Naïve pluripotency is associated with global DNA hypomethylation (original) (raw)
. Author manuscript; available in PMC: 2013 Sep 1.
Published in final edited form as: Nat Struct Mol Biol. 2013 Feb 17;20(3):311–316. doi: 10.1038/nsmb.2510
Abstract
Naïve pluripotent embryonic stem (ESCs) cells and embryonic germ (EGCs) cells are derived from the preimplantation epiblast and primordial germ cells (PGCs), respectively. We investigated whether differences exist between ESCs and EGCs in view of their distinct developmental origins. PGCs are programmed to undergo global DNA demethylation; however we find that EGCs exhibit equivalent levels of global DNA methylation to ESCs. Importantly, inhibition of MEK and Gsk3b by 2i conditions leads to a pronounced reduction in DNA methylation in both cell types. This is driven by Prdm14 and is associated with downregulation of Dnmt3a and Dnmt3b. However, genomic imprints are maintained in 2i and we report derivation of EGCs with intact genomic imprints. Collectively, our findings establish that culture in 2i instils a naïve pluripotent state with a distinctive epigenetic configuration that parallels molecular features observed in both the preimplantation epiblast and nascent PGCs.
INTRODUCTION
Pluripotency can be defined as the ability at the single cell level to engender all somatic cell lineages as well as germ cells. In the preimplantation embryo pluripotency is established in the epiblast of the late inner cell mass (ICM)1,2. These cells can be captured and maintained in culture as embryonic stem cells (ESCs)3-5. Both ICM cells and ESCs can contribute to chimeras and colonize the germline following reintroduction to the embryo, providing functional proof of their naïve pluripotency6-8. Conversely, neither postimplantation epiblast nor the primed pluripotent stem cells (EpiSCs) derived from this tissue have the capacity to contribute efficiently to chimeras following blastocyst integration9-11. Pluripotency is lost in the embryo upon somatic differentiation12 and can only be reinstated experimentally by reprogramming strategies13. However, in the developing postimplantation embryo, primordial germ cells (PGCs) can give rise to embryonic germ cells (EGCs)14,15 which exhibit all the properties of naïve pluripotent stem cells including contribution to chimeras16,17. Thus, preimplantation epiblast and PGCs share the distinctive capacity to give rise to naïve pluripotent stem cells under permissive conditions ex vivo.
During their development PGCs undergo a unique epigenetic reprogramming process that entails global chromatin rearrangement and DNA demethylation18,19. This includes erasure of DNA methylation marks at imprinted loci at E11.520. Consequently, EGC lines derived from PGCs at or after this stage lack DNA methylation imprint marks17,21 and this loss of imprinting leads to skeletal abnormalities in high contribution chimeras21. Previous reports have documented that EGC lines derived from early PGCs prior to E11.5 also exhibit imprint erasure, although in some cases less extensively than lines derived from later stages22. Consequently, to date, there has been no report of EGC lines derived with intact genomic imprints and this feature is widely considered to distinguish them from ESCs17,21.
Besides the loss of genomic imprints, it has been hypothesized that in connection with their PGC origin, EGCs are also characterized by global DNA hypomethylation23. To investigate what differences, if any, exist between ESCs and EGCs we measured gene expression and global DNA methylation levels in a large cohort of genetically matched cell lines. We show that ESCs and EGCs are highly similar at the transcriptome level. We further demonstrate that, contrary to previous assumptions, both ESCs and EGCs contain comparable levels of DNA methylation and that EGCs can be derived with imprints intact. We additionally show that global DNA methylation levels of both ESCs and EGCs are directly responsive to the culture environment and identify Prdm14 as a key factor underlying this epigenetic regulation.
RESULTS
Culture over origin defines pluripotent transcriptome
In order to assess similarities and differences between ESCs and EGCs that would reflect their distinct embryonic origin, we derived a large cohort of genetically identical EGC and ESC lines. We derived EGC lines from E8.5 mouse embryos using three different protocols: FCS+LIF on MEFs, FCS+LIF on MEFs for 48 hours followed by 2i+LIF and direct derivation into 2i+LIF24,25 (Fig. 1a). For direct comparison, ESC lines were derived in either FCS+LIF on MEFs or in 2i+LIF conditions and once established the cell lines were also switched between the two culture environments (hereafter referred to as FCS and 2i, respectively; Fig. 1a).
Figure 1. Culture environment defines the transcriptional profile of pluripotent stem cells.

(a) Summary of derivation procedure and culture conditions for ESC and EGC lines analyzed in this study; (b) Unsupervised hierarchical clustering of ESC and EGC lines demonstrates the dominant effect of culture condition on the transcriptional profile of pluripotent cells; (c) Transcript levels of germline marker genes, microarray log2 expression intensity. * FDR < 0.05, fold change > 1.5 for FCS versus 2i (see online methods for details), ≥ four independent cell lines for each condition (see Fig. 1a), error bars represent standard error of mean (SEM).
First, we set out to compare ESCs and EGCs at the transcriptional level. Unsupervised hierarchical clustering of Affymetrix gene expression data revealed the major distinction between all cell lines is culture condition and not the embryonic origin (Fig. 1b). We observed a dramatic effect of the culture environment on the pluripotent transcriptome in both EGCs and ESCs with 2016 genes differentially expressed between FCS and 2i (FDR < 0.05, fold change > 1.5, see online methods for details; Supplementary Table 1 and Supplementary Fig. 1). Additionally, EGCs and ESCs clustered based on the maintenance culture condition rather than the derivation procedure (Fig. 1b), supporting the inter-convertibility of the two molecular states defined by FCS or 2i26. These results thus indicate that the culture environment in which cells are maintained has a substantial and dominant effect over cellular origin with respect to global gene expression.
