Landscape of Somatic Retrotransposition in Human Cancers (original) (raw)
. Author manuscript; available in PMC: 2013 May 17.
Published in final edited form as: Science. 2012 Jun 28;337(6097):967–971. doi: 10.1126/science.1222077
Abstract
Transposable elements (TEs) are abundant in the human genome, and some are capable of generating new insertions through RNA intermediates. In cancer, the disruption of cellular mechanisms that normally suppress TE activity may facilitate mutagenic retrotranspositions. We performed single-nucleotide resolution analysis of TE insertions in 43 high-coverage whole-genome sequencing data sets from five cancer types. We identified 194 high-confidence somatic TE insertions, as well as thousands of polymorphic TE insertions in matched normal genomes. Somatic insertions were present in epithelial tumors but not in blood or brain cancers. Somatic L1 insertions tend to occur in genes that are commonly mutated in cancer, disrupt the expression of the target genes, and are biased toward regions of cancer-specific DNA hypomethylation, highlighting their potential impact in tumorigenesis.
Transposable elements (TEs) have proliferated in mammalian genomes by integrating new copies primarily through RNA-mediated mechanisms. Whereas most TEs are inactive remnants fixed within the human population, younger TEs account for much of the structural variation among individual genomes (1). TE activity in somatic tissues is normally repressed through epigenetic and post-transcriptional mechanisms (2–4), but some TEs escape repression and generate new polymorphic insertions during the transient release of these repression mechanisms in germ cells and during early embryonic development (5–7). Some of these insertions can occur later in life (8) and result in disease-causing gene alterations (9), such as the few examples reported in colon and lung cancers (10, 11). However, despite its mutagenic potential, the extent to which somatic retrotransposition contributes to tumorigenesis in various tissues remains largely unexplored. Here, we identify TE integration sites at single-nucleotide resolution, characterizing the insertional mechanisms and distinguishing retrotransposition from other types of genomic rearrangements common in cancer genomes.
Analysis of TEs with short-read sequencing is challenging, because numerous, often nearly identical TE instances make it difficult to discern the true source of the sequenced fragments. Detection of somatic TE insertions in cancer genomes is further complicated by heterozygosity, cellular and genetic heterogeneity of tumor samples, and complex genomic rearrangements found in many cancers. We developed Tea (TE analyzer), a computational method that detects the exact position and mechanism of TE insertions from paired-end whole-genome sequencing data with high accuracy (Fig. 1).
Fig. 1.

(A) To detect somatic insertions of TEs, paired-end sequencing data from tumor and matched normal samples are aligned to both the reference genome and a custom repeat assembly of canonical and divergent TE sequences. Two types of supporting reads are identified: (i) repeat-anchored mate (RAM) reads, in which one of the paired-end reads is mapped to a unique location in the genome, whereas the other is associated with a TE (reads 1 to 4), and (ii) clipped reads, which span the TE insertion breakpoints and show partial alignment to the reference or the repeat assembly (reads 5 to 8). The distances between the clipping positions and the clipped sequences are used to infer the insertion mechanism. For instance, duplicated sequences at the insertion site (TSD) and the poly-A tail of the inserted TE are characteristics of an endonuclease-mediated target-primed retrotransposition. (B) Example: a validated somatic L1 insertion in the 3′ UTR of GPATCH2 in colorectal cancer (CR3518). The top chart displays two clusters of RAM reads (green) whose mate pairs (not shown) are associated with L1 repeat sequences. Clipped (partially aligned) reads spanning the insertion breakpoint are shown underneath, with each nucleotide in a different color (nucleotides matching the reference are not shown). The consecutive red bases to the right of the insertion come from the poly-T tail of the inserted L1 in the negative orientation. The separation of clipped read positions between the strands reveals a 19-bp TSD (bottom). No RAMs or clipped reads are observed in the matched normal (blood) sample.
