Role of Transglutaminase 2 in Celiac Disease Pathogenesis (original) (raw)
. Author manuscript; available in PMC: 2013 Jul 16.
Published in final edited form as: Semin Immunopathol. 2012 Mar 22;34(4):513–522. doi: 10.1007/s00281-012-0305-0
Introduction to Transglutaminase 2 (TG2) Chemistry and Biology
Biochemical properties
Transglutaminase 2 (TG2) is a 78 kDa, calcium dependent enzyme in mammals. A member of the transglutaminase family, it catalyzes the post-translational modification of selected glutamine residues on the surface of its protein or peptide substrates. The catalytic mechanism of transglutaminase 2 shares some similarities with cysteine proteases (Figure 1), where a thiol residue (Cys277 in human TG2) in the active site attacks the glutamine carboxamide, releasing ammonia. The resulting thioester intermediate can then be attacked by a variety of biological nucleophiles, typically an ε-amino group from a surface lysine residue of a second protein or peptide substrate. The overall reaction results in the formation of a stable ε(γ-glutamyl)-lysine isopeptide bond (a.k.a. “cross-link”) that is resistant to proteolysis [1]. Alternatively, the acyl-enzyme intermediate can be hydrolyzed, leading to the net conversion of a glutamine residue into a glutamic acid; this transformation has relevance to celiac disease (vide infra).[2] Like most other mammalian transglutaminases, TG2 is a multifunctional protein with other biochemical properties. Perhaps most notably, it binds to guanine nucleotides and functions as a G-protein in signaling processes involving selected 7-transmembrane receptors [3]. It has also been reported to exhibit protein disulfide isomerase and serine/threonine kinase activities.[4, 5]
Figure 1.
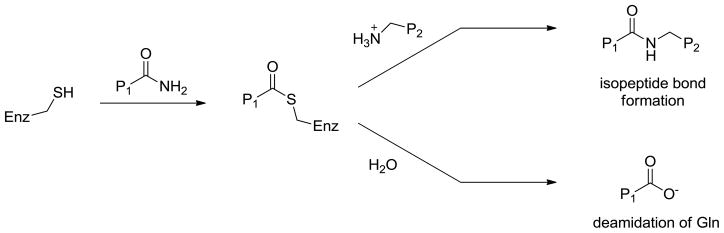
In the catalytic mechanism of transglutaminases, the active site cysteine (Enz-SH) attacks the glutamine of the acyl donor substrate P1, forming an intermediate thioester that can either be attacked by the amine of an acyl acceptor substrate P2, yielding a peptide bond or be hydrolyzed, corresponding to the net deamidation of P1.[107] The acyl donor substrate P1 bearing a glutamine is typically a protein but small peptides are recognized as well. The acyl acceptor substrate P2 is typically a lysine sidechain on a protein or small peptide but can be replaced by small molecule aliphatic amines. [108–110]
Localization
TG2 is abundantly expressed in many organs including, for example, the liver, heart, intestine, as well as blood cells such as erythrocytes [6]. It is found in both intracellular and extracellular locations [7]. Inside the cell, TG2 is especially abundant in the cytosol, but can also be found in mitochondria [8, 9] and the nucleus [10, 11]. It should be noted that, notwithstanding its localization in the mitochondria and the extracellular matrix, TG2 does not possess an N-terminal export sequence. Instead, it is thought to be externalized as a complex with β1-integrin via recycling endosomes [12] and/or cell surface shedding of heparan sulfates [13]. In the extracellular matrix, TG2 binds tightly to fibronectin via a 42 kDa gelatin binding domain. Because TG2 contributes to gluten immunotoxicity by modifying selected peptides, the pathogenesis of celiac disease is presumed to depend upon the activity of extracellular TG2 (vide infra).
Biological function
A variety of physiological roles have been ascribed to TG2, and have been extensively reviewed elsewhere [7, 6, 14–19]. In the intracellular environment, TG2 is thought to play a pro-apoptotic as well as an anti-apoptotic role, depending on the cellular context and biological cues [20]. Extracellular TG2 is believed to be involved in cell adhesion [21, 22], matrix assembly [18, 23], wound healing [15, 24, 25], receptor signaling [26], and a variety of cellular behaviors including proliferation, invasion, motility and survival [27, 28]. Some of these biological functions have been shown to depend upon the cross-linking activity of the enzyme; others are not. Notwithstanding this diversity of putative biological roles, TG2 knockout mice are developmentally and reproductively normal [29–31]. In this review we will necessarily limit our discussion to only those functions that have compelling relevance to celiac disease pathogenesis.
Post-translational regulation
The expression of TG2 is elaborately regulated in response to a number of transcriptional signals, as reviewed elsewhere [28]. In light of its ubiquity, abundance, and relatively non-specific recognition of glutamine-bearing substrates, the transglutaminase activity of TG2 must also be tightly controlled at the post-translational level. From the perspective of understanding celiac disease pathogenesis, it must be understood that TG2 in the small intestinal mucosa is predominantly inactive under normal physiological conditions [32–35]. It must therefore be activated. A full understanding of the post-translational mechanisms underlying TG2 activation is therefore critical.
The best understood post-translational regulators of cross-linking activity are Ca2+ ions, guanine nucleotides (GTP and GDP), and the redox environment of the protein. Transglutaminase activity of full-length TG2 requires Ca2+ ion binding. Up to six Ca2+ ions can bind to each molecule of TG2 with an apparent overall dissociation constant of 90 μM [36]. In contrast, TG2 activity is inhibited upon binding to one molecule of GTP or GDP, the dissociation constant being 1.6 μM [37]. The GTP-bound form has one residual high-affinity calcium binding site, and requires high (> 1 mM) concentrations of Ca2+ to regain activity [38, 39]. Activation is accompanied by a large conformational change from a compact (“closed”) to an extended (“open”) structure (Figures 2 and 3) [40, 41]. These regulatory features presumably explain why TG2 is ordinarily inactive inside cells, where the free Ca2+ concentration is tightly controlled at very low levels (< 1 μM) and GTP is abundant. However, they fail to explain the inactivity of extracellular TG2, which resides in an environment of relatively abundant Ca2+ ions and low GTP/GDP concentration [32]. The latter property is accounted for by the reversible formation of a disulfide bond between vicinal Cys370 and Cys371 [33, 34]. Because the redox potential is ordinarily high in the extracellular matrix, extracellular TG2 is predominantly maintained in a disulfide bonded, inactive state. Its enzymatic activity can be transiently activated by the protein cofactor thioredoxin-1 (TRX). Whereas the spectrum of biological cues that trigger redox-mediated activation of extracellular TG2 remains to be understood, interferon-γ has been shown to elicit a burst of TRX release from monocytic cells [34]. Interferon-γ also activates extracellular TG2 associated with enterocytic T84 monolayers via a PI3-kinase dependent mechanism [35]. As discussed below, this observation has implications for celiac disease pathogenesis. The transglutaminase activity of intracellular TG2 can also be induced in response to certain stresses [17], although the mechanisms by which this occurs remain poorly understood. Short splice variants of TG2 with C-terminal modifications have been detected in various cells and tissues [42–45]. As these variants appear to be independent of GTP and have a lower calcium requirement than full-length TG2 [43, 46, 47], they may be the sources of intracellular catalytic activity of TG2.
