Defining Clinical Leptin Resistance - Challenges and Opportunities (original) (raw)
. Author manuscript; available in PMC: 2012 Feb 17.
Abstract
The lack of a universally accepted definition of the term “leptin resistance” led the National Institutes of Health to hold a workshop, “Toward a Clinical Definition of Leptin Resistance”. Leptin resistance is generally defined as the failure of endogenous or exogenous leptin to promote anticipated salutary metabolic outcomes in states of over-nutrition or obesity, although the hormone's inability to promote desired responses in specific situations results from multiple molecular, neural, behavioral, and environmental mechanisms. Thus, the term “leptin resistance” does not imply a single specific mechanism, but rather connotes distinct meanings across investigators and in different contexts. Clinically, exploiting behavioral and metabolic sensitivity to the hormone, rather than elaborating a universal, quantifiable definition of “leptin resistance”, is the goal and specific predictors of sensitivity should be established. It is clear that the availability of relevant human data is limited, however, and a substantial amount of new information must be acquired and disseminated to accomplish these goals.
Keywords: Leptin, obesity, genetic models, humans, therapy
Overview
The phrase “_leptin resistance_” arose not long after the discovery of this adipose tissue-derived hormone in late 1994 (1, 2). Found in more than 4000 citations since (Figure 1), “leptin resistance” appears to connote diverse meanings in distinct contexts and to different investigators, including obesity in the face of hyperleptinemia to the failure of pharmacologic leptin to suppress feeding (3-5). The disparate and confusing usage of the term “leptin resistance” led the National Institute of Diabetes, Digestive and Kidney diseases at the National Institutes of Health to hold a workshop, “Toward a Clinical Definition of Leptin Resistance,” on February 25th, 2011. The workshop goal was to explore current usage of the phrase “_leptin resistance_” and to work toward a quantifiable, clinically useful definition. This article provides background about the field, and reviews the proceedings and main themes generated at the workshop.
Figure 1.
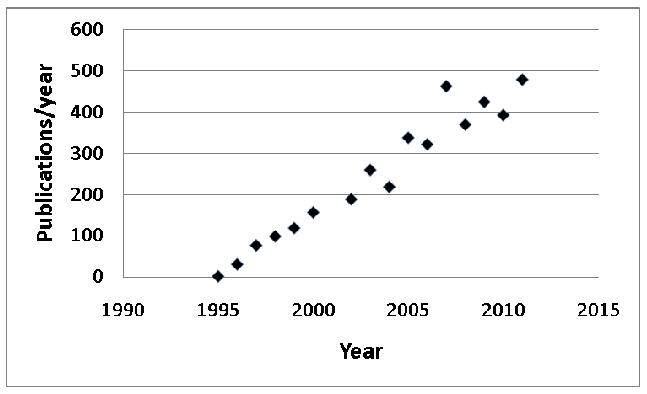
Number of publications per year citing “leptin resistance” since the hormone's discovery. Data from PubMed.
Leptin Biology
Leptin, a 16 kDa cytokine, is produced by adipose tissue in approximate proportion to adipose tissue mass (1, 6, 7). Thus, circulating leptin concentrations generally reflect the status of long-term adipose tissue energy stores (1, 6, 7). Leptin modulates a host of physiologic processes and behaviors in line with the need for these reserves (8, 9). For instance, adequate fat stores/leptin concentrations diminish the drive to feed, while enabling energy expenditure via a variety of neuroendocrine axes, autonomic outputs, and activities. Conversely, inadequate leptin (e.g., during caloric restriction or stable weight reduction) increases the desire to eat, decreases energy utilization, and promotes a variety of behavioral adaptations to diminished energy stores. Leptin also supports reproductive competence and immune function and contributes to the regulation of metabolic homeostasis (by modulating insulin secretion, hepatic glucose production, and lipid metabolism), as well as some aspects of bone biology (10-18). The biological effects of leptin action suggest its potential therapeutic utility in a variety of pathologic states, including obesity, diabetes, and their comorbid diseases. Indeed, leptin effectively treats the metabolic complications of lipodystrophy syndromes and restores menstruation in hypothalamic amenorrhea (19). The finding that most forms of obesity are associated with apparently diminished metabolic efficacy of leptin (“leptin resistance,” see below) has, however, complicated its potential use to treat obesity.
