-targeting siRNA. In contrast to cIAP1, knockdown of cIAP2 does not enhance, and may in fact attenuate, recruitment of procaspase-8 (Fig. 4B). These data indicate that cIAP1 degradation by SM-164 greatly promotes TRAIL-DISC formation.
Ablation of cIAP1 facilitates the interaction between RIP1 and caspase-8
Ablation of cIAP1 by Smac mimetics was shown to greatly enhance recruitment of RIP1 to the TNFα–receptor complex (25, 26), which plays a major role in activation of caspase-8 and apoptosis induction by Smac mimetics as a single agent. We therefore examined the recruitment of RIP1 to the TRAIL-receptor complex and the role of RIP1 in apoptosis induction by TRAIL alone and in combination with SM-164.
In contrast to the marked enhancement of RIP1 recruitment to TNFα–receptor complex by Smac mimetics (25, 26), ablation of cIAP1 by SM-164 or siRNA has little or no effect on the recruitment of RIP1 to TRAIL-receptor complex at all the time-points examined (Fig. 4A-B and SI Fig. S10).
We next investigated the interaction between RIP1 and caspase-8 in the cytoplasm. Upon TRAIL treatment, RIP1 forms a complex with caspase-8 within 10 min upon TRAIL treatment, which is markedly increased at the 30 min time-point (Fig. 4A lower panel) and TRAIL-stimulation also leads to an interaction of RIP1 with cIAP1 within 5 min. At each time point examined, degradation of cIAP1 by SM-164 markedly enhances the interaction between RIP1 and caspase-8 (Fig. 4A lower panel). To further investigate the role of XIAP, cIAP1, and cIAP2 on the interaction of RIP1 with caspase-8, RIP1-immunoprecipitation was performed using the TRAIL-receptor complex-depleted lysates from cells transfected with siRNA against each IAP (Fig. 4B). While knockdown of cIAP1 markedly enhances the interaction between RIP1 and caspase-8, knockdown of XIAP has a minimal effect and knockdown of cIAP2 appears even to inhibit the interaction (Fig. 4B). Western blotting showed the absence of DR4 and DR5 in the RIP1-immunoprecipitation, indicating that the interaction of RIP1 with cIAP1 and caspase-8 occurs primarily in the cytoplasm (Fig. 4A lower panel). Collectively, these data indicate that cIAP1 markedly inhibits the interaction of RIP1 and caspase-8 upon TRAIL-stimulation and that degradation of cIAP1 greatly enhances this interaction in the cytoplasm and promotes the activation of caspase-8.
RIP1 plays an essential role in TRAIL sensitization by SM-164 but not in TRAIL as a single agent
RIP1 plays a key role in TNFα-mediated apoptosis induction by Smac mimetics (29, 47). However, critical differences exist between TNFα- and TRAIL-mediated apoptosis induction (48). We therefore examined the role of RIP1 in the activity of TRAIL alone or its combination with SM-164 in TRAIL-sensitive 2LMP, MDA-MB-436 and MDA-MB-468 and TRAIL-resistant MDA-MB-453 cancer cell lines (Fig. 5 and SI Fig. S11-13).
Figure 5.