Figure 2.
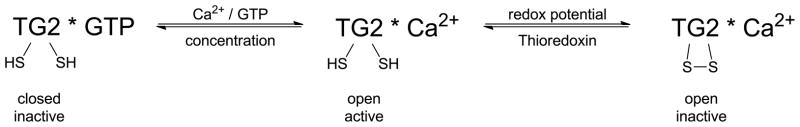
States of TG2 under different physiological conditions and their activity.
Figure 3.

Crystal structures of TG2 in closed (A, PDB 1K3V)[41] and open (B, PDB 2Q3Z)[40] conformations. Domains are colored as follows: N-terminal domain (blue), catalytic domain (green), first C-terminal β-barrel (yellow), second C-terminal β-barrel (red). A: In the closed conformation, GDP is bound to the nucleotide binding site (insert A.1) at the interface between the catalytic domain and the first C-terminal β-barrel. TG2 is inactive, because a loop from the first C-terminal β-barrel (yellow) protrudes towards the acyl donor site tunnel. The tunnel is lined by Trp241, Gln276, Trp278, Trp332 and Phe334 (green spheres), and harbors the active site Cys277 at its bottom (insert A.2). B: In the open conformation, the loop from the first C-terminal β-barrel has moved to allow substrate access to the active site (insert B.1), which is occupied by an inhibitor irreversibly bound to Cys277. The vicinal disulfide bond between Cys370 and Cys371 is depicted in insert B.2.
Is the Transglutaminase Activity of TG2 Required for Celiac Disease Pathogenesis?
At present, a causative link between the transglutaminase activity of TG2 and celiac disease pathogenesis has not yet been definitively established. Nonetheless, a large body of evidence supports this hypothesis. First, TG2 has remarkably high substrate specificity for immunopathogenic gluten peptides. Second, recognition of these gluten peptides by the disease associated HLA-DQ2 or -DQ8 is strongly enhanced by TG2-catalyzed deamidation. Third, interferon-γ is the predominant cytokine secreted when DQ2-restricted, gluten-responsive T cells derived from the celiac intestine are activated [2]. As discussed above, interferon-γ also triggers TRX-mediated activation of extracellular TG2, thereby plausibly establishing an auto-amplificatory loop for gluten-mediated inflammation. Last but not least, chronic exposure of celiac patients to dietary gluten is invariably accompanied by the production of autoantibodies against TG2. As elaborated below, whereas the mechanistic underpinnings of some of these phenomena are relatively well understood, others are not so. A deeper understanding of these TG2-related events will not only enable definitive verification of the hypothesis that transglutaminase activity is necessary for gluten induced pathogenesis in celiac disease, but could also cast fundamentally new light on the biological function of TG2.
Much is known today about the structural basis for gluten recognition by TG2. Although glutamine residues are abundant in this family of polypeptides, and non-enzymatic (acid-promoted) deamidation occurs non-specifically [48], TG2 catalyzes highly selective deamidation of glutamine residues occurring in specific sequence motifs [49–51]. In particular, the repetitive sequence motif QXPZ, where X is any residue and Z is a hydrophobic residue, is a favored site of TG2-catalyzed deamidation. The X-ray crystal structure of TG2 bound covalently to a pentapeptide mimic of this substrate provides a clear rationale for such preference [40]. It also sets the stage for structure-based design of small molecule TG2 inhibitors that could prove pivotal in pharmacologically testing the causal link between TG2 activity and pathogenesis in human patients.
The discovery of the remarkable concordance between TG2 specificity for gluten peptides and the high affinity of the resulting deamidated peptides for HLA-DQ2 (or – DQ8) inaugurated a fundamentally new chapter in our understanding of celiac disease pathogenesis [52–55]. With the solution of high-resolution X-ray crystal structures of representative deamidated gluten peptides in complex with DQ2 [56] and DQ8 [57] (Figure 4), the pathogenic significance of the distinctive pattern of proline residues in proteolytically resistant gluten peptides has been clarified. Equally important is the TG2-catalyzed introduction of a negative charge at a precise location in a gluten peptide (Table 1). Negatively charged carboxylate residues at P4 or P6 in a DQ2 ligand or P9 in a DQ8 ligand anchor these peptides tightly in the corresponding HLA binding grooves (Table 2). The engineering of a synthetic T cell antigen that is capable of triggering a TG2 dependent autoimmune response via an alternative class II major histocompatibility complex in an experimental animal would represent a powerful, albeit challenging, test of the importance of TG2 activity in celiac disease pathogenesis.
Figure 4.

A: Crystal structure of HLA-DQ2 (A; PDB 1S9V; α-chain in green, β-chain in blue) in complex with the αI-gliadin peptide (stick representation). The P4 and the P6 sites preferentially bind deamidated gliadin peptides. B: The P4 glutamine of αI-gliadin is shown interacting with Lysβ71. Deamidation of this residue by TG2 would stabilize this interaction. C: The deamidated P6 glutamate of αI-gliadin is shown hydrogen bonding with Tyrβ9 and Serβ30 and interacting with Lysβ71 through two water molecules.[56] D and E: Crystal structure of HLA-DQ8 (PDB 2NNA; α-chain in green, β-chain in blue) in complex with α2-gliadin:223–240. The P9 site of HLA-DQ8 binds a deamidated glutamine residue through two salt bridges with Argα76 and a hydrogen bond with Tyrβ37.[57] See also Table 1.
Table 1.
Amino acid sequences of immunotoxic gluten peptides and their specificity for TG2 mediated deamidation [51, 104]
| Amino Acid Sequence Immunotoxic Gluten Peptides | TG2 Specificity kcat/KM (min−1mM−1) |
|---|---|
| LQLQPF(PQPQLPY)3PQPQPF | 440 |
| QLQPFPQPQLPYPQPQS | 260 |
| PQPQLPYPQPQLPY | 300 |
| QLQPFPQPQLPY | 66 |
| PQQPQQSFPQQQRP | 61 |
| γ-Fibrinogen Peptide | |
| TIGEGQQHHLG | 63 |
Table 2.