Mechanisms of Action
Leptin acts via its cell surface receptor (LEPR): While there exist multiple LEPR isoforms that result from alternative splicing of the Lepr mRNA, the LEPR-B form of the receptor mediates essentially all known physiologic effects of leptin (20-22). LEPR-B is a type 1 cytokine receptor that mediates intracellular signaling via an associated Janus family tyrosine kinase (Jak2) (23, 24). Major intracellular signaling pathways emanating from activated LEPR-B include the phosphorylation and activation of the signal transducer and activator of transcription (STAT) pathways mediated by STAT3 and STAT5, the PTPN11→ extracellular regulated kinase (ERK) pathway, and the feedback inhibition pathway mediated by SOCS3 (24, 25). Another inhibitory pathway involves PTP1B (26). Both SOCS3 and PTP1B decrease leptin sensitivity and response in vitro and in vivo (25-29) (Figure 2).
Figure 2.
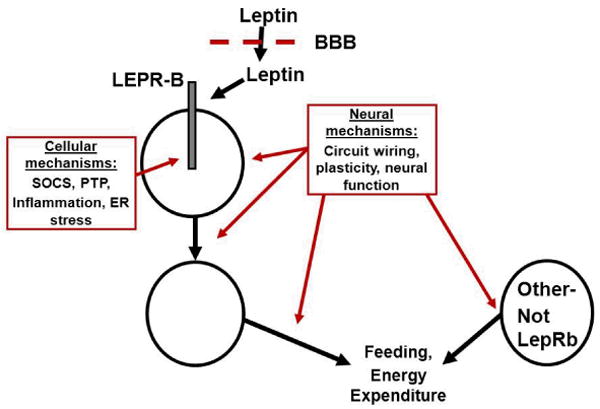
Potential sites/mechanisms interfering with the regulation of energy balance to produce obesity. Leptin is transported across the blood brain barrier (BBB) to access first order, LEPR-B expressing neurons, which connect with downstream neurons to eventually modulate food intake and energy expenditure. Other circuits, including those not modulated by leptin, also contribute to the control of feeding and energy expenditure. Interference (red) with any of these processes, including BBB transport of leptin, cellular processes in LEPR-B neurons (such as LEPR-B signaling), or alterations in the neural function, “wiring”, or plasticity in any of the leptin-regulated or leptin-independent circuits that contribute to energy balance are expected to promote obesity (and increase circulating leptin concentrations).
Most known leptin action is attributable to its effects in the brain, where LEPR-B is predominantly expressed (9, 30-32). Leptin action and LEPR-B expression have also been reported in a variety of tissues outside of the brain, including some cells of the immune system (33). There is little evidence to support a role for peripheral LEPR-B expression in the control of whole body energy balance or glucose homeostasis, however. CNS leptin has been shown to mediate most leptin actions, including control of neuroendocrine axes, the autonomic nervous system, satiety, and the limbic system, as well as numerous behaviors. A great deal of research in this area has focused on important neurons of the hypothalamic arcuate nucleus that express proopiomelanocortin or Agouti-related peptide, but direct leptin action on these neurons only accounts for a fraction of total leptin action, and LEPR-B is highly expressed in several other brain regions associated with the control of feeding-related behaviors and energy expenditure- including multiple hypothalamic, midbrain and brainstem nuclei (9, 34, 35).
Leptin Resistance
Definitions
The biology of leptin, as outlined above, was elucidated by studying leptin deficiency and examining the effects of leptin repletion on low-leptin states in animal models and humans. These leptin-deficient states include not only genetic leptin deficiency (very rare in humans) and caloric restriction (including anorexia), but also other conditions associated with diminished fat stores (including lipodystrophy syndromes and untreated insulin deficiency) (2, 8, 12, 36-40). Administration of exogenous leptin mitigates the increased appetite, decreased energy expenditure and neuroendocrine dysfunction associated with each of these states of leptin insufficiency. Leptin also attenuates the hyperglycemia caused by uncontrolled insulin-deficient diabetes and lipodystrophy syndromes (12, 19).
The ability of exogenous leptin to reduce food intake/body weight (and to modulate metabolism and endocrine function) in the face of replete adipose stores, especially in obese individuals, has proven to be more limited, however (41, 42). Indeed, commensurate with their large adipose mass, obese individuals exhibit elevated circulating leptin concentrations relative to lean subjects (6), and these elevated leptin concentrations (obviously) fail to return body adiposity to within the normal range. In rare instances, genetic mutations may abrogate LEPR function (here, circulating leptin is increased to concentrations well above those observed in “normal” obese humans of similar fat mass) (43). These instances are similar to classical hormone resistance/insensitivity syndromes (growth hormone insensitivity, Type A insulin resistance, etc.), in which genetic alterations of the hormone receptor prevent hormone action (44, 45).
