Aestuaramides, a natural library of cyanobactin cyclic peptides resulting from isoprene-derived Claisen rearrangements (original) (raw)
. Author manuscript; available in PMC: 2014 May 17.
Published in final edited form as: ACS Chem Biol. 2013 Feb 22;8(5):877–883. doi: 10.1021/cb300614c
Abstract
We report 12 cyanobactin cyclic peptides, the aestuaramides, from the cultivated cyanobacterium Lyngbya aestuarii. We show that aestuaramides are synthesized enzymatically as reverse _O_-prenylated tyrosine ethers that subsequently undergo a Claisen rearrangement to produce forward _C_-prenylated tyrosine. These results reveal that a non-enzymatic Claisen rearrangement dictates isoprene regiochemistry in a natural system. They also reveal one of the mechanisms that organisms use to generate structurally diverse compound libraries starting from simple ribosomal peptide pathways (RiPPs).
Natural products are important starting points in the process of drug discovery.1 In part, this is because they present a diversity of scaffolds and other chemical features. A major goal of natural products research has been to understand how enzymes dictate regioselective chemical modifications that underlie this structural diversity. For instance, many natural products are alkylated by isoprene equivalents, such as dimethylallylpyrophosphate (DMAPP), which is installed by a large enzyme class known as prenyltransferases.2 Prenyltransferases are known to carry out regioselective prenylation of aromatic carbons, hydroxyl groups, amines, and thiols, leading to diverse, bioactive products.
Mechanistically, prenyltransferases function by ionization of prenyl diphosphate, followed by electrophilic alkylation of the substrates.3 A longstanding hypothesis has been that prenyltransferases dictate regioselectivity by positioning substrates. However, recent biochemical work supports the hypothesis that the final regioselectivity observed may not always be enzymatically controlled. Instead, in some cases a non-enzymatic Claisen rearrangement may dictate the isolated chemistry.4 In other cases, regioselectivity is at least partly substrate controlled, where different substrates exhibit differences in prenylation pattern by the same enzyme.5,6
We have been studying this phenomenon using a family of small, cyclic peptides known as cyanobactins, which present a diverse series of prenylated metabolites.7–10 Cyanobactins begin as small proteins synthesized on the ribosome. Subsequently, they are posttranslationally modified in a variety of ways. For example, cyanobactins are often prenylated. The isoprene unit usually results from DMAPP, which is installed in a ‘forward’ or ‘reverse’ orientation on the amino acids Ser, Thr, Tyr, and possibly Trp by a series of prenyltransferases exemplified by LynF (Figure 1).4
Figure 1. Prenylation in cyanobactin cyclic peptides.

(a) Previously identified cyanobactin natural products are reverse _O_-prenylated on Ser/Thr or forward _O_-prenylated on Tyr. (b) LynF reverse _O_-prenylates Tyr, and the product undergoes a spontaneous Claisen rearrangement in vitro.
Purified LynF accepts many different Tyr-containing substrates, including cyclic peptides with diverse amino acid sequences and simple Tyr derivatives, such as boc-Tyr.4 For all of the substrates attempted in in vitro reactions, LynF _O_-prenylates Tyr in the reverse orientation, which is an otherwise unprecedented biochemical modification. Surprisingly, we found that this reverse-prenylated intermediate undergoes a spontaneous Claisen rearrangement, even in the absence of enzyme, to yield a forward C-prenylated moiety that is very common in natural products (Figure 1B). This result supported earlier hypotheses that, perhaps, spontaneous Claisen rearrangements might underlie many of the observed prenylated natural products.11 To the best of our knowledge, this may be the only biological relevant Claisen rearrangement outside of the well-studied shikimate pathway.12
A weakness of our previous study was that the actual natural products made by LynF, from the cyanobacterium Lyngbya aestuarii, were not known. Instead, we predicted the structures of the natural products based upon the L. aestuarii genome sequence, and on that basis synthesized putative substrate analogs.4,7 Because it is now known that the regioselectivity of at least some prenyltransferases is substrate controlled,5,6 it remained possible that this Claisen rearrangement was substrate-dependent and not naturally occurring. Therefore, we sought to isolate these putative new compounds from their natural source. Here, we report isolation of novel compounds, aestuaramides A (1) and B (2), and we demonstrate that the spontaneous Claisen rearrangement of the natural products is important in their natural structural diversity. These are the first novel compounds reported from L. aestuarii, a globally abundant cyanobacterium.13–15
RESULTS AND DISCUSSION
The L. aestuarii genome sequence contained a cyanobactin pathway that we predicted would build cyclic peptides using the ribosomally derived core sequences, ACMPCYP and VCMPCYP (Figure 2).7 We obtained the sequenced strain, L. aestuarii PCC 8106, and cultivated it over a six-month period. This led to gradual accumulation of a cyanobacterial broth (20 L) from continuous culture, which was rapidly extracted with methanol to repress isolation artifacts. The crude extract was analyzed by Fourier transform ion cyclotron resonance (FTICR) MS/MS, leading to the identification of 12 cyclic peptides, which we named aestuaramides A-L (1–12) (Figure 2). Two of these, aestuaramides A and B (1 and 2), resulted from the core sequence VCMPYCP and were present in sufficient quantity to enable purification and NMR analysis.
