Neurobiology of Resilience (original) (raw)
. Author manuscript; available in PMC: 2013 Nov 1.
Published in final edited form as: Nat Neurosci. 2012 Oct 14;15(11):1475–1484. doi: 10.1038/nn.3234
Abstract
Humans exhibit a remarkable degree of resilience in the face of extreme stress, with most resisting the development of neuropsychiatric disorders. Over the past 5 years, there has been increasing interest in the active, adaptive coping mechanisms of resilience; however, in humans, the majority of published work focuses on correlative neuroendocrine markers that are associated with a resilient phenotype. In this review, we highlight a growing literature in rodents that is starting to complement the human work by identifying the active behavioral, neural, molecular, and hormonal basis of resilience. The therapeutic implications of these findings are important and can pave the way for an innovative new approach to drug development for a range of stress–related syndromes.
Keywords: depression, anxiety, major depressive disorder (MDD), post–traumatic stress disorder (PTSD), stress disorders, sex differences, stress inoculation, stress mastery, mesolimbic dopamine system, synaptic plasticity, structural plasticity, glutamatergic neurotransmission, hypothalamic–pituitary–adrenal (HPA) axis, hypothalamic–pituitary–gonadal (HPG) axis, cortisol, corticosterone, estrogen, testosterone, progesterone, social defeat stress, chronic unpredictable stress, learned helplessness, early intermittent stress, neuroendocrine, glucocorticoid
Introduction
Over the past decade, there has been increasing attention paid to the phenomenon of resilience, the fact that most people, when exposed even to extraordinary levels of stress and trauma, manage to maintain normal psychological and physical functioning and avoid serious mental illness. While resilience has been identified across the spectrum of psychiatric disorders, we focus here on resilience as it relates to posttraumatic stress disorder (PTSD) and major depressive disorder (MDD). In this context, resilience refers to the capacity of an individual to avoid negative social, psychological, and biological consequences of extreme stress that would otherwise compromise their psychological or physical well being. Recent reports indicate that resilience in humans represents an active, adaptive process, and not simply the absence of pathological responses that occur in more susceptible individuals1, 2 (Fig. 1). The concept of resilience is difficult to operationalize, since it encapsulates many divergent behavioral phenotypes. Indeed, the study of human resilience is still a mostly phenomenological literature which has only begun to characterize biological factors in resilient individuals that are associated with more successful coping responses. As will be seen, most of these studies have focused, by necessity, on peripheral neuroendocrine changes that are predictive of resilience or on genetic variations that are linked—albeit still preliminarily—with resilient outcomes.
Figure 1. Schematic of gene x environment interactions that promote resilience.

The scheme describes how behavioral strategies through stress inoculation can interact with an individual’s genetic constitution to control expression of key genes—via epigenetic processes—in the brain’s limbic regions to mount active, adaptive molecular and cellular changes that mediate resilience.
Over the past ~5 years, neural and molecular mechanisms related to stress resilience have been investigated in laboratory animals. This work has provided more causal information about neuroadaptations in brain and their neuroendocrine output that contribute to resilience (Fig. 1). Work to date has demonstrated that such resilience—the ability to avoid deleterious behavioral changes in response to chronic stress—is mediated not only by the absence of key molecular abnormalities which occur in susceptible animals to impair their coping ability, but also by the presence of novel molecular adaptations which occur uniquely in resilient individuals to help promote normal behavioral function. The former can be seen as mechanisms of passive resilience; the latter as mechanisms of active resilience. Certain active resilience mechanisms have been shown to counteract maladaptive molecular changes seen in susceptible animals, thus providing mechanistic insight into the biological basis of active coping.
Here we review the evolving biological understanding of resilience by integrating findings from humans and animals and identify key areas for future investigation. We emphasize known active processes correlated with resilience in humans and provide newer evidence in rodent models for the molecular and cellular mechanisms that underlie active resilience. The reader is referred elsewhere for the vast literature of stress–induced changes that have not been explicitly related to active resilience per se. Finally, we conclude with a discussion of how this growing body of knowledge can guide the development of novel treatments for a range of stress–related disorders by enhancing such natural mechanisms of resilience.
Neuroendocrine Findings in Human Resilience
Hypothalamic–pituitary–adrenal (HPA) axis
A major mediator of the impact of stress on brain and behavior is activation of the HPA axis, which results in widespread hormonal, neurochemical, and physiological alterations3. Glucocorticoids, released from the adrenal cortex as a consequence of HPA axis activation, interact with steroid receptors expressed throughout brain that function primarily as transcription factors to regulate cellular function beyond the time scale of acute stress effects. In particular, glucocorticoid receptors (GRs) and mineralocorticoid receptors (MRs), which also respond to glucocorticoids, are expressed at high levels in hippocampus, amygdala, prefrontal cortex (PFC), and other limbic and midbrain structures where they modulate the neural circuitry and neuroendocrine systems that underlie behavioral responses to stress.
The effects of stress on the HPA axis depend upon the developmental timing of the stress, as well as other critical factors such as stress magnitude, type, and duration (see “stress inoculation” and “stress mastery” below). Most research on the effects of stress on HPA axis function in humans has concentrated on MDD and PTSD. Numerous studies have reported elevated blood glucocorticoid levels in roughly two–thirds of individuals with MDD, although a smaller subset of depressed individuals show reduced glucocorticoid levels and typically display less severe symptoms4. Hypocortisolemia has also been reported widely in PTSD; however, here too the findings have been mixed5.
The variable findings of glucocorticoid levels in MDD and PTSD have posed challenges for understanding the role of the HPA axis in risk or resistance to the development of stress–related disorders. Indeed, the distinction between MDD and PTSD is not clear—there are no objective biological measures that differentiate these syndromes—and stress exposure and adverse life events are important risk factors for both disorders. Increased cerebrospinal fluid levels of corticotropin releasing hormone (CRH) have been documented more generally in adults who report a history of childhood abuse6, and thus does not always correlate with the development of MDD or PTSD7, 8. While the HPA axis is central to normal stress responses, the relationship between HPA axis function and resilience to stress–related affective illness is still unclear. Recent studies suggest that exogenous glucocorticoid replacement can protect against PTSD in trauma–exposed humans, however, it is unknown whether these mechanisms occur naturally to promote stress resilience. Nevertheless, as will be seen below, human and animal studies have recently identified active biological responses that can blunt stress–induced HPA activation to promote resilience.
Dehydroepiandrosterone (DHEA)
DHEA is a precursor for the synthesis of anabolic steroids and is co–released with cortisol (corticosterone in rodents) from the adrenal cortex in response to stress. It may also act directly on several steroid hormone receptors. While the physiological consequences of DHEA are not completely understood, DHEA might counter the actions of cortisol as well as exert anti–oxidant and anti–inflammatory effects. Reports have shown that blood DHEA levels increase under acute stress and that a higher level of DHEA, or a higher DHEA to cortisol ratio, is associated with less dissociative symptoms and superior performance in healthy subjects undergoing military survival training9.