As separation between ESCs and EGCs may be eclipsed by the prominent difference between FCS and 2i, we performed unsupervised hierarchical clustering of cell lines cultured in FCS only (Supplementary Fig. 1a) or in 2i only (Supplementary Fig. 1b). While some separation was observed, the two cell types did not cluster discretely into two groups. Upon statistical analysis, only 83 genes were significantly different between ESC and EGC lines (FDR < 0.05, fold change > 1.5; Supplementary Table 1). Interestingly, as a group germline markers did not show a significant difference in expression between ESC and EGC lines (P = 0.300, Gene Set Enrichment Analysis (GSEA), normalized enrichment score (NES) = 1.11), although we noted an impact of the culture condition on the expression of this class of genes (Fig. 1c). Collectively our data shows that ESCs and EGCs are near identical at the transcriptional level and indicates a lack of appreciable transcriptional memory of the germline origin in EGCs.
Culture in 2i leads to global DNA hypomethylation
PGCs are programmed to undergo genome-wide erasure of DNA methylation including genomic imprints27. Therefore, we asked whether EGCs are also characterized by global DNA hypomethylation in comparison to ESCs. We analyzed global levels of 5-methylcytosine (5mC) by liquid chromatography-mass spectrometry (LC-MS) and detected no difference between ESCs and EGCs. However, to our surprise we found that all pluripotent cell lines (ESC and EGC) grown in 2i conditions have dramatically decreased global 5mC content (P < 0.0001, unpaired t-test, n ≥ 6; Fig. 2a and Fig. 2b). To evaluate this loss of DNA methylation further, we used thin layer chromatography (TLC) which specifically profiles 5mC present in CCGG sites, a typical feature of CpG islands. A large drop in 5mC was observed under 2i conditions (Fig. 2c), implying a major proportion of the reduction in DNA methylation occurred at these genomic regions.
Figure 2. Global DNA hypomethylation in 2i.

a) Analysis of global 5mC content by LC-MS in EGC and ESC lines cultured in FCS or 2i. Each data point represents the average of two technical replicates for an independent cell line. * P < 0.05, *** P < 0.001, as determined by unpaired t-tests; (b) Immunofluorescence staining for 5mC and 5hmC in EGC line EG-2i-1 cultured in FCS or 2i. DNA is counterstained with propidium iodide (PI). Scale bar, 10 μm; (c) Thin layer chromatography (TLC) analysis of methylation levels in DNA of ESC and EGC lines. Each cell line was grown side-by-side in 2i or FCS; Control oligonucleotides (oligo) containing 5mC or C are shown (d) Summary of bisulphite sequencing of repetitive elements in ESC line ES-2i-4 grown in FCS or 2i. * P < 0.05, ** P < 0.01, Mann-Whitney U-tests, ≥ eight clones for each condition; (e) Summary of bisulfite sequencing for Dazl promoter DNA methylation in ESC line ES-2i-4 and EGC line EG-3, both of which were cultured side-by-side in FCS or 2i.
To assess DNA methylation at both repetitive elements and single copy sequences under FCS and 2i conditions, we employed bisulphite sequencing. In agreement with global DNA hypomethylation, we observed significantly lower methylation levels in 2i at LINE1s (long interspersed nuclear element 1; P < 0.05, Mann-Whitney U-test) and minor and major satellites (P < 0.01, Mann-Whitney U-test), but not at IAPs (Fig. 2d and Supplementary Fig. 2a). Of interest, despite lower DNA methylation levels, the expression of these repetitive elements was not altered between culture conditions (Supplementary Fig. 2b). This implies that an alternative mechanism other than DNA methylation acts to control the expression of these repetitive elements in pluripotent cells, consistent with previous reports pointing towards the critical role of histone modifications in transcriptional repression of these sequences28.
In contrast, the expression of a number of genes has previously been shown to be regulated by DNA methylation in ESCs29,30. Consistent with this, we found reduced methylation in 2i in both ESCs and EGCs at the promoter of Dazl (Fig. 2e and Supplementary Fig. 2c,d) and Gstp2 (Supplementary Fig. 2d), both of which were upregulated under 2i culture conditions.
To examine the effect of reduced DNA methylation in 2i on global gene expression in pluripotent cells, we compared our microarray results with Dnmt1, Dnmt3a and Dnmt3b triple knockout ESC expression data29. We found that the group of genes affected by the triple Dnmt knockout were significantly affected by culture condition (P < 0.001, GSEA, NES = 1.23; Supplementary Fig. 2e). This overlap suggests that a proportion, but not all, of the transcriptional differences between FCS and 2i may be due to altered DNA methylation.
In order to elucidate the molecular grounds for the observed global DNA hypomethylation in 2i we assessed the expression of DNA methyltransferases. Using our microarray data, we observed a significant decrease in the expression of the de novo DNA methyltransferases Dnmt3a and Dnmt3b and their co-factor Dnmt3l in 2i conditions (FDR < 0.05, fold change > 1.5), while the expression of the maintenance DNA methyltransferase Dnmt1 remained unchanged. We confirmed these findings by quantitative real-time PCR (qPCR) and found that the Dnmt3a2 variant was specifically altered whereas Dnmt3a1 was not (Fig. 3a). These changes were also observed at the protein level, with low expression of Dnmt3a2 and Dnmt3b in cell lines cultured in 2i, regardless of their embryonic origin (Fig. 3b and Fig. 3c).
Figure 3. Characteristics of DNA methylation changes between culture conditions.