In the vast majority of human obesity however, Lepr is unperturbed, and obesity and any diminished responsiveness to leptin must result from other mechanisms. The poor efficacy of endogenous leptin to promote leanness in obese subjects has given rise to the notion of functional “leptin resistance,” which is based loosely upon and often compared to the concept of insulin resistance in type 2 diabetes - where diminished cellular and metabolic insulin responsiveness coexist with the hypersecretion of insulin (46, 47). Implicit in the concept of leptin resistance is the idea that processes that promote and/or result from obesity impair leptin action, thereby facilitating the occurrence of obesity and attenuating the potential efficacy of therapy with exogenous leptin. Understanding mechanisms that may underlie “leptin resistance” is thus crucial both for determining the causes of obesity and identifying potential mechanisms that can be targeted for therapy.
While the goal of this NIH conference was to generate a universal, quantifiable definition of leptin resistance, the common application of “leptin resistance” to label both the presence of hyperleptinemia in obesity and the failure of exogenous leptin administration to provide therapeutic benefit suggests the difficulty of identifying such a universal definition. Regarding the use of “leptin resistance” to label hyperleptinemia in obesity: Due to their increased adipose tissue mass, virtually all obese humans and animal models (except some animal strains and extraordinarily rare patients with genetic leptin deficiency) display elevated circulating leptin concentrations relative to lean controls (1, 6, 7). Additionally, processes other than those attendant to leptin action and/or LEPR-B signaling per se likely contribute to the determination of adiposity and etiology of obesity in each person- including the function and/or anatomic variation of numerous neural circuits: While some of these circuits may be directly modulated and/or developmentally programmed by leptin, many are not (47-53) (Figure 2). Furthermore, the finding that even very obese individuals exhibit changes in hunger and energy expenditure upon moderate weight loss, and that these changes are blunted by the administration of leptin, suggests that the elevated levels of leptin in obese individuals may be functionally relevant (54). Thus, defining leptin resistance as hyperleptinemia in obesity merely serves to note the elevated concentration of a single hormone whose production is expected to be increased in this condition, alterations in the action of which may or may not be primary to the genesis of obesity, and where levels of the hormone retain some physiologic relevance. Thus, we do not favor referring to the coexistence of hyperleptinemia in obesity as “leptin resistance.”
When focusing on the potential therapeutic utility of leptin, the limited ability of exogenous leptin to promote desired outcomes represents the central issue, and it is this aspect of the leptin resistance concept that we will focus on in this review. Even restricting use of “leptin resistance” to states poorly responsive to exogenous leptin, the term covers a multitude of situations, however. It refers, for example, to assays of leptin action as diverse as the induction of STAT3 phosphorylation in the brain (in animal models) and other tissues, to the attenuation of feeding and the restraint of body weight and adiposity (in either animals or humans). These may be assayed following acute or long-term treatment with leptin doses ranging from the approximately physiologic to the heroically pharmacologic. Practically speaking, therefore, “leptin resistance” is a term so broadly-applied and context-dependent that there can be no universal, quantifiable, clinically useful definition of “leptin resistance.”
Potential Mechanisms
Studies in animal models have suggested a number of basic mechanisms that may underlie attenuated responsiveness to leptin. Many of these represent processes engaged during or promoted by overnutrition and obesity, including changes in circulating leptin-binding proteins, reduced transport of leptin across the blood-brain barrier and/or the provocation of processes that diminish cellular LEPR-B signaling (inflammation, ER stress, feedback inhibition, etc.) (25-27, 29, 55-59). Alterations in the development of leptin-regulated neurons and other components of the circuitry that controls leptin action could also blunt leptin action throughout life (50). While differing in their specifics, these potential explanations for the decreased efficacy of leptin all postulate that nutritional alterations or obesity (or increased ambient leptin concentrations themselves (60, 61)) impair leptin action. Indeed, interference with many of the cellular mechanisms that attenuate LEPR-B signaling improves leptin action in cells and animal models, revealing that these mechanisms decrease leptin action in vivo, as well as suggesting the potential utility of these processes as points of therapeutic intervention (26, 28, 55, 62). Thus, while it is not possible to precisely define leptin resistance in a universal, precise, and quantifiable manner, it is clearly useful to identify and understand mechanisms that may attenuate leptin action in vivo.