Initial research in PTSD supported the hypothesis that DHEA or the DHEA to cortisol ratio may represent a resilience factor. It has been reported that DHEA responses to adrenocorticotropic hormone (ACTH) were elevated in PTSD and negatively correlated with the severity of symptoms, suggesting that DHEA release during stress may buffer the severity of PTSD 9. Consistent with this interpretation, a separate study found that DHEA levels were elevated in PTSD but that higher levels were correlated with symptom improvement and better coping while a lower DHEA to cortisol ratio was positively correlated with the severity of PTSD symptoms10. However, a contrary report linked elevated DHEA to increased suicidality in male veterans with PTSD11, and an initial randomized trial of DHEA supplementation in men undergoing military survival training was negative12. Future work is therefore needed to determine whether DHEA is indeed a causative factor in positive coping.
Testosterone
Testosterone has been strongly linked to social rank and aggression. In both men and woman, testosterone increases in “winners” following an athletic competition and, on average, athletes with the highest saliva testosterone levels have higher rankings within the team13. Testosterone levels are also positively correlated with the degree of social connectedness with teammates, greater feelings of personal success and dominance14. However, more causative studies are needed to demonstrate this definitively. Given its role in social behavior and positive mood, it is not surprising that blood and saliva testosterone levels decrease following stress15 and that lower circulating levels are often found in individuals with PTSD or MDD16, 17. Early studies in men suggest that testosterone may be effective in treatment–resistant depression and as an adjunct to SSRI treatment17. Although much future work is needed, testosterone may serve as a pro–resilience factor by promoting positive mood and social connectedness.
Neuropeptide Y
Neuropeptide Y (NPY), a peptide neurotransmitter, modulates stress responses in animals, and studies in humans support the possibility that NPY may represent a protective factor in the face of stress18, 19. In one study, higher blood NPY levels were shown to predict better performance under stress during military survival training and were found in Special Forces soldiers, compared to their non–Special Forces military counterparts18. A follow–up study reported that higher levels of NPY in response to acute stress predicted less psychological distress and fewer symptoms of dissociation19. These studies suggest a protective role for NPY under conditions of high stress, which is consistent with tentative human genetic studies that weakly implicate variations in the NPY gene in emotional behavior and stress responses (discussed below)20, 21.
Early Genetic Findings in Human Resilience
Genetic factors are important determinants for the risk or resilience to psychiatric disorders. Most work thus far has focused on candidate genes with relatively weak associations reported. Some recent examples of genes related to the HPA axis, serotonergic systems, or neuropeptide Y that show weak to moderate associations with resilient phenotypes are listed in Table 1. The field is now pivoting increasingly to genome–wide studies on large numbers of people to parse the complex genetic contributions to mood or anxiety disorders. We anticipate exciting findings in the coming years as the genetic basis of resilience becomes better understood.
Table 1.
Early genetic findings in resilience
| Genetic findings | Role in resilience | Reference |
|---|---|---|
| GR–heterocomplex co–chaperone gene FKBP5 | 4 SNPs increase PTSD risk in children experiencing severe abuse. | 95 |
| ADCYAP1R1 gene encodes for PACAP. | SNP associated lower levels of pituitary adenylyl cyclase–activating polypeptide (PACAP) and rates of PTSD in woman. | 96 |
| CRH receptor–1 gene (CRHR1) | 3 SNPs associated with lower rates of MDD in women reporting childhood abuse. | 97 |
| Serotonin transporter (5–HTTLRP) | Variable reports that the short allele is associated with negative emotions and vulnerability to stress–related disorders.Long allele associated with higher levels of resilience as measured by self–report. | 98, 99 |
| Neuropeptide Y (NPY) | Long form variant of NPY is related to reduced PTSD susceptibility and negative emotional states in MDD. | 20, 100 |
Animal Models of Resilience
Definition of resilience in animals
As with humans, chronic stress leads to the development of depression– or anxiety–like behaviors in only a subset of laboratory animals22–27. The remaining animals, which have been termed resilient in some studies, usually exhibit some deleterious symptoms in response to the stress, but do not exhibit deficits in key behavioral domains. For example, following chronic social defeat stress, all genetically inbred C57BL6/J male mice exhibit a constellation of symptoms including heightened reactivity of the HPA axis, deficits in exploratory–based behavior that are interpreted as increased anxiety, and stress–induced polydipsia24. However, ~35% of the stressed mice, considered “resilient,” do not exhibit social avoidance, hyperthermia elicited by social interactions, anhedonia–like symptoms (reduced interest in sucrose, high fat food, or sex), or a metabolic syndrome characterized by over–eating, obesity, and central leptin resistance24, 28. Using this classification, resilient animals are not devoid of symptoms and, in fact, exhibit some behavioral adaptations that appear maladaptive, but they exhibit clear resistance to many other maladaptive sequelae of the chronic social stress.
Other stress paradigms have been used to study resilience in animals. It has long been known that inbred rodents subjected to learned helplessness models display a range of responses. Depending on the severity and duration of exposure to inescapable foot shock, a subset of animals, ~30% in some studies, develop learned helplessness—they fail to escape when escape becomes possible, while another subset (termed resilient) escapes with latencies seen in unstressed animals29. Maier and colleagues view the risk of learned helplessness to be highly dependent upon the animal’s own control over the stress, with resilience promoted by control over cessation of the stress (For review see 30). Cohen et al.23 recently utilized a predator odor to induce a stress response and then classified rats into 3 groups; one-third each that was extremely disrupted, partially disrupted, or minimally disrupted. The classifications were based largely on the number and type of behavioral deficits and the degree of change in brain NPY levels. The animals classified as extremely disrupted exhibited anxiety–like behaviors and increased acoustic startle responses, as well as large reductions in NPY across multiple brain regions. Both the partially and minimally disrupted groups exhibited mixed deficits within these domains. In another recent study, Delgado et al.26 utilized a chronic “mild” stress (CMS) paradigm (where rats were exposed to varying physical and psychosocial stresses) and defined resilient animals as those that did not exhibit significant anhedonia measured by reduced sucrose consumption. This form of stress induced anhedonia–like symptoms in 70% of exposed rats. They went on to use structural magnetic resonance imaging (MRI) and spectroscopy to show that CMS did not reduce hippocampal volume or alter glutamate metabolism in resilient mice as seen in susceptible mice.
An equally important literature has focused on strain differences in relative risk or resilience, studies that have shed light on potential genetically controlled traits that make an animal more or less susceptible to stress. In the social defeat stress model, the relative distribution of resilience differs across mouse strains. For example, 10 days of social defeat in C57BL6/J mice results in ~35% resilient mice, while other strains such as CD1 or FVB are closer to 100% resilient 27. These ratios are also a function of the severity and duration of the defeat episodes. In a related study, Vidal et al. (2011) found that better coping strategies might make Sprague Dawley rats more resilient to social defeat stress than Wistar rats as measured in fear– and anxiety–based domains31. Strain–dependent responses to other physical stressors such as restraint or CMS have been shown, underscoring the role of complex genetics in regulating risk and resilience to chronic stress32–34.