(a) qPCRs of Dnmt and Tet expression in EGCs maintained in 2i or FCS. ** P < 0.01, *** P < 0.001, unpaired t-tests, ≥ four independent cell lines for each condition, error bars represent SEM; (b) Western blot of Dnmt expression in ESCs and EGCs cultured in 2i or FCS for five passages; (c) Immunofluorescence staining for Dnmt3a and Dnmt3b in EGC line EG-2i-1 cultured in FCS or 2i. DNA counterstained with DAPI. Scale bar, 10 μm; (d) Global 5hmC content as a proportion of dG (left) or 5mdC (right) by LC-MS in a subset of ESC and EGC lines grown in FCS or 2i. Each data point represents the average of two technical replicates for an independent cell line. ** P < 0.01, unpaired t-tests; (e) 5mC abundance by LC-MS in ESCs and EGCs swapped from FCS to 2i conditions and vice versa for five passages. Each data point represents the average of two technical replicates for each cell line. * P < 0.05, ** P < 0.01, paired t-tests for swaps, unpaired t-test for 2i comparison; (f) qPCRs of Dnmt expression during embryoid body differentiation of cells grown in FCS or 2i. Oct4 and Sox17 expression shown for progression of differentiation; (g) Global 5mC levels by LC-MS in undifferentiated (D0) and day 4 embryoid bodies (D4) derived from pluripotent cells grown in FCS or 2i. Each data point represents the average of two technical replicates for one sample derived from an independent cell line. ** P < 0.01, unpaired t-tests.
The prominent changes in 5mC content in 2i may be linked to loss of 5mC through Tet driven hydroxylation31,32. Indeed, our microarray analysis as well as qPCR data showed changes in expression of Tet hydroxylases (Fig. 3a and Supplementary Fig. 3a). However, the hypomethylated cells grown under 2i conditions contained significantly reduced amounts of 5hmC (P < 0.01, unpaired t-test; Fig. 3d and Fig. 2b). Furthermore, the 5hmC/5mC ratio remained similar between cells grown in the different conditions suggesting that the reduced 5hmC abundance reflects the lower amount of available 5mC substrate (Fig. 3d).
Transcriptional profiles are reversible between culture conditions26, however DNA methylation is considered to be a relatively stable epigenetic mark. Surprisingly, we found a complete switch in global 5mC levels upon shifting the cells from 2i to FCS (P < 0.01, paired t-test) and vice versa (P < 0.05, paired t-test; Fig. 3e). This occurred within five passages, despite constant levels of Dnmt1 and increased expression of Uhrf1, a factor required for the maintenance methylation function of Dnmt133,34, in 2i conditions (Fig. 3a and Supplementary Fig. 3a). In this context we note that a group of factors implicated in the regulation of DNA methylation were differentially expressed between FCS and 2i culture conditions (P = 0.004, GSEA, NES = 1.36; Supplementary Fig. 3a). We thus suggest that the cumulative effect of downregulation of Dnmt3a, Dnmt3b and Dnmt3l in combination with changes in abundance of factors implicated in targeting of Dnmts may be responsible for the global DNA methylation changes upon transferring pluripotent cells from serum containing into 2i culture conditions.
Global DNA methylation is restored upon differentiation
Previous studies have documented that DNA methylation is dispensable for ESC self-renewal35-38, but is essential for the ability of cells to differentiate. As ESCs grown in 2i are not compromised in their ability to differentiate24,26, we investigated the dynamics of Dnmt expression during embryoid body differentiation. We found that hypomethylated EGCs and ESCs grown in 2i efficiently upregulated both Dnmt3a2 and Dnmt3b upon differentiation to levels similar to those seen after differentiation of cells grown in FCS (Fig. 3f). In agreement with this, global 5mC levels were equivalent in embryoid bodies derived from cells cultured in 2i or FCS (Fig. 3g).
Prdm14 is a critical regulator of DNA hypomethylation
Dnmt3b has been described as a direct target of Prdm14 in mouse ESCs39. We found that Prdm14 was highly upregulated in 2i as a response to both inhibitors (Fig. 4a and Fig. 1c), suggesting a plausible mechanism leading to Dnmt3b suppression and subsequent reduced DNA methylation in 2i. To test this hypothesis, we derived _Prdm14_−/− ESCs from heterozygous Prdm14+/− intercrosses using 2i (N.G, J. Tischler, H.G.L, M.A.S, unpublished data). We established by qPCR that Dnmt3b was highly upregulated in _Prdm14_−/− ESCs compared to wildtype ESCs grown in 2i conditions (P < 0.05, unpaired t-test; Fig. 4b). Additionally, we observed a slight upregulation of Dnmt3a1 (P < 0.01, unpaired t-test), but not of Dnmt1 or Dnmt3a2 (Fig. 4b). LC-MS analysis revealed that despite being maintained in 2i conditions, _Prdm14_−/− ESCs had global levels of DNA methylation comparable to wildtype ESCs cultured in FCS (Fig. 4c). These findings thus demonstrate that Prdm14 is a key driver of the DNA hypomethylation observed in 2i cultures.
Figure 4. Prdm14 regulates DNA methylation in pluripotent stem cells.
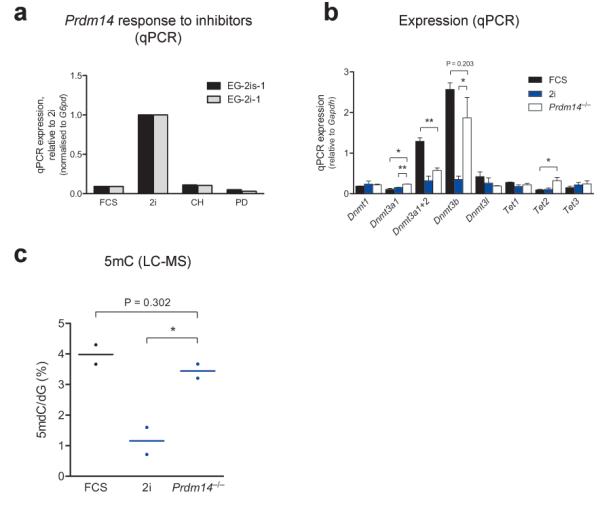
(a) Prdm14 expression as assessed by qPCR in cells grown in FCS, 2i or in the presence of a single Erk (PD) or Gsk3b (CH) inhibitor (LIF was added in all conditions); (b) qPCR analysis of Dnmt and Tet expression levels in _Prdm14_−/− ESCs (two independent cell lines) in comparison to representative ESC lines grown in FCS or 2i (three independent cell lines for each condition). * P < 0.05, ** P < 0.01, results of unpaired t-tests are shown for comparison to _Prdm14_−/− ESCs only, error bars represent SEM; (c) Analysis of global 5mC levels in _Prdm14_−/− ESCs in comparison to representative ESC lines grown in FCS or 2i. Each data point represents the average of two technical replicates for an independent cell line. * P < 0.05, unpaired t-tests.