The “normal” response to exogenous leptin, against which leptin resistance is often defined (especially in rodents), most often results from the attainment of circulating leptin concentrations thousands of times higher than physiological (63), and the anorectic response to these doses is modest and subject to relatively rapid tachyphylaxis. During evolution, undernutrition presumably represented a greater threat to survival than did overnutrition, with the result that the defense against starvation (low leptin) produces stronger responses than does the defense against nutritional surfeit (high leptin). A similar line of reasoning suggests that the efficacy of leptin may be near maximal at the concentrations found in most obese people at baseline, and that the addition of exogenous leptin may, therefore, raise circulating leptin concentrations without substantially increasing leptin action. While a related hypothesis suggests that obesity was also selected against by factors including the likelihood of predation (64), the ability of palatable calorically-dense diets to promote obesity in most animals and the failure of elevated leptin levels to reduce body weight in obese animals suggests that leptin may not play a major role at this end of the spectrum. The possibility that leptin action may be near its physiological maximum in states of obesity and the notion that obesity attenuates leptin action are not mutually exclusive- both mechanisms may contribute.
Clinical Issues
In human subjects, it is generally not possible to examine the molecular mechanisms associated with or underlying “leptin resistance” experimentally; rather, the state must be operationally defined as decreased responsiveness to exogenous leptin by certain criteria. As noted, however, there is no standard for the definition of human “leptin resistance.” Is leptin resistance defined by the response to a high or low dose of leptin, given once for a short time or chronically over weeks to months, in patients at baseline or following some amount of weight reduction? What is the attenuated response by which we define leptin resistance: decreased food intake, weight loss, alterations in blood glucose or lipids, hepatic triglycerides, immune function, etc.?
Since the efficacy of leptin for the control of many metabolic parameters (e.g., weight, glucose, lipids, hepatic steatosis, etc.) is likely to be of interest, leptin responsiveness must be defined in terms of the response of specific parameters, individually, to leptin treatment.
Leptin sensitivity, not defining “leptin resistance,” is what really matters
It is more practical in humans to define sensitivity rather than resistance to leptin's actions. While clinical “leptin resistance” may be captured in a general way as poor responsiveness to exogenous leptin, for the reasons outlined above, it is not possible to define clinical leptin resistance in a precise manner that can be assessed with a single, universal assay. A pragmatic approach to leptin resistance and therapeutic leptin action thus focuses not on defining clinical leptin resistance in a universal manner, but rather on assessing leptin sensitivity: Which individuals are likely to respond to leptin and/or can be sensitized to exogenous leptin? While this may seem, on the surface, to be a semantic argument, it acknowledges that there can be no universal definition of leptin resistance and that defining who we can effectively treat and the parameters of effective treatment represent the crucial issues.
How to assess leptin sensitivity effectively in the clinical setting is not currently clear. The available information suggests that, in general, leptin sensitivity is greatest in those with low adiposity and low endogenous circulating leptin in the non-weight reduced state (65). Body adiposity and leptin concentration are unlikely to represent the only predictors for leptin sensitivity, however, as each individual is likely to exhibit an idiosyncratic response to overnutrition or obesity (i.e., in terms of genetic differences or ER stress or other responses that might limit leptin action). Indeed, the sexual dimorphism in circulating leptin concentrations suggest underlying differences in leptin production and/or action by sex (6). Similarly, circulating free leptin concentrations are determined not only by leptin production, but by its clearance and by levels of circulating soluble LEPR (59). Furthermore, adiponectin and other circulating factors may predict aspects of the therapeutic efficacy of leptin (66). Thus, it will be crucial to assess and report measures of leptin responsiveness in large samples of individuals in the context of a variety of parameters that may affect leptin sensitivity (e.g., age, sex, BMI, adiposity, fat distribution, circulating levels of leptin and other factors, etc.). Also, leptin is manufactured in multiple forms, each with different pharmacodynamics parameters and potentially distinct determinants of efficacy.
In addition to the modulation of body weight and adiposity, the examination of other potentially useful outcomes of leptin therapy (e.g., glucose homeostasis, lipid metabolism, hepatic lipid content, etc.) will be important. Furthermore, as the relationship between potential measures of acute leptin action (e.g., 24-hour food intake, energy expenditure, leptin-stimulated STAT3 phosphorylation in peripheral blood monocytes, or brain imaging) and the potential therapeutic chronic effects of leptin are unclear, it would be useful to examine both acute and long-term responsiveness of individuals to determine the potential predictive value of acute studies.