The central question concerning all animal studies of resilience is how they relate to resilience as defined in humans. As seen from the above examples, the definition of resilience, and the percent of animals exhibiting resilience, varies across several stress paradigms. Most studies of resilience to date have focused on the absence of some behavioral or molecular abnormality in a subset of stressed animals, and only more recently have turned to more active mechanisms of resilience—protective changes that occur uniquely in resilient individuals. The crucial criterion for defining resilience in animals, however, as noted earlier, is the ability to avoid some or all of the deleterious behavioral effects of chronic stress. The ability to avoid such deleterious consequences of stress in turn depends on both passive and active resilience mechanisms. Experimentally, it is thus essential to evaluate whether the absence (passive mechanism) or presence (active mechanism) of a given stress–induced molecular, cellular, or circuit change exerts a positive effect on the animal’s behavior, that is, whether it makes the animal less prone to exhibit maladaptive behavioral traits. Such a definition would be analogous to the Diagnostic and Statistical Manual of Mental Disorders–IV (DSM–IV), where behavioral symptoms reach threshold for diagnosis only when they cause significant distress or impairment in the individual’s ability to function. Even this is complex, since there is an ongoing debate that questions whether anxiety and depression symptoms are maladaptive from a ethological perspective or whether they serve an evolutionary purpose such as avoidance in the face of threat or by reducing expenditure of resources35. Our group has defined resilience to chronic social defeat stress as the absence of social avoidance, anhedonia, and metabolic syndrome, which are highly correlated with one another24, 27, 28, 36. The anhedonic and metabolic symptoms are clearly maladaptive and indeed are reversed by chronic administration of antidepressants to susceptible mice36, 37. Interpretation of social avoidance is more complicated, even though it too is reversed by chronic antidepressant treatment, since one could argue that avoiding an unfamiliar mouse after 10 days of aggressive encounters is adaptive, not maladaptive. However, we have shown that susceptible mice also avoid non–aggressive C57BL6/J littermates, and that such social avoidance is essentially permanent—it persists for >6 months—despite housing with non–aggressive littermates, suggesting that it does reflect a maladaptive response36, 38. Therefore, when defining resilience in non–human species, it is essential to utilize a more nuanced definition, considering not only the presence or absence of depression– or anxiety–like behaviors, but also their ethological relevance to the psychological and physical health of the organism. In the following sections, we highlight recent work that has begun to identify active, adaptive coping mechanisms—behavioral, neural, molecular, and hormonal, which promote such behavioral resilience.
Stress over the life cycle in the development of resilience
Developmental and psychological research in humans over the last two decades has demonstrated that, rather than being a rare trait, resilience in both children and adults is a common outcome following adversity – representing successful, adaptive coping in the face of stress39, 40. Within the general population, between 50–60% experience a severe trauma, yet the prevalence of illness is estimated to be only 7.8%41. Children in particular display remarkable resilience across a range of negative environmental stressors39. One important set of observations concerns the potential pro–resilience effect of encountering and overcoming stress–inducing situations during development. According to this model, coping with moderate amounts of stress leads to an individual sense of mastery and promotes resilience in the future. It might be that more moderate exposure to stress allows for a sense of stress mastery enhancing one’s perception of control. This inverted U shape relationship between stress and coping can be observed in all organisms throughout their lifetime and suggests that maintaining optimal stress exposure might prevent the development of major psychiatric dysfunction.
A potentially related phenomenon observed in laboratory animals, sometimes referred to as “stress inoculation,” was first described by Levine et al. who showed that infant rats exposed to intermittent foot shocks subsequently respond more effectively when confronted with novel situations compared to their non–stressed counterparts42. Subsequent work has examined in detail the long–term effects of brief intermittent mother–offspring separations in squirrel monkeys (for review see43). In a common experimental design, socially housed monkeys are randomized to either brief intermittent separations or a non–separated control condition at 17 weeks of age. Over 10 separation sessions, a monkey is removed from the group for one hour per week. At nine months of age, behavioral and hormonal parameters are measured in separated and non–separated monkeys in a novel environment stress test44. Compared to non–separated monkeys, previously separated monkeys showed fewer signs of anxiety and increased exploration of the environment coupled with diminished plasma levels of cortisol and ACTH44. Separated monkeys also demonstrated enhanced response inhibition to previously rewarding stimuli, suggesting improved cognitive control of behavior45. These and related findings highlight the critical window of stress exposure and suggest that early intermittent separations enhance stress tolerance, i.e., promotes resilience.
The development of standardized rodent models of early life stress will greatly increase our ability to study the mechanisms of stress inoculation. A recent study showed that maternal deprivation during early periods combined with chronic unpredictable stress (CUS; exposure to varying physical stresses) during juvenile periods promoted a greater degree of stress resilience than maternal deprivation alone or when combined with adult CUS46. This work points to critical developmental mechanisms engaged during early stress exposure, however, much future work is needed to make these paradigms more routine, particularly in mice.
While a relatively large animal and human literature has shown that chronic exposure of adults to high levels of stress is usually associated with increased susceptibility to mood, anxiety, and addiction disorders6, 24, 36, 47–49, there is some evidence that more graded exposure of adults to stress might reduce such vulnerabilities, that is, promote resilience. Over several decades, McEwen and colleagues systematically measured the type, length, and quality of stress experience on multiple rat behaviors and found that stress affects most behavioral domains with an inverted U–shape curve, where low and high levels of stress both impair behavior, while intermediate levels actually promote positive coping responses (for review see50). The shape of the inverted U differs by many factors including sex, strain, and behavioral domain in question (Fig. 2). For example, while female rodents tend to be more susceptible to stress–induced dysfunction in emotional domains (i.e., sucrose preference and forced swim test), they tend to show a greater degree of stress resilience in cognitive domains (i.e., object placement and recognition) (for review see51). These findings suggest that some level of stress and the context in which it is experienced may help adult animals to develop better coping responses to future stress experiences.
Figure 2. Stress inoculation shifts the inverted U shape curve to promote resilience.
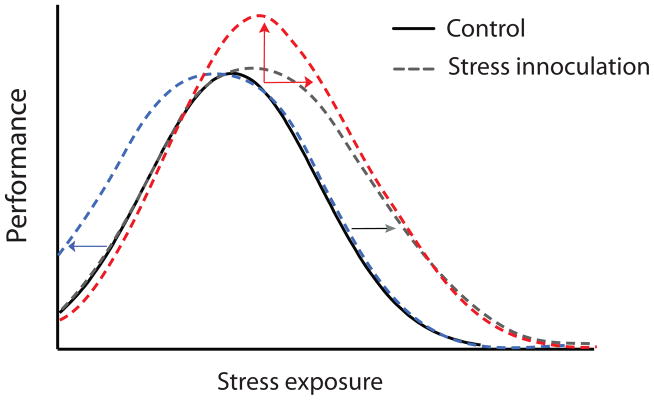
Graded or controlled stress experience can promote better performance on several behavioral tasks. Included are 3 hypothetical curves describing how stress inoculation might affect responses to future stress. 1) A leftward shift shows that inoculation might make lower levels of stress promote better performance; 2) An upward shift shows that inoculation might promote higher maximal performance in response to stress; and 3) A rightward shift shows that inoculation might enable the maintenance of optimal performance at higher levels of stress.