Figure 2. Ribosomal synthesis of aestuaramides.

The LynE precursor peptide is shown at top, with the sequences encoding products underlined and bold. Compounds identified in this study are numbered. Based upon biochemical precedent, LynADG would lead to production of cyclic peptides 2 and 8. Subsequently, prenylation by LynF would lead to reverse _O_-prenylated Tyr products 1 and 7.
Aestuaramide A (1) was assigned a molecular formula of C40H51N7O6S3 (m/z 822.3160 [M+H]+), as determined by FTICR and MS/MS analyses. This formula, in tandem with the predicted linear sequence VCMPCYP, revealed that 1 was likely isoprenylated. NMR spectroscopy in DMSO-_d_6 confirmed the presence of five spin systems that could be assigned to the amino acids (Val, Met, Pro, Tyr, Pro) and one that could be assigned to isoprene (Figure 3, Table 1). Additionally, two sharp singlets (δ=7.77, 8.03) in the aromatic region were consistent with the presence of two Cys-derived thiazoles. The amino acid sequence was confirmed by a combination of ROESY, MS/MS, and chemical degradation. ROESY data showed that Tyr was adjacent to Pro1, while Met was adjacent to Pro2. MS/MS confirmed that the peptide was cyclic and matched the proposed peptide sequence precisely (Figure 4). Further evidence was obtained by carefully hydrolyzing related peptide 2 and analyzing it by Marfey’s method. We optimized hydrolysis following a protocol that leaves thiazole intact.16 All of the predicted amino acids were present in the molecule, according to this analysis. In particular, the thiazole-Pro and thiazole-Val connectivities were confirmed from this result. Additionally, this method revealed that the configurations of all amino acids were L.
Figure 3.
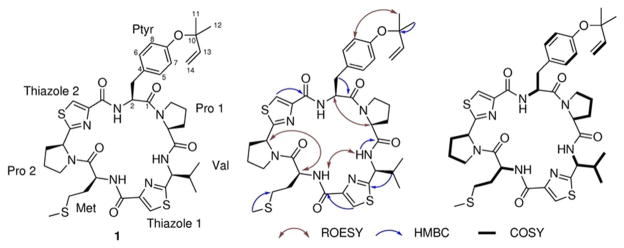
Summary of key NMR data for 1.
Table 1.
NMR data for 1 and 2 in DMSO-d6.
| 1 | 2 | |||
|---|---|---|---|---|
| 13C | 1H (J in Hz) | 1H (J in Hz) | ||
| Met | NH | - | 7.02, d (8.6) | 7.04, d (8.3) |
| 1 | ND | - | - | |
| 2 | 49.4, CH | 4.78, m | 4.77, m | |
| 3 | 32.3, CH2 | 1.95, m; 1.75 m | 1.97, m; 1.73 m | |
| 4 | 30.4, CH2 | 2.40, m | 2.41, m | |
| 5 | 14.4, CH3 | 2.04, s | 2.08, s | |
| Thiazole 1 | 1a | 169.4, C | - | |
| 2 | ND | - | - | |
| 3a | 123.8, CH | 7.76, s | 7.77, s | |
| Val | NH | - | 9.08, s | 9.10, d (9.5) |
| 1 | 172.8, C | - | - | |
| 2 | 54.9, CH | 4.75, m | 4.75, m | |
| 3 | 30.6, CH | 2.34, m | 2.38, m | |
| 4 | 19.6, CH3 | 0.58, d (6.5) | 0.60, d (6.7) | |
| 5 | 19.2, CH3 | 0.78, d (6.5) | 0.82, d (6.7) | |
| Pro 1 | 1 | 170.4, C | - | - |
| 2 | 61.3, CH | 3.08, m | 3.10, m | |
| 3 | 27.8, CH2 | 1.72, m; 1.50, m | 1.74, m; 1.56, m | |
| 4 | 23.4, CH2 | 1.64, m; 1.57, m | 1.65, m; 1.58, m | |
| 5 | 48.5, CH2 | 3.90, m; 3.11, m | 3.93, m; 3.11, m | |
| Ptyr | NH | - | 7.50, d (7.0) | 7.51, d (7.0) |
| 1 | 174.5, C | - | ||
| 2 | 52.4, CH | 4.03, m | 3.98, m | |
| 3 | 38.5, CH2 | 2.98, m; 2.52, m | 2.94, dd (13.0, 4); 2.52, m | |
| 4 | 133.2, C | - | ||
| 5/6 | 129.6, CH | 6.91, d (8.0) | 6.85, d (8.0) | |
| 7/8 | 121.6, CH | 6.88, d (8.0) | 6.69, d (8.0) | |
| 9 | 158.1, C | - | - | |
| 9-OH | - | - | 9.33, s | |
| 10 | 79.3, C | - | - | |
| 11/12 | 26.4, CH3 | 1.36, s | - | |
| 13 | 143.9, CH | 6.07, dd (18.0, 11.4) | - | |
| 14 | 113.6, CH2 | 5.12, m | - | |
| Thiazole 2 | 1 a | 170.7, C | - | - |
| 2 | ND | - | - | |
| 3 a | 124.7, CH | 8.05, s | 8.05, s | |
| Pro 2 | 1 | ND | - | - |
| 2 | 58.5, CH | 5.86, brd (7.3) | 5.85, d (7.3) | |
| 3 | 28.8, CH2 | 2.41, m; 1.96, m | 2.44, m; 2.04, m | |
| 4 | 21.4, CH2 | 1.92, m; 1.74, m | 1.92, m; 1.73, m | |
| 5 | 46.4, CH2 | 3.67, m; 3.52, m | 3.70, m; 3.52, m |
Figure 4. Summary of key MS data.