Can Leptin Sensitivity be Increased?
Some investigators have suggested that pharmacologic interruption of the cellular mechanisms apparently attenuating LEPR-B signaling could increase leptin sensitivity (55). Indeed, early attempts have met with preliminary success in animal models. Furthermore, the finding that some compounds (e.g., the amylin derivative, pramlintide) augment leptin action in some individuals suggests that it may be possible to increase the sensitivity of some individuals to therapeutic leptin administration (67). Whether such a result is a consequence of the initial weight loss induced by a non-leptin drug, by direct impact of such a drug on leptin receptor signaling, or mediated by other phenomena remains unclear. Certainly however, combinatorial approaches appear to hold some promise for clinical leptin therapy. As above for responsiveness to leptin alone, it will be important to determine traits and/or acute assays and endpoints that predict the responsiveness of patients to many types of potential leptin-sensitizing therapies.
Summary
- The term “leptin resistance” does not refer to a primary Lepr defect, but is commonly used as a catch-all phrase for states of obesity where hyperleptinemia and/or decreased responsiveness to leptin administration is observed.
- Defining leptin resistance as hyperleptinemia in obesity merely serves to highlight the concentration of a single hormone whose production is expected to be increased in this condition, alterations in the action of which may or may not be primary to the genesis of obesity, and where levels of the hormone retain some physiologic relevance. Thus, we do not favor referring to the coexistence of hyperleptinemia in obesity as “leptin resistance.”
- Despite some utility of this term for describing relative insensitivity to exogenous leptin, “leptin resistance” implies neither a single specific mechanism nor metric of leptin action. Because of the pluripotent effects of leptin and the numerous ways in which its action is studied, the universal application of a narrow definition of leptin resistance appears unlikely to be practical or useful in most contexts.
- Pragmatically, when considering leptin as a potential clinical therapeutic agent, predictors of therapeutic responsiveness to exogenous leptin (rather than a precise universal definition of “leptin resistance”) represent the most important consideration.
- We currently lack good predictors of, and acute assays to define, leptin responsiveness in humans. Such tools are needed to identify those who might be helped by leptin administration. The performance of studies to define potentially sensitive groups, with rapid dissemination of the resultant data, represents a high priority.
- Further clinical studies are also required to examine mechanisms by which leptin sensitivity may potentially be increased, and to determine predictors that identify individuals who may benefit most from such sensitizing treatments.
- Many of the points raised here with regard to assessing the physiological efficacy of leptin are useful in considering the pharmacology of other molecules of potential use in the measurement of obesity.
Acknowledgments
Meeting Support: This conference was supported by the National Institutes of Diabetes and Digestive and Kidney Diseases.
| Meeting Organizers |
|---|
| Carol Haft |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Steven Heymsfield |
| Pennington Biomedical Research Center |
| Maren Laughlin |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Martin Myers |
| University of Michigan |
| Participant List |
|---|
| Michael Appel |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| William Banks |
| VA Medical Center, St. Louis University |
| Olivier Blondel |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Tom Carroll |
| Amylin Pharmaceuticals, Inc. |
| Streamson Chua |
| Albert Einstein College of Medicine |
| Alex DePaoli |
| InteKrin Therapeutics, Inc. |
| Ralph DiLeone |
| Yale University |
| James Everhart |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Judith Fradkin |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Phillip Gorden |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Carol Haft |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Joan Han |
| Eunice Kennedy Shriver National Institute of Child Health and Human Development |
| Steven Heymsfield |
| Pennington Biomedical Research Center |
| Mary Horlick |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Barbara Kahn |
| Beth Israel Deaconess Medical Center/Harvard Medical School |
| George Kunos |
| National Institute on Alcohol Abuse and Alcoholism |
| Maren Laughlin |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Rudolph Leibel |
| Columbia University |
| Barbara Linder |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Bradford Lowell |
| Beth Israel Deaconess Medical Center/Harvard Medical School |
| David Maggs |
| Amylin Pharmaceuticals, Inc. |
| Saul Malozowski |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Christos Mantzoros |
| Harvard Medical School |
| Eleftheria (Terry) Maratos-Flier |
| Beth Israel Deaconess Medical Center/Harvard Medical School |
| Heike Muenzberg-Gruening |
| Pennington Biomedical Research Center |
| Martin Myers |
| University of Michigan |
| Elif Oral |
| University of Michigan |
| Umut Ozcan |
| Children's Hospital Boston/Harvard Medical School |
| Aaron Pawlyk |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Matthew Rechler |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Marc Reitman |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Michael Rosenbaum |
| Columbia University College of Physicians and Surgeons |
| Sheryl Sato |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Philipp Scherer |
| The University of Texas Southwestern Medical Center at Dallas |
| Corinne Silva |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Philip Smith |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Myrlene Staten |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Yossef Tam |
| National Institute on Alcohol Abuse and Alcoholism |
| Matthias Tschöp |
| University of Cincinnati |
| Allison Xu |
| University of California, San Francisco |
| Jack Yanovski |
| Eunice Kennedy Shriver National Institute of Child Health and Human Development |
| Susan Yanovski |
| National Institute of Diabetes and Digestive and Kidney Diseases |
| Lori Zeltser |
| Columbia University College of Physicians and Surgeons |
Footnotes
*
a listing of meeting organizers and attendees is presented in the acknowledgement section.