Immunotoxic deamidated gluten epitopes and their EC50 for binding to the HLA-DQ2 and –DQ8 antigen receptors [57, 105, 106]
| Gluten Peptide Immunotoxic Core | HLA-DQ2 Binding | HLA-DQ8 Binding | |
|---|---|---|---|
| Gluten Epitope | Sequence | EC50 (μM) | EC50 (μM) |
| Gliadin-α2 | PQPELPYPQ | 15 | - |
| Gliadin-α9 | PFPQPELPY | 8 | - |
| Gliadin-α20 | FRPEQPYPQ | 30 | - |
| Glu-5 | EXPEQPQQF | 100 | - |
| Gliadin-γ2 | PYPEQPEQP | 65 | - |
| Gliadin-γ1 | PQQSFPEQE | 14 | - |
| Gliadin-γ30 | IIQPEQPAQ | 10 | - |
| Glt-17 | PFSEQEQPV | 25 | - |
| Gliadin-α1 | EGSFQPSQE | - | ~2 |
In contrast to the mechanistic clarity summarized above for gluten antigen recognition by TG2 and recognition of deamidated antigens by DQ2 or DQ8, very little is known thus far about the structural basis for recognition of oxidized TG2 by TRX. Because the high-resolution structures of both proteins have been solved, understanding this selective protein-protein interaction at an atomic level is entirely feasible. In turn, such insights could enable the design of chemical tools to modulate the efficiency extracellular TG2 activation, thereby attenuating gluten mediated T cell inflammation in the small intestines of celiac patients as well as animal models of the disease.
Perhaps most intriguingly, the role of serum IgA and IgG antibodies to TG2 represents a major frontier for molecular investigations into celiac disease pathogenesis. Such autoantibodies are hallmarks of CD, and are in fact used as diagnostic markers [58, 59]. However, our understanding of the role of these autoantibodies in the onset and progression of disease pathogenesis is very limited. Anti-TG2 antibodies are prolifically secreted in the small intestinal mucosa of celiac patients in response to dietary gluten [60]. They are preferentially localized in the sub-epithelial layer, where they adhere tightly to extracellular TG2 on fibroblasts and on the basement membrane of the small intestine [61–63]. Patient derived anti-TG2 autoantibodies induce enterocyte proliferation [64, 65], inhibit enterocyte differentiation [64], and modulate epithelial barrier function [60, 66, 67]. Thus, it is entirely possible that anti-TG2 antibodies promote intestinal crypt hyperplasia and villous blunting, two hallmarks of a celiac lesion. Proof of this hypothesis awaits elucidation of the docking mode(s) of disease-specific autoantibodies on TG2 and the biochemical implications of these antigen-antibody interactions. The recent discovery of a celiac disease-specific conformational epitope on the surface of TG2 may provide an important starting point for such investigations [68].
Physiological and Toxicological Consequences of TG2 Activation
As summarized above, arguably the most important toxicological consequence of extracellular TG2 activity in the celiac small intestine is the activation of gluten-dependent inflammatory T cells. However, it has also been reported that a certain gluten peptide (designated p31–43) activates intracellular TG2, leading to the degradation of the anti-inflammatory peroxisome proliferator-activated receptor (PPARγ)[69, 70]. PPARγ is a hormone receptor produced by several cell types, including epithelial cells, which negatively regulates inflammatory responses [71] and modulates oxidative stress [72, 73]. Therefore, activation of intracellular TG2 in the celiac small intestine can also be expected to contribute towards gut inflammation. However, because this phenomenon neither involves DQ2/8 nor does it involve disease-specific T cells, it remains to be explained if and how gluten mediated activation of intracellular TG2 by this mechanism is specific to the celiac small intestine.
More generally, activation of intracellular and/or extracellular TG2 can be expected to have multifactorial effects on mucosal biology. The connection between these biological consequences of TG2 activation and the wide range of intestinal and extra-intestinal symptoms observed in celiac patients represents an important direction for future research. Furthermore, if TG2 inhibition does prove to be an effective way to control the disease, then it will also be important to understand the physiological and toxicological consequences of inhibiting this ubiquitous but mostly inactive enzyme.
Models for Investigating the Causes and Consequences of TG2 Activation
It should be noted that, until now, the causes and consequences of gluten-dependent TG2 activation could only have been investigated in the celiac patient, a daunting option. Recently however, encouraging steps have been taken towards the development of engineered mice in which dietary gluten consumption, DQ8-restricted T cell response, intestinal villous atrophy, and autoantibody formation are phenotypically linked.
Intestinal epithelial cell culture models have also been used to investigate fundamental aspects of TG2 activity as it relates to celiac disease. For example, exposure to IFN-γ increases both gluten peptide permeability [74] and the transglutaminase activity of extracellular TG2 [35] in a simple T84 enterocytic cell culture model. More advanced studies along these lines could benefit enormously from the application of emerging methods to culture primary intestinal epithelial cells [75, 76] and especially methods to co-culture enterocytes with fibroblasts and cells derived from the immune system.
An essential aspect of modeling the role of TG2 in celiac disease is the requirement for HLA-DQ2 or -DQ8. Towards this end, transgenic mice have been generated that express both these human class II MHC receptors [77, 78]. Already, these humanized mice are providing critical insights into the role of TG2 in celiac disease pathogenesis. For example, in HLA-DQ8 mice, whereas TG2-catalyzed deamidation facilitates the recruitment of a broad repertoire of inflammatory T cells in response to gluten antigens, a limited range of T cells is also provoked in response to native gluten peptides both in mice and humans [79]. Nonetheless, even though these humanized mice react strongly to an intravenous gluten antigen challenge, the hallmark of celiac disease - villous atrophy - is not observed [77, 78, 80]. Moreover, oral gluten is not an effective antigen in either mouse strain. In contrast, transgenic mice that constitutively over-express IL-15 in the gut show many of the features associated with the human condition, including gluten-dependent defects in the regulation of intraepithelial lymphocytes, spontaneous inflammation of the small intestine, villous atrophy and crypt hyperplasia [81–83]. While these animals are the most comprehensive models for celiac disease thus far, the status of TG2 activity in their small intestinal mucosa remains to be elucidated. If it can be shown that mucosal TG2 activity is upregulated constitutively or in response to gluten in these animals, then many of the above questions could be powerfully addressed using this model.
TG2 as a drug target for celiac disease
With its ubiquitous nature and its diverse functions, there might be concern that pharmacological inhibition of TG2 would give rise to undesired target-related effects. While the physiological consequences of TG2 inhibition have not yet been studies in detail, the absence of a strong phenotype in TG2 knockout mice suggests that TG2 inhibition should be tolerated [29–31]. In addition, in the context of CD, the location of the pathological enzymatic activity in the lamina propria would present an opportunity for engineering TG2 inhibitors for a localized action to mediate any undesired consequences of systemic pharmacological inhibition [84].