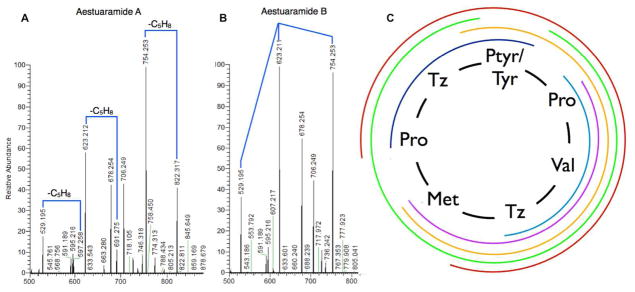
FTICR-MS/MS shows that (a) 1 is _O_-prenylated, while (b) 2 is not; the MS/MS spectra are otherwise identical. C. MS/MS data provide evidence for the fragments shown, providing unambiguous evidence of peptide sequence (see Supporting Information for full details).
NMR spectroscopy unambiguously showed that 1 contained a reverse _O_-Tyr isoprene moiety. The ROESY spectrum revealed correlations between the methyl groups of the reverse-prenyl moiety and the Tyr ε protons. In addition, the 1H and 13C chemical shifts of the Tyr aromatic moiety and isoprene were very similar to those previously reported for reverse-_O_-prenylated boc-Tyr.4 Finally, MS/MS data confirmed this assignment. We previously showed using multiple substrates that reverse _O_-prenylated Tyr readily loses isoprene in the course of mass spectrometry, while the _C_-prenylated Tyr does not lose isoprene under most conditions.4 Similarly, 1 readily lost isoprene even under mild fragmentation conditions (Figure 4).
Based upon FTICR-MS, aestuaramide B (2) had a molecular formula of C35H43N7O6S3 (m/z 754.2153 [M+H]+), indicating that it was likely an unprenylated analog of 1. Indeed, comparative analysis of 1H NMR and MS data revealed that these compounds were nearly identical, except that peaks corresponding to isoprene were absent in 2 (Figure 4, Table 1). Although the 1H NMR spectra of 1 and 2 were largely identical, the greatest chemical shift differences observed were localized to the phenol ring of tyrosine (1 δ=6.91, 6.88; 2 δ=6.83, 6.67). Moreover, the 1H NMR spectrum of 2 contained a sharp singlet consistent with a phenolic OH (δ=9.33), while this signal was clearly absent in the 1H spectrum of 1. To further confirm the amino acid sequences and position of prenylation of these peptides, we carefully examined their MS/MS data, which were consistent with the proposed structures (Figure 4). A comparison of MS/MS spectra for 1 and 2 was particularly telling, since the fragments were similar, but 2 lacked isoprene.
In 1:1 D2O:CD3OD at 50 °C, 1 was underwent the Claisen rearrangement over a 21-hour period (Figure 5, Table 2). This rearrangement could be clearly observed by following the chemical shifts of the aromatic residues and comparing them to those for authentic standards of forward _C_-prenylated and reverse _O_-prenylated Tyr that we had previously characterized. The rate of rearrangement was slower than what we had previously observed in aqueous enzyme reaction buffers with LynF,4 indicating either an important effect of aqueous buffers, or substrate dependence in the rearrangement itself. An identical, _C_-prenylated compound was also found in the freshly harvested L. aestuarii extract, and therefore this compound was named aestuaramide C (3).
Figure 5. Claisen rearrangement converting compound 1 to 3.