Conflicts: None
References
- 1.Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 3.Miyazaki M, Sampath H, Liu X, Flowers MT, Chu K, Dobrzyn A, Ntambi JM. Stearoyl-CoA desaturase-1 deficiency attenuates obesity and insulin resistance in leptin-resistant obese mice. Biochem Biophys Res Commun. 2009;380:818–822. doi: 10.1016/j.bbrc.2009.01.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stradecki HM, Jaworski DM. Hyperphagia and leptin resistance in tissue inhibitor of metalloproteinase-2 deficient mice. J Neuroendocrinol. 2011;23:269–281. doi: 10.1111/j.1365-2826.2010.02105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Froy O, Sherman H, Bhargava G, Chapnik N, Cohen R, Gutman R, Kronfeld-Schor N, Miskin R. Spontaneous caloric restriction associated with increased leptin levels in obesity-resistant alphaMUPA mice. Int J Obes (Lond) 2011;35:226–235. doi: 10.1038/ijo.2010.125. [DOI] [PubMed] [Google Scholar]
- 6.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, Caro JF. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 7.Frederich RC, Lollmann B, Hamann A, Napolitano-Rosen A, Kahn BB, Lowell BB, Flier JS. Expression of ob mRNA and its encoded protein in rodents: Impact of nutrition and obesity. J Clin Invest. 1995;96:1658–1663. doi: 10.1172/JCI118206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahima RS, Prabakaran D, Mantzoros CS, Qu D, Lowell BB, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 9.Myers MG, Jr, Munzberg H, Leinninger GM, Leshan RL. The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab. 2009;9:117–123. doi: 10.1016/j.cmet.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pocai A, Morgan K, Buettner C, Gutierrez-Juarez R, Obici S, Rossetti L. Central leptin acutely reverses diet-induced hepatic insulin resistance. Diabetes. 2005;54:3182–3189. doi: 10.2337/diabetes.54.11.3182. [DOI] [PubMed] [Google Scholar]
- 11.Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, Depaoli AM, O'Rahilly S. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang MY, Chen L, Clark GO, Lee Y, Stevens RD, Ilkayeva OR, Wenner BR, Bain JR, Charron MJ, Newgard CB, Unger RH. Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci U S A. 2010;107:4813–4819. doi: 10.1073/pnas.0909422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kulkarni RN, Wang ZL, Wang RM, Hurley JD, Smith DM, Ghatei MA, Withers DJ, Gardiner JV, Bailey CJ, Bloom SR. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J Clin Invest. 1997;100:2729–2736. doi: 10.1172/JCI119818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, Shen J, Vinson C, Rueger JM, Karsenty G. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100:197–207. doi: 10.1016/s0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- 15.Nogueiras R, Wiedmer P, Perez-Tilve D, Veyrat-Durebex C, Keogh JM, Sutton GM, Pfluger PT, Castaneda TR, Neschen S, Hofmann SM, Howles PN, Morgan DA, Benoit SC, Szanto I, Schrott B, Schurmann A, Joost HG, Hammond C, Hui DY, Woods SC, Rahmouni K, Butler AA, Farooqi IS, O'Rahilly S, Rohner-Jeanrenaud F, Tschop MH. The central melanocortin system directly controls peripheral lipid metabolism. J Clin Invest. 2007;117:3475–3488. doi: 10.1172/JCI31743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stafford JM, Yu F, Printz R, Hasty AH, Swift LL, Niswender KD. Central nervous system neuropeptide Y signaling modulates VLDL triglyceride secretion. Diabetes. 2008;57:1482–1490. doi: 10.2337/db07-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang CP, Tall AR. Transcriptional profiling reveals global defects in energy metabolism, lipoprotein, and bile acid synthesis and transport with reversal by leptin treatment in ob/ob mouse liver. J Biol Chem. 2001;276:49066–49076. doi: 10.1074/jbc.M107250200. [DOI] [PubMed] [Google Scholar]
- 18.Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 19.Kelesidis T, Kelesidis I, Chou S, Mantzoros CS. Narrative review: the role of leptin in human physiology: emerging clinical applications. Ann Intern Med. 2010;152:93–100. doi: 10.1059/0003-4819-152-2-201001190-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chua SC, Jr, Chung WK, Wu-Peng XS, Zhang Y, Liu SM, Tartaglia L, Leibel RL. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science. 1996;271:994–996. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- 21.Chua SC, Jr, Koutras IK, Han L, Liu SM, Kay J, Young SJ, Chung WK, Leibel RL. Fine structure of the murine leptin receptor gene: Splice site suppression is required to form two alternatively spliced transcripts. Genomics. 1997;45:264–270. doi: 10.1006/geno.1997.4962. [DOI] [PubMed] [Google Scholar]
