Early Origins of Adult Disease: Approaches for Investigating the Programmable Epigenome in Humans, Nonhuman Primates, and Rodents (original) (raw)
Abstract
According to the developmental origins of health and disease hypothesis, in utero experiences reprogram an individual for immediate adaptation to gestational perturbations, with the sequelae of later-in-life risk of metabolic disease. An altered gestational milieu with resultant adult metabolic disease has been observed in instances of both in utero constraint (e.g., from famine or uteroplacental insufficiency) and overt caloric abundance (e.g., from a maternal high-fat, caloric-dense diet). The commonality of the adult metabolic phenotype begs the question of how diverse in utero experiences (i.e., reprogramming events) converge on common metabolic pathways and how the memory of these events is maintained across the lifespan. We and others have investigated the molecular mechanisms underlying fetal programming and observed that epigenetic modifications to the fetal and placental epigenome accompany these reprogramming events. Based on several lines of emerging data in human and nonhuman primates, it is now felt that modified epigenetic signature—and the histone code in particular—underlies alterations in postnatal gene expression and metabolic pathways central to accurate functioning and maintenance of health. Because of the tissue lineage specificity of many of these modifications, nonhuman primates serve as an apt model system for the capacity to recapitulate human gene expression and regulation during development. This review summarizes recent epigenetic advances using rodent and primate (both human and nonhuman) models during in utero development and contributing to adult diseases later in life.
Keywords: behavioral epigenetics, developmental origins of health and disease (DOHaD), environmental epigenetics, microbiome, nonhuman primate (NHP)
Overview
Genomic DNA is the template of all organisms’ heredity, and the coordination and regulation of its expression result in the wide complexity and diversity of those organisms. The realization that factors other than the DNA sequence per se regulate gene expression resulted in a burgeoning of studies on epigenetics. The epigenome can be considered as a code written (or rewritten and revised) as a reflection of a modified genomic code, which in turn regulates gene expression as a result of its essential role in regulating accessibility of the transcriptional machinery. Epigenetics has had a tremendous impact on the molecular understanding of gene regulation during development. The widely studied correlation between diet, environment, and genetics has enabled scientists to move to the next step of understanding how the gestational milieu affects our genome and programs for health and disease (Figure 1). This review summarizes the seminal studies on in utero factors affecting the epigenetic regulation and programming for adult diseases such as obesity, hypertension, type 2 diabetes, and cardiovascular disease. We emphasize the unique importance of human and nonhuman primate (NHP1) models in understanding the role of epigenomic modifications in unraveling the molecular mysteries underlying the developmental origins of adult disease.
Figure 1.
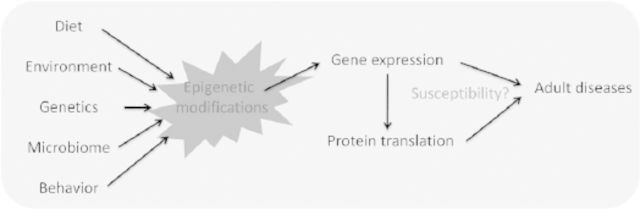
Schematic overview of potential developmental exposures rendering risk of adult metabolic disease.
Epidemiological Studies Linking Early Origins with Adult Diseases
Early Retrospective Cohort Studies
Among the population-based studies on the developmental origins of health and disease, the most extensively documented and well-studied epidemiological studies employ subjects from the Dutch hunger winter. Unlike subsequent famines (e.g., the African famines of the 20th century), the Dutch famine occurred during a very short and well-defined period, enabling fairly precise exposure modeling.
In the winter of 1944–1945, the Axis alliance blocked all food convoys and confiscated stored supplies throughout the western and northern Netherlands, leading to a famine (daily intake of <1000 kcal and, for many, 500 calories/day) that resulted in the death from starvation of more than 18,000 women and children (Lumey et al. 2007). Despite significant poverty and tremendous hardship, the healthcare and data records were preserved, with the maintenance of meticulous records on periconception, early- and late-gestation maternal weight, relative nutrition, and fetal birthweight and gestation. Follow-up studies on children born during this period provided clear evidence that maternal undernutrition during the critical development phase was associated with the children's eventual development of schizophrenia (Susser and Lin 1992), elevated cholesterol and triglyceride levels (Lumey et al. 2009), impaired glucose tolerance (de Rooij et al. 2006; Ravelli et al. 1998), higher body mass index (Roseboom et al. 2000a, 2000c), and higher risk of coronary heart disease (Ravelli et al. 1999; Roseboom et al. 2000b). Barker's hypothesis emerged from these early cohort descriptions and has served as a cornerstone of developmental origins of health and disease; it posits that disturbances during critical gestational windows are associated with, and likely account for, the development of certain metabolic diseases later in life (Miller et al. 2010; Tice 2010).
Numerous subsequent epidemiological studies have been conducted to understand and support these initial observations, and several paramount studies are underway. Similar findings of a predisposition to adult metabolic disease have been found in studies of individuals exposed in utero and through early childhood to famine in Nigeria. Famine struck millions of Biafrans during the Nigerian Civil War of 1967–1970. During the first year of the civil war, Biafra lost most of its territory and access to airports and seaports, its oil refineries, and most of its oilfields, resulting in mass starvation of its population. From late 1968 until the final surrender in January 1970, Biafran refugees in packed, fetid camps were kept alive through one of the largest privately organized relief operations in African history. Akin to the Dutch winter famine, this short interval of mass starvation was documented clearly and was unequivocal in its devastating scope and long-term implications. Forty years since the famine, individuals exposed to it prenatally show hypertension, impaired glucose tolerance, increased waist circumference, and overweight phenotypes compared with individuals born after the famine (Hult et al. 2010).