Although further research is necessary to confirm some of these hypotheses, it seems clear that moderate degrees of stress exposures during early life, adolescence, and adulthood can shift an individual’s stress–vulnerability curve to the right or broaden the curve by increasing the range of tolerable stress for the organism. The ability to harness such approaches might have important therapeutic applications.
Neurobiological Findings in Animal Models of Resilience
In recent years, the aforementioned animal models have just begun to be used to define the neural circuits and molecular adaptations within these circuits that contribute to resilience. In several cases, key findings in animals have been demonstrated in human postmortem brain, which provides an important measure of validation. As mentioned above, resilience is often defined as the absence of behavioral symptoms in a subset of animals subjected to chronic stress. Indeed, numerous studies have identified pro–susceptibility neural and molecular factors, which, if removed, promote stress resilience52–54. However, this approach suggests that stress resilience is solely a passive process, whereby an animal’s lack of response is adaptive. While this is no doubt true for certain conditions and biological pathways, there is increasing evidence that stress resilience also arises from active coping strategies, both behavioral and molecular. For example, many have argued that, during chronic exposure to stress, behavioral strategies that limit the stress experience could promote resilience. During social defeat stress, animals that engage in less submissive posturing during the attack show less social avoidance, suggesting that this behavioral coping strategy may affect the aggressive interaction and thereby lessen the effects of the stress55. While other examples of behavioral coping strategies have been described56, only recently have neurobiological mechanisms of active resilience been characterized. In turns out that resilience to chronic social defeat stress is associated with large numbers of unique changes in gene expression and chromatin modifications in specific brain regions that are not seen in susceptible animals24, 57. In this section, we focus on such active mechanisms of resilience, first considering the evolving neural circuitry implicated in resilience and then presenting several specific active molecular processes that have been shown recently to mediate a resilient phenotype in rodent models.
Glutamatergic signaling and synaptic connectivity
Depression and anxiety are heterogeneous disorders marked by deficits in many behavioral domains and controlled by multiple brain structures (Fig. 3). Numerous studies suggest that depression and anxiety in humans result in part from hypo–activation and reduced volume of frontal cortical and hipppocampal regions that control subcortical structures such as nucleus accumbens (NAc) and amygdala, although hyper–activation of certain PFC regions (e.g., subgenual area of anterior cingulate cortex) is also involved58–61. In addition, fMRI studies have shown that both depression and anxiety are associated with hyperactivity of amygdala 62. While imaging studies in NAc are less clear, most would argue for hypoactivity of NAc in depression, which is supported by deep brain stimulation studies that show electrical stimulation of the anterior limb of the internal capsule (which includes NAc) alleviates symptoms of depression and anxiety. fMRI blood oxygen level dependent (BOLD) signals in humans provide an understanding of which structures are more or less active based on oxygen utilization, however, it is unclear whether these adaptations occur in inhibitory or excitatory neurons, or for that matter glia, and thus whether they reflect an overall increase or decrease in net circuit activity. To address this question, investigators are studying rodent models to understand circuit–level synaptic changes in glutamate systems with far greater precision. In general, the literature supports the idea that chronic stress reduces dendritic arborization and glutamatergic dendritic spine density of pyramidal neurons in PFC and hippocampus, and reduces hippocampus neurogenesis, while increasing dendritic spine number or branching in amygdala and NAc (for review see63). Hypoactive PFC and hippocampal inputs to neurons in these subcortical structures may mediate their activation and the subsequent activity–dependent structural plasticity of their dendrites, although further research is needed in this area.
Figure 3. Brain circuitry implicated in resilience to depression and anxiety disorders.

Depicted are the major brain structures in mood–related circuits that are altered by stress in animal models of depression or implicated in human depression. The red solid lines represent excitatory glutamatergic afferents to NAc from mPFC, amygdala, and hippocampus, and glutamatergic innervation of VTA by amygdala. GABAergic afferents, shown in purple, are inhibitory circuits, and include connections from NAc to VTA and hypothalamus. Dopamine neurons (shown in green solid lines) project from VTA to a range of limbic targets, including NAc, mPFC, amygdala, and hippocampus. Peptidergic pathways though which the hypothalamus (e.g., ARC, arcuate nucleus, and LH, lateral hypothalamus) alters neurotransmission in NAc and VTA are shown in solid black lines. Each structure contains specialized neuronal cell types thought to regulate stress responses, including resilience. These cell types, color–coded to reflect the transmitter signal they convey, include amygdala, PFC and hippocampal glutamatergic neurons (red), GABAergic NAc medium spiny neurons (purple), hypothalamic peptidergic neurons (black), and VTA dopaminergic neurons (red). CP, caudate-putamen; DMT, dorsomedial thalamus; SC, superior colliculus; IC, inferior colliculus; VP, ventral pallidum; SNr, substantia nigra; PAG, periaqueductal gray; DR, dorsal raphe; and LC, locus ceruleus.
Despite this work focused on stress susceptibility, few reports have to date examined whether resilience is associated with any active mechanisms within this defined brain circuitry. Recent studies have shown a greater degree of c–Fos, FosB, or ΔFosB expression in glutamatergic neurons of mPFC (infralimbic, paralimbic PFC) of resilient mice following chronic predator or social defeat stress25, 64, 65. Increased expression of these immediate early genes would suggest increased neuronal activation within this brain region, which might represent a pro–resilience adaptation. Consistent with this hypothesis, Covington et al. showed that direct optogenetic stimulation of mPFC neurons with channel rhodopsin (ChR2) promotes resilience to social defeat stress65. While this initial work did not distinguish between glutamatergic vs. GABAergic neurons, more recent work using a viral vector that specifically targets ChR2 to glutamatergic neurons in mPFC shows that optogenetic stimulation of the glutamatergic microcircuit from PFC to NAc is antidepressant (Christoffel, Soc. Neurosci. Abstr., 2012). Future studies examining the contribution of other glutamatergic microcircuits to resilient responses, using this powerful methodology, will further delineate the circuit basis of resilience.
Early intermittent maternal separations that promote resilience increase cortical volume within ventromedial PFC (VMPFC)66, changes opposite in direction to those seen in depressed humans and stressed rodents. Understanding the molecular basis of these active restructuring processes may shed light on the hypofrontality observed in depression or anxiety disorders67, 68, as well as the circuit mechanisms underlying stress inoculation. Notably, environmental enrichment similarly increases the complexity of the dendritic tree, dendritic spine density, and synaptic protein levels of pyramidal neurons in hippocampus and PFC, suggesting that this may be a shared feature of resilience under these two distinct conditions69. Likewise, in rodents, enrichment increases expression of FosB and ΔFosB in mPFC and is reported to confer resilience to stress–induced increases in depression– and anxiety–like behaviors across multiple domains25 (Fig. 4). These data provide an important contrast to earlier studies of the deleterious effects of early life stress on neural structures involved in emotional behavior and stress responses. Rats exposed to severe chronic stress during gestation, which promotes depression–like behaviors later in life, exhibit decreased dendritic spine density in the anterior cingulate gyrus and orbitofrontal cortex70. The striking contrasts between the neural and behavioral effects observed in the context of different stress paradigms highlights the markedly divergent consequences resulting from differences in the developmental timing and the type, magnitude, and duration of stress exposure, as stated above.