We applied Tea to whole-genome sequencing data from tumor and matched normal blood samples from a total of 43 colorectal, prostate, ovarian, multiple myeloma, and glioblastoma cancer patients (table S1). Our analysis revealed 194 high-confidence somatic TE insertions (183 L1s, 10 Alus, and 1 ERV) with the average per tumor type ranging from 0 to 29 (Fig. 2A and table S2). All of the somatic L1 and Alu insertions were observed in the cancers of epithelial cell origin (colorectal, prostate, and ovarian), with colorectal tumors showing the highest frequency of somatic L1 insertions, but not in the blood or brain cancers. Some of the TE insertions detected by our method may be generated by mechanisms other than classical retrotransposition. Although none of the ERV1 family elements are thought to be active in humans, we observed an ERV somatic insertion (PABL_A repeat, ERV1 family) in a myeloma sample in the intron of DAPK1 (death-associated protein kinase 1), later validated by polymerase chain reaction (PCR) and Sanger sequencing (fig. S1 and tables S3 and S4). Given the small insertion size [375 base pairs (bp)] and 2-bp microhomology at both breakpoint junctions, the ERV1 insertion was likely generated via the microhomology-mediated break-induced repair mechanism (12).
Fig. 2.

(A) Frequency of high-confidence somatic L1 insertions varies across 5 colorectal, 7 prostate, 8 ovarian, 7 multiple myeloma, and 16 glioblastoma tumors. Three epithelial cancers (colorectal, prostate, and ovarian) show frequent somatic L1 insertions, whereas no insertions are observed in the blood and brain cancers. One colorectal tumor (CR3518) contains 102 L1 insertions, increasing the average somatic event frequency for colorectal tumors from 9 to 28 when this sample is included. (B) The genes affected by somatic TE insertions are significantly enriched for genes with high mutation rates as estimated from the exome sequencing data of 228 additional colorectal tumors (P < 1 × 10−15). The mutation frequency of each gene was adjusted for its total exon size. (Inset) The top 15 genes with nonsilent mutations. (C) The transcript levels of 45 genes with somatic TE insertions in colorectal tumors were compared with those from 28 normal colorectal tissues, and the expression fold changes are shown. Overall, the genes with a TE insertion were significantly down-regulated in tumors (P = 6.3 × 10−4, background distribution based on randomly sampled gene sets). KCNIP1 appears twice because of two somatic insertions in two different samples. The dashed line marks 50% reduction in expression.
One colorectal tumor (CR3518) showed a high somatic TE insertion frequency (106 events) along with microsatellite instability, a high nonsilent single-nucleotide or short insertion/deletion (indel) mutation rate (430 mutations), altered DNA repair pathways via MLH1 epigenetic silencing and a POLE mis-sense mutation and belonged to the high CpG island methylator phenotype (CIMP-high) (table S5). In contrast, the other colorectal tumors (13) were microsatellite-stable, had a low simple mutation rate (45 to 60 mutations), lacked detectable aberrations among the DNA mismatch repair genes tested, and belonged to CIMP-low or non-CIMP methylation subgroups (table S5). Taken together, this suggests the presence of tumor subtypes with respect to TE activity.
A representative set of predicted insertions was selected for PCR-based validation (supplementary text S1). We confirmed 25 out of 26 somatic L1 insertions examined in colorectal tumors and all of the 13 L1 insertions examined in ovarian cancers, resulting in an overall accuracy of 97% (table S3). Six junctions and two entire L1 insertions were further examined by using Sanger sequencing, confirming the presence of the L1 sequences, insertion breakpoints, target-site duplication (TSD) sequences, and poly-A tails (table S4). The sensitivity of detection depends on the purity of the tumor samples and clonality of the events. Our approach was able to detect heterozygous insertions in samples with tumor purity as low as 49% (fig. S2). Events present at lower frequency, such as insertions accumulated at later stages of tumorigenesis, were likely missed. Our approach cannot detect insertions landing in highly repetitive regions, so the true number of events is likely to be higher.
Of the 194 detected somatic TE insertions, 64 were located within 62 annotated genes (table S2), including those implicated for tumor suppressor functions through deletions or epigenetic silencing (supplementary text S2): for example, NELL1, DBC1, ROBO2, and PARK2 (14–18). The affected set was enriched for genes associated with cell-adhesion functions (P = 9.8 × 10−5; false discovery rate = 0.04), including CDH12, ROBO2, NRXN3, FPR2, COL11A1, NEGR1, NTM, and CTNNA2. We examined nucleotide mutation frequencies (single nucleotide or small indel) across 232 additional colorectal tumors and found that the TE target genes are significantly enriched for frequently mutated genes (Fig. 2B, bootstrap Kolmogorov-Smirnov test, P < 10−15 for both nonsilent and all mutations). A separate analysis of 40 genes targeted by the somatic TE insertions only in colorectal tumors also showed significant enrichment of frequently mutated genes (P < 10−15; fig. S3). Because recurrently mutated genes are likely to be important drivers of tumorigenesis, our results suggest potential contribution of TE insertions to cancer development (19, 20).