ESCs and EGCs can maintain DMR methylation in 2i
Genomic imprinting is regulated by DNA methylation at imprinting control regions40. Using bisulphite sequencing, we found that ESCs grown in 2i maintain methylation at imprinted differentially methylated regions (DMRs) despite global DNA hypomethylation (Fig. 5a and Supplementary Fig. 4a). This is consistent with unchanged Dnmt1 levels between culture conditions and the described involvement of Dnmt1 in maintenance of DNA methylation at imprinted DMRs40. While Dnmt3a and Dnmt3b double knockout ESCs do show abnormalities in imprinted DNA methylation at high passage number36, low but detectable levels of Dnmt3a and Dnmt3b enzymes, combined with unaltered Dnmt1 levels, appear sufficient to prevent loss of methylation at these loci in 2i conditions.
Figure 5. Imprinted DMR methylation status of ESC and EGC lines.

(a) Summary of bisulphite sequencing of imprinted DMRs in ESC line ES-2i-4 cultured in 2i or FCS; (b) Summary of bisulphite analyses of imprinted DMRs in EGCs derived in 2i or FCS. Note that EGCs derived in 2i exhibit two distinct methylation patterns; (c) Bisulphite sequencing of imprinted DMRs in an EGC line derived in 2i following an interspecific cross, allowing the parental origin of each allele to be determined. Blue line represents the paternal allele and red the maternal allele. Filled and open circles represent methylated and unmethylated CpGs, respectively; (d) Image of chimeras generated by injection of EG-2i-1 (agouti) into C57BL/6 blastocysts (black).
Previous reports have indicated that EGCs derived at E8.5 show variability in the extent of erasure of DNA methylation marks at imprinted DMRs 17,22. However, most lines exhibit a high degree of imprint erasure and Kcnq1ot1 DMR methylation is invariably erased in all EGC lines tested thus far22. Consistent with this we found that our EGCs derived using the traditional protocol exhibited erasure of methylation at the Kcnq1ot1 and Peg3 DMRs, but retained methylation at the IG-DMR (Fig. 5b and Supplementary Fig. 4a). Our data hence confirms that the derivation of EGC lines using traditional conditions leads to the loss of genomic imprints. Surprisingly, EGC lines derived using 2i exhibited two distinct methylation patterns: two lines displayed almost complete imprint erasure and two lines retained a completely intact imprint methylation pattern (Fig. 5b and Supplementary Fig. 4a). Moreover, using EGCs derived in 2i from an interspecific cross we confirmed the presence of allele specific DMR methylation (Fig. 5c). Thus in total, we have derived three EGC lines with intact imprint marks using 2i, identifying a fundamental difference to historically derived EGC lines. The observed methylation profiles were retained through differentiation in embryoid bodies and were also generally stable upon switching the culture condition (data not shown).
The connection between loss of genomic imprinting in EGCs and compromised viability of high contribution chimeras has been well established21. Consistently, blastocyst injection of our EGCs derived and maintained in FCS produced very few live born pups (3 from 79 total blastocysts) and no chimeras (Supplementary Fig. 4b). Although heavily pregnant females were observed, the pregnancies were either spontaneously aborted or the pups immediately cannibalized. EG-2i-4, an EGC line derived in 2i which lacks imprints, similarly failed to produce chimeras (Supplementary Fig. 4b). To the contrary, and similar to our control ESC line (derived and maintained in 2i), we obtained robust contribution to chimeras upon injection of EG-2i-1, which showed normal methylation imprints (Fig. 5d and Supplementary Fig. 4a,b). The chimeras generated from EG-2i-l were also test-mated and produced germline transmission (Supplementary Fig. 4c). Our data thus clearly shows that 2i culture conditions allow for derivation of EGC lines with intact genomic imprints and hence neither the global level of DNA methylation nor the lack of genomic imprinting can be considered distinguishing features of pluripotent EGCs.
DISCUSSION
Our results reveal that both EGCs and ESCs reach a transcriptionally similar identity despite their distinct embryonic origins. We furthermore show that despite previous assumptions, EGCs and ESCs show comparable levels of global DNA methylation and EGC lines can be derived with intact genomic imprints. We thus establish that neither global levels of DNA methylation nor presence of genomic imprints are distinguishing features of pluripotent cells derived from PGCs or the ICM.
Our findings document that different culture conditions are associated with profound changes in global levels of DNA methylation. We demonstrate that pluripotent cells (both ESCs and EGCs) grown under 2i conditions are characterized by DNA hypomethylation and downregulated expression of the de novo DNA methyltransferases Dnmt3a and 3b, as well as Dnmt3l. As the naïve pluripotent state in 2i is characterized by global DNA hypomethylation and subsequent differentiation is accompanied by the accumulation of DNA methylation, the higher levels of methylation observed in FCS cultures might be indicative of a more advanced differentiation status8. The observed pronounced effect of Dnmt3a and Dnmt3b on the global levels of DNA methylation (Fig. 2 and Fig. 3) supports previous suggestions regarding the importance of a combination of both de novo and maintenance Dnmts to sustain genome-wide DNA methylation41 and might be indicative of an as yet unappreciated dynamic nature of DNA methylation.