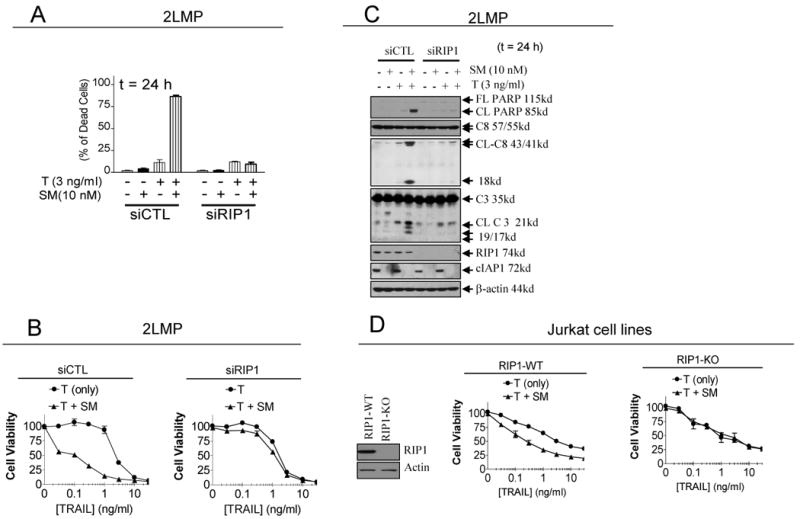
RIP1 plays an essential role in TRAIL-sensitization by SM-164 but not in the single-agent activity of TRAIL. (A)-(C). 2LMP cells were transfected with control siRNA, and siRNA against RIP1 for 48 h. (A). Transfected cells were treated for a further 24 h and cell death induction was determined with trypan blue exclusion assay; (B) Transfected cells were treated for a further 24 h and cell lysates were subjected to western blotting analysis as indicated; (C). Transfected cells were treated for a further 60 h and cell viability was determined by a WST-8 assay. (D) Jurkat RIP1 wild type (RIP1-WT) and RIP1 knockout (RIP1-KO) cells were treated with SM-164 alone, TRAIL alone or their combination for 48 h and cell viability was determined by a WST-8 assay.
2LMP cells transfected with RIP1 siRNA were treated with TRAIL, SM-164 alone, or their combination, followed by determination of cell death induction and cell viability inhibition and examination of PARP cleavage and processing of caspase-8, -3 and -9 by Western blot (Fig. 5A-C). Efficient RIP1 knockdown has only a modest effect on cell death induction, cell viability inhibition, activation of caspases and cleavage of PARP by TRAIL as a single agent in 2LMP cells. In contrast, RIP1 knockdown effectively blocks the robust sensitization to TRAIL by SM-164 in 2LMP cells (Fig. 5A-C). Similarly, efficient RIP1 knockdown in MDA-MB-436 and MDA-MB-468 cell lines has little or no effect in cell viability inhibitory activity by TRAIL but effectively blocks the sensitization by SM-164 in both cell lines (SI Fig. S11-12). In the TRAIL-resistant MDA-MB-453 cancer cell line, efficient RIP1 knockdown has no effect on the activity of TRAIL and SM-164 as a single agent, but it blocks the robust cleavage of PARP and processing of caspase-8 and -3 and cell viability inhibition by the combination (SI Fig. S13A-B).
To complement the siRNA experiments, we next employed the Jurkat cell line and its RIP1 knock-out counterpart to further investigate the role of RIP1 (49). While SM-164 clearly sensitizes TRAIL in inhibition of cell viability in the Jurkat parental cell line, SM-164 is completely ineffective in enhancing the activity of TRAIL in the Jurkat RIP1 knock-out cell line (Fig. 5D).
These data show that while RIP1 plays a minimal role in the activity of TRAIL as a single agent, it is required for the synergistic interaction between SM-164 and TRAIL.
SM-164 enhances apoptosis induction by TRAIL in xenograft tumor tissues and their combination achieves tumor regression
To further evaluate the therapeutic potential of SM-164 in combination with TRAIL, we tested the in vivo activity for TRAIL and SM-164 as single agents and their combination using the 2LMP xenograft model.
A single dose of SM-164 at 5 mg/kg i.v. is highly effective in induction of cIAP1 degradation (Fig. 6A), but fails to induce caspase-3 activation, PARP cleavage or apoptosis over a 6-24 h time period in tumor tissues (Fig. 6A-B). Although TRAIL is very effective as a single agent in cell viability inhibition in vitro in the 2LMP cell line, a single dose of TRAIL at 10 mg/kg induces only modest caspase-3 activation, PARP cleavage and minimal apoptosis in tumor tissues (Fig. 6A-B). In contrast, their combination induces robust caspase-3 activation, PARP cleavage and strong apoptosis in tumor tissues (Fig. 6A-B); 50% of tumor cells are in fact TUNEL positive in tumor tissues at the 6 h time-point (Fig. 6B, right panel, SI Fig. S14). H&E staining further showed that the combination causes extensive damage to tumor tissues (Fig. 6C). In comparison, SM-164, TRAIL alone and their combination have no effect on all normal mouse tissues examined, including highly proliferative tissues such as spleen, small intestine and bone marrow (SI Fig. S15).