Although none of the somatic events hit coding regions, insertions of TE sequence in untranslated regions (UTRs) or intronic regions can disrupt gene expression (21). Indeed, over a quarter of the identified disease-causing TE insertions are located in introns or UTRs (9). We thus compared the mRNA levels of the 45 genes hit by somatic TEs in colorectal cancer between the affected tumor and normal samples. We found that expression of the targeted genes is typically altered in the sample carrying the TE insertion, resulting in significantly lower expression levels on average (P = 6.3 × 10−4; Fig. 2C and fig. S4). The impact of L1 insertions may depend on the orientation of the L1 insertion relative to the target gene, with antisense insertions being less disruptive (21). Indeed, expression of the 27 genes targeted by sense insertions showed significant decrease (P = 3.9 × 10−4), whereas expression reduction of the 18 genes targeted in the antisense direction did not reach statistical significance (P = 0.17). Two somatic L1 insertions were found in the 3′ UTRs of F13B and GPATCH2 in one colorectal tumor (CR3518) (Fig. 1B and fig. S5). The insertion in GPATCH2, a gene that has been implicated in breast cancer growth (22), coincided with significant reduction of its expression level (P = 3.3 × 10−5; Fig. 2C and fig. S4).
To contrast the features of somatic TE insertions in cancer genomes with polymorphic insertions in human populations, we analyzed 44 normal genomes (41 normal blood samples from cancer patients and three healthy individuals from the HapMap project) and identified a total of 7449 TE insertions (5531 Alus, 1645 L1s, 225 SVAs, 31 ERVKs, and 17 ERVL-MaLRs) that are absent in the reference genome (tables S6 to S8). Because the majority of such polymorphic events are passed down through gametes or generated during early embryonic development, we refer to them as germline insertions. Among the detected germline insertions, 3521 (47%) were polymorphisms not reported in earlier studies (11, 23–27) or the Database of Retrotransposition Insertion Polymorphisms (dbRIP) (28) (fig. S6). Although ERVK family polymorphisms are rare (10 records in dbRIP) and none have been reported for the ERVL-MaLR family, we identified 31 distinct LTR5 and 17 distinct THE1 polymorphic sites (two LTR5 and one THE1 insertions were experimentally validated, supplementary text S1). Each individual genome contained an average of 791 Alu, 169 L1, 33 SVA, and 8 ERV insertions not found in the reference assembly (fig. S6).
On the basis of a partial reconstruction of the inserted L1 sequences (tables S2 and S6), we find that most insertions are not full-length L1 instances but fragments substantially truncated at the 5′ end (Fig. 3A). The trend is more pronounced for somatic insertions, which are truncated more often (Fisher’s exact test, P = 1.3 × 10−12), and the truncated sequences are on average significantly shorter (545 versus 1050 bp; Wilcoxon test, P = 4.1 × 10−7). Although most of the somatic L1 insertions originated from young L1Ta subfamilies that are known to be active (table S2 and supplementary text S3), the truncations indicate that the vast majority (>98%) of the somatic insertions would not be competent of further retrotransposition.
Fig. 3.
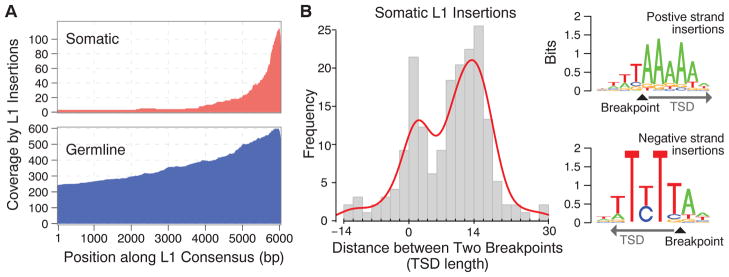
(A) Most of the identified insertions do not contain a full L1 sequence (6 kbp) but are truncated at the 5′ end. The parts of the L1 sequence found within the identified somatic and germline insertions are illustrated as a coverage plot. (B) A positive distance between the clipping positions of clipped reads with negative-and positive-strand mapping (Fig. 1B) corresponds to the length of the duplicated sequence at a TSD, whereas a negative or zero distance corresponds to a microdeletion or lack of duplication at the insertion site. The major TSD peak at ~15 bp is characteristic of an endonuclease-dependent L1 retrotransposition. Sequence analysis around the insertion breakpoints revealed the 5′-TTTT/A-3′ (where the slash indicates the insertion breakpoint) motif, consistent with a canonical sequence for L1-endonuclease target sites (31). The insertions belonging to the minor TSD peak (0 to 2 bp) did not show a significant sequence motif.