The prospect of TG2 as a potential target for celiac disease has motivated the development of several classes of TG2 inhibitors as tools and potential lead compounds (Figure 5). Three general inhibition strategies have emerged for blocking the catalytic activity of TG2, targeting the active site from either the acyl donor or the acyl acceptor substrate binding pockets or targeting an allosteric site [85].
Figure 5.

Structures of selected TG2 inhibitors
Acyl-donor pocket active site inhibitors
The most widely utilized approach to inhibiting TG2 activity relies on blocking the active site from the acyl donor substrate binding pocket. These inhibitors are substrate competitive; both substrate-mimetic peptidic scaffolds and small molecules have been developed [85].
The peptidic scaffolds that have been developed usually bear a glutamine-isostere with a reactive functionality, such as epoxy or diazo-ketones or Michael acceptor systems (A; B) that covalently modify the active site Cys277 [40, 86–88]. A class of irreversible inhibitors around the mildly electrophilic 3-bromo-4,5-dihydroisoxazole (DHI) warhead motif on a single amino acid scaffold has been extensively developed (C)[89–91]. Small molecule inhibitors binding to the active site include the cinnamoyl ketones (D)[92, 93], the acrylamido-arylsulfonamides (E)[94] the irreversible thioimidazolium derivatives (F) [88, 95] and the aminoethyl-arylketones (G;H) [96, 97]
Acyl-acceptor pocket active site inhibitors
A number of pseudo-substrate inhibitors that compete with acyl-acceptor substrates have been disclosed, mostly aliphatic amines mimicking lysine.[85]. However, it is unclear whether the acyl-acceptor binding pocket of human TG2 represents an adequate hot-spot for targeting small molecule inhibitors.
Allosteric inhibitors
Although TG2 is physiologically regulated by allosteric binding of GTP, this approach has not been extensively used in the design of TG2 inhibitors. It appears that, unlike in the case of kinases, where the nucleotide binding pocket is the site most site frequently targeted by inhibitors mimicking the nucleobase interactions, the respective pocket in TG2 only tolerates close GTP analogues [98]. Two classes of TG2 inhibitors have been reported to target an allosteric site on TG2, the thienopyrimidinone hydrazides (I) [99] and the acylidene oxindoles (J)[100]. The former class appears to bind to either the GTP site or a site that is coupled to it. However, for neither of these two inhibitor classes has the binding site been definitively identified [101].
Notwithstanding the plethora of potential TG2 inhibitors, only few have been validated in living cell or animal systems. Inhibitors based on the DHI warhead (C) have been used in cell culture to probe and inhibit TG2 activity, as well as in mice to block poly(I:C) mediated TG2 activation [32, 90, 91, 102]; in neither case were pronounced toxic effects observed, even in response to multiple dosing regimens. The thioimidazolium-derivative (F) has also been used in experiments with celiac patients’ biopsies [103]. Further studies are warranted to understand target-associated and compound-specific toxicological effects.
Conclusion
A number of lines of evidence suggest that TG2 may be one of the earliest disease-relevant proteins to encounter immunotoxic gluten in the celiac gut. These and other investigations also suggest that the reaction catalyzed by TG2 on dietary gluten peptides is essential for the pathogenesis of celiac disease. If so, several questions are of critical significance. How is TG2 activated in the celiac gut? What are the disease-specific and general consequences of activating TG2? Can local inhibition of TG2 in the celiac intestine suppress gluten induced pathogenesis in a dose-responsive manner? And what are the long-term consequences of suppressing TG2 activity in the small intestinal mucosa? Answers to these questions will depend upon the development of judicious models and chemical tools. They also have the potential of yielding powerful next-generation drug candidates for treating this widespread but overlooked chronic disease.
Bibliography
- 1.Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature’s biological glues. Biochem J. 2002;368(Pt 2):377–96. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol. 2002;2(9):647–655. doi: 10.1038/nri885. [DOI] [PubMed] [Google Scholar]
- 3.Nakaoka H, Perez DM, Baek KJ, Das T, Husain A, Misono K, Im MJ, Graham RM. Gh: a GTP-binding protein with transglutaminase activity and receptor signaling function. Science. 1994;264(5165):1593–6. doi: 10.1126/science.7911253. [DOI] [PubMed] [Google Scholar]
- 4.Hasegawa G, Suwa M, Ichikawa Y, Ohtsuka T, Kumagai S, Kikuchi M, Sato Y, Saito Y. A novel function of tissue-type transglutaminase: protein disulphide isomerase. Biochem J. 2003;373(Pt 3):793–803. doi: 10.1042/BJ20021084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mishra S, Murphy LJ. Tissue transglutaminase has intrinsic kinase activity: identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J Biol Chem. 2004;279(23):23863–8. doi: 10.1074/jbc.M311919200. [DOI] [PubMed] [Google Scholar]
- 6.Fesus L, Piacentini M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci. 2002;27(10):534–9. doi: 10.1016/s0968-0004(02)02182-5. [DOI] [PubMed] [Google Scholar]
- 7.Park D, Choi SS, Ha K-S. Transglutaminase 2: a multi-functional protein in multiple subcellular compartments. Amino Acids. 2010;39(3):619–31. doi: 10.1007/s00726-010-0500-z. [DOI] [PubMed] [Google Scholar]
- 8.Rodolfo C, Mormone E, Matarrese P, Ciccosanti F, Farrace MG, Garofano E, Piredda L, Fimia GM, Malorni W, Piacentini M. Tissue transglutaminase is a multifunctional BH3-only protein. J Biol Chem. 2004;279(52):54783–92. doi: 10.1074/jbc.M410938200. [DOI] [PubMed] [Google Scholar]
- 9.Malorni W, Farrace MG, Matarrese P, et al. The adenine nucleotide translocator 1 acts as a type 2 transglutaminase substrate: implications for mitochondrial-dependent apoptosis. Cell Death Differ. 2009;16(11):1480–92. doi: 10.1038/cdd.2009.100. [DOI] [PubMed] [Google Scholar]