Interestingly, downregulation of both Dnmt3a and Dnmt3b is observed in cells of early preimplantation epiblast42,43 and the blastocyst represents the nadir in global DNA methylation during early development44,45 (Fig. 6a). Aspects of this molecular state, such as downregulation of Dnmt3a and Dnmt3b and upregulation of pluripotency genes, are also recapitulated upon PGC specification46. We therefore propose that a characteristic feature of cells with the inherent capacity to generate pluripotency is the protection from de novo methylation. This naïve state is sustained or recapitulated in vitro under 2i conditions (Fig. 6b).
Figure 6. Naïve pluripotency is characterized by DNA hypomethylation.
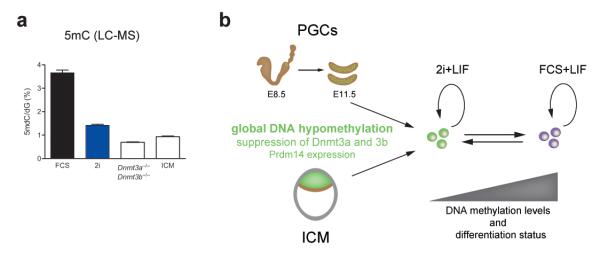
(a) LC-MS analysis of global 5mC levels in E4.0 inner cell mass (ICM) cells in comparison to ESC lines grown in FCS or 2i, and Dnmt3a and 3b double knockout ESCs. Two technical replicates were analyzed for each sample, error bars represent SEM; (b) Model depicting the capture of the naïve pluripotent state in 2i culture conditions. This state is characterized by DNA hypomethylation, suppression of de novo methyltransferases and expression of Prdm14, and thus has clear mechanistic parallels to the in vivo sources of naïve pluripotent stem cells. Although pluripotent cells cultured in FCS conditions are still categorized as naïve we propose that they are somewhat more advanced in their differentiation status. This is evidenced by decreased Prdm14 levels, increased expression of the de novo methyltransferases and the accumulation of DNA methylation.
Our data demonstrates that the PGC determinant Prdm14 is upregulated in 2i conditions and plays a critical role in maintaining the observed DNA hypomethylation in vitro. Prdm14 is expressed in both the ICM of the preimplantation embryo and in nascent PGCs47 and it is thus conceivable that it influences global DNA methylation also in these contexts. We also note that Prdm14 might act through multiple downstream effectors; almost half (106/231) of the Prdm14 target genes that are responsive to Prdm14 knockdown in ESCs39 are significantly affected by culture condition (FDR < 0.05, fold change > 1.5; Supplementary Fig. 3c). Prdm14 targets include numerous epigenetic modifiers. Consistent with these findings, we observe altered expression of several of these factors in 2i conditions alongside changes in the abundance of relevant histone modifications, some of which have been directly implicated in the regulation of de novo DNA methylation (Supplementary Fig. 3). Similar changes in epigenetic regulators and histone modifications are observed in both ICM and nascent PGCs expressing Prdm14 in vivo, suggesting a common epigenetic signature in cells with the capacity to generate pluripotency.
Finally, we demonstrate that similar to the ICM in vivo, imprints can be maintained in pluripotent cells in 2i even in the context of global DNA hypomethylation. In fact, the derivation of EGCs with normal imprints is actually facilitated by 2i and, to our knowledge, this is the first report of EGCs with intact genomic imprint marks. The difference in imprint status between EGC lines that were obtained from the same derivation experiment reflects either molecular heterogeneity of the starting PGC population at E8.5 or emerges during the derivation process. In either case, our results demonstrate that loss of imprints is not a prerequisite for EGC derivation. As imprint instability has been demonstrated in some ESC lines48,49 we suggest that imprint status is not a reliable method to discriminate between ESCs and EGCs.
Collectively our results provide new insights into the unique epigenetic status of naïve pluripotency with distinct mechanistic parallels to the epigenetic changes occurring during developmental acquisition of pluripotent capacity in the mouse embryo (Fig. 6b).
ONLINE METHODS
Animal studies were authorized by the Cambridge University ethics review committee and a UK Home Office Project License, and carried out in a Home Office-designated facility.
Cell culture
FCS medium consists of DMEM-F12 (Gibco) supplemented with 15% FCS, 0.1 mM MEM non-essential amino acids, 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM 2-mecaptanethanol and mouse LIF 10 μg/ml (prepared in house). Cells cultured in serum were maintained on a mouse embryonic fibroblast (MEF) feeder layer. For downstream analysis MEFs were removed by serial panning and the purity of ESCs or EGCs confirmed by alkaline phosphatase staining. 2i medium consists of the MEK inhibitor PD0325901 (PD) 1 μM, the GSK3 inhibitor CHIR99021 (CH) 3 μM and mouse LIF 10 μg/ml (as above) in N2B27 medium50. Cells cultured in 2i were maintained on laminin (10 μg/ml, Sigma). Cells were passaged by dissociation with trypsin and replating every 2-3 days. ESCs were derived in serum conditions or in 2i as described previously51,52. EGCs were derived as described previously25. All cell lines were derived from embryos produced by crossing mixed background Oct4ΔPE-GFP transgenic males53 with strain-129SvEv female mice.
Passage numbers
The passage number for all the cell lines shown in Fig. 1a ranges between 7 and 15. The ESCs which were split into different conditions were cultured side-by-side for 5 passages in 2i and FCS. In Fig. 3e, the cells were either maintained in their usual culture condition (for instance, 2i) or swapped to the alternative condition (FCS, and vice versa) for 5 passages. Therefore, the passage numbers for this experiment range between 17 and 22.
Chimeras and embryos
Mouse chimeras were produced by micro-injection of EGCs or ESCs (agouti) into E3.5 C57BL/6 blastocysts. Chimerism was assessed by agouti coat colour. Inner cell masses (ICMs) were isolated from embryonic day (E) 4.0 blastocysts by immunosurgery as described previously54.