Recent Longitudinal Cohort Studies
Other longitudinal cohort and population-based analyses have been undertaken in the past two decades and are summarized in Table 1. Initiated in 1998, the Southampton Women's Survey is a prospective cohort study that has assessed the diet, body composition (attained anthropometric measurements), physical activity, and social circumstances of a large cohort of pregravid women aged 20 to 34 years living in Southampton, United Kingdom. Women who subsequently became pregnant were studied at 11, 19, and 34 weeks of gestation, and ongoing follow-up of their offspring during infancy and childhood is underway (Inskip et al. 2006). Although their work is ongoing, initial data gathered from principle component analysis revealed the presence of two distinct occurring cohorts: “prudent diet” and “high-energy diet” (Crozier et al. 2006). Of note, when analyzed by maternal weight gain in pregnancy, “appropriate” (per 2009 Institute of Medicine guidelines) gain was linked to lower levels of adiposity in the offspring, whereas excessive weight gain was associated with greater neonatal fat mass and marginally associated with fat mass at 6 years of age (Crozier et al. 2010). OBELIX, a second large European cohort, aimed to study whether prenatal and early postnatal exposure to endocrine-disrupting chemicals can be a risk factor for obesity and metabolic syndrome–related diseases later in life (Legler et al. 2011).
Table 1.

In a similar vein, Project Viva is a US-based initiative to determine the relationship between maternal diet and behavior with offspring health through infancy and early childhood. The Project Viva cohort arises from slightly more than 2100 gravidae recruited at the time of their first prenatal visit (<22 weeks gestation) from community-based practice groups in eastern Massachusetts. Studies conducted on Project Viva cohorts have established the association between enhanced maternal–fetal n-3 polyunsaturated fatty acid status and lower childhood adiposity (Donahue et al. 2011). Finally, the heretofore unparalleled efforts of the National Children's Study2 will offer comprehensive, true population-based analyses among household-based enrollment of pregravidae with planned follow-up through 21 years of age. Ultimately, it is our opinion that the National Children's Study has the potential to become one of the richest and vibrant research efforts geared toward studying children's health and development through critical windows in development.
Epigenomic Modifications That Contribute to the Developmental Landscape and Fetal Programming
The aforementioned epidemiological studies collectively establish an association between perturbations during human gestation and altered risk of disease later in life. However, as with all observational studies, it is crucial to decipher the biological underpinnings and molecular mechanisms to establish a causal relationship between so-called fetal programming and later-in-life disease.
A fundamental tenet of modern biology is that the environment shapes the organism. At the species level, the process of evolution involves selection of the fittest DNA specimens over time according to the principles outlined by Charles Darwin. The mechanism involved is the inheritance of genetic traits first described by Gregor Mendel and later understood to be attributable to differences in DNA sequence. Since these early days of understanding that genes and chromosomes function as the backbone of our genetic heredity, the view of heredity has been written in the language of DNA, with the assumption that genetic mutations and changes in the nucleotide backbone have driven most descriptions of how phenotypic traits and diseases are handed down from one generation to another.
It is now known that this mechanism is only partially correct. Although selective pressures certainly can result in population genetic drift (generally through either single nucleotide polymorphisms or copy number variants), evidence from the past decade suggests that the environment largely shapes the organism through epigenetic mechanisms without effecting changes in DNA sequence (Bocock and Aagaard-Tillery 2009; Suter and Aagaard-Tillery 2009). Epigenetic mechanisms such as DNA methylation, histone acetylation, and RNA interference and their effects in gene activation and inactivation are increasingly understood to have a profound effect on alteration of an individual's appearance, transmission of a specific congenital anomaly, and even one's lifetime risk of common diseases such as obesity. Thus we have arrived at the point in our current understanding of genetics whereby we acknowledge that, although genomic DNA is the template of our heredity, it is the orchestration and regulation of its expression by epigenetic mechanisms in chromatin remodeling, DNA methylation, and microRNA (miRNA1) variation that ultimately result in the complexity and diversity among individuals observed in nature.
So what are epigenetic modifications? As schematically outlined in Figure 2, the nucleosome serves as the fundamental unit structuring chromatin. Two copies of histone proteins H3 and H4 form the tetramer, and two copies of H2A and H2B dimers join to form the histone octamer. Around this octamer, 147 bases of DNA are wrapped approximately 1.7 times in a superhelix to form the nucleosome (Luger et al. 1997). Adenosine triphosphate–dependent enzymes remodel the chromatin to provide access for transcription machinery (Ho and Crabtree 2010). Several histone modifications, such as acetylation, methylation, ubiquitylation, phosphorylation, and sumolyation, have been studied (Bernstein et al. 2005; Jenuwein and Allis 2001). DNA methylation is addition of methyl groups to the cytosine bases mediated through DNA methyltransferases (DNMTs1). DNMTs are classified as maintenance (DNMT1) and de novo methyltransferases (DNMT3a and DNMT3b), but all of them can be active in both states (Bestor 1992; Bestor et al. 1988; Chen et al. 2003). DNA methylation is inversely correlated with gene expression. Histone modifications are thought to act as guides for DNA methylation, thereby affecting the gene expression (Reik 2007; Vire et al. 2006). During preimplantation, the genome is demethylated and, after implantation, gets remethylated progressively in patterns structured ultimately by species and tissue type (cellular differentiation). Thus, in utero_,_ maternal, and environmental factors have the unequivocal potential to program the fetal epigenomic code. However, to what extent this is largely proscribed and prescribed by species, degree of pluripotency, and lineage of differentiation is still being deciphered.
Figure 2.
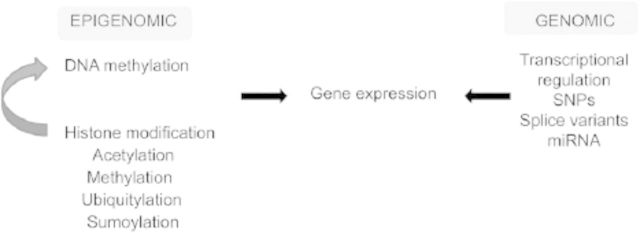
Schematic representation of both genomic and epigenomic mechanisms of regulating gene expression. miRNA, microRNA; SNP, single nucleotide polymorphisms.
Genomic Imprinting
One of the most exciting attributes of epigenetic inheritance is the potential to meaningfully alter the pace of change. The in utero environment can theoretically lead to early potentially adaptive changes in the phenotype of the organism, which then bear the potential to be passed on to subsequent generations. Ergo, epigenetic mechanisms of inheritance offer potential plasticity to the organism's response to environmental challenges and are particularly responsive to environmental challenges during reproduction and development. However, to avoid “epigenomic entropy” as the methylome is actively demethylated and remethylated during early embryogenesis and development, there must be structure to this process. One such form of structure is differentially methylated regions and imprinted loci that maintain their methylation marks from the maternal and paternal gametes.