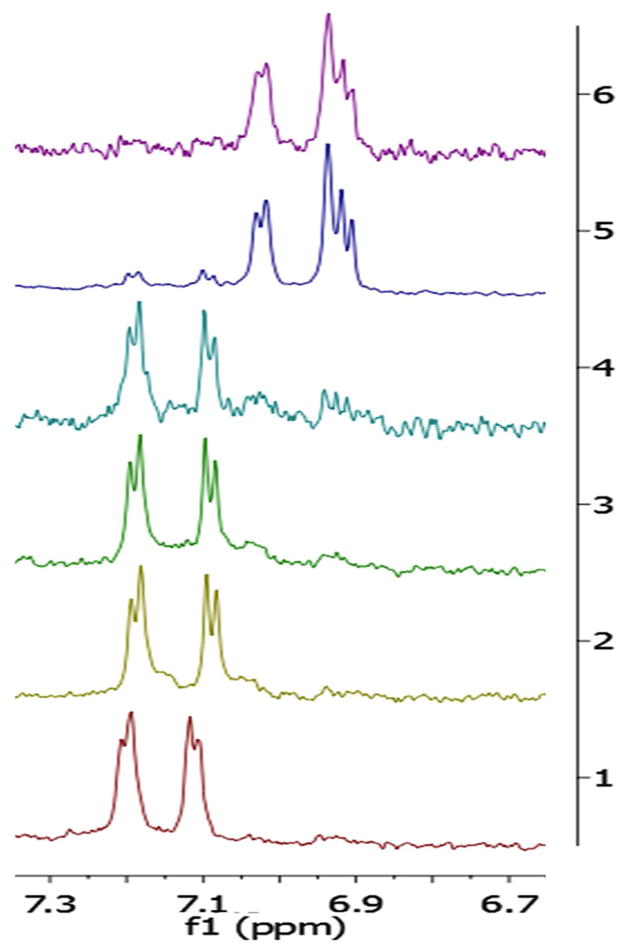
Shown is the Tyr aromatic region of the 1H NMR spectrum as compound 1 was subjected to heating as described in materials and methods; y-axis: 1: time 0, room temperature; 2: time 0, 50 °C; 3: 0.5 h, 50 °C; 4: 2 h, 50 °C; 5: 19 h, 50 °C; 6: 21 h, 50 °C. At time points 1–4, the starting material 1 dominates the spectrum, while by time period 6 1 is completely absent and has been replaced by 3 (see Figure S3K for more details).
Table 2.
Key NMR data for spectra shown in Figure 5.
| No. | 1 | 3 |
|---|---|---|
| 5 | 7.20, d (8.0) | 6.94, s |
| 6 | 7.20, d (8.0) | 7.02, d (7.6) |
| 7 | 7.11, d (8.0) | - |
| 8 | 7.11, d (8.0) | 6.91. d (7.6) |
| 11 | 1.54, s | 1.82, s |
| 12 | 1.54, s | 1.79, s |
| 13 | 6.24, dd (17.6, 10.9) | - |
| 14 | 5.28, m | 5.35, brs |
Compound 3 had a nearly identical 1H NMR spectrum to that of 1 in 1:1 D2O:CD3OD (Figure S3K). The major differences observed were in the chemical shifts of the aromatic moiety and in the isoprene unit. In contrast to the _p_-phenol of 1, 3 had three 1H signals in the aromatic region, representing a 1,3,4-trisubstituted aromatic ring. The chemical shifts of these protons (δ=7.02 (H-6), 6.91 (H-5), 6.94 (H-8)) were consistent with a dialkylated phenol. The observed coupling constants showed that an additional alkyl group was present at H-7 (Figure S3J). Chemical shifts of the isoprene moiety had changed from those for a reverse-prenylated compound (1) to a forward-prenylated compound. In particular, the isoprene methyls were at δ=1.54 in 1 and δ=1.79, 1.82 in 3. By FTICR-MS analysis, 3 and its forward-_C_-prenylated relatives exhibit strikingly different fragmentation patterns than the reverse-_O_-prenylated series. While the _O_-prenylated 1 readily loses C5H8, this loss is not observed in 3 or in any other member of the _C_-prenylated series. In the course of analysis, the rearrangement product 3 was spontaneously oxidized. By LC-FT-ICR and MS/MS the C-prenylated and Met-oxidized 3 was identical with one of the compounds in the crude extract. The rearrangement and single oxidation product of 1 was identical by LC-FTICR-MS/MS to compound 6 (Figure S5 and see below). Aside from these differences, the only obvious difference in the 1H NMR spectra was that the thiazole-2 proton exhibited a downfield shift from δ=8.04 to δ=8.08 during the conversion of 1 to 3, while the thiazole-1 proton was invariant.
A limitation of the above data is that, owing to the amount of material recovered, 13C data was only available for compound 1 and not 2 or 3. Assignments thus relied on 1H NMR data and FTICR-MS/MS. The MS/MS spectra of these compounds were identical except for differences directly attributable to prenylation of tyrosine. Moreover, by NMR this prenylation could be clearly observed to occur on tyrosine in both 1 and 3.
In addition to 1–3, nine other related products could be observed in the L. aestuarii crude extract by MS analysis (Figures 1 and S2). Aestuaramides A–C (1–3) resulted from posttranslational modification of the precursor peptide sequence, VCMPCYP. In addition to these reverse _O_-prenylated, unprenylated, and forward _C_-prenylated compounds, we also observed three further products, aestuaramides D-F (4–6), corresponding to the sulfoxide derivative of 1–3, leading to six total cyclic peptides derived from the VCMPCYP sequence. FTICR-MS provided high-resolution masses. Further, the MS/MS fragmentation patterns were virtually identical to those for the non-sulfoxide analogs, except fragments containing Met are larger by 16 Da. We have previously observed a similar pattern of oxidized and non-oxidized Met in other cyanobactins.17
Similarly, six related products resulting from the ACMPCYP precursor peptide sequence were identified. These compounds (aestuaramides G-L, 7–12) were reverse _O_-prenylated, unprenylated, or forward _C_-prenylated, with or without Met oxidation. The structures were assigned using genome sequencing-based prediction and very similar MS/MS spectra to those of their NMR-characterized relatives, 1 and 2.