Affymetrix microarrays and data analysis
100 ng of RNA isolated using the Qiagen RNeasy Mini kit was processed with the Ambion WT Expression kit. Samples were fragmented, labelled and hybridized to Affymetrix GeneChip Mouse Gene 1.0 ST Arrays as per the Affymetrix GeneChip WT Terminal Labelling and Hybridisation kit and scanned with the GeneChip scanner. At least four biological replicates were included for each condition (see Fig. 1a). Quality control was undertaken using Affymetrix Expression Console software. Preprocesing by robust multiarray averaging (RMA) was performed in R using the Bioconductor package linear models for microarray data (limma)55 and batches were corrected using Combat56. Statistical testing was performed using limma using log2 transformed data; differentially expressed genes were identified using thresholds of Benjamini-Hochberg FDR < 0.05 and fold change > 1.5. Gene Set Enrichment Analysis (GSEA)57 was used for statistical testing of differential expression of gene sets. Hierarchical clustering and heat maps were generated using Partek software version 6.6.
Real time quantitative PCR
Random primed reverse transcription was performed using the Invitrogen Superscript III kit on 1 μg RNA for cell line samples or 250 ng RNA for embryoid body samples. 3 μl of 1:40 dilutions (1:10 for embryoid body samples) was added to a 10 μL reaction mix (Bioline Sensimix SYBR No-Rox) and qPCR was performed using the standard curve method. Cycling conditions were 95°C 10 min, 40 cycles of 95°C 15 sec, 60°C 15 sec, 72°C 15 sec, followed by a melting curve from 55°C to 95°C (1°C increments). Four genes were tested for their use as a normalisation gene (glucose-6-phosphate dehydrogenase X-linked (G6pd), glyceraldehyde-3-phosphate dehydrogenase (Gapdh), beta-actin and hypoxanthine guanine phosphoribosyl transferase); G6pd was selected due to its consistency of expression between the sample sets shown in Fig. 1a; Gapdh was selected for Prdm14 knockout sample comparisons. Major satellites were assessed by semi-quantitative PCR due to the presence of multiple PCR products. All primer pairs (sequences available upon request) were annealed at 60°C. Two-tailed unpaired t-tests were performed using Graphpad Prism software.
Immunofluorescence staining
For 5mC and 5hmC staining cells grown on Labtech slides were briefly washed in PBS and fixed in 4% paraformaldehyde (PFA) (prepared in PBS) for 15 min at room temperature. The cells were subsequently permeabilized using PBS, 1% BSA, 0.5% Triton X-100 (30 min), washed in PBS and incubated with RNAse A (20 mg/ml in PBS) for 1 hr at 37°C. Following subsequent PBS washes, the DNA was denatured in 4 M HCl for 10 min at 37°C, the slides neutralized using extensive PBS washes, blocked in PBS, 1% BSA, 0.1% Triton X-100 (30 min) and incubated with the antibody (5hmC Active Motif 39791 (polyclonal) 1:400; 5mC Diagenode 081100 (monoclonal) 1:4,000) in the same buffer at 4°C overnight. The slides were subsequently washed three times in PBS, 1% BSA, 0.1% Triton X-100 and incubated with Alexa fluorophore conjugated secondary antibodies (Invitrogen) for 1 hr at room temperature in the dark, washed in PBS, 1% BSA, 0.1% Triton X-100 and twice in PBS. Finally, the slides were mounted in Vectashield containing DAPI (Vector Laboratories) and imaged using Leica SP5 confocal microscope. Immunofluorescence detection of Dnmts and Oct4 expression was carried out as above without the RNAse and HCl denaturation steps (Dnmt3a: Imgenex IMG-268A (monoclonal) 1:10; Dnmt3b: Imgenex IMG-268A (monoclonal) 1:400; Oct4: Abcam Ab19857 (polyclonal) 1:100). DNA was visualized by DAPI.
Western hybridisation
For DNA methyltransferase western hybridisations, cells were lysed in buffer containing 150 mM NaCl, 50 mM Tris-HCl pH 7.5, 0.5% Triton X-100 (v/v), and protease inhibitor cocktail (Roche). 10 μg of homogenized protein was loaded onto an 8% acrylamide/bis gel and transferred to a PVDF membrane after electrophoresis. After blocking with 5% non-fat milk for 1 hr, membranes were incubated at 4°C overnight with primary antibodies at the following dilutions: Dnmt1 Imgenex IMG-261 (monoclonal) 1:500, Dnmt3a Imgenex IMG-268A (monoclonal) 1:500, Dnmt3b Imgenex IMG-184A (monoclonal) 1:20,000, laminB (C-20) Santa Cruz sc-6216 (polyclonal) 1:10,000. HRP conjugated secondary antibodies were incubated for 1 hr at room temperature. Amersham ECL western blotting analysis systems were used for detection on a GE ImageQuant LAS 4000 mini.
For histone modification western hybridisations, histones were extracted using Triton extraction buffer (PBS containing 0.5% Triton X-100 (v/v), 2 mM phenylmethylsulfonyl fluoride (PMSF), 0.02% (w/v) NaN3). After 10 min on ice, samples were centrifuged, washed in Triton extraction buffer then incubated in 0.2 N HCl overnight at 4°C. Supernatants were measured by Bradford assay following centrifugation. 0.5 μg of histone protein was loaded on 20% acrylamide/bis gels and after electrophoresis were transferred to PVDF membranes. Blocking was performed with 5% BSA for 1 hr at room temperature. Membranes were then incubated for 1 hr with primary antibodies at the following dilutions: H3K9me2 Upstate 07-441 (polyclonal) 1:5,000, H3K9me3 Abcam ab8898 (polyclonal) 1:5,000 , H4K20me3 Upstate 07-463 (polyclonal) 1:10,000, H3K27me3 Cell Signalling 97565 (polyclonal) 1:10,000, H3K4me2 Upstate 04-030 (polyclonal) 1:10,000, H3K4me3 Abcam ab8580 (polyclonal) 1:10,000, H3K36me3 Upstate 05-801 (monoclonal) 1:10,000. Secondary antibodies were incubated for 1 hr at room temperature.. Blots were developed using Amersham ECL and signal was detected using standard chemiluminescent Kodak BioMax light film.