- 10.Lesort M, Attanavanich K, Zhang J, Johnson GVW. Distinct nuclear localization and activity of tissue transglutaminase. J Biol Chem. 1998;273(20):11991–11994. doi: 10.1074/jbc.273.20.11991. [DOI] [PubMed] [Google Scholar]
- 11.Peng X, Zhang Y, Zhang H, Graner S, Williams JF, Levitt ML, Lokshin A. Interaction of tissue transglutaminase with nuclear transport protein importin-alpha3. FEBS Lett. 1999;446(1):35–9. doi: 10.1016/s0014-5793(99)00018-6. [DOI] [PubMed] [Google Scholar]
- 12.Zemskov EA, Mikhailenko I, Hsia R-C, Zaritskaya L, Belkin AM. Unconventional secretion of tissue transglutaminase involves phospholipid-dependent delivery into recycling endosomes. PloS One. 2011;6(4):e19414. doi: 10.1371/journal.pone.0019414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z, Collighan RJ, Pytel K, Rathbone DL, Li X, Griffin M. Characterization of the heparin binding site of tissue transglutaminase: its importance in the enzyme’s cell surface targeting, matrix deposition and cell signalling. J Biol Chem. 2012 doi: 10.1074/jbc.M111.294819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh US, Pan J. Transglutaminase and cell-survival signaling. Prog Exp Tumor Res. 2005;38:75–88. doi: 10.1159/000084234. [DOI] [PubMed] [Google Scholar]
- 15.Verderio EAM, Johnson TS, Griffin M. Transglutaminases in wound healing and inflammation. Prog Exp Tumor Res. 2005;38:89–114. doi: 10.1159/000084235. [DOI] [PubMed] [Google Scholar]
- 16.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Biol. 2003;4(2):140–56. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 17.Ientile R, Caccamo D, Griffin M. Tissue transglutaminase and the stress response. Amino Acids. 2007;33(2):385–94. doi: 10.1007/s00726-007-0517-0. [DOI] [PubMed] [Google Scholar]
- 18.Collighan RJ, Griffin M. Transglutaminase 2 cross-linking of matrix proteins: biological significance and medical applications. Amino Aids. 2009;36(4):659–70. doi: 10.1007/s00726-008-0190-y. [DOI] [PubMed] [Google Scholar]
- 19.Belkin AM. Extracellular TG2: emerging functions and regulation. FEBS J. 2011;278(24):4704–16. doi: 10.1111/j.1742-4658.2011.08346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fésüs L, Szondy Z. Transglutaminase 2 in the balance of cell death and survival. FEBS Lett. 2005;579(15):3297–302. doi: 10.1016/j.febslet.2005.03.063. [DOI] [PubMed] [Google Scholar]
- 21.Gaudry CA, Verderio E, Aeschlimann D, Cox A, Smith C, Griffin M. Cell surface localization of tissue transglutaminase is dependent on a fibronectin-binding site in its N-terminal beta-sandwich domain. J Biol Chem. 1999;274(43):30707–14. doi: 10.1074/jbc.274.43.30707. [DOI] [PubMed] [Google Scholar]
- 22.Gaudry CA, Verderio E, Jones RA, Smith C, Griffin M. Tissue transglutaminase is an important player at the surface of human endothelial cells: evidence for its externalization and its colocalization with the beta(1) integrin. Exp Cell Res. 1999;252(1):104–13. doi: 10.1006/excr.1999.4633. [DOI] [PubMed] [Google Scholar]
- 23.Zemskov E, Janiak A, Hang J, Waghray A. The role of tissue transglutaminase in cell-matrix interactions. Front Biosci. 2006;(1):1057–1076. doi: 10.2741/1863. [DOI] [PubMed] [Google Scholar]
- 24.Telci D, Griffin M. Tissue transglutaminase (TG2) - a wound response enzyme. Front Biosci. 2006;11(4):867–82. doi: 10.2741/1843. [DOI] [PubMed] [Google Scholar]
- 25.Haroon ZA, Hettasch JM, Lai TS, Dewhirst MW, Greenberg CS. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999;13(13):1787–95. doi: 10.1096/fasebj.13.13.1787. [DOI] [PubMed] [Google Scholar]
- 26.Nunes I, Gleizes PE, Metz CN, Rifkin DB. Latent transforming growth factor-beta binding protein domains involved in activation and transglutaminase-dependent cross-linking of latent transforming growth factor-beta. J Cell Biol. 1997;136(5):1151–63. doi: 10.1083/jcb.136.5.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehta K, Fok JY, Mangala LS. Tissue transglutaminase: from biological glue to cell survival cues. Front Biosci. 2006;11(3):173–85. doi: 10.2741/1789. [DOI] [PubMed] [Google Scholar]
- 28.Gundemir S, Colak G, Tucholski J, Johnson GVW. Transglutaminase 2: A molecular Swiss army knife. Biochim Biophys Acta. 2011;1823(2):406–419. doi: 10.1016/j.bbamcr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Laurenzi V, Melino G. Gene disruption of tissue transglutaminase. Mol Cell Biol. 2001;21(1):148. doi: 10.1128/MCB.21.1.148-155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001;276(23):20673–8. doi: 10.1074/jbc.M010846200. [DOI] [PubMed] [Google Scholar]
- 31.Sarang Z, Tóth B, Balajthy Z, Köröskényi K, Garabuczi E, Fésüs L, Szondy Z. Some lessons from the tissue transglutaminase knockout mouse. Amino Acids. 2009;36(4):625–31. doi: 10.1007/s00726-008-0130-x. [DOI] [PubMed] [Google Scholar]
- 32.Siegel M, Strnad P, Watts RE, Choi K, Jabri B, Omary MB, Khosla C. Extracellular Transglutaminase 2 Is Catalytically Inactive, but Is Transiently Activated upon Tissue Injury. PLoS ONE. 2008;3(3):e1861. doi: 10.1371/journal.pone.0001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stamnaes J, Pinkas DM, Fleckenstein B, Khosla C, Sollid LM. Redox regulation of transglutaminase 2 activity. J Biol Chem. 2010;285(33):25402–9. doi: 10.1074/jbc.M109.097162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin X, Stamnaes J, Kloeck C, Diraimondo TR, Sollid LM, Khosla C. Activation of extracellular transglutaminase 2 by thioredoxin. J Biol Chem. 2011;286(43):37866–37873. doi: 10.1074/jbc.M111.287490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiRaimondo TR, Klöck C, Khosla C. Interferon-γ Activates Transglutaminase 2 via a Phosphatidylinositol-3-Kinase Dependent Pathway: Implications for Celiac Sprue Therapy. J Pharmacol Exp Ther. 2012 doi: 10.1124/jpet.111.187385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergamini CM. GTP modulates calcium binding and cation-induced conformational changes in erythrocyte transglutaminase. FEBS Lett. 1988;239(2):255–8. doi: 10.1016/0014-5793(88)80928-1. [DOI] [PubMed] [Google Scholar]