Characterization of RiPP products remains a challenge in part because low yields are sometimes obtained. Countering this problem, an advantage of working with RiPPs is that the peptide sequence is genomically encoded, so that if a fragment containing the predicted sequence is found with high resolution by mass spectrometry, then based upon the size of the genome of the producer it is probable (or in some cases virtually certain) that the compound in question is encoded by the RiPP sequence.18,19 With aestuaramides, standard amino acids Pro, Tyr, and Met were clearly observed. The main problem lies in the assignment of posttranslational modifications. In the case of aestuaramides, biochemical precedent indicated that the products should be N-C cyclic (presence of LynA and LynG) and that they should contain thiazoles (LynD and oxidase). Indeed, in the MS spectra, the major compounds lacked H10O3 in comparison with the predicted linear peptides. The fragmentation methods used here cannot break the thiazole-amino acid linkage, such that Val-thiazole, Ala-thiazole, and Pro-thiazole could be inferred. Fragments for these predictions were present in all spectra. Moreover, when a peptide is cyclic rather than linear, the number of possible fragments greatly increases,20 and indeed the evidence here favors cyclization. However, these are hypotheses that require further testing. Fortunately, in the case of 1, a complete NMR data set confirmed the amino acid assignments and provided additional strong support in favor of the presence of thiazoles and N-C circularization. Additional 1H NMR spectra of 2 and 3 served to further support these structures. Comparison of the fragmentation pattern of 1 to those for 2–12 showed that they were very similar to 1, with the differences being explained by the minor change in peptide sequence and in prenylation pattern, as described above. Because compounds 4–12 were elucidated solely on the basis of MS and genome sequence, they are tentatively assigned with a lower level of evidence.
Previously, we showed that LynF reverse _O_-prenylates Tyr in diverse artificial substrates, and that these intermediates spontaneously rearrange to form the forward _C_-prenylated derivatives. 4 We proposed that this Claisen rearrangement might possibly underlie many different types of _C_-prenylated phenolic natural products. Here, we show that LynF reverse _O_-prenylates its natural substrates, and that these rearrange to form forward _C_-prenylated products within their natural environments. With these specific substrates, the Claisen rearrangement is quite slow, and in fact much slower than with the substrate analogs that we previously used,4 meaning that all intermediates could be directly observed. The identification of these natural compounds confirms that this spontaneous Claisen rearrangement is important in natural isoprenoid chemistry. Our initial work with purified enzyme itself suggests that it can be difficult to directly observe this transformation,4 and that the possibility should be considered more seriously in future studies of phenol prenylation.
The presence of both forward- and reverse-prenylated cyclic peptides in a single organism adds to the chemical complexity of ribosomal peptide natural products, which are now referred to as RiPPs.21 In the aestuaramide biosynthetic gene cluster (lyn), a single precursor peptide, LynE, encodes the sequences for both the VCMPCYP and ACMPCYP series of compounds (Figure 2). A heterocyclase, LynD, was proposed to synthesize thiazole from Cys, with the help of an oxidase found in LynG.22,23 Subsequently, LynA and LynG proteases cleave and circularize the compounds, in reactions that have been characterized in related cyanobactin systems.24–26 Based upon our work with purified LynF, it is only at this late stage, when peptides have already been macrocyclized, that prenylation would take place.4 We previously showed that prenylation is perhaps the least efficient enzymatic step in cyanobactin pathways.27 In heterologous expression experiments in Escherichia coli, the natural cyanobactin trunkamide was prenylated by 0, 1, or 2 DMAPP units. Our results here show that these prenyltransferases can be quite slow even in their native hosts. Indeed, LynF and relatives exhibit exceptionally relaxed substrate specificity,24 which is likely directly related to this slow rate. Finally, Met autoxidation17 leads to a further six derivatives, for 12 total compounds resulting from a single precursor peptide. Autoxidation is probably non-enzymatic (spontaneous), since oxidation increases over time even in extracted or purified materials.