Figure 6.
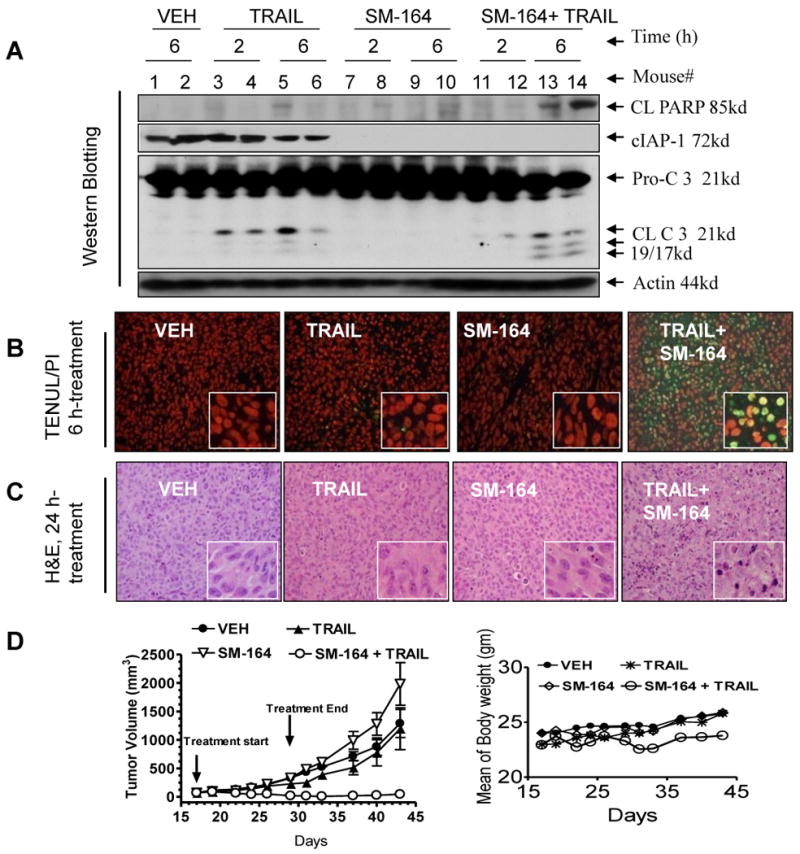
SM-164 induces cIAP1 degradation in tumor tissues and dramatically enhances the in vivo antitumor activity of TRAIL and the combination of SM-164 and TRAIL achieves tumor regression without toxicity to animals (A) Nude mice, bearing established 2LMP xenograft tumors (200-300 mm3), were treated with a single dose of SM-164 alone, TRAIL alone, or their combination, or vehicle (VEH). Tumor tissues were harvested at the time points indicated. Activation of caspases and PARP cleavage, and cIAP1 degradation in tumor tissues were analyzed by Western blot. (B) Apoptosis in tumor tissues was examined by TUNEL staining (left panels) and scored under microscope (right panel). At least 1,000 cells were counted. (C) Tumor tissues were harvested at 24 h time point and examined by H&E staining. Xenograft tumor tissues treated with SM-164 were characterized with cell shrinkage, nuclear pyknosis and chromatin condensation. (D) Antitumor activity of SM-164 alone, TRAIL alone and their combination in the 2LMP xenograft model. Nude mice (8-12 per group), bearing established 2LMP xenograft tumors, were treated with SM-164 at 5 mg/kg, i.v. alone, TRAIL at 10 mg/kg, i.p. alone, their combination, or VEH, daily, 5 days a week for 2 weeks. The mean of tumor volume ± SEM (left panel) and mean animal body ± SEM (right panel) were shown for each group, respectively.