- 37.Begg GE, Holman SR, Stokes PH, Matthews JM, Graham RM, Iismaa SE. Mutation of a critical arginine in the GTP-binding site of transglutaminase 2 disinhibits intracellular cross-linking activity. J Biol Chem. 2006;281(18):12603–9. doi: 10.1074/jbc.M600146200. [DOI] [PubMed] [Google Scholar]
- 38.Bergamini CM, Signorini M, Poltronieri L. Inhibition of erythrocyte transglutaminase by GTP. Biochim Biophys Acta. 1987;916(1):149–151. doi: 10.1016/0167-4838(87)90222-6. [DOI] [PubMed] [Google Scholar]
- 39.Bergamini CM, Dondi A, Lanzara V, et al. Thermodynamics of binding of regulatory ligands to tissue transglutaminase. Amino acids. 2010;39(1):297–304. doi: 10.1007/s00726-009-0442-5. [DOI] [PubMed] [Google Scholar]
- 40.Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 Undergoes a Large Conformational Change upon Activation. PLoS Biol. 2007;5(12):2788–2796. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci USA. 2002;99(5):2743–7. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Antonyak MA, Jansen JM, Miller AM, Ly TK, Endo M, Cerione Ra. Two isoforms of tissue transglutaminase mediate opposing cellular fates. Proc Natl Acad Sci USA. 2006;103(49):18609–14. doi: 10.1073/pnas.0604844103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai T-S, Liu Y, Li W, Greenberg CS. Identification of two GTP-independent alternatively spliced forms of tissue transglutaminase in human leukocytes, vascular smooth muscle, and endothelial cells. FASEB J. 2007;21(14):4131–43. doi: 10.1096/fj.06-7598com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Citron BA, Suo Z, SantaCruz K, Davies PJA, Qin F, Festoff BW. Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem Int. 2002;40(1):69–78. doi: 10.1016/s0197-0186(01)00062-6. [DOI] [PubMed] [Google Scholar]
- 45.Kojima S, Kuo T-F, Tatsukawa H. Regulation of transglutaminase-mediated hepatic cell death in alcoholic steatohepatitis and non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2012;27(Suppl 2):52–7. doi: 10.1111/j.1440-1746.2011.07009.x. [DOI] [PubMed] [Google Scholar]
- 46.Monsonego A, Friedmann I, Shani Y, Eisenstein M, Schwartz M. GTP-dependent conformational changes associated with the functional switch between Galpha and cross-linking activities in brain-derived tissue transglutaminase. J Mol Biol. 1998;282(4):713–20. doi: 10.1006/jmbi.1998.2052. [DOI] [PubMed] [Google Scholar]
- 47.Monsonego A, Shani Y, Friedmann I, Paas Y, Eizenberg O, Schwartz M. Expression of GTP-dependent and GTP-independent tissue-type transglutaminase in cytokine-treated rat brain astrocytes. J Biol Chem. 1997;272(6):3724–32. doi: 10.1074/jbc.272.6.3724. [DOI] [PubMed] [Google Scholar]
- 48.Sjöström H, Lundin KE, Molberg O, et al. Identification of a gliadin T-cell epitope in coeliac disease: general importance of gliadin deamidation for intestinal T-cell recognition. Scand J Immunol. 1998;48(2):111–5. doi: 10.1046/j.1365-3083.1998.00397.x. [DOI] [PubMed] [Google Scholar]
- 49.Fleckenstein B, Molberg Ø, Qiao S-W, Schmid DG, von der Mülbe F, Elgstøen K, Jung G, Sollid LM. Gliadin T cell epitope selection by tissue transglutaminase in celiac disease. Role of enzyme specificity and pH influence on the transamidation versus deamidation process. J Biol Chem. 2002;277(37):34109–16. doi: 10.1074/jbc.M204521200. [DOI] [PubMed] [Google Scholar]
- 50.Vader LW, de Ru A, van der Wal Y, Kooy YMC, Benckhuijsen W, Mearin ML, Drijfhout JW, van Veelen P, Koning F. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J Exp Med. 2002;195(5):643–9. doi: 10.1084/jem.20012028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piper JL, Gray GM, Khosla C. High selectivity of human tissue transglutaminase for immunoactive gliadin peptides: implications for celiac sprue. Biochemistry. 2002;41(1):386–93. doi: 10.1021/bi011715x. [DOI] [PubMed] [Google Scholar]
- 52.Molberg O, Mcadam SN, Körner R, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4(6):713–7. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 53.van de Wal Y, Kooy Y, van Veelen P, Peña S, Mearin L, Papadopoulos G, Koning F. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. 1998;161(4):1585–8. [PubMed] [Google Scholar]
- 54.Anderson RP, Degano P, Godkin AJ, Jewell DP, Hill AVS. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant A-gliadin T-cell epitope. Nat Med. 2000;6(3):337–42. doi: 10.1038/73200. [DOI] [PubMed] [Google Scholar]
- 55.Arentz-Hansen H, Körner R, Molberg O, et al. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med. 2000;191(4):603–12. doi: 10.1084/jem.191.4.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim C-Y, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci USA. 2004;101(12):4175–9. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henderson KN, Tye-Din JA, Reid HH, et al. A structural and immunological basis for the role of human leukocyte antigen DQ8 in celiac disease. Immunity. 2007;27(1):23–34. doi: 10.1016/j.immuni.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 58.Dieterich W, Laag E, Schöpper H, Volta U, Ferguson A, Gillett H, Riecken EO, Schuppan D. Autoantibodies to tissue transglutaminase as predictors of celiac disease. Gastroenterol. 1998;115(6):1317–21. doi: 10.1016/s0016-5085(98)70007-1. [DOI] [PubMed] [Google Scholar]
- 59.Sulkanen S, Halttunen T, Laurila K, Kolho KL, Korponay-Szabó IR, Sarnesto A, Savilahti E, Collin P, Mäki M. Tissue transglutaminase autoantibody enzyme-linked immunosorbent assay in detecting celiac disease. Gastroenterol. 1998;115(6):1322–8. doi: 10.1016/s0016-5085(98)70008-3. [DOI] [PubMed] [Google Scholar]
- 60.Caja S, Mäki M, Kaukinen K, Lindfors K. Antibodies in celiac disease: implications beyond diagnostics. Cell Mol Immunol. 2011;8(2):103–9. doi: 10.1038/cmi.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mäki M. The humoral immune system in coeliac disease. Baillieres Clin Gastroenterol. 1995;9(2):231–49. doi: 10.1016/0950-3528(95)90030-6. [DOI] [PubMed] [Google Scholar]