This represents a striking structural diversity from a single RiPP precursor peptide. Most commonly, only a single product results from a single precursor peptide, although there are a few cases in which up to four products result. Twelve natural products from a single precursor peptide may represent a record for RiPPs. The production of numerous derivatives from a single pathway is one of the ways in which variable tailoring steps allow RiPP pathways contribute to chemical diversity in bacteria. Of note, there are two copies of the Val-containing sequence and one of the Ala-containing sequence encoded on LynE. As shown above, we noted a greater amount of Val-containing products in comparison to Ala-containing products. One possibility is that this ratio directly correlates to a different abundance of the two species. In cyanobactin precursor peptides, there are several cases with multiple repeats of product-coding sequences in a single peptide, although repetition of the same coding sequence within a peptide appears to be relatively rare.17 There are also many cases in which only a single coding sequence is found in a cyanobactin precursor peptide. Our current working hypothesis is that multiple coding sequences within a single peptide result from a duplication of the entire precursor peptide, followed by recombination to eliminate nonidentical sequences. In support of this hypothesis, we have documented several cases in which multiple precursor peptides (up to nine) are found in a single cyanobactin operon.17 In cases that we have examined, it appears that only some of these precursors lead to isolable products.17 It seems likely that this repetition may be more common than generally realized, since it creates an assembly challenge that can lead to artifacts in shotgun-sequenced genomes. Alternatively, we have previously documented between-strain cyanobactin pathway recombination.28 This indicates that the environment also serves as a reservoir for coding sequence diversity. This is very similar to the duplication-recombination scenario above, except that multiple precursor peptides would be created in multiple strains, rather than in a single strain. Interestingly, the phenomenon of multiple coding sequences in a single precursor peptide has also been found in N-C cyclic peptides from plants.29
L. aestuarii is a biologically important, dominant benthic (bottom-dwelling) component of shallow bays in oceans around the world.13–15 To the best of our knowledge, the only previously reported products of L. aestuarii are widely occurring UV-shielding pigments.30,31 Therefore, aestuaramides represent the first novel compounds from this widespread source. It has been shown that other cyanobactins are components of numerous types of cyanobacteria, including species that are dominant components of the plankton.7,8,10,17 Cyclic peptides such as aestuaramides may be exceptionally widespread metabolites in natural ecosystems. The natural roles of the compounds are largely unknown, but their abundance, structural diversity, and occasionally potent biological activity in pharmaceutical screens hints that they may be important in the biology of cyanobacteria and other organisms.
METHODS
General Methods
1H NMR, 13C NMR and 1H-1H COSY spectra were recorded on a Varian INOVA 500 (1H 500 MHz, 13C 125MHz) NMR spectrometer with a 3 mm Nalorac MDBG probe at 300 K. HSQC and HMBC spectra were recorded on a Varian INOVA 600 (1H 600 MHz, 13C 150 MHz) NMR spectrometer equipped with a 5 mm 1H[13C,15N] triple resonance cold probe with a z-axis gradient, utilizing residual solvent signals for referencing. High-resolution mass spectra (HRMS) were obtained using a Bruker APEXII FTICR mass spectrometer equipped with an actively shielded 9.4 T superconducting magnet, an external Bruker APOLLO ESI source, and a Synrad 50W CO2 CW laser. Onyx monolithic C18 semipreparative columns were used for HPLC, as conducted on a Hitachi Elite Lachrom System equipped with a Diode Array L-2455 detector. C18 flash chromatography was performed using silica gel 90 (Fluka).
Cultivation
L. aestuarii PCC 8106 was obtained from the Pasteur Culture Collection and maintained in MN marine medium. To produce aestuaramides, the cyanobacteria were cultivated in MN marine medium (~10 mL) in plastic 100 × 15 mm Petri dishes on a windowsill at room temperature. MN medium (1 L): artificial seawater to 75% strength; MgSO4•7H2O (40 mg); CaCl2 (20 mg); NaNO3 (750 mg); K2HPO4•3H2O (20 mg); citric acid (3 mg); ferric ammonium citrate (3 mg); EDTA disodium potassium salt (0.5 mg); NaHCO3 (2 mg); trace metal mix (1 mL). Trace metal mix (1 L): H3BO3 (2.86 g); MnCl2•4H2O (1.81 g); ZnSO4•7H2O (222 mg); NaMoO4•2H2O (390 mg); CuSO4•5H2O (79 mg); Co(NO3)2•6H2O (49.4 mg). Seed cultures of L. aestuarii were transferred into the Petri dishes, which were then allowed to grow for a 2-month period.
Extraction and Purification
L. aestuarii was removed from the media by filtration to yield a cell mass (33.8 g wet weight), which was flash frozen with liquid N2 and ground with a mortar and pestle. This material was extracted with methanol (4 × 500 mL). Wet HP-20 resin (Supelco) was added to 75% of the initial crude extract (1.5 L), and the extract was dried onto the resin by rotary evaporation. The resin was extensively washed with water and eluted with a methanol gradient (50%, 75%, 100%), followed by 75% acetone in water. Fractions eluting in 100% methanol or 75% acetone were dried and resuspended in methanol, to which DMSO was then added yielding 80/20 DMSO/methanol mixtures that were purified by C18 flash chromatography. Columns were equilibrated in 1/1 methanol/water, the applied extract was washed with the same mixture, and the desired products were eluted with 75% acetonitrile. Finally, C18 HPLC was performed with a flow rate of 3 mL min−1 and a gradient from 1% to 60% acetonitrile over 60 min, to yield 1 (0.4 mg, 1.6×10−3% of wet weight) and 2 (0.1 mg, 3.9×10−4% of wet weight). For FTICR of the crude extract, a small-scale methanol extract was used with freshly flash-frozen material.