We next examined the combination efficacy, as well as potential toxicity with systemic administration of these two agents against established 2LMP tumors (Fig. 6D). SM-164 (5 mg/kg, i.v. daily, five days/week and TRAIL (10 mg/kg i.p. daily, five days/week) were administered alone or in combination for two weeks. Although neither SM-164 nor TRAIL as single agents results in any signs of toxicity in mice, they fail to achieve any significant antitumor activity. In contrast, their combination induces tumor regression. At the end of the treatment, the combination reduces the mean tumor volume by 80% and the tumors in the combination treatment group continue to shrink after treatment ended and are undetectable on day 33 in 6 out of 8 cases. This strong antitumor activity by SM-164 is persistent and three of the 8 tumors remain completely regressed three months after the treatment concluded. In comparison, the mean tumor volume in the control and the treatment groups with TRAIL alone or SM-164 alone are over 1000 mm3 on day 43 (Fig. 6D, left panel).
The combination treatment causes no gross abnormalities or other signs of toxicity. Mice treated with the combination experience a slight but statistically insignificant body weight loss at the end of 2 weeks of treatment and all regain their weight by day 33 (Fig. 6D, right panel).
Our in vivo data thus demonstrate that while neither SM-164 nor TRAIL shows appreciable antitumor activity as a single agent, their combination is highly effective in induction of apoptosis and achieves tumor regression. The combination is also selectively toxic to tumor tissues over normal mouse tissues. Taken together, our in vivo data further suggest that the combination of SM-164 and TRAIL may have considerable therapeutic potential for the treatment of human cancers.
Discussion
Both TRAIL and Smac mimetics are being developed in the clinic as new anticancer drugs. While TRAIL is well tolerated in patients, its single-agent efficacy is very limited (31-33). Similarly, extensive preclinical and early clinical data also suggest that Smac mimetics may have very limited single-agent activity (24, 27, 30). Therefore, rational combination strategies are clearly needed for the successful development of both TRAIL and small-molecule Smac mimetics as new anticancer drugs.
Earlier observations using Smac-based peptides and subsequent studies using potent, cell-permeable small-molecule Smac mimetics have indicated that there is a very strong synergy between Smac-based compounds and TRAIL against human cancer cell lines originating from different tissues (34-40). The data obtained from our present study using SM-164, a highly potent Smac mimetic, with TRAIL also clearly shows that SM-164 is capable of dramatically enhancing the anticancer activity of TRAIL in vitro in >70% of human breast, prostate and colon cancer cell lines and in both TRAIL-sensitive and TRAIL-resistant cancer cell lines. For the first time, we have demonstrated that although both TRAIL and SM-164 alone have no single-agent activity in a xenograft model of human breast cancer, their combination can achieve rapid tumor regression, while showing no toxicity to animals. Our in vitro and in vivo data, together with those from previous studies (34-40), strongly suggest that combination of Smac mimetics with TRAIL warrants clinical investigation as an attractive new cancer therapeutic strategy.
The design of Smac mimetics was based upon the interaction between XIAP and Smac and previous investigations on the combination of Smac mimetics with TRAIL have focused on XIAP as the primary molecular target for Smac mimetics (34-39). However, Smac mimetics also bind to cIAP1 and cIAP2 with very high affinities and induce rapid degradation of these cIAP proteins. Degradation of cIAP1/2 is an early and key event in TNFα-mediated apoptosis induction by Smac mimetics as single agents. However, the role of cIAP1/2 proteins in the combination of Smac mimetics with TRAIL has not been defined. Using siRNA technology and XIAP knock-out cells, our data show that XIAP and cIAP1 are two non-redundant inhibitors of TRAIL-induced apoptosis. While ablation of either XIAP or cIAP1 can modestly sensitize TRAIL in both TRAIL-sensitive and TRAIL-resistant cancer cell lines, targeting both XIAP and cIAP1 can achieve a much stronger effect. Furthermore, while knock-down of cIAP1 by siRNA can robustly upregulate cIAP2, knock-down of cIAP2 does not further sensitize cancer cells to TRAIL. Moreover, ablation of cIAP1 by siRNA or by SM-164 greatly enhances the recruitment of caspase-8 and FADD to the TRAIL death-receptor complex, promoting the interaction of RIP1 with caspase-8 in the cytoplasm and activation of caspase-8, but ablation of cIAP2 fails to lead to any of these outcomes, indicating a differential role for cIAP1 and cIAP2 in TRAIL sensitization. The strong sensitization of TRAIL achieved by SM-164 is closely mimicked by concurrent ablation of both cIAP1 and XIAP. In cancer cells, when both XIAP and cIAP1 are removed, SM-164 is unable to further sensitize the cells to TRAIL. Collectively, these data provide strong evidence that XIAP and cIAP1 are the primary cellular targets for SM-164 in its synergistic interaction with TRAIL. Since both XIAP and cIAP1 proteins are over-expressed in tumor cells, the ability of SM-164 to concurrently and potently target XIAP and cIAP1 may prove to be a unique advantage in achieving a strong synergy with TRAIL.