The local sequence properties around the identified breakpoints for 64% of the somatic L1 insertions showed both TSDs (≥5 bp) and poly-A tails, which together with the strong 3′ bias of the inserted sequences suggest that the majority of somatic TE insertions in cancers are retrotransposition events driven by the endonuclease-mediated pathway (5). The distribution of TSD lengths showed a peak around 15 bp, characteristic of endonuclease-mediated retrotransposition, for both somatic and germline insertions (Fig. 3B and fig. S6). However, somatic insertions exhibited an additional peak at 0 bp, indicating that some may have been generated by an alternative mechanism such as the one mediated by DNA breaks that does not result in TSDs (29, 30). Consistent with this, somatic L1 insertions with TSDs show sequence motifs at the breakpoints that correspond to the canonical L1-endonuclease recognition sequence (31), whereas the insertions lacking TSDs do not show such a clear recognition sequence (Fig. 3B).
Somatic and germline L1 insertion sites differ in their genomic distribution and epigenetic characteristics. The germline L1 insertions are significantly depleted from genes (22% depletion, P = 1.0 × 10−12), likely because of strong negative selection acting on such events (32). The somatic L1 insertions do not show such notable depletion (11% depletion, P = 0.28) but are nevertheless biased away from transcriptionally active regions (housekeeping genes and common open chromatin regions, figs. S7 to S9). DNA methylation suppresses both TE RNA expression and integration (33), and genome-wide disruption of DNA methylation has been documented in cancers (34). Examination of whole-genome DNA methylation profiles in colorectal cancer (35) shows that somatic L1 insertion sites are significantly overrepresented within regions of DNA hypomethylation (P = 4.2 × 10−9; Fig. 4A). The L1 insertion bias toward common hypomethylation domains suggests that loss of DNA methylation promotes integration of L1 instances. Supporting this hypothesis, we find that the germline L1 insertions are significantly enriched (P = 3.0 × 10−4) in sperm-specific hypomethylation regions (7), whereas somatic insertions are biased toward cancer-specific hypomethylation regions (Fig. 4).
Fig. 4.
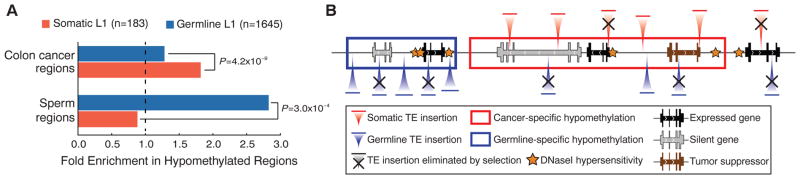
(A) Somatic L1 insertions are biased toward hypomethylated regions in cancer cells. Colon cancer regions were assessed in independent samples. (B) A model of TE insertion preferences and subsequent selection process that bias genomic distribution of TE insertions is illustrated. Somatic insertions are strongly biased toward cancer-specific DNA hypomethylation regions (red box) and encounter selection that depletes them from transcriptionally active genes unless such insertions promote tumorigenesis. By contrast, germline insertions are biased toward germline-specific DNA hypomethylation domains (blue box) and are depleted from all genes.
Our analysis suggests that some TE insertions provide a selective advantage during tumorigenesis, rather than being merely passenger events that precede clonal expansion. We observed differential deregulation of TE activity across and within different cancer types. We also found that such insertions preferentially occur at genes commonly mutated in cancer, including tumor suppressors, substantially disrupting their expression. Although a more extensive panel of matched genomic and epigenetic data is needed to investigate the functional impact of retrotransposition events and the pathways involved, our analysis reveals the extent of TE insertions in human tumors and lays the foundation for determining the role of these events in human neoplasia.