- 22.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Woolf EA, Monroe CA, Tepper RI. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 23.Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim H, Lai CF, Tartaglia LA. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci USA. 1996;93:8374–8378. doi: 10.1073/pnas.93.16.8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robertson SA, Leinninger GM, Myers MG., Jr Molecular and neural mediators of leptin action. Physiol Behav. 2008;94:637–642. doi: 10.1016/j.physbeh.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Molecular Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 26.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, Neel BG. PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 27.Bjorbaek C, El Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- 28.Bjorbaek C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG., Jr SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem. 2000;275:40649–40657. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- 29.Bjornholm M, Munzberg H, Leshan RL, Villanueva EC, Bates SH, Louis GW, Jones JC, Ishida-Takahashi R, Bjorbaek C, Myers MG., Jr Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J Clin Invest. 2007;117:1354–1360. doi: 10.1172/JCI30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, Chua SC., Jr Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest. 2005;115:3484–3493. doi: 10.1172/JCI24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395:535–547. [PubMed] [Google Scholar]
- 33.Matarese G, Moschos S, Mantzoros CS. Leptin in immunology. J Immunol. 2005;174:3137–3142. doi: 10.4049/jimmunol.174.6.3137. [DOI] [PubMed] [Google Scholar]
- 34.Patterson CM, Leshan RL, Jones JC, Myers MG., Jr Molecular mapping of mouse brain regions innervated by leptin receptor-expressing cells. Brain Res. 2011 doi: 10.1016/j.brainres.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy [In Process Citation] Nature. 1999;401:73–76. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 37.Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, Depaoli AM, Reitman ML, Taylor SI, Gorden P, Garg A. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346:570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 38.Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O'Rahilly S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–884. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 39.German JP, Wisse BE, Thaler JP, Oh IS, Sarruf DA, Ogimoto K, Kaiyala KJ, Fischer JD, Matsen ME, Taborsky GJ, Jr, Schwartz MW, Morton GJ. Leptin Deficiency Causes Insulin Resistance Induced by Uncontrolled Diabetes. Diabetes. 2010 doi: 10.2337/db09-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, Karalis A, Mantzoros CS. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351:987–997. doi: 10.1056/NEJMoa040388. [DOI] [PubMed] [Google Scholar]
- 41.Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, McCamish M. Jama-Journal of the American Medical Association. Vol. 282. 1999. Recombinant leptin for weight loss in obese and lean adults - A randomized, controlled, dose-escalation trial; pp. 1568–1575. [DOI] [PubMed] [Google Scholar]
- 42.Mantzoros CS, Flier JS. Editorial: leptin as a therapeutic agent--trials and tribulations. J Clin Endocrinol Metab. 2000;85:4000–4002. doi: 10.1210/jcem.85.11.7062. [DOI] [PubMed] [Google Scholar]
- 43.Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougneres P, leBouc Y, Froguel P, Guy-Grand B. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 44.Moller DE, Flier JS. Insulin resistance: mechanisms, syndromes, and implications. N Engl J Med. 1991;325:938–948. doi: 10.1056/NEJM199109263251307. [DOI] [PubMed] [Google Scholar]