Our data supported that similar to TNFα, TRAIL induces apoptosis through both Rip1-dependent and –independent pathways (47). As a single agent, TRAIL induces apoptosis in a RIP1-independent manner. The synergy between TRAIL and Smac mimetics, however, relies on the interaction of procaspase-8 with RIP1 and is RIP1-dependent. Knockdown of RIP1 abrogates or dramatically attenuates cell viability inhibition by the combination (Fig.4B-C, Fig.5A-D). This RAP1-depedndent event takes place once cIAP1 is removed by a Smac mimetic or siRNA (Fig.4A, C, Fig.5A-D). The interaction of procaspase-8 and RIP1 is detected in the cytoplasm (Fig.4B-C), demonstrating that RIP1-dependent caspase-8 activation primarily takes place in cytoplasm. However, removal of cIAP1 also markedly increases the procaspase-8 recruitment to TRAIL-receptor complex (Fig.4A).
Although SM-164 potently antagonizes XIAP and induces efficient down-regulation of cIAP1 in all the cancer cell lines we have evaluated in the present study, SM-164 enhances the anticancer activity of TRAIL in the majority but not all of cancer cell lines examined. These data show that while XIAP and cIAP1effectively inhibit the activity of TRAIL, they are not the only proteins that mediate the resistance of cancer cells to TRAIL. For example, cFLIP (40, 50) and Mcl-1 (51) can also effectively attenuate the activity of TRAIL and may play a role in mediating the resistance of cancer cells to the combination of Smac mimetics and TRAIL.
In summary, our present study furthers our understanding on the underlying molecular mechanism of the strong synergy between Smac mimetics and TRAIL and provides strong support that the combination of Smac mimetics and TRAIL should be evaluated in the clinic as a new cancer therapeutic strategy for the treatment of a variety of human cancers.
Supplementary Material
1
Acknowledgments
Grant Support: the Breast Cancer Research Foundation (S. W.), the Susan G. Komen Foundation (H. S.), National Cancer Institute Grants R01CA109025 (S. W.) and R01CA127551 (S. W.), and University of Michigan Cancer Center Core grant from the National Cancer Institute P30CA046592.
Footnotes
Conflict of Interest: S.W. serves as a consultant for Ascenta Therapeutics and owns stocks and stock options in Ascenta Therapeutics, which is developing a Smac mimetic for cancer treatment.