LC-MS Analysis of D/L-FDLA Derivatives
Compound 2 (20 _μ_g) was dissolved in 6 N HCl (200 _μ_L) and heated in sealed ampule vials at 110 °C for 8 h. The solvent was removed in vacuo. The acid hydrolysate of 2 was dissolved in H2O. To a 50 _μ_L aliquot was added 1 N NaHCO3 (20 _μ_L) and 1-fluoro-2,4-dinitrophenyl-5-L-leucinamide (1% solution in acetone, 100 _μ_L), and the mixture was heated to 40 °C for 50 min. The solution was cooled to room temperature, neutralized with 1 N HCl (20 _μ_L), and then dried in vacuo. The residue was dissolved in MeOH and then analyzed by LC-MS. The analysis of the L- and D-FDLA derivatives was performed on a EclipseXDB-18 column (150 × 4.6 mm, 5 _μ_m) employing a linear gradient of from 20% to 80% CH3CN in 0.01 M ammonium formate at 0.5 mL min−1over 50 min. The retention times of the D- and L-FDLA derivatives, respectively, were as follows: L-Ala: 17.90, 16.50 min, m/z 428 [M − H]−; L-Val-Tzl: 31.80, 29.50 min, m/z 493 [M − H]−; L-Pro: 24.18, 20.61 min, m/z 408 [M − H]−; L-Tyr: 17.90, 17.05 min, m/z 474 [M − H]−; L-Pro-Tzl: 26.38, 26.28 min, m/z 491 [M − H]−.
Claisen Rearrangement
Compound 1 (0.4 mg) in 1:1 CD3OD:D2O was heated to 50 °C in the NMR probe and followed by 1H NMR for 21 h.
Supplementary Material
1_si_001
Acknowledgments
This work was funded by NIH GM071425 and GM092009. We thank K. Parsawar, C. Nelson, and J. Cox (U. Utah) for obtaining the mass spectra used in this study.
Footnotes
References
- 1.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat PRod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heide L. Prenyl transfer to aromatic substrates: genetics and enzymology. Curr Op Chem Biol. 2009;13:171–179. doi: 10.1016/j.cbpa.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 3.Haug-Schifferdecker E, Arican D, Brückner R, Heide L. A new group of aromatic prenyltransferases in fungi, catalyzing a 2,7-dihydroxynaphthalene 3-dimethylallyl-transferase reaction. J Biol Chem. 2010;285:16487–16494. doi: 10.1074/jbc.M110.113720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McIntosh JA, Donia MS, Nair SK, Schmidt EW. Enzymatic basis of ribosomal peptide prenylation in cyanobacteria. J Am Chem Soc. 2011;133:13698–13705. doi: 10.1021/ja205458h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wollinsky B, Ludwig L, Hamacher A, Yu X, Kassack MU, Li SM. Prenylation at the indole ring leads to a significant increase of cytotoxicity of tryptophan-containing cyclic dipeptides. Bioorg Med Chem Lett. 2012;22:3866–3869. doi: 10.1016/j.bmcl.2012.04.119. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Morita H, Wakimoto T, Mori T, Noguchi H, Abe I. Prenylation of a nonaromatic carbon of indolylbutenone by a fungal indole prenyltransferase. Org Lett. 2012;14:3080–3083. doi: 10.1021/ol301129x. [DOI] [PubMed] [Google Scholar]
- 7.Donia MS, Ravel J, Schmidt EW. A global assembly line for cyanobactins. Nat Chem Biol. 2008;4:341–343. doi: 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leikoski N, Fewer DP, Sivonen K. Widespread occurrence and lateral transfer of the cyanobactin biosynthesis gene cluster in cyanobacteria. Appl Environ Microbiol. 2009;75:853–857. doi: 10.1128/AEM.02134-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt EW, Donia MS. In: Chapter 23 Cyanobactin Ribosomally Synthesized Peptides-A Case of Deep Metagenome Mining. Hopwood DA, editor. 2009. pp. 575–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sivonen K, Leikoski N, Fewer DP, Jokela J. Cyanobactins-ribosomal cyclic peptides produced by cyanobacteria. Appl Microbiol Biotechnol. 2010;86:1213–1225. doi: 10.1007/s00253-010-2482-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quillinan AJ, Scheinmann F. Application of the Claisen rearrangement to the synthesis of heterocyclic bicyclo2,2,2octenones: an approach to the morellins based on new biogenetic suggestions. J Chem Soc D Chem Commun. 1971:966–967. [Google Scholar]
- 12.Andrews PR, Smith GD, Young IG. Transition state stabilization and enzymic catalysis. Kinetic and molecular orbital studies of the rearrangement of chorismate to prephenate. Biochemistry. 1973;12:3492–3498. doi: 10.1021/bi00742a022. [DOI] [PubMed] [Google Scholar]
- 13.Martínez-Alonso M, Mir J, Caumette P, Gaju N, Guerrero R, Esteve I. Distribution of phototrophic populations and primary production in a microbial mat from the Ebro Delta, Spain. Int Microbiol. 2004;7:19–25. [PubMed] [Google Scholar]