Genomic imprinting is an epigenetic phenomenon that silences expression of paternally or maternally derived alleles. It is postulated that the imprint is established in the parental germ lines and then maintained during early zygote formation when the remainder of the methylome is demethylated to regulate expression of imprinted genes that are essential to support proper embryonic development (reviewed in Feng 2010). Of the 120 or so identified imprinted loci in primates, the majority are essential for embryonic development, especially placental formation, and others regulate metabolism, behavior, and physiological functions. In humans, disruption of genomic imprinting causes several diseases, including cancer and (of interest herein) metabolic disorders.
Striking evidence for epigenetic mechanisms in the etiology of obesity is observed in individuals with the imprinting disorder Prader-Willi Syndrome (Jiang et al. 1998). Prader-Willi Syndrome is a development disorder characterized by low birth weight followed by hyperphagia and obesity in childhood. Prader-Willi Syndrome arises from a disruption of a parent-of-origin expression of alleles located within a 2 megabase region of chromosome 15 (15q11q13), and mapping and characterization of this region has helped to elucidate the epigenetic mechanisms involved in the etiology of Prader-Willi Syndrome (Lee et al. 2008).
The region on chromosome 15 associated with Prader-Willi Syndrome is not the only imprinted region implicated in the etiology of obesity. Aberrant DNA methylation within the differentially methylated region of the GNAS gene on chromosome 20 (20q13.11) renders pseudohypoparathyroidism type 1b (Kelsey 2010). Familial pseudohypoparathyroidism type 1b is a maternally transmitted syndrome characterized by obesity and resistance to parathyroid hormone and thyroid stimulating hormone. However, sporadic cases of pseudohypoparathyroidism type 1b caused by a loss of methylation within the differentially methylated region of the maternally derived chromosome with no apparent genetic changes (Linglart et al. 2007; Liu et al. 2000) have been reported. Again, alterations of DNA methylation patterns (imprinting) without a known genetic abnormality are associated with an obesogenic phenotype. However, it is not known if aberrant imprinting is implicated in the current obesity epidemic (Stoger 2008). Moreover, evolutionary genomic imprinting patterns and expression in mammals are highly conserved and observed only in marsupials and placental mammals, although imprinting in the human placenta has been shown to have limited evolutionary conservation (Monk et al. 2006). To date, an NHP model to study genomic imprinting in association with obesity syndromes has not been identified.
Primate Models That Recapitulate Population-Based Studies to Provide Mechanistic Insight
What advantages do primate animal models offer over rodent models? Rodent models have historically served as primary tools in studies of human diseases because of their fast generation time, large litter size, easier handling, and small size and because of the ability to perform transgenic studies (i.e., over at least three generations) using them. However, these advantages are accompanied by significant limitations. For example, because of their faster generation time, the evolutionary rate appears more rapid than has occurred among hominids, and more recent data suggest that there are wide structural and physiological differences between humans and rodents (Raaum et al. 2005). As a second pertinent example, maternal and fetal metabolic physiology comparatively differs among rodents and primates, as demonstrated by the regulation of satiety and appetite being developed postnatally in rodents and prenatally in humans and NHPs (Bouret et al. 2004; Grayson et al. 2006; Grove et al. 2003; Grove and Smith 2003; Koutcherov et al. 2002). As a third example, placental structure and function are distinct among primates, which is of importance in light of the essential role of placentation in regulation of fetal nutrient transport and fatty acid and lipid metabolism (Maltepe et al. 2010). The overall aim of NHP models is to generate an animal model that is robust and that both mimics the gravid physiology unique to primates (generally one uterus, one placenta, and one fetus) and recapitulates human gene function and regulation.
The closest genomic relatives to humans belong to Hominidae, with clades of humans, chimpanzee, and gorilla being of closest genomic relation (human to chimp bearing greatest sequence similarity), and the more distant clades of orangutan and rhesus still sharing 98% or more identity. The remarkable phenotypic variance is highlighted by recalling the significant eras of divergence, with human to chimpanzee having diverged in the neighborhood of 3.5 to 5 million years ago, whereas orangutan and macaque diverged 14 million years ago and 23 million years ago (Raaum et al. 2005). Thus, it is intuitive that these NHP models would serve as excellent animal models to study developmental origins of adult diseases because, despite our genomic similarity and recent divergence, our phenotypic variance is profound.
The landscape of fetal programming and the role of epigenomic modifications differ spatially and temporally with tissue and development stage-specific gene expression. For example, investigators delving into the evolution of cognitive capacity have observed well-conserved and predictive patterns of gene expression in the human brain compared with the chimpanzee brain (Caceres et al. 2003; Enard and Paabo 2004; Gu and Gu 2003; Khaitovich et al. 2006), with early studies suggesting hypermethylation of the cerebrum and thymus occurred with species specificity (Gama-Sosa et al. 1983). Subsequent studies capable of interrogating discriminate changes have demonstrated that the methylation pattern is conserved between humans and chimpanzees, with prior observed differences in gene expression being at least in part explained by differential methylation (Pai et al. 2011).
Which cellular pathways seem to be primarily affected across primate species? Cell cycle–related kinase (CCRK ) gene is involved in cell cycle growth, and transcriptional regulation has been identified to be differentially methylated and expressed in human and chimpanzee brain cortices. The intraspecific differential promoter methylation of CCRK in humans and chimpanzees has been postulated to be connected to interindividual differences in brain development. Thus, the divergence of DNA methylation in CpG islands can be considered as an epigenetic “footprint” of genes crucial for human development compared with NHPs (Farcas et al. 2009). In a baboon model of reduced maternal nutrient availability, it has been observed that the fetal hepatic PCK1 gene encoding phosphoenolpyruvate caroxykinase 1 has a hypomethylated promoter (Nijland et al. 2010). In the same model, it has been demonstrated that the juvenile offspring of mothers with reduced maternal nutrient availability exhibit increased fasting glucose, increased fasting insulin, and b cell responsiveness (Choi et al. 2011). Interestingly, Rodriguez and colleagues (2011) have demonstrated that exposure to prenatal betamethasone (synthetic glucocorticoid administered in case of premature labor to pregnant women) in maternal baboon model results in sex-specific effects in reversal learning and attention in the juvenile offspring. Maternal nutrient restriction during pregnancy in baboons altered fetal hepatic insulin-like growth factor (Li et al. 2009). Comparative epigenetic regulation with the closest relative of humans (NHPs) could be the crucial element in improving our understanding of phenotypic differences, cognition development, and susceptibility to adult diseases such as obesity, metabolic syndrome, and cardiovascular disease (Table 2).