References
- 1.Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21:485–95. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 3.Reed JC. Apoptosis-based therapies. Nature Reviews Drug Discovery. 2002;1:111–21. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 4.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108(2):153–64. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 5.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nature Reviews Molecular Cell Biology. 2002;3:401–10. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 6.Deveraux QL, Reed JC. IAP family proteins—suppressors of apoptosis. Genes & Development. 1999;13:239–52. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 7.Tamm I, Kornblau SM, Segall H, et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clinical Cancer Research. 2000;6:1796–803. [PubMed] [Google Scholar]
- 8.Yang L, Cao Z, Yan H, Wood WC. Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: implication for cancer specific therapy. Cancer Research. 2003;63:6815–24. [PubMed] [Google Scholar]
- 9.Parton M, Krajewski S, Smith I, et al. Coordinate expression of apoptosis-associated proteins in human breast cancer before and during chemotherapy. Clinical Cancer Research. 2002;8:2100–8. [PubMed] [Google Scholar]
- 10.Jaffer S, Orta L, Sunkara S, Sabo E, Burstein DE. Immunohistochemical detection of antiapoptotic protein X-linked inhibitor of apoptosis in mammary carcinoma. Human Pathology. 2007;38:864–70. doi: 10.1016/j.humpath.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 11.Krajewska M, Krajewski S, Banares S, et al. Elevated expression of inhibitor of apoptosis proteins in prostate cancer. Clinical Cancer Research. 2003;9:4914–25. [PubMed] [Google Scholar]
- 12.Fulda S. Inhibitor of apoptosis proteins as targets for anticancer therapy. Expert Review of Anticancer Therapy. 2007;7:1255–64. doi: 10.1586/14737140.7.9.1255. [DOI] [PubMed] [Google Scholar]
- 13.Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12:1543–68. doi: 10.1007/s10495-007-0087-3. [DOI] [PubMed] [Google Scholar]
- 14.Vucic D, Fairbrother WJ. The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clinical Cancer Research. 2007;13(20):5995–6000. doi: 10.1158/1078-0432.CCR-07-0729. [DOI] [PubMed] [Google Scholar]
- 15.LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene. 2008;27(48):6252–75. doi: 10.1038/onc.2008.302. [DOI] [PubMed] [Google Scholar]
- 16.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000 Jul 7;102(1):33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 17.Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000 Jul 7;102(1):43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 18.Shiozaki EN, Shi Y. Caspases, IAPs and Smac/DIABLO: mechanisms from structural biology. Trends in Biochemical Sciences. 2004;29:486–94. doi: 10.1016/j.tibs.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Sun H, Nikolovska-Coleska Z, Yang CY, et al. Design of small-molecule peptidic and nonpeptidic Smac mimetics. Accounts of Chemical Research. 2008;41(10):1264–77. doi: 10.1021/ar8000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mannhold R, Fulda S, Carosati E. IAP antagonists: promising candidates for cancer therapy. Drug Discovery Today. 2010;15(5-6):210–9. doi: 10.1016/j.drudis.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 21.AEG40826/HGS1029. http://www.aegera.com/aeg40826.php. 2008.
- 22.AT-406. http://www.ascenta.com/development/index.php#at406. Ascenta and University of Michigan 2009.
- 23.Genetech. IAP antagonist GDC-0152 in Phase Ia. 2009 http://wwwclinicaltrialsgov/ct2/show/NCT00977067?term=IAP+antagonist&rank=1.
- 24.Infante JR, Dees EC, Burris I, HA, et al. A phase I study of LCL161, an oral IAP inhibitor, in patients with advanced cancer. American Association for Cancer Research 101st Annual Meeting 2010; 2010. Abstract # 2775. [Google Scholar]
- 25.Varfolomeev E, Blankenship JW, Wayson SM, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007 Nov 16;131(4):669–81. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 26.Vince JE, Wong WW, Khan N, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007 Nov 16;131(4):682–93. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 27.Petersen SL, Wang L, Yalcin-Chin A, et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. 2007 Nov;12(5):445–56. doi: 10.1016/j.ccr.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J, Bai L, Sun H, et al. SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Research. 2008 Nov 15;68(22):9384–93. doi: 10.1158/0008-5472.CAN-08-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertrand MJ, Milutinovic S, Dickson KM, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Molecular Cell. 2008 Jun 20;30(6):689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 30.Oost TK, Sun C, Armstrong RC, et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. Journal of Medicinal Chemistry. 2004;47:4417–26. doi: 10.1021/jm040037k. [DOI] [PubMed] [Google Scholar]