- 62.Rantala I, Mäki M, Laasonen A, Visakorpi JK. Periodate-lysine-paraformaldehyde as fixative for the study of duodenal mucosa. Morphologic and immunohistochemical results at light and electron microscopic levels. Acta Pathol Microbiol Immunol Scand A. 1985;93(4):165–73. doi: 10.1111/j.1699-0463.1985.tb03936.x. [DOI] [PubMed] [Google Scholar]
- 63.Korponay-Szabó IR, Halttunen T, Szalai Z, Laurila K, Király R, Kovács JB, Fésüs L, Mäki M. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut. 2004;53(5):641–8. doi: 10.1136/gut.2003.024836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Halttunen T, Mäki M. Serum immunoglobulin A from patients with celiac disease inhibits human T84 intestinal crypt epithelial cell differentiation. Gastroenterol. 1999;116(3):566–72. doi: 10.1016/s0016-5085(99)70178-2. [DOI] [PubMed] [Google Scholar]
- 65.Barone MV, Caputo I, Ribecco MT, Maglio M, Marzari R, Sblattero D, Troncone R, Auricchio S, Esposito C. Humoral immune response to tissue transglutaminase is related to epithelial cell proliferation in celiac disease. Gastroenterol. 2007;132(4):1245–53. doi: 10.1053/j.gastro.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 66.Zanoni G, Navone R, Lunardi C, et al. In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes. PLoS Med. 2006;3(9):e358. doi: 10.1371/journal.pmed.0030358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med. 2008;205(1):143–54. doi: 10.1084/jem.20071204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simon-Vecsei Z, Király R, Bagossi P, et al. A single conformational transglutaminase 2 epitope contributed by three domains is critical for celiac antibody binding and effects. Proc Natl Acad Sci USA. 2012;109(2):431–6. doi: 10.1073/pnas.1107811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Re V, Simula MP, Notarpietro A, Canzonieri V, Cannizzaro R, Toffoli G. Do gliadin and tissue transglutaminase mediate PPAR downregulation in intestinal cells of patients with coeliac disease? Gut. 2010;59(12):1730–1. doi: 10.1136/gut.2010.209395. [DOI] [PubMed] [Google Scholar]
- 70.Luciani A, Villella VR, Vasaturo A, et al. Lysosomal accumulation of gliadin p31–43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut. 2010;59(3):311–9. doi: 10.1136/gut.2009.183608. [DOI] [PubMed] [Google Scholar]
- 71.Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2(10):748–59. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 72.Rizzo G, Fiorucci S. PPARs and other nuclear receptors in inflammation. Curr Opin Pharmacol. 2006;6(4):421–7. doi: 10.1016/j.coph.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 73.Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C, Danni O, Thiemermann C, Fantozzi R. Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol. 2006;530(1–2):70–80. doi: 10.1016/j.ejphar.2005.11.049. [DOI] [PubMed] [Google Scholar]
- 74.Bethune MT, Siegel M, Howles-Banerji S, Khosla C. Interferon-gamma released by gluten-stimulated celiac disease-specific intestinal T cells enhances the transepithelial flux of gluten peptides. J Pharmacol Exp Ther. 2009;329(2):657–68. doi: 10.1124/jpet.108.148007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ootani A, Li X, Sangiorgi E, et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med. 2009;15(6):701–6. doi: 10.1038/nm.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Snippert HJ, van der Flier LG, Sato T, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143(1):134–44. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 77.Black KE, Murray JA, David CS. HLA-DQ determines the response to exogenous wheat proteins: a model of gluten sensitivity in transgenic knockout mice. J Immunol. 2002;169(10):5595–600. doi: 10.4049/jimmunol.169.10.5595. [DOI] [PubMed] [Google Scholar]
- 78.Chen Z, Dudek N, Wijburg O, Strugnell R, Brown L, Deliyannis G, Jackson D, Koentgen F, Gordon T, McCluskey J. A 320-kilobase artificial chromosome encoding the human HLA DR3-DQ2 MHC haplotype confers HLA restriction in transgenic mice. J Immunol. 2002;168(6):3050–6. doi: 10.4049/jimmunol.168.6.3050. [DOI] [PubMed] [Google Scholar]
- 79.Hovhannisyan Z, Weiss A, Martin A, et al. The role of HLA-DQ8 beta57 polymorphism in the anti-gluten T-cell response in coeliac disease. Nature. 2008;456(7221):534–8. doi: 10.1038/nature07524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.de Kauwe AL, Chen Z, Anderson RP, Keech CL, Price JD, Wijburg O, Jackson DC, Ladhams J, Allison J, McCluskey J. Resistance to celiac disease in humanized HLA-DR3-DQ2-transgenic mice expressing specific anti-gliadin CD4+ T cells. J Immunol. 2009;182(12):7440–50. doi: 10.4049/jimmunol.0900233. [DOI] [PubMed] [Google Scholar]
- 81.Ohta N, Hiroi T, Kweon M-N, Kinoshita N, Jang MH, Mashimo T, Miyazaki J-I, Kiyono H. IL-15-dependent activation-induced cell death-resistant Th1 type CD8 alpha beta+NK1.1+ T cells for the development of small intestinal inflammation. J Immunol. 2002;169(1):460–8. doi: 10.4049/jimmunol.169.1.460. [DOI] [PubMed] [Google Scholar]
- 82.DePaolo RW, Abadie V, Tang F, et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature. 2011;471(7337):220–4. doi: 10.1038/nature09849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yokoyama S, Watanabe N, Sato N, Perera P-Y, Filkoski L, Tanaka T, Miyasaka M, Waldmann TA, Hiroi T, Perera LP. Antibody-mediated blockade of IL-15 reverses the autoimmune intestinal damage in transgenic mice that overexpress IL-15 in enterocytes. Proc Natl Acad Sci USA. 2009;106(37):15849–54. doi: 10.1073/pnas.0908834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sollid LM, Khosla C. Future therapeutic options for celiac disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2(3):140–7. doi: 10.1038/ncpgasthep0111. [DOI] [PubMed] [Google Scholar]