Table 2.

NHPs have been evaluated to study different aspects of reproductive biology. For instance, baboons, rhesus macaques, pig-tailed macaques, and cynomolgus monkeys have been characterized to study endometriosis, which is a common cause of infertility in women (Hastings et al. 2006). Leveraging the baboon model to study endometriosis, it has been observed that together the inhibition of histone deacetylase and progesterone lead to expression of glycodelin gene (decreased expression observed in endometriosis) (Jaffe et al. 2007). Placental methylation for several genes, including APC, SFRP2, CYP24A1, and DNMT1, was conserved in primate species (with variation in distribution and absolute methylation levels) as opposed to nonprimate species (Ng et al. 2010).
NHPs have been also been explored as models to study metabolism with respect to enzymatic methylation of arsenic (carconigenic) compounds, which acts as a detoxification mechanism. Out of 17 NHPs, only four species demonstrated hepatic arsenite methyltransferase activity, thereby suggesting further evaluation of alternative detoxification mechanisms in these primates (Wildfang et al. 2001). The summary of studies performed in other arenas, such as metabolism, reproductive biology, and neurology, are summarized in Table 3.
Table 3.

Maternal Factors That Shape the Fetal Epigenome
Malnutrition
Malnutrition results from either under- or over-intake of nutrients. We have understood for decades that there are fetal consequences to relative paucity of maternal intake of folate, vitamin B6, and vitamin B12 (neural tube malformations) and protein restriction (muscle wasting and intrauterine growth restriction). In the past decade, the implications of a caloric-dense, high-fat diet (namely, metabolic syndrome and obesity) have gained attention because of the alarming increase in the rate of adult and childhood obesity. Obesity results from higher caloric intake than energy expenditure. We will now look at the epigenetic effects on the fetus caused be maternal one-carbon supplementation, protein restriction, and high-fat diet (HFD1) exposure.
Maternal One-Carbon Metabolism
In recent years, more detailed information that suggests a crucial role for the one-carbon pathway intermediates in mediating epigenomic modifications, primarily methylation patterning, has emerged. Briefly, methionine is an essential amino acid that is converted to S-adenosyl methionine, which acts as a single-carbon donor and gets converted to S-adenosyl homocysteine. S-adenosyl homocysteine then gets converted to homocysteine. Folate, vitamin B6, and vitamin B12 are involved in the one-carbon metabolism pathway, and thus under- and/or over-intake of nutrients can affect the one-carbon metabolism pathway. Increased levels of plasma homocysteine levels have been associated with cardiovascular disease, placental abruptions, and neural tube defects (Ananth et al. 2007; Eskes 1998; Hague 2003; Mills et al. 1995; Refsum et al. 1998; Steegers-Theunissen 1991). Carbon donor supplementation during pregnancy is felt to bear beneficial effects toward the offspring. Waterland and colleagues (2008) have demonstrated that methyl donor supplementation in maternal diet prevents inheritance of obesity. Using the well-characterized yellow agouti (Avy) model, which contains a transposable element in agouti gene, they observed that maternal dietary methyl intake alters the phenotype of offspring by the transposable element, resulting in increased DNA methylation at the Avylocus (Waterland and Jirtle 2003). Histone modifications have also been reported at the proximal metastable epiallele, resulting in epigenetic alteration and gene expression (Dolinoy et al. 2010). Taken together, these lines of evidence support the notion that supplementation with one-carbon intermediates has the potential to alter methylation patterns in utero.
Maternal Protein Restriction: Rodent Models
In the rodent, several models of maternal low protein (MLP1) diet exposure have been useful in characterizing epigenetic and metabolic changes in the offspring. In utero exposure to MLP leads to decreased body weight of the offspring, predisposition to hypertension, and altered glucose metabolism (Langley and Jackson 1994; Langley-Evans et al. 1996). Bertram and colleagues (2001) have reported that glucocorticoid receptor (GR1) gene expression is increased at least twofold in the liver, kidney, lung, and brain of fetuses exposed to MLP in utero. This increase in expression is maintained into adult life compared with unexposed controls. Peroxisome proliferator-activated receptor (PPARa) expression is also altered in rat liver with MLP exposure (Lillycrop et al. 2005). It is now established that the GR and PPARa are epigenetically regulated by MLP diet exposure. PPARa is important for lipid and carbohydrate homeostasis, and GR is important for blood pressure regulation. Lillycrop and colleagues (2005, 2007) reported that in the rat liver, the promoter region of the GR, GR110, shows a 33% decrease in methylation and expression of GR is increased by 84% in offspring exposed to MLP. Expression of the DNA methyltransferase Dnmt1 is also lowered in MLP offspring. Further characterization of GR110 revealed altered histone modification patterns in MLP offspring, with an increase in H3K9 acetylation and a decrease in H3K9 dimethylation and trimethylation. PPARa has a 21% decrease in promoter methylation (Lillycrop et al. 2005).
Interventions to alleviate the adverse phenotypes of MLP exposure have similarly been studied. In rodent models, offspring exposed to MLP diet supplemented with folic acid do not show a propensity for hypertension in adulthood (Torrens et al. 2006). On a molecular level, the offspring also did not show a difference in promoter methylation in the GR or PPARa promoters compared with control diet animals (Lillycrop et al. 2005). It appears that the supplementation reverted both the phenotypic and epigenetic changes of MLP. Timing of folic acid supplementation is important. If folic acid is given to protein-restricted offspring in the juvenile period (postnatal days 35–40), changes in metabolism and epigenotype are not normalized (Burdge et al. 2009). MLP exposure increases the expression of the transcription factor CCAAT/enhancer-binding protein in the skeletal muscle of female offspring but not male offspring (Zheng et al. 2011). Analysis of the CCAAT/enhancer-binding protein promoter revealed increased acetylated histones H3 and H4 in the female offspring but not the male offspring. A study of genes that are involved in predisposition to hypertension showed gene-specific changes in expression accompanied by altered promoter methylation (Bogdarina et al. 2007). The angiotensin receptor, AT1b, shows increased messenger RNA and protein levels in the adrenal gland in 1-week-old rats exposed to MLP. This increased expression is accompanied by a decrease in promoter methylation. However, changes in gene expression caused by MLP exposure are not always concomitant with changes in DNA methylation. The glucokinase gene in rat is reduced in 6-month-old MLP rat liver, but there is no differential promoter methylation between control animals and MLP-exposed animals (Bogdarina et al. 2004).