Thin layer chromatography (TLC)
Genomic DNA (500 ng) was digested with the restriction enzyme MspI overnight at 37°C. Oligonucleotides containing only mdC or dC were used as controls and subjected to the same treatment as genomic DNA. Following incubation for 1 hr at 37°C with 2.5 U of antarctic phosphatase, samples were purified using the Qiagen QIAquick PCR purification kit. 80 ng of the eluate was end-labelled for 1 hr at 37°C with 2 μl (approximately 0.02 uCi) of P32-ATP using 10 U T4 polynucleotide kinase. Unincorporated isotopes were removed by Qiagen column purification. DNA quantity equivalent of 50,000 counts was aliquoted and denatured by heating at 99°C. The reaction was incubated with 2 U of Nuclease P1 and 10 mM ammonium acetate (pH 5.3) for 2 hr at 45°C in a 5 μL reaction volume. 1.5 μL of digested DNA was spotted onto a cellulose TLC plate and developed for approximately 8 hr in isobutyric acid-water-30% ammonium hydroxide (66🔞3) in a sealed TLC tank. The TLC plate was dried and incubated in an autoradiography cassette overnight. Radioactivity levels were recorded using a Fujifilm FLA-5100 phosphoimager.
Mass spectrometry
Up to 500 ng genomic DNA was denatured by heating at 100°C for 3 min. Samples were incubated with 1/10 volume of 0.1 M ammonium acetate (pH 5.3) and 2 U of nuclease P1 for 2 hr at 45°C. 1/10 volume of 1 M ammonium bicarbonate and 0.002 units of phosphodiesterase I were added, followed by incubation for 2 hr at 37°C. Finally, samples were incubated for 1 hr at 37°C with 0.5 U alkaline phosphatase. Samples were subsequently diluted in 2 mM ammonium formate (pH 5.5). The nucleosides were separated on an Agilent RRHD Eclipse Plus C18 2.1 × 100 mm 1.8u column using the HPLC system 1200 (Agilent) and analyzed using Agilent 6490 triple quadrupole mass spectrometer. To calculate the concentrations of individual nucleosides within the samples analyzed, standard curves with known amounts of synthetic nucleosides were generated and used to convert the peak area values to corresponding concentrations.
Additional methods are provided in the Supplementary Note.
Supplementary Material
1
2
ACKNOWLEDGEMENTS
We thank members of the Smith, Surani and Hajkova labs for stimulating discussions and C. Mulas for comments on the manuscript. We are also grateful to B. Snijders from the MRC Clinical Sciences Centre Proteomics facility and Agilent Technologies for their support regarding the LC-MS analysis. Affymetrix array hybridisation and quality control was carried out by the Genomics Laboratory at the MRC Clinical Sciences Centre. A.S. is a Medical Research Council Professor. This work was supported by Medical Research Council funding to P.H. (MC A652 5PY70), as well as a Wellcome Trust grant to M.A.S. (RG49135) and Medical Research Council funding to A.S. (G1001028)
Footnotes
REFERENCES
- 1.Gardner RL, Beddington RS. Multi-lineage ‘stem’ cells in the mammalian embryo. J. Cell Sci. Suppl. 1988;10:11–27. doi: 10.1242/jcs.1988.supplement_10.2. [DOI] [PubMed] [Google Scholar]
- 2.Nichols J, Smith A. Pluripotency in the Embryo and in Culture. Cold Spring Harbor Perspectives in Biology. 2012;4:a008128–a008128. doi: 10.1101/cshperspect.a008128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 4.Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci USA. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brook FA, Gardner RL. The origin and efficient derivation of embryonic stem cells in the mouse. Proc Natl Acad Sci USA. 1997;94:5709–5712. doi: 10.1073/pnas.94.11.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardner RL, Rossant J. Investigation of the fate of 4-5 day post-coitum mouse inner cell mass cells by blastocyst injection. J Embryol Exp Morphol. 1979;52:141–152. [PubMed] [Google Scholar]
- 7.Bradley A, Evans M, Kaufman MH, Robertson E. Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature. 1984;309:255–256. doi: 10.1038/309255a0. [DOI] [PubMed] [Google Scholar]
- 8.Nichols J, Smith A. Naive and Primed Pluripotent States. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 9.Rossant J, Gardner RL, Alexandre HL. Investigation of the potency of cells from the postimplantation mouse embryo by blastocyst injection: a preliminary report. J Embryol Exp Morphol. 1978;48:239–247. [PubMed] [Google Scholar]
- 10.Brons IGM, et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- 11.Tesar PJ, et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- 12.Osorno R, et al. The developmental dismantling of pluripotency is reversed by ectopic Oct4 expression. Development. 2012;139:2288–2298. doi: 10.1242/dev.078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010;465:704–712. doi: 10.1038/nature09229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsui Y, Zsebo K, Hogan BL. Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture. Cell. 1992;70:841–847. doi: 10.1016/0092-8674(92)90317-6. [DOI] [PubMed] [Google Scholar]
- 15.Resnick JL, Bixler LS, Cheng L, Donovan PJ. Long-term proliferation of mouse primordial germ cells in culture. Nature. 1992;359:550–551. doi: 10.1038/359550a0. [DOI] [PubMed] [Google Scholar]
- 16.Stewart CL, Gadi I, Bhatt H. Stem cells from primordial germ cells can reenter the germ line. Dev Biol. 1994;161:626–628. doi: 10.1006/dbio.1994.1058. [DOI] [PubMed] [Google Scholar]
- 17.Labosky PA, Barlow DP, Hogan BL. Mouse embryonic germ (EG) cell lines: transmission through the germline and differences in the methylation imprint of insulin-like growth factor 2 receptor (Igf2r) gene compared with embryonic stem (ES) cell lines. Development. 1994;120:3197–3204. doi: 10.1242/dev.120.11.3197. [DOI] [PubMed] [Google Scholar]
- 18.Hajkova P, et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452:877–881. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hajkova P, et al. Genome-Wide Reprogramming in the Mouse Germ Line Entails the Base Excision Repair Pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hajkova P, et al. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 21.Tada T, et al. Epigenotype switching of imprintable loci in embryonic germ cells. Dev Genes Evol. 1998;207:551–561. doi: 10.1007/s004270050146. [DOI] [PubMed] [Google Scholar]