- 45.Savage MO, Attie KM, David A, Metherell LA, Clark AJ, Camacho-Hubner C. Endocrine assessment, molecular characterization and treatment of growth hormone insensitivity disorders. Nat Clin Pract Endocrinol Metab. 2006;2:395–407. doi: 10.1038/ncpendmet0195. [DOI] [PubMed] [Google Scholar]
- 46.Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–543. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 47.Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–651. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hill JW, Elmquist JK, Elias CF. Hypothalamic pathways linking energy balance and reproduction. Am J Physiol Endocrinol Metab. 2008;294:E827–E832. doi: 10.1152/ajpendo.00670.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berthoud HR. Interactions between the “cognitive” and “metabolic” brain in the control of food intake. Physiol Behav. 2007 doi: 10.1016/j.physbeh.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 50.Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- 51.Ring LE, Zeltser LM. Disruption of hypothalamic leptin signaling in mice leads to early-onset obesity, but physiological adaptations in mature animals stabilize adiposity levels. J Clin Invest. 2010;120:2931–2941. doi: 10.1172/JCI41985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 53.Horvath TL, Bruning JC. Developmental programming of the hypothalamus: a matter of fat. Nat Med. 2006;12:52–53. doi: 10.1038/nm0106-52. [DOI] [PubMed] [Google Scholar]
- 54.Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE, Leibel RL. Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. J Clin Endocrinol Metab. 2002;87:2391–2394. doi: 10.1210/jcem.87.5.8628. [DOI] [PubMed] [Google Scholar]
- 55.Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Jr, Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 56.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levin BE, Dunn-Meynell AA, Banks WA. Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling before obesity onset. Am J Physiol Regul Integr Comp Physiol. 2004;286:R143–R150. doi: 10.1152/ajpregu.00393.2003. [DOI] [PubMed] [Google Scholar]
- 58.Lou PH, Yang G, Huang L, Cui Y, Pourbahrami T, Radda GK, Li C, Han W. Reduced body weight and increased energy expenditure in transgenic mice over-expressing soluble leptin receptor. PLoS One. 2010;5:e11669. doi: 10.1371/journal.pone.0011669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J, Scarpace PJ. The soluble leptin receptor neutralizes leptin-mediated STAT3 signalling and anorexic responses in vivo. Br J Pharmacol. 2009;158:475–482. doi: 10.1111/j.1476-5381.2009.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia is required for the development of leptin resistance. PLoS One. 2010;5:e11376. doi: 10.1371/journal.pone.0011376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qiu J, Ogus S, Lu R, Chehab FF. Transgenic mice overexpressing leptin accumulate adipose mass at an older, but not younger, age. Endocrinology. 2001;142:348–358. doi: 10.1210/endo.142.1.7909. [DOI] [PubMed] [Google Scholar]
- 62.Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004 doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- 63.Faouzi M, Leshan R, Bjornholm M, Hennessey T, Jones J, Munzberg H. Differential Accessibility of Circulating Leptin to Individual Hypothalamic Sites. Endocrinology. 2007 doi: 10.1210/en.2007-0655. [DOI] [PubMed] [Google Scholar]
- 64.Krol E, Speakman JR. Regulation of body mass and adiposity in the field vole, Microtus agrestis: a model of leptin resistance. J Endocrinol. 2007;192:271–278. doi: 10.1677/JOE-06-0074. [DOI] [PubMed] [Google Scholar]
- 65.Bluher S, Mantzoros CS. Leptin in humans: lessons from translational research. Am J Clin Nutr. 2009;89:991S–997S. doi: 10.3945/ajcn.2008.26788E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iannucci CV, Capoccia D, Calabria M, Leonetti F. Metabolic syndrome and adipose tissue: new clinical aspects and therapeutic targets. Curr Pharm Des. 2007;13:2148–2168. doi: 10.2174/138161207781039571. [DOI] [PubMed] [Google Scholar]
- 67.Turek VF, Trevaskis JL, Levin BE, Dunn-Meynell AA, Irani B, Gu G, Wittmer C, Griffin PS, Vu C, Parkes DG, Roth JD. Mechanisms of amylin/leptin synergy in rodent models. Endocrinology. 2010;151:143–152. doi: 10.1210/en.2009-0546. [DOI] [PubMed] [Google Scholar]