- 14.Paerl HW, Prufert LE, Ambrose WW. Contemporaneous N2 fixation and oxygenic photosynthesis in the nonheterocystous mat-forming cyanobacterium Lyngbya aestuarii. Appl Environ Microbiol. 1991;57:3086–3092. doi: 10.1128/aem.57.11.3086-3092.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rath J, Mandal S, Adhikary SP. Salinity induced synthesis of UV-screening compound scytonemin in the cyanobacterium Lyngbya aestuarii. J Photochem Photobiol B. 2012;115:5–8. doi: 10.1016/j.jphotobiol.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Fujii K, Yahashi Y, Nakano T, Imanishi S, Baldia SF, Harada KI. Simultaneous detection and determination of the absolute configuration of thiazole-containing amino acids in a peptide. Tetrahedron. 2002;58:6873–6879. [Google Scholar]
- 17.Donia MS, Schmidt EW. Linking chemistry and genetics in the growing cyanobactin natural products family. Chem Biol. 2011;18:508–519. doi: 10.1016/j.chembiol.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sudek S, Haygood MG, Youssef DT, Schmidt EW. Structure of trichamide, a cyclic peptide from the bloom-forming cyanobacterium Trichodesmium erythraeum, predicted from the genome sequence. Appl Environ Microbiol. 2006;72:4382–4387. doi: 10.1128/AEM.00380-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kersten RD, Yang YL, Xu Y, Cimermancic P, Nam SJ, Fenical W, Fischbach MA, Moore BS, Dorrestein PC. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat Chem Biol. 2011;7:794–802. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ng J, Bandeira N, Liu WT, Ghassemian M, Simmons TL, Gerwick WH, Linington R, Dorrestein PC, Pevzner PA. Dereplication and de novo sequencing of nonribosomal peptides. Nat Meth. 2009;6:596–599. doi: 10.1038/nmeth.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Goransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Muller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJT, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Sussmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McIntosh JA, Donia MS, Schmidt EW. Insights into heterocyclization from two highly similar enzymes. J Am Chem Soc. 2010;132:4089–4091. doi: 10.1021/ja9107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McIntosh JA, Schmidt EW. Marine molecular machines: Heterocyclization in cyanobactin biosynthesis. Chem Bio Chem. 2010;11:1413–1421. doi: 10.1002/cbic.201000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McIntosh JA, Robertson CR, Agarwal V, Nair SK, Bulaj GW, Schmidt EW. Circular logic: Nonribosomal peptide-like macrocyclization with a ribosomal peptide catalyst. J Am Chem Soc. 2010;132:15499–15501. doi: 10.1021/ja1067806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J, McIntosh J, Hathaway BJ, Schmidt EW. Using marine natural products to discover a protease that catalyzes peptide macrocyclization of diverse substrates. J Am Chem Soc. 2009;131:2122–2124. doi: 10.1021/ja8092168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koehnke J, Bent A, Houssen WE, Zollman D, Morawitz F, Shirran S, Vendome J, Nneoyiegbe AF, Trembleau L, Botting CH, Smith MCM, Jaspars M, Naismith JH. The mechanism of patellamide macrocyclization revealed by the characterization of the PatG macrocyclase domain. Nat Struc Molec Biol. 2012;19:767–772. doi: 10.1038/nsmb.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tianero MDB, Donia MS, Young TS, Schultz PG, Schmidt EW. Ribosomal route to small-molecule diversity. J Am Chem Soc. 2012;134:418–425. doi: 10.1021/ja208278k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donia MS, Fricke WF, Ravel J, Schmidt EW. Variation in tropical reef symbiont metagenomes defined by secondary metabolism. PloS one. 2011;6:e17897. doi: 10.1371/journal.pone.0017897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Condie JA, Nowak G, Reed DW, Balsevich JJ, Reaney MJ, Arnison PG, Covello PS. The biosynthesis of Caryophyllaceae-like cyclic peptides in Saponaria vaccaria L. from DNA-encoded precursors. Plant J. 2011;67:682–690. doi: 10.1111/j.1365-313X.2011.04626.x. [DOI] [PubMed] [Google Scholar]
- 30.Abed RMM, Kohls K, Schoon R, Scherf AK, Schacht M, Palinska KA, Al-Hassani H, Hamza W, Rullkötter J, Golubic S. Lipid biomarkers, pigments and cyanobacterial diversity of microbial mats across intertidal flats of the arid coast of the Arabian Gulf (Abu Dhabi, UAE) FEMS Microbiol Ecol. 2008;65:449–462. doi: 10.1111/j.1574-6941.2008.00537.x. [DOI] [PubMed] [Google Scholar]
- 31.Balskus EP, Case RJ, Walsh CT. The biosynthesis of cyanobacterial sunscreen scytonemin in intertidal microbial mat communities. FEMS Microbiol Ecol. 2011;77:322–332. doi: 10.1111/j.1574-6941.2011.01113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1_si_001