One intriguing aspect of the MLP rat model system was the discovery that the adverse phenotypes are transmitted transgenerationally even when the exposure is eliminated in subsequent gestations. Exposure to the MLP diet during gestation is associated with both hypertension and insulin resistance in the subsequent F1 generation, even when the F1 rat is on a control diet postweaning. Their offspring (the F2 generation) have also been reported to have high blood pressure and insulin resistance in adulthood, and disruptions in glucose homeostasis have been reported in the F3 generation (Benyshek et al. 2006; Martin et al. 2000; Zambrano et al. 2005). Study of the adult male F2 liver showed decreased PPARa and GR promoter methylation (Burdge et al. 2007). The methylation status of the hepatic promoters was passed through a transgenerational model even though the F2 generation was never exposed in utero to the low protein diet and consumed only a control diet in adulthood. Transgenerational inheritance patterns are provocative evidence for a highly heritable, epigenetic-mediated mode of phenotypic transmission.
Maternal HFD Exposure: A Model For Understanding Epigenomic Modifications and In Utero Programming
Obesity is a well-characterized, multifactorial disease associated with higher risk for hypertension, insulin sensitivity, and cardiovascular disease. These risk factors together are known as metabolic syndrome. Epigenetics is one of the factors that explain the differential gene expression contributing to higher risk for disease state. For example, increased DNA methylation in peripheral blood leukocytes has been correlated with cardiovascular risk (Kim et al. 2010). Similarly, DNA methylation and histone acetylation have been shown to influence the regulation of somatic angiotensin converting enzyme regulation, which is crucial for maintaining cardiovascular homeostasis (Riviere et al. 2011). Histone modifications (e.g., histone lysine methylation) leads to dysregulation of genes involved in diabetes and related complications (Villeneuve et al. 2011).
It has long been postulated that obesity begets obesity. However, distinguishing factors associated with obesity (i.e., caloric intake) from obesity per se is not an easy undertaking. Along with reports of the effects of maternal undernutrition, protein restriction, and intrauterine growth restriction, studies of maternal overnutrition and high caloric density are becoming of immense importance for their potential elucidations on the current clinical epidemic of obesity. Animal models of maternal HFD exposure on offspring outcome show that, indeed, obesity begets obesity. In an NHP model of maternal obesity, fetal offspring exhibit the pathology of nonalcoholic fatty liver disease in utero (McCurdy et al. 2009). In a rat model of maternal obesity, although pups from obese dams did not weigh more than those from lean dams, they gained greater body weight and body fat when fed an HFD postweaning than those from lean dams (Shankar et al. 2008). Exposure to HFD in utero in animal models has been shown to make offspring more susceptible to type 2 diabetes, altered leptin sensitivity, hypertension, and obesity (Armitage et al. 2005; Chang et al. 2008; Ferezou-Viala et al. 2007; Gniuli et al. 2008; Kirk et al. 2009; Mitra et al. 2009; Samuelsson et al. 2008).
Histone Code Variations
Various epigenetic changes have been associated with exposure to a maternal HFD, with histone modifications serving as a cornerstone to these observations. As one such example, we have demonstrated in an NHP model of maternal obesity that fetuses exposed to a maternal HFD in utero have increased levels of hepatic H3K14 acetylation, concomitant with decreased levels of HDAC1 protein, messenger RNA, and activity; these changes are accompanied by persistent changes postnatally (Aagaard-Tillery et al. 2008). Employing techniques of chromatin immunoprecipitation followed by differential display polymerase chain reaction, we have identified the peripheral circadian regulator Npas2 (a transcription factor essential in maintaining oscillatory circadian rhythms) as a fetal gene reprogrammed by H3K14ac in HFD-exposed animals but not by maternal obesity per se. Of interest, although no differences were seen in Npas2 promoter DNA methylation, promoter H3K14ac occupation was increased in HFD-exposed fetal animals; this was not true in obese animals reverted to a control diet. This alteration in promoter occupancy resulted in alterations in gene expression of Npas2 in HFD-exposed animals (Suter et al. 2011). It was also reported that a postweaning HFD significantly disrupted Npas2 expression even without HFD exposure in utero.
Our observations are of interest to the discussion herein because alterations and disruptions of circadian rhythms are correlated with obesity and cardiovascular disease; the contribution of maternal diet to reprogramming the fetal circadian rhythm is of interest in furthering our understanding of current childhood obesity trends. Glucose and lipid homeostasis are known to display circadian variation, and an HFD in a murine model amplifies the diurnal variation in glucose tolerance in a _Clock_-dependent fashion. Similar to glucose homeostasis, characterization of peripheral Clock family members in liver and adipose tissue demonstrates robust and coordinated expression of the circadian oscillation system as well as _Clock_-controlled downstream effectors (Zvonic et al. 2006). Temporally restricted feeding or alterations in diet content (caloric dense/high fat) results in a coordinated phase shift in circadian expression of the major oscillator genes and their downstream targets in adipose tissues (Turek et al. 2005; Zvonic et al. 2006). Our evidence served as a fundamental description that the peripheral circadian metabolic clock in the developing primate fetus can be altered in utero by virtue of maternal dietary exposure, as evidenced by our observed changes in circadian gene expression patterns in fetal liver (Shankar et al. 2008).
Differential Methylation
Although we did not observe a role for differential methylation in the regulation of Npas2, other investigators have shown that DNA methylation changes have been reported in a mouse model of offspring exposed to an HFD in utero. Male mice aged 18 to 24 weeks exposed in utero to a maternal HFD showed an increased preference for glucose and fat. Gene expression changes were reported in the brain, including alterations in dopamine reuptake transporter, µ-opiod receptor, and preproenkephalin. Global and gene-specific DNA methylation patterns in the brain were altered in HFD-exposed animals (Vucetic et al. 2010). HFD exposure altered gene expression, epigenetic marks, and food preference in animals maintained on a control diet.