Figure 4. Active molecular mechanisms in limbic brain circuits that promote resilience in animal models.

Four examples of active adaptive molecular processes that confer resilience to chronic social defeat stress are shown. In mPFC (infralimbic and paralimbic), resilient animals display molecular evidence of increased neural activity, which has been shown via optogenetic techniques to promote resilience. The cell type exhibiting this hyperactivity is not known (GABAerigc versus glutamatergic). VTA dopamine neurons of resilient animals show increased transcription of K+ channel subunits, which normalizes the stress–induced increase in VTA firing rate that drives deleterious responses to stress. In NAc, resilience is associated with increased ΔFosB–mediated transcription of GluA2, a Ca2+–impermeable AMPA glutamate receptor subunit that counteracts glutamate hyperactivity found in susceptible mice. In hypothalamus, resilience is associated with hypermethylation of the Crh gene to suppress its transcription and reduce HPA hyperactivity found in susceptible mice.
While susceptibility to chronic social defeat stress is associated with increased glutamatergic tone, including greater frequency of excitatory currents and number of glutamatergic synapses, on medium spiny neurons in NAc, there is recent evidence that resilience is mediated in part via an active adaptation that opposes this susceptibility mechanism52, 71. ΔFosB is induced in medium spiny NAc neurons preferentially in resilient animals, where it promotes resilience partly by inducing expression of GluA2 (GluR2), an AMPA glutamate receptor subunit that reduces the Ca2+ permeability and overall conductance of AMPA channels71. Gene expression arrays have identified many other targets for ΔFosB in NAc of resilient animals, which now warrant examination for their possible role in mediating resilience as well. Indeed, reduced levels of ΔFosB and GluA2 have been documented in NAc of depressed humans examined postmortem71 (Fig. 4). The selective induction of ΔFosB in NAc of resilient animals is mediated via the activation of serum response factor (SRF) under these conditions72, although the mechanism responsible for SRF’s selective induction in resilient individuals remains unknown.
K+ channel driven intrinsic excitability of neurons
Another active neural mechanism of resilience is the normalization of firing rate of ventral tegmental area (VTA) dopamine neurons in the chronic social defeat stress paradigm24 (Fig. 4). Our initial analysis showed that the firing rate of VTA dopamine neurons was normal in resilient animals and increased in susceptible animals, which would appear to reflect the absence of a stress–induced change in resilience. However, upon closer examination, there is an independent, active process occurring in resilient mice that normalizes VTA firing to control levels and thus prevents social avoidance and sucrose preference deficits. Thus, the hyper–excitability of VTA dopamine neurons in susceptible mice is mediated in part by induction of hyperpolarization–activated cation current (_I_h), which increases the intrinsic excitability of these neurons73, Friedman Soc. Neurosci. Abstr., 907.26, 2011 Surprisingly, this current is similarly increased in VTA neurons of resilient mice, suggesting the existence of an additional ionic mechanism, which counteracts the increased _I_h function and normalizes firing rate in resilient mice. Our early microarray analyses identified large increases in a cluster of K+ channel subunits, including KCNF1, KCNH3, KCNK4 and KCNQ3, in the resilient VTA24. This finding was functionally confirmed by electrophysiological studies, and such induction of K+ channels functionally occludes the increased _I_h current and thereby actively promotes behavioral resilience24, 73.
It is surprising that increased excitability of VTA dopamine neurons mediates susceptibility, with normalization mediating resilience, since such firing is generally seen as promoting reward and motivation, both implicated in resilience2. However, increased firing of VTA dopamine neurons has long been documented in response to rewards as well as to aversive stimuli, and it has been suggested that they increase salience for both types of stimuli24, 73–76. One possible explanation of this paradox is that different subsets of VTA dopamine neurons may show activation by rewarding vs. aversive stimuli77. This highlights the fact that, across numerous laboratories and experimental conditions, there is not a one–to–one correspondence between reward and resilience. Not all molecular changes in the VTA or NAc that increase drug or natural reward promote resilience, and vice versa. Optogenetic studies, which now make it possible to selectively activate subsets of VTA dopamine neurons, or their efferent or afferent connections, should help our understanding of the circuit mechanisms by which VTA dopamine neuron firing regulates responses to rewarding and aversive stimuli and determine stress susceptibility vs. resilience78, Chaudhury, Soc. Neurosci. Abstr., 907.27, 2011.
K+ channel–mediated adaptations have also been observed in NAc after prolonged social isolation of adult mice or rats, which induces depression– and anxiety–like behavioral abnormalities49 and promotes susceptibility to chronic social defeat stress71. Induction of particular K+ channel subunits in NAc in response to adult social isolation was shown to contribute to depression–like symptoms seen in this paradigm, while normalization of K± channel function contributes to antidepressant responses49.
Consistent with this work in VTA and NAc, there is increasing evidence that several types of K+ channels function as general gatekeepers of neuronal excitability in numerous experimental systems79, 80. The findings centered around K+ channels underscore the value of focusing on the molecular mechanisms of resilience and warrant attention as potential targets for the development of new treatments of stress–associated mental disorders.
Neuroendocrine mechanisms
As mentioned above, the HPA axis plays a critical role in mediating stress responses, with disruption of normal HPA function (up or down) associated with both depressive and anxiety syndromes in humans. Rodent models have largely supported this literature. For example, prenatal exposure to glucocorticoids increases CRH levels in the central nucleus of amygdala and reduces volume of PFC structures in adulthood70. Elevated glucocorticoid levels may mediate the ability of stress to reduce the dendritic spine density of PFC pyramidal neurons in parallel with hypertrophic effects in basolateral amygdala, as described above70. There is also evidence for a possible role for the HPA axis in resilience. For example, work from Meaney and coworkers have characterized the effects of early life maternal care in rats on glucocorticoid receptor expression in hippocampus and on emotional behavior. They find that high levels of maternal care are associated with decreased DNA methylation of the GR gene, higher levels of GR expression, greater feedback inhibition of the HPA axis, and resilient stress responses in adulthood (for review see 81). The GR gene is likely just one of many epigenetic targets of greater maternal care that promote resilience later in life.
Less is known about HPA axis adaptations in adult animals that might contribute to resilience82. One recent paper found an epigenetic mechanism, induced by chronic stress in resilient mice only, that controls HPA axis hyperactivity83 (Fig. 4). The authors showed that, after chronic social defeat stress, Crh gene expression is increased in the paraventricular nucleus (PVN) of hypothalamus of susceptible animals and that this adaptation is necessary for the development of social avoidance83. Notably, the Crh gene is hypermethylated and silenced to prevent Crh induction in the subset of animals termed resilient for their lack of social avoidance. Expression of a small interfering RNA (RNAi) to decrease CRH expression was sufficient to prevent social avoidance in susceptible mice. Consistent with these observations, environmental enrichment paradigms that promote resilience in rodents also reduce ACTH and corticosterone responses to stress, suggesting an interesting link among genetic, experience–based, and epigenetic factors84.