Supplementary Material
Supplementary Materials for Landscape of Somatic Retrotransposition in Human Cancers
Table S2-S4, S6, S7
Table S8
Acknowledgments
We thank A. Langdon and U. Samarakoon for technical assistance and T. Golub and the Broad Institute for a myeloma sample obtained under a materials transfer agreement (MTA) from the Multiple Myeloma Research Consortium. This work was supported by NIH grants R01GM082798 and RC1HG005482 (P.J.P.), K25AG037596 (P.V.K.), U01HG005209 and U01HG005725 (C.L.), F32AG039979 (R.I.), and U24CA144025 (R.K.). P.J.P. is a Sloan Research Fellow. R.K. is on the board of directors of AVEO, Metamark, Enlight, and KEW Group. The sequencing data are available at dbGaP (www.ncbi.nlm.nih.gov/gap) with the following accessions: TCGA ovarian cancer, glioblastoma multiforme, and colorectal cancer (phs000178.v5.p5); multiple myeloma (phs000348.v1.p1); and prostate cancer (phs000330.v1.p1). The Tea software is available at http://compbio.med.harvard.edu/Tea.
Footnotes
References and Notes
- 1.Kidd JM, et al. Cell. 2010;143:837. doi: 10.1016/j.cell.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maksakova IA, Mager DL, Reiss D. Cell Mol Life Sci. 2008;65:3329. doi: 10.1007/s00018-008-8494-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slotkin RK, Martienssen R. Nat Rev Genet. 2007;8:272. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- 4.Yang N, Kazazian HH., Jr Nat Struct Mol Biol. 2006;13:763. doi: 10.1038/nsmb1141. [DOI] [PubMed] [Google Scholar]
- 5.Prak ETL, Kazazian HH., Jr Nat Rev Genet. 2000;1:134. doi: 10.1038/35038572. [DOI] [PubMed] [Google Scholar]
- 6.Bourc’his D, Bestor TH. Nature. 2004;431:96. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 7.Molaro A, et al. Cell. 2011;146:1029. doi: 10.1016/j.cell.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baillie JK, et al. Nature. 2011;479:534. doi: 10.1038/nature10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hancks DC, Kazazian HH., Jr Curr Opin Genet Dev. 2012;22:191. doi: 10.1016/j.gde.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miki Y, et al. Cancer Res. 1992;52:643. [PubMed] [Google Scholar]
- 11.Iskow RC, et al. Cell. 2010;141:1253. doi: 10.1016/j.cell.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hastings PJ, Ira G, Lupski JR. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.CRA00R was excluded from this assessment because the event frequency in this tumor was underestimated as a result of a data quality issue in the matched normal sample (see materials and methods).
- 14.Poulogiannis G, et al. Proc Natl Acad Sci USA. 2010;107:15145. doi: 10.1073/pnas.1009941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin Z, et al. Oncogene. 2007;26:6332. doi: 10.1038/sj.onc.1210461. [DOI] [PubMed] [Google Scholar]
- 16.Slovak ML, et al. Clin Cancer Res. 2011;17:3443. doi: 10.1158/1078-0432.CCR-10-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dickinson RE, Fegan KS, Ren X, Hillier SG, Duncan WC. PLoS One. 2011;6:e27792. doi: 10.1371/journal.pone.0027792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Izumi H, et al. Hum Mol Genet. 2005;14:997. doi: 10.1093/hmg/ddi092. [DOI] [PubMed] [Google Scholar]
- 19.Beroukhim R, et al. Proc Natl Acad Sci USA. 2007;104:20007. [Google Scholar]
- 20.Zheng H, et al. Cancer Cell. 2010;17:497. doi: 10.1016/j.ccr.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han JS, Szak ST, Boeke JD. Nature. 2004;429:268. doi: 10.1038/nature02536. [DOI] [PubMed] [Google Scholar]
- 22.Lin ML, et al. Cancer Sci. 2009;100:1443. doi: 10.1111/j.1349-7006.2009.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beck CR, et al. Cell. 2010;141:1159. doi: 10.1016/j.cell.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ewing AD, Kazazian HH., Jr Genome Res. 2010;20:1262. doi: 10.1101/gr.106419.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ewing AD, Kazazian HH., Jr Genome Res. 2011;21:985. doi: 10.1101/gr.114777.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials for Landscape of Somatic Retrotransposition in Human Cancers
Table S2-S4, S6, S7
Table S8