Noncoding miRNA levels have also been reported to be altered by HFD exposure. Fifteen-week-old mice maintained on a postweaning chow diet exposed in utero to HFD were studied for alterations in hepatic gene expression and miRNA levels. HFD-exposed animals had reduced hepatic let-7c. Expression of approximately 5.7% of all miRNAs was altered with HFD exposure. Furthermore, expression levels of metabolic regulators PPARa and CPT-1a were altered (Zhang et al. 2009). Aberrant DNA methylation and miRNA regulation have also been observed in diabetic nephropathy (Villeneuve et al. 2011).
In sum, the recent epidemic of childhood obesity has directed efforts toward understanding contributing factors. Extensive molecular studies from the Dutch hunger winter demonstrated that siblings exposed to the famine had decreased DNA methylation at the imprinted insulin-like growth factor 2 locus decades after the famine (Heijmans et al. 2008). The fetuses of obese mothers had greater body fat percentage, cord leptin, and interleukin 6 involved in inflammation than fetuses of lean mothers (Catalano et al. 2009). Maternal HFD in NHPs is also associated with decreased plasma n-3 fatty acids and fetal hepatic apoptosis (Grant et al. 2011). Thus, as established by above and many other studies, obesity begets obesity.
Environment
In addition to alternations in maternal nutrition, other in utero exposures modify and reprogram the developing fetus. The best characterized of these include alcohol, tobacco, and nicotine, alongside bisphenol A and organophosphates. Adverse effects of these substances are well documented, but the involvement of epigenetic mechanisms in these effects is not yet understood completely. Waddington hypothesized that the development path tends to follow a pathway termed as “canalization.” In the more modern version of Waddington's sentinel hypotheses, it is generally assumed that because of developmental plasticity, the fetus adapts to multiple environmental signals in utero and programs for the later life (reviewed in Burdge and Lillycrop 2010).
Alcohol Exposure
Maternal alcohol exposure results in fetal alcohol syndrome, which is associated with aberrant epigenetic programming. Altered DNA methylation leading to phenotypic changes has been demonstrated by several investigators (Haycock 2009; Kaminen-Ahola et al. 2010; Liu et al. 2009; Miranda et al. 2010; Ouko et al. 2009; Pandey et al. 2008; Zhou et al. 2011). Alcohol interferes with one-carbon metabolism in a manner similar to vitamin B12 deficiency causing disruption of DNA methylation by inhibiting methionine synthase, an enzyme that converts homocysteine to methionine (Cravo and Camilo 2000). Mice that were administered alcohol during midgestation showed increased levels of acetylated histone H3K9/18, followed by apoptosis in fetal lungs (Wang et al. 2010). Another study demonstrated genome-wide hypomethylation in fetuses of pregnant mice administered alcohol (Garro et al. 1991).
Tobacco Exposure
As a second example of a common exposure and the effects on fetal programming, significant advances have been made in understanding the underlying genomic and epigenomic mechanisms with maternal tobacco exposure. In more than four decades of research, epidemiological studies have established maternal tobacco consumption during pregnancy as the paramount risk factor for intrauterine growth restriction and small-for-gestational-age births in developed and developing nations. Nicotine, a compound present in tobacco smoke, is known to cause adverse fetal development issues by mediating intrauterine vessel constriction, leading to decreased placental blood flow (reviewed in Suter and Aagaard-Tillery 2009). In utero tobacco exposure is known to upregulate CYP1A1 placental expression in association with differential methylation at the xenobiotic response element transcription factor site (Suter et al. 2010). Methylation of PKCε gene promoter occurs because of prolonged nicotine exposure, resulting in aberrant fetal heart development (Lawrence et al. 2011). Children exposed in utero to tobacco were observed to have a 2.3% increase in DNA methylation in AXL, a tyrosine kinase that is highly heritable and is important in cancer and immune function (Breton et al. 2011).
Maternal smoking is also associated with poor fetal outcomes and aberrant miRNA expression. A study on maternal smoking in humans found that the development-associated miRNAs (miR16, miR21, and miR146a) were downregulated in placenta and miR146 downregulation was dose dependent (Maccani et al. 2010). Prenatal exposure to maternal cigarette smoking is also known to affect fetal brain and behavior development. For example, a study by Toledo-Rodriguez and colleagues (2010) found that adolescents who were exposed to maternal smoking in utero had increased DNA methylation in the brain-derived neurotrophic factor gene. Maternal smoking exposure in utero affects global and gene-specific DNA methylation (Breton et al. 2009). Although the detrimental effects of maternal tobacco smoke exposure to the developing fetus have been well documented, multiple population-based studies have identified maternal tobacco use as the strongest modifiable risk factor for intrauterine growth restriction. It is also implicated with other adverse pregnancy outcomes such as preterm birth, placental abruption, placenta previa, amniotic fluid emboli, and maternal deep vein thrombi. In spite of increased public awareness for more than three decades, the prevalence of smoking during pregnancy is reported to be persistently as high as 12–20%.
Numerous reports have found an association between maternal smoking during pregnancy and being overweight or obese in childhood (Gorog et al. 2011; Griffiths et al. 2010; Kleiser et al. 2009; Mangrio et al. 2010; Mizutani et al. 2007). Studies have shown that children born to smoking mothers trend toward higher serum glucose and have a predisposition to develop diabetes (Huang et al. 2007; Montgomery and Ekbom 2002). Exposure to environmental tobacco smoke is also a risk factor for childhood obesity. Numerous reports have shown that childhood obesity is more prevalent in children who were exposed in utero to environmental tobacco smoke through having a father who smokes (Kwok et al. 2010; Leary et al. 2006; von Kries et al. 2008).