- 31.Bellail AC, Qi L, Mulligan P, Chhabra V, Hao C. TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials. 2009 Jan;4(1):34–41. doi: 10.2174/157488709787047530. [DOI] [PubMed] [Google Scholar]
- 32.Carlo-Stella C, Lavazza C, Locatelli A, Vigano L, Gianni AM, Gianni L. Targeting TRAIL agonistic receptors for cancer therapy. Clinical Cancer Research. 2007 Apr 15;13(8):2313–7. doi: 10.1158/1078-0432.CCR-06-2774. [DOI] [PubMed] [Google Scholar]
- 33.Hall MA, Cleveland JL. Clearing the TRAIL for cancer therapy. Cancer Cell. 2007;12:4–6. doi: 10.1016/j.ccr.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 34.Fulda S, Wick W, Weller M, Debatin KM. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med. 2002 Aug;8(8):808–15. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004 Sep 3;305(5689):1471–4. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 36.Bockbrader KM, Tan M, Sun Y. A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL- and etoposide-induced apoptosis in breast cancer cells. Oncogene. 2005;24:7381–8. doi: 10.1038/sj.onc.1208888. [DOI] [PubMed] [Google Scholar]
- 37.Fakler M, Loeder S, Vogler M, et al. Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance. Blood. 2009 Feb 19;113(8):1710–22. doi: 10.1182/blood-2007-09-114314. [DOI] [PubMed] [Google Scholar]
- 38.Vogler M, Walczak H, Stadel D, et al. Targeting XIAP bypasses Bcl-2-mediated resistance to TRAIL and cooperates with TRAIL to suppress pancreatic cancer growth in vitro and in vivo. Cancer Research. 2008 Oct 1;68(19):7956–65. doi: 10.1158/0008-5472.CAN-08-1296. [DOI] [PubMed] [Google Scholar]
- 39.Vogler M, Walczak H, Stadel D, et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Research. 2009 Mar 15;69(6):2425–34. doi: 10.1158/0008-5472.CAN-08-2436. [DOI] [PubMed] [Google Scholar]
- 40.Cheung HH, Mahoney DJ, LaCasse EC, Korneluk RG. Down-regulation of c-FLIP Enhances Death of Cancer Cells by Smac Mimetic Compound. Cancer Research. 2009;69(19):7730–8. doi: 10.1158/0008-5472.CAN-09-1794. [DOI] [PubMed] [Google Scholar]
- 41.Sun H, Nikolovska-Coleska Z, Lu J, et al. Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent Smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. Journal of the American Chemical Society. 2007 Dec 12;129(49):15279–94. doi: 10.1021/ja074725f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006 Sep;58(3):621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 43.Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res. 2004 Dec 1;10(23):7994–8004. doi: 10.1158/1078-0432.CCR-04-1087. [DOI] [PubMed] [Google Scholar]
- 44.Okumura K, Huang S, Sinicrope FA. Induction of Noxa sensitizes human colorectal cancer cells expressing Mcl-1 to the small-molecule Bcl-2/Bcl-xL inhibitor, ABT-737. Clin Cancer Res. 2008 Dec 15;14(24):8132–42. doi: 10.1158/1078-0432.CCR-08-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheung HH, Plenchette S, Kern CJ, Mahoney DJ, Korneluk RG. The RING domain of cIAP1 mediates the degradation of RING-bearing inhibitor of apoptosis proteins by distinct pathways. Mol Biol Cell. 2008;19(7):2729–40. doi: 10.1091/mbc.E08-01-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cummins JM, Kohli M, Rago C, Kinzler KW, Vogelstein B, Bunz F. X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Research. 2004 May 1;64(9):3006–8. doi: 10.1158/0008-5472.can-04-0046. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008 May 16;133(4):693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 48.Jin Z, El-Deiry WS. Distinct signaling pathways in TRAIL- versus tumor necrosis factor-induced apoptosis. Mol Cell Biol. 2006;26(21):8136–48. doi: 10.1128/MCB.00257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O'Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT. Ubiquitination of RIP1Regulates an NF-kB-Independent Cell-Death Switch in TNF Signaling. Current Biology. 2007;17:418–24. doi: 10.1016/j.cub.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murtaza I, Saleem M, Adhami VM, Hafeez BB, Mukhtar H. Suppression of cFLIP by lupeol, a dietary triterpene, is sufficient to overcome resistance to TRAIL-mediated apoptosis in chemoresistant human pancreatic cancer cells. Cancer Research. 2009 Feb 1;69(3):1156–65. doi: 10.1158/0008-5472.CAN-08-2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim SH, Ricci MS, El-Deiry WS. Mcl-1: a gateway to TRAIL sensitization. Cancer Research. 2008;68(7):2062–4. doi: 10.1158/0008-5472.CAN-07-6278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1