- 85.Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol Ther. 2007;115(2):232–245. doi: 10.1016/j.pharmthera.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marrano C, de Macédo P, Keillor JW. Evaluation of novel dipeptide-bound alpha, beta-unsaturated amides and epoxides as irreversible inhibitors of guinea pig liver transglutaminase. Bioorg Med Chem. 2001;9(7):1923–1928. doi: 10.1016/s0968-0896(01)00101-8. [DOI] [PubMed] [Google Scholar]
- 87.Wodzinska JM. Transglutaminases as targets for pharmacological inhibition. Mini Rev Med Chem. 2005;5(3):279–92. doi: 10.2174/1389557053175416. [DOI] [PubMed] [Google Scholar]
- 88.Hausch F, Halttunen T, Mäki M, Khosla C. Design, synthesis, and evaluation of gluten peptide analogs as selective inhibitors of human tissue transglutaminase. Chem Biol. 2003;10(3):225–231. doi: 10.1016/s1074-5521(03)00045-0. [DOI] [PubMed] [Google Scholar]
- 89.Chittur SV, Klem TJ, Shafer CM, Davisson VJ. Mechanism for acivicin inactivation of triad glutamine amidotransferases. Biochemistry. 2001;40(4):876–87. doi: 10.1021/bi0014047. [DOI] [PubMed] [Google Scholar]
- 90.Choi K, Siegel M, Piper JL, Yuan L, Cho E, Strnad P, Omary B, Rich KM, Khosla C. Chemistry and Biology of Dihydroisoxazole Derivatives: Selective Inhibitors of Human Transglutaminase 2. Chem Biol. 2005;12(4):469–475. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 91.Watts RE, Siegel M, Khosla C. Structure-Activity Relationship Analysis of the Selective Inhibition of Transglutaminase 2 by Dihydroisoxazoles. J Med Chem. 2006;49(25):7493–7501. doi: 10.1021/jm060839a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pardin C, Pelletier JN, Lubell WD, Keillor JW. Cinnamoyl Inhibitors of Tissue Transglutaminase. J Org Chem. 2008;73(15):5766–5775. doi: 10.1021/jo8004843. [DOI] [PubMed] [Google Scholar]
- 93.Pardin C, Roy I, Chica RA, Bonneil E, Thibault P, Lubell WD, Pelletier JN, Keillor JW. Photolabeling of tissue transglutaminase reveals the binding mode of potent cinnamoyl inhibitors. Biochemistry. 2009;48(15):3346–53. doi: 10.1021/bi802021c. [DOI] [PubMed] [Google Scholar]
- 94.Prime ME, Andersen OA, Barker J, et al. Discovery and SAR of Potent and Selective Covalent Inhibitors of Transglutaminase 2 for Huntington’s Disease. J Med Chem. 2012 doi: 10.1021/jm201310y. [DOI] [PubMed] [Google Scholar]
- 95.Freund KF, Doshi KP, Gaul SL, Claremon DA, Remy DC, Baldwin JJ, Pitzenberger SM, Stern AM. Transglutaminase inhibition by 2-[(2-oxopropyl) thio] imidazolium derivatives: mechanism of factor XIIIa inactivation. Biochemistry. 1994;33(33):10109–10119. doi: 10.1021/bi00199a039. [DOI] [PubMed] [Google Scholar]
- 96.Ozaki S, Ebisui E, Hamada K, Goto J-I, Suzuki AZ, Terauchi A, Mikoshiba K. Potent transglutaminase inhibitors, aryl beta-aminoethyl ketones. Bioorg Med Chem Lett. 2010;20(3):1141–4. doi: 10.1016/j.bmcl.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 97.Lai T-S, Liu Y, Tucker T, Daniel KR, Sane DC, Toone E, Burke JR, Strittmatter WJ, Greenberg CS. Identification of chemical inhibitors to human tissue transglutaminase by screening existing drug libraries. Chem Biol. 2008;15(9):969–78. doi: 10.1016/j.chembiol.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schaertl S, Prime M, Wityak J, Dominguez C, Munoz-Sanjuan I, Pacifici RE, Courtney S, Scheel A, Macdonald D. A Profiling Platform for the Characterization of Transglutaminase 2 (TG2) Inhibitors. J Biomol Screen. 2010;15(5):478–87. doi: 10.1177/1087057110366035. [DOI] [PubMed] [Google Scholar]
- 99.Duval E, Case A, Stein RL, Cuny GD. Structure-activity relationship study of novel tissue transglutaminase inhibitors. Bioorg Med Chem Lett. 2005;15(7):1885–9. doi: 10.1016/j.bmcl.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 100.Klöck C, Jin X, Choi K, Khosla C, Madrid PB, Spencer A, Raimundo BC, Boardman P, Lanza G, Griffin JH. Acylideneoxoindoles: a new class of reversible inhibitors of human transglutaminase 2. Bioorg Med Chem Lett. 2011;21(9):2692–6. doi: 10.1016/j.bmcl.2010.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Case A, Stein RL. Kinetic analysis of the interaction of tissue transglutaminase with a nonpeptidic slow-binding inhibitor. Biochemistry. 2007;46(4):1106–15. doi: 10.1021/bi061787u. [DOI] [PubMed] [Google Scholar]
- 102.Dafik L, Albertelli M, Stamnaes J, Sollid LM, Khosla C. Activation and inhibition of transglutaminase 2 in mice. PloS one. 2012;7(2):e30642. doi: 10.1371/journal.pone.0030642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Rispo A, Griffin M, Issekutz T, Quaratino S, Londei M. Unexpected role of surface transglutaminase type II in celiac disease. Gastroenterol. 2005;129(5):1400–13. doi: 10.1053/j.gastro.2005.07.054. [DOI] [PubMed] [Google Scholar]
- 104.Shan L, Molberg Ø, Parrot I, Hausch F, Filiz F, Gray GM, Sollid LM, Khosla C. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297(5590):2275–9. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- 105.Vader W, Stepniak D, Kooy Y, Mearin L, Thompson A, van Rood JJ, Spaenij L, Koning F. The HLA-DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten-specific T cell responses. Proc Natl Acad Sci USA. 2003;100(21):12390–5. doi: 10.1073/pnas.2135229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kooy-Winkelaar Y, van Lummel M, Moustakas AK, et al. Gluten-specific T cells cross-react between HLA-DQ8 and the HLA-DQ2α/DQ8β transdimer. J Immunol. 2011;187(10):5123–9. doi: 10.4049/jimmunol.1101179. [DOI] [PubMed] [Google Scholar]
- 107.Nemes Z, Petrovski G, Csosz E, Fésüs L. Structure-function relationships of transglutaminases - a contemporary view. Prog Exp Tumor Res. 2005;38:19–36. doi: 10.1159/000084231. [DOI] [PubMed] [Google Scholar]
- 108.Lorand L, Conrad SM. Transglutaminases. Mol Cell Biochem. 1984;58(1–2):9–35. doi: 10.1007/BF00240602. [DOI] [PubMed] [Google Scholar]
- 109.Facchiano A, Facchiano F. Transglutaminases and their substrates. Prog Exp Tumor Res. 2005;38:37–57. doi: 10.1159/000084232. [DOI] [PubMed] [Google Scholar]
- 110.Schrode J, Folk JE. Transglutaminase-catalyzed cross-linking through diamines and polyamines. J Biol Chem. 1978;253(14):4837–40. [PubMed] [Google Scholar]