Animal models have similarly been used to determine the molecular mechanism involved in these adverse effects to smoke exposure. Of the 4000 chemicals found in tobacco smoke, most studies have focused on the effects of prenatal nicotine exposure on the offspring development. The results have not been clear cut. Using a rat model of maternal nicotine exposure, it has been reported that there is no effect on offspring growth (Franke et al. 2008; Navarro et al. 1989); increased propensity to adiposity, b-cell dysfunction, and insulin resistance caused by maternal nicotine exposure has also been reported (Bruin et al. 2007, 2008; Holloway et al. 2005). The discrepancy may be caused by length of exposure (prenatal and/or lactation), dose, or a combination of both. More studies are necessary to determine the underlying etiology of tobacco smoke exposure and predisposition to obesity.
There is a fundamental dearth of studies on the effects of tobacco smoke associated with epigenetic changes that may be a key in understanding the origins of childhood and adult disease. Although there are numerous studies on changes in DNA methylation in a cancer paradigm with smoke exposure, little has been done on the fetal origins of disease. However, studies have shown that some morbidities associated with prenatal smoke exposure are transgenerational, suggesting an epigenetic mechanism of inheritance (Holloway et al. 2007; Li et al. 2005). A study of buccal cell DNA methylation in kindergarten and first-grade children who had been exposed prenatally to tobacco smoke showed a decrease in methylation of AluYb8 repetitive elements (Breton et al. 2009). We have demonstrated that placental promoter methylation of an important enzyme necessary for processing the polycyclic aromatic hydrocarbons found in tobacco smoke, CYP1A1, is decreased by 10% in mothers who smoke, concomitant with a fourfold increase in CYP1A1 gene expression (Suter et al. 2010).
As we move forward in the ability to derive meaningful molecular and phenotypic associations associated with the epidemic of obesity, in utero tobacco exposure may serve as an ideal disease model with which to explore the role of epigenomics in relation to pertinent environmental exposures. We now have more than 40 years of clinical evidence suggesting a causal relationship between tobacco use and delivery of infants who are small for gestational age. Studies dating back to 1957 by Simpson and Linda showed a significant 200 g difference in birth weight among infants who were exposed to as few as 10 cigarettes per day. Yet the mechanisms leading to the attenuation of fetal birth weight and adverse pregnancy outcomes are complex, involving multiple epidemiologic, genetic or epigenetic, and sociodemographic factors. Current proposed efforts aimed at understanding the potential genetic, epigenetic, and metabolic basis for susceptibility to the more common exposures, such as tobacco, are of paramount importance in clinical and translational medicine.
Maternal Behavioral and Developmental Outcome
Decades of research on maternal behavior and psychological state have provided evidence for an association between transmission of maternal behavior and characteristic of the offspring. For example, licking and grooming behavior in rodents has been characterized as maternal behavior toward pups within the first week of life (Francis et al. 1999; Liu et al. 1997; Weaver et al. 2004a, 2004b). The adult offspring born to low licking and grooming mothers demonstrated enhanced stress reactivity (Francis et al. 1999; Liu et al. 1997; Weaver et al. 2004a). It has been established that maternal care affects expression levels of neuron-specific GR by altering DNA methylation (McGowan et al. 2008).
Differential DNA methylation has been characterized in candidate genes involved in brain and cognition development. Schizophrenic patients are known to have differential DNA methylation of genes involved in the GABAergic system, serotonin system, dopaminergic system, and brain-derived neurotrophic factor (BDNF) gene (reviewed in Roth et al. 2009). Glutamic acid decarboxylase, an enzyme involved in GABA synthesis, is decreased in schizophrenic patients (Akbarian et al. 1995; Heckers et al. 2002). Understanding the histone modifications in schizophrenic patients is a less-studied arena; however, seminal studies have demonstrated histone modifications in schizophrenic patients compared with healthy individuals (Akbarian et al. 2005). Increased HDAC1 activity has also been observed in schizophrenic patients (Sharma et al. 2008). The synapsin 3 gene promoter has differential DNA methylation in humans, cynomolgous monkeys, and baboons but was not found to be associated with schizophrenic condition in humans (Murphy et al. 2008). In infant rhesus macaques, it has been demonstrated that higher CpG methylation of 5-HTT (serotonin transporter) but not the rh5-HTTLPR promoter polymorphism is associated with exacerbating effects caused by exposure of early life stress on behavioral reactivity in rhesus infants (Kinnally et al. 2010).
Depression morbidity rates have increased in the past decades. Dysregulation of DNA methylation of genes such as BDNF has been observed in depressive patients (Angelucci et al. 2005). Epigenetic regulation of the BDNF gene has been shown in fear learning (Lubin et al. 2008). Maternal mood and mental state is related to fetal brain development. Prenatal exposure to maternal depression leads to methylation of the GR gene in neonates and infant cortisol stress responses (Oberlander et al. 2008). Substance abuse during pregnancy is known to affect fetal development and epigenetic programming. Repeated exposure to cocaine is known to affect histone modifications (reviewed in Maze and Nestler 2011; Suter and Aagaard-Tillery 2009.
Maternal Microbiome
In terms of emerging arenas of likely paramount interest in understanding fetal programming, studies on the establishment of the perinatal microbiome are emerging as potential high impact. By way of overview, the microbiome (our so-called second genome) is associated with states of health and well-being throughout our lifetime. To date, the dominant paradigm in Western medicine has been to consider the majority of microbes as “foreign” and has led to the prevailing view that the treatment of overtly dominant microorganisms will result in disease cures. Such a view seemingly ignores long-standing observations that humans are remarkable hosts to microbes that have, in fact, coevolved as highly plethoric communities (Srinivasan et al. 2009; Turnbaugh and Gordon 2009; Xu and Gordon 2003). Human-associated microorganisms are present in numbers exceeding the quantities of human cells by at least 10-fold beginning in the neonatal period, and the collective genome (the microbiome) exceeds the human genome in terms of gene content by more than 100-fold. Scientists have recently come to appreciate that the microbiota are a metabolically and antigenically vibrant diverse community that may function as mutualists (symbiotically beneficial), commensals (of neither harm nor benefit), or pathogens (of host detriment) (Ley et al. 2005; Ley, Hamady, et al. 2008; Ley, Lozupone, et al. 2008; Turnbaugh et al. 2007, 2009; Vael and Desager 2009).