As alluded to earlier, male rodents are more resilient than females with respect to the effects of chronic stress on emotional aspects of depression– and anxiety–like behavioral domains. For example, we have shown that sub–chronic unpredictable stress induces anhedonia (decreased sucrose preference), increased immobility on the forced swim test, and increased anxiety (greater latency to feed in a novel environment and decreased time grooming in the splash test). The data clearly show that females are more sensitive than males in these domains85, Hodes, Soc. Neurosci. Abstr. 219.01, 2011. However, as mentioned above, stressed females tend to perform better on non–aversive cognitive or memory tasks compared to males. Stress enhances the performance of female rodents on the radial arm maze, Morris water maze, Y–maze, non–associative learning, and object placement tasks, whereas stress impairs male performance in these assays86–88. Conversely, in tests of acute stress or aversive conditioning, stress enhances learning in males and impairs it in females89, 90. These data highlight the possibility that males and females may use different coping strategies in the face of stress, which has led to the hypothesis that gonadal hormones, such as testosterone in males, might promote resilience to deficits on emotional domains, while estrogen or progesterone may promote stress resilience in females within cognitive domains. Moreover, the literature suggests that on cognitive domains females cope better with chronic forms of stress, whereas males tend to cope better with acute stress.
While most human work is limited to correlative studies, there is evidence that testosterone in males promotes resilience in MDD and PTSD, potentially consistent with epidemiological data showing that woman are significantly more vulnerable to developing these disorders than men 16, 17. Based on the animal work stated above, future studies in humans should investigate sex differences in vulnerability to stress–related deficits in cognitive and emotional domains. Indeed, evidence from women across the reproductive lifespan suggests that fluctuating ovarian hormones are likely a biological source of increased prevalence for these disorders. Work in rodents largely confirms this, showing that removal of ovarian hormones decreases the pro–depressant or anxiogenic effects of stress, while simultaneously increasing the negative effects of stress on spatial and non–spatial memory85, 91, 92. As well, maternal experience in female rodents seems to promote resilience to the effects of stress on cognition, which is likely in part through hormonal mechanisms regulating oxytocin93. While much future work is needed to understand these interesting roles for gonadal hormones in promoting or opposing resilience, studies of the underlying mechanisms of sex differences in stress responses can provide us with unique biological information about the mechanisms of coping in depression and anxiety disorders.
Therapeutic Implications
An important finding from animal studies of neurobiological and neuroendocrine mechanisms of resilience–like behavioral adaptations is that resilience is likely mediated, in large part, via active adaptations that occur selectively in resilient individuals (Fig. 1). Indeed, genome–wide studies in the chronic social defeat paradigm have identified a range of gene expression changes and chromatin modifications in VTA and NAc that occur uniquely in resilience24, 57. Examples include ΔFosB and SRF, discussed above, as well as HDAC2 (histone deacetylase–2) and the WNT (wingless)–DVL (disheveled)–GSK3β (glycogen synthase kinase–3β) signaling cascade71, 72, 94 In fact, there is significant overlap between genes that are regulated in resilience and those that are regulated by chronic antidepressant treatment of susceptible individuals57, raising the possibility that one way in which current antidepressants work is by inducing in depressed individuals some of the same adaptations that occur naturally in inherently resilient individuals. These insights thus suggest a new path forward for the development of new treatments of stress–related disorders: in addition to looking for ways to prevent or reverse the deleterious effects of stress, it should be possible to induce natural mechanisms of resilience, distinct from current antidepressant actions, in more vulnerable populations.
There is already considerable behavioral evidence for this approach. Stress resilience is enhanced in specific populations, such as military personnel and rescue workers, through controlled exposure to stress–related stimuli. Similarly, behavior therapy uses controlled stress exposure as one means to treat symptoms of mood and anxiety disorders. For example, exposure therapy and cognitive behavioral therapy can aid individuals with PTSD through cognitive restructuring and relaxation techniques following a traumatic event to promote recovery. These effects are well documented to reverse hyperactivity of PFC–amygdala microcircuits shown to be overactive in PTSD (for review see62). It is possible that similar behavioral approaches might be adopted in at risk populations to enhance resilience to subsequent stressful life events and prevent the development of these disorders.
Such treatments might be understood as being analogous to stress inoculation in that experiencing more moderate levels of stress, combined with techniques that reduce physiological and psychological perception of the trauma, can promote positive coping responses. The observation that such approaches significantly reduce PTSD severity supports the view that targeting underlying biological mechanisms of stress inoculation may provide us with novel protein targets for new medications that further promote resilience in at–risk populations. Likewise, identification of pro–resilience factors should make it possible to identify predictive biomarkers of resilience, which should greatly aid in recognizing at–risk populations.
Future Directions
As we learn more about the neurobiological mechanisms that confer resilience on an individual, the goal to develop treatment strategies to restore or enhance coping resources should improve the efficacy of treatment. However, we are just beginning to identify such resilience factors. A major gap in the field is the lack of coordination between human and animal studies. Human research has identified several tentative neuroendocrine concomitants of resilience (e.g., testosterone, NPY), which have not yet been adequately investigated mechanistically in animal models, while it remains challenging to experimentally interrogate the vast majority of neurobiological mechanisms discovered in animals (e.g., ΔFosB, K+ channels) in living humans.
Nevertheless, work to date identifies several important areas for future investigation. First, there is a great need for human brain imaging studies to determine the brain structures and circuits that mediate stress resilience. Deep brain stimulation in humans60 could potentially be used to provide highly valuable causal information about dysfunction of brain structures and circuits in depression and anxiety. This information could then be used in conjunction with optogenetic studies in rodent models, where we can more definitively describe the neural circuitry of resilience. Second, it is crucial to identify the range of heritable factors that help determine an individual’s capacity for resilience. Extrapolating from genetic studies of other complex human traits, it is likely that complex combinations of perhaps hundreds of genetic variations, rare and common in the population, comprise this genetic basis of resilience. Third, we must characterize the epigenetic mechanisms that control the degree to which this genetic predilection for resilience become manifest. Part of this epigenetic control of gene expression will occur in response to a host of environmental stimuli throughout life, but a portion may occur through random events during brain development. Fourth, far more insight is needed into the genetic, epigenetic, neurobiological, and neuroendocrine basis of sex differences in stress susceptibility vs. resilience. Finally, we need to better define how just the right type and level of stress inoculation, through this complex interplay of mechanisms, can promote resilience.
In the end, studies of resilience have unleashed a fundamentally novel way of understanding an individual’s responses to adverse life events, and have ushered in an exciting new era in studies of MDD, PTSD, and other stress–related disorders.
Acknowledgments
Preparation of this review was supported by grants from the National Institute of Mental Health: R01 MH090264 (SJR), K23 MH094707 (JWM), R01 MH092306 (MHH), and R01 MH51399, P50 MH66172, and P50 MH96890 (EJN).