- 22.Shovlin TC, Durcova-Hills G, Surani A, Mclaren A. Heterogeneity in imprinted methylation patterns of pluripotent embryonic germ cells derived from pre-migratory mouse germ cells. Dev Biol. 2008;313:674–681. doi: 10.1016/j.ydbio.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 23.Tada M, Tada T, Lefebvre L, Barton SC, Surani MA. Embryonic germ cells induce epigenetic reprogramming of somatic nucleus in hybrid cells. EMBO J. 1997;16:6510–6520. doi: 10.1093/emboj/16.21.6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ying Q-L, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leitch HG, et al. Embryonic germ cells from mice and rats exhibit properties consistent with a generic pluripotent ground state. Development. 2010;137:2279–2287. doi: 10.1242/dev.050427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marks H, et al. The Transcriptional and Epigenomic Foundations of Ground State Pluripotency. Cell. 2012;149:590–604. doi: 10.1016/j.cell.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hajkova P. Epigenetic reprogramming - taking a lesson from the embryo. Curr Opin Cell Biol. 2010;22:342–350. doi: 10.1016/j.ceb.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 28.Hutnick LK, Huang X, Loo T-C, Ma Z, Fan G. Repression of retrotransposal elements in mouse embryonic stem cells is primarily mediated by a DNA methylation-independent mechanism. Journal of Biological Chemistry. 2010;285:21082–21091. doi: 10.1074/jbc.M110.125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karimi MM, et al. DNA Methylation and SETDB1/H3K9me3 Regulate Predominantly Distinct Sets of Genes, Retroelements, and Chimeric Transcripts in mESCs. Cell Stem Cell. 2011;8:676–687. doi: 10.1016/j.stem.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hackett JA, et al. Germline DNA Demethylation Dynamics and Imprint Erasure Through 5-Hydroxymethylcytosine. Science. 2012 doi: 10.1126/science.1229277. doi:10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bostick M, et al. UHRF1 Plays a Role in Maintaining DNA Methylation in Mammalian Cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 34.Sharif J, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 35.Lei H, et al. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 36.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson M, et al. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol. 2004;24:8862–8871. doi: 10.1128/MCB.24.20.8862-8871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsumura A, et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006;11:805–814. doi: 10.1111/j.1365-2443.2006.00984.x. [DOI] [PubMed] [Google Scholar]
- 39.Ma Z, Swigut T, Valouev A, Rada-Iglesias A, Wysocka J. Sequence-specific regulator Prdm14 safeguards mouse ESCs from entering extraembryonic endoderm fates. Nat Struct Mol Biol. 18:120–7. doi: 10.1038/nsmb.2000. [DOI] [PubMed] [Google Scholar]
- 40.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 41.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–811. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirasawa R, et al. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008;22:1607–1616. doi: 10.1101/gad.1667008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirasawa R, Sasaki H. Dynamic transition of Dnmt3b expression in mouse pre- and early post-implantation embryos. Gene Expr Patterns. 2009;9:27–30. doi: 10.1016/j.gep.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Borgel J, et al. Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet. 2010;42:1093–1100. doi: 10.1038/ng.708. [DOI] [PubMed] [Google Scholar]
- 45.Smith ZD, et al. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–344. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurimoto K, et al. Complex genome-wide transcription dynamics orchestrated by Blimp1 for the specification of the germ cell lineage in mice. Genes Dev. 2008;22:1617–1635. doi: 10.1101/gad.1649908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamaji M, et al. Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nat Genet. 2008;40:1016–1022. doi: 10.1038/ng.186. [DOI] [PubMed] [Google Scholar]
- 48.Dean W, et al. Altered imprinted gene methylation and expression in completely ES cell-derived mouse fetuses: association with aberrant phenotypes. Development. 1998;125:2273–2282. doi: 10.1242/dev.125.12.2273. [DOI] [PubMed] [Google Scholar]
- 49.Humpherys D, et al. Epigenetic instability in ES cells and cloned mice. Science. 2001;293:95–97. doi: 10.1126/science.1061402. [DOI] [PubMed] [Google Scholar]
- 50.Ying Q-L, Stavridis M, Griffiths D, Li M, Smith A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotech. 2003;21:183–186. doi: 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
- 51.Tang F, et al. Tracing the Derivation of Embryonic Stem Cells from the Inner Cell Mass by Single-Cell RNA-Seq Analysis. Cell Stem Cell. 2010;6:468–478. doi: 10.1016/j.stem.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nichols J, Silva J, Roode M, Smith A. Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development. 2009;136:3215–3222. doi: 10.1242/dev.038893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshimizu T, et al. Germline-specific expression of the Oct-4/green fluorescent protein (GFP) transgene in mice. Dev Growth Differ. 1999;41:675–684. doi: 10.1046/j.1440-169x.1999.00474.x. [DOI] [PubMed] [Google Scholar]
- 54.Nichols J, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 55.Smyth GK. In: Bioinformatics and Computational Biology Solutions using R and Bioconductor. Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- 56.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2006;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 57.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2