Although current major national research efforts (i.e., the National Institutes of Health Road Map initiative known as the Human Microbiome Project) will enable sequence-based comprehensive characterization of the adult human microbiota (Turnbaugh et al. 2007), how and when these diverse microbiota communities take up residence in the host and how they vary in reproductive life are unexplored at a population-wide level (Ley et al. 2005, 2006; Ley, Hamady, et al. 2008; Ley, Lozupone et al. 2008; Vael and Desager 2009). In other words, although researchers may soon know what constitutes the adult human microbial guild, how it is established will not be known. Moreover, the impact of the microbiome on the development of common perinatal disorders, such as preterm birth, will also not be known. Characterization of the pregnancy and neonatal microbiomes will allow a number of crucial gaps in understanding to begin to be addressed. For example, does this core microbiome change in association with the mode of delivery or timing of birth? What is the impact of the environment on the microbiome, and what is the impact of the microbiome at birth on the lifelong health status of the individual? Can microbial specimens acquired longitudinally from a human perinatal population base be interrogated to analyze microbiomes with efficient and reliable sequencing and computational methodologies in the future?
In an attempt to characterize the core gut microbiome, studies have demonstrated varying effects of diet, ethnicity, and physiological state on the core microbiome of an individual. An established link is present between the maternal immune status and the development of the fetal immune system (Figure 3). Other investigators working in a murine model of colitis have shown that maternal methyl donor supplementation increased susceptibility to colitis in offspring, and this occurred in association with both an altered microbiome in the offspring and epigenetic modifications in key immune response genes (Schaible et al. 2011). Intrauterine growth restriction also affects the offspring intestinal microbiome and leads to persistent alterations in the developing offspring gut microbiome (Fança-Berthon et al. 2010). Obesity has also been linked to human microbiomic composition and short-chain fatty acid metabolism (Dzielinska et al. 2010). Maternal diabetes is also linked to changes in microbiome composition in the offspring, although future studies are critical to corroborate the association of maternal diet versus diabetes and the microbiome and immunity of the offspring (Kaplan et al. 2011; Vassallo and Walker 2008). All of these data should be taken with a note of caution because a definite causal relationship has not been established between variations in the microbiome profile and lifelong risk of obesity and diabetes due to confounding factors such as ethnicity, age, diet, sex, and other lifestyle factors (reviewed in Musso et al. 2010). Undoubtedly, significant data will emerge in coming years and shed light on the essential emerging role of the metagenomics community in the establishment of lifelong metabolic health.
Figure 3.

Potential means of influencing the establishment of the microbiome in infants
Conclusion
In summary, we have argued that in accordance with the developmental origins of adult disease hypothesis, perturbations in the in utero environment influence the development of diseases later in life. This was first observed to occur in response to maternal nutritional constraints, which resulted in growth-restricted infants who later were observed to develop profound changes, including obesity, insulin resistance, hypertension, heart disease, and lipid disorders. Working in animal models, multiple and converging lines of evidence have shown that these lifelong disease risks occur through the static reprogramming of gene expression by epigenetic modifications to the regulation of gene expression.
The field of epigenetic effects on reproduction and development is young but robust. Scientists have discovered a few of the earliest clues in this complex epigenomic code, with many more left to be unraveled. Primates as a whole and NHPs in particular will continue to serve as apt models to accurately identify the crucial temporal, spatial, and developmental landscape alterations in the epigenome. The employment of recent advances in metagenomics-based research to now adapt “gene, epigene, germ, and environment” interactions into our emerging understanding on what shapes the fetal program will be integral to defining the role of gene and environment in shaping this landscape.
Acknowledgments
This work was supported by a National Institutes of Health Director New Innovator Pioneer award (DP21DP2OD001500-01 to K.M. Aagaard) and the Burroughs Wellcome Fund Preterm Birth Initiative 1008819.01. We thank the members of the Aagaard laboratory—Dr. Melissa Suter, Dr. Min Hu, Ms. Lori Showalter, Ms. Cynthia Shope, and Dr. Aishe Chen—for helpful discussions regarding the manuscript.
Biography
Radhika S. Ganu, PhD, is a postdoctoral associate in the Department of Obstetrics and Gynecology; R. Alan Harris, PhD, is assistant professor in the Department of Human and Molectular Genetics; Kiara Collins is a project intern; and Kjersti M. Aagaard, MD, PhD, is assistant professor in the Department of Obstetrics and Gynecology and the Department of Human and Molecular Genetics, all at Baylor College of Medicine, Houston, Texas.
Footnotes
1
Abbreviations that appears ≥3x throughout this article: DNMT, DNA methyltransferase; GR, glucocorticoid receptor; HFD, high fat diet; miRNA, microRNA; MLP, maternal low protein; NHP, nonhuman primate; PPARa, peroxisome proliferator-activated receptor.
References
- Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, Lane RH. 2008. Developmental origins of disease and determinants of chromatin structure: Maternal diet modifies the primate fetal epigenome. J Mol Endocrinol 41:91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE, Jr, Jones EG. 1995. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry 52:258–266 [DOI] [PubMed] [Google Scholar]
- Akbarian S, Ruehl MG, Bliven E, Luiz LA, Peranelli AC, Baker SP, Roberts RC, Bunney WE, Jr, Conley RC, Jones EG, Tamminga CA, Guo Y. 2005. Chromatin alterations associated with down-regulated metabolic gene expression in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry 62:829–840 [DOI] [PubMed] [Google Scholar]
- Ananth CV, Elsasser DA, Kinzler WL, Peltier MR, Getahun D, Leclerc D, Rozen RR. 2007. Polymorphisms in methionine synthase reductase and betaine-homocysteine S-methyltransferase genes: Risk of placental abruption. Mol Genet Metab 91:104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci F, Brene S, Mathe AA. 2005. BDNF in schizophrenia depression and corresponding animal models. Mol Psychiatry 10:345–352 [DOI] [PubMed] [Google Scholar]
- Armitage JA, Lakasing L, Taylor PD, Balachandran AA, Jensen RI, Dekou V, Ashton N, Nyengaard JR, Poston L. 2005. Developmental programming of aortic and renal structure in offspring of rats fed fat-rich diets in pregnancy. J Physiol 565:171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyshek DC, Johnston CS, Martin JF. 2006. Glucose metabolism is altered in the adequately-nourished grand-offspring (F3 generation) of rats malnourished during gestation and perinatal life. Diabetologia 49:1117–1119 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, Schreiber SL, Lander ES. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120:