References
- 1.Charney DS. Am J Psychiatry. 2004;161:195–216. doi: 10.1176/appi.ajp.161.2.195. [DOI] [PubMed] [Google Scholar]
- 2.Feder A, Nestler EJ, Charney DS. Nat Rev Neurosci. 2009;10:446–457. doi: 10.1038/nrn2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herman JP, Cullinan WE. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- 4.Stetler C, Miller GE. Psychosom Med. 2011;73:114–126. doi: 10.1097/PSY.0b013e31820ad12b. [DOI] [PubMed] [Google Scholar]
- 5.Meewisse ML, Reitsma JB, de Vries GJ, Gersons BP, Olff M. Br J Psychiatry. 2007;191:387–392. doi: 10.1192/bjp.bp.106.024877. [DOI] [PubMed] [Google Scholar]
- 6.Heim C, Newport DJ, Mletzko T, Miller AH, Nemeroff CB. Psychoneuroendocrinology. 2008;33:693–710. doi: 10.1016/j.psyneuen.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Heim C, Newport DJ, Miller AH, Nemeroff CB. JAMA. 2000;284:2321. [PubMed] [Google Scholar]
- 8.Yehuda R, Golier JA, Kaufman S. Am J Psychiatry. 2005;162:998–1000. doi: 10.1176/appi.ajp.162.5.998. [DOI] [PubMed] [Google Scholar]
- 9.Rasmusson AM, Vythilingam M, Morgan CA., 3rd CNS Spectr. 2003;8:651–656. 665–657. doi: 10.1017/s1092852900008841. [DOI] [PubMed] [Google Scholar]
- 10.Yehuda R, Brand SR, Golier JA, Yang RK. Acta Psychiatr Scand. 2006;114:187–193. doi: 10.1111/j.1600-0447.2006.00801.x. [DOI] [PubMed] [Google Scholar]
- 11.Butterfield MI, et al. Am J Psychiatry. 2005;162:380–382. doi: 10.1176/appi.ajp.162.2.380. [DOI] [PubMed] [Google Scholar]
- 12.Taylor MK, et al. Stress. 2011 [Google Scholar]
- 13.Oliveira T, Gouveia MJ, Oliveira RF. Psychoneuroendocrinology. 2009;34:1056–1064. doi: 10.1016/j.psyneuen.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Edwards DA, Wetzel K, Wyner DR. Physiol Behav. 2006;87:135–143. doi: 10.1016/j.physbeh.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 15.Morgan CA, 3rd, et al. Biol Psychiatry. 2000;47:891–901. doi: 10.1016/s0006-3223(99)00307-8. [DOI] [PubMed] [Google Scholar]
- 16.Mulchahey JJ, et al. Psychoneuroendocrinology. 2001;26:273–285. doi: 10.1016/s0306-4530(00)00052-4. [DOI] [PubMed] [Google Scholar]
- 17.Pope HG, Jr, Cohane GH, Kanayama G, Siegel AJ, Hudson JI. Am J Psychiatry. 2003;160:105–111. doi: 10.1176/appi.ajp.160.1.105. [DOI] [PubMed] [Google Scholar]
- 18.Morgan CA, 3rd, et al. Biol Psychiatry. 2000;47:902–909. doi: 10.1016/s0006-3223(99)00239-5. [DOI] [PubMed] [Google Scholar]
- 19.Morgan CA, 3rd, et al. Biol Psychiatry. 2002;52:136–142. doi: 10.1016/s0006-3223(02)01319-7. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Z, et al. Nature. 2008;452:997–1001. doi: 10.1038/nature06858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mickey BJ, et al. Arch Gen Psychiatry. 2011;68:158–166. doi: 10.1001/archgenpsychiatry.2010.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taliaz D, et al. J Neurosci. 2011;31:4475–4483. doi: 10.1523/JNEUROSCI.5725-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen H, et al. Neuropsychopharmacology. 2011 [Google Scholar]
- 24.Krishnan V, et al. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 25.Lehmann ML, Herkenham M. J Neurosci. 2011;31:6159–6173. doi: 10.1523/JNEUROSCI.0577-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delgado y Palacios R, et al. Biol Psychiatry. 2011;70:449–457. doi: 10.1016/j.biopsych.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 27.Golden SA, Covington HE, 3rd, Berton O, Russo SJ. Nat Protoc. 2011;6:1183–1191. doi: 10.1038/nprot.2011.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lutter M, et al. Nat Neurosci. 2008;11:752–753. doi: 10.1038/nn.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berton O, et al. Neuron. 2007;55:289–300. doi: 10.1016/j.neuron.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 30.Fleshner M, Maier SF, Lyons DM, Raskind MA. Stress. 2011;14:498–502. doi: 10.3109/10253890.2011.596865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vidal J, Buwalda B, Koolhaas JM. Behav Processes. 2011;88:76–80. doi: 10.1016/j.beproc.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Uchida S, et al. Neuron. 2011;69:359–372. doi: 10.1016/j.neuron.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 33.Mozhui K, et al. J Neurosci. 2010;30:5357–5367. doi: 10.1523/JNEUROSCI.5017-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andrus BM, et al. Mol Psychiatry. 2012;17:49–61. doi: 10.1038/mp.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nesse RM. Arch Gen Psychiatry. 2000;57:14–20. doi: 10.1001/archpsyc.57.1.14. [DOI] [PubMed] [Google Scholar]
- 36.Berton O, et al. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 37.Covington HE, 3rd, et al. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Covington HE, 3rd, Vialou VF, LaPlant Q, Ohnishi YN, Nestler EJ. Neurosci Lett. 2011;493:122–126. doi: 10.1016/j.neulet.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Masten AS. Am Psychol. 2001;56:227–238. doi: 10.1037//0003-066x.56.3.227. [DOI] [PubMed] [Google Scholar]
- 40.Bonanno GA. Am Psychol. 2004;59:20–28. doi: 10.1037/0003-066X.59.1.20. [DOI] [PubMed] [Google Scholar]
- 41.Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Arch Gen Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- 42.Levine S. Science. 1962;135:795–796. doi: 10.1126/science.135.3506.795-a. [DOI] [PubMed] [Google Scholar]
- 43.Lyons DM, Parker KJ, Schatzberg AF. Dev Psychobiol. 2010;52:616–624. doi: 10.1002/dev.20500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parker KJ, Buckmaster CL, Schatzberg AF, Lyons DM. Arch Gen Psychiatry. 2004;61:933–941. doi: 10.1001/archpsyc.61.9.933. [DOI] [PubMed] [Google Scholar]
- 45.Parker KJ, Buckmaster CL, Justus KR, Schatzberg AF, Lyons DM. Biol Psychiatry. 2005;57:848–855. doi: 10.1016/j.biopsych.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 46.Ricon T, Toth E, Leshem M, Braun K, Richter–Levin G. Stress. 2011 doi: 10.3109/10253890.2011.572207. [DOI] [PubMed] [Google Scholar]
- 47.Bradley RG, et al. Arch Gen Psychiatry. 2008;65:190–200. doi: 10.1001/archgenpsychiatry.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE. Am J Psychiatry. 2010;167:509–527. doi: 10.1176/appi.ajp.2010.09101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wallace DL, et al. Nat Neurosci. 2009;12:200–209. doi: 10.1038/nn.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McEwen BS, Gianaros PJ. Annu Rev Med. 2011;62:431–445. doi: 10.1146/annurev-med-052209-100430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luine V. Stress. 2002;5:205–216. doi: 10.1080/1025389021000010549. [DOI] [PubMed] [Google Scholar]
- 52.Christoffel DJ, et al. J Neurosci. 2011;31:314–321. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsankova NM, et al. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 54.Christoffel DJ, et al. Neuropsychopharmacology. 2012 doi: 10.1038/npp.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wood SK, Walker HE, Valentino RJ, Bhatnagar S. Endocrinology. 2010;151:1795–1805. doi: 10.1210/en.2009-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ono Y, et al. Stress. 2011 [Google Scholar]
- 57.Wilkinson MB, et al. J Neurosci. 2009;29:7820–7832. doi: 10.1523/JNEUROSCI.0932-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Price JL, Drevets WC. Neuropsychopharmacology. 2010;35:192–216. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murrough JW, Iacoviello B, Neumeister A, Charney DS, Iosifescu DV. Neurobiol Learn Mem. 2012 doi: 10.1016/j.nlm.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 60.Mayberg HS. J Clin Invest. 2009;119:717–725. doi: 10.1172/JCI38454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Tol MJ, et al. Biol Psychiatry. 71:593–602. doi: 10.1016/j.biopsych.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 62.Linden DE. Mol Psychiatry. 2006;11:528–538. doi: 10.1038/sj.mp.4001816. [DOI] [PubMed] [Google Scholar]
- 63.Christoffel DJ, Golden SA, Russo SJ. Rev Neurosci. 2011;22:535–549. doi: 10.1515/RNS.2011.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adamec R, Toth M, Haller J, Halasz J, Blundell J. Physiol Behav. 2012 doi: 10.1016/j.physbeh.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 65.Covington HE, 3rd, et al. J Neurosci. 2010;30:16082–16090. doi: 10.1523/JNEUROSCI.1731-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Katz M, et al. Dev Neurosci. 2009;31:293–299. doi: 10.1159/000216540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Milad MR, Orr SP, Pitman RK, Rauch SL. Psychophysiology. 2005;42:456–464. doi: 10.1111/j.1469-8986.2005.00302.x. [DOI] [PubMed] [Google Scholar]
- 68.Rauch SL, et al. Neuroreport. 2005;16:1909–1912. doi: 10.1097/01.wnr.0000186599.66243.50. [DOI] [PubMed] [Google Scholar]
- 69.Kozorovitskiy Y, et al. Proc Natl Acad Sci U S A. 2005;102:17478–17482. doi: 10.1073/pnas.0508817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lupien SJ, McEwen BS, Gunnar MR, Heim C. Nat Rev Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- 71.Vialou V, et al. Nat Neurosci. 2010;13:745–752. doi: 10.1038/nn.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vialou V, et al. J Neurosci. 2011;30:14585–14592. doi: 10.1523/JNEUROSCI.2496-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cao JL, et al. J Neurosci. 2010;30:16453–16458. doi: 10.1523/JNEUROSCI.3177-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goto Y, Otani S, Grace AA. Neuropharmacology. 2007;53:583–587. doi: 10.1016/j.neuropharm.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grace AA, Floresco SB, Goto Y, Lodge DJ. Trends Neurosci. 2007;30:220–227. doi: 10.1016/j.tins.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 76.Brischoux F, Chakraborty S, Brierley DI, Ungless MA. Proc Natl Acad Sci U S A. 2009;106:4894–4899. doi: 10.1073/pnas.0811507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lammel S, Ion DI, Roeper J, Malenka RC. Neuron. 70:855–862. doi: 10.1016/j.neuron.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shumake J, Ilango A, Scheich H, Wetzel W, Ohl FW. J Neurosci. 2010;30:5876–5883. doi: 10.1523/JNEUROSCI.3604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luscher C, Slesinger PA. Nat Rev Neurosci. 2010;11:301–315. doi: 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Balana B, et al. Proc Natl Acad Sci U S A. 2011;108:5831–5836. doi: 10.1073/pnas.1018645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weaver IC, et al. J Neurosci. 2005;25:11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meaney MJ, Szyf M. Dialogues Clin Neurosci. 2005;7:103–123. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Elliott E, Ezra–Nevo G, Regev L, Neufeld–Cohen A, Chen A. Nat Neurosci. 2011;13:1351–1353. doi: 10.1038/nn.2642. [DOI] [PubMed] [Google Scholar]
- 84.Moncek F, Duncko R, Johansson BB, Jezova D. J Neuroendocrinol. 2004;16:423–431. doi: 10.1111/j.1365-2826.2004.01173.x. [DOI] [PubMed] [Google Scholar]
- 85.LaPlant Q, et al. Biol Psychiatry. 2009;65:874–880. doi: 10.1016/j.biopsych.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Conrad CD, Grote KA, Hobbs RJ, Ferayorni A. Neurobiol Learn Mem. 2003;79:32–40. doi: 10.1016/s1074-7427(02)00018-7. [DOI] [PubMed] [Google Scholar]
- 87.Galea LA, et al. Neuroscience. 1997;81:689–697. doi: 10.1016/s0306-4522(97)00233-9. [DOI] [PubMed] [Google Scholar]
- 88.Bowman RE, Beck KD, Luine VN. Horm Behav. 2003;43:48–59. doi: 10.1016/s0018-506x(02)00022-3. [DOI] [PubMed] [Google Scholar]
- 89.Wood GE, Shors TJ. Proc Natl Acad Sci U S A. 1998;95:4066–4071. doi: 10.1073/pnas.95.7.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wood GE, Beylin AV, Shors TJ. Behav Neurosci. 2001;115:175–187. doi: 10.1037/0735-7044.115.1.175. [DOI] [PubMed] [Google Scholar]
- 91.Autry AE, Adachi M, Cheng P, Monteggia LM. Biol Psychiatry. 2009;66:84–90. doi: 10.1016/j.biopsych.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bowman RE, Ferguson D, Luine VN. Neuroscience. 2002;113:401–410. doi: 10.1016/s0306-4522(02)00156-2. [DOI] [PubMed] [Google Scholar]
- 93.Douglas AJ, Brunton PJ, Bosch OJ, Russell JA, Neumann ID. Endocrinology. 2003;144:5268–5276. doi: 10.1210/en.2003-0461. [DOI] [PubMed] [Google Scholar]
- 94.Wilkinson MB, et al. J Neurosci. 2011;31:9084–9092. doi: 10.1523/JNEUROSCI.0039-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Binder EB, et al. JAMA. 2008;299:1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ressler KJ, et al. Nature. 2011;470:492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Polanczyk G, et al. Arch Gen Psychiatry. 2009;66:978–985. doi: 10.1001/archgenpsychiatry.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stein MB, Campbell–Sills L, Gelernter J. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:900–906. doi: 10.1002/ajmg.b.30916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Murrough JW, Charney DS. Biol Psychiatry. 2011;69:510–512. doi: 10.1016/j.biopsych.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 100.Domschke K, et al. Eur Neuropsychopharmacol. 2010;20:301–309. doi: 10.1016/j.euroneuro.2009.09.006. [DOI] [PubMed] [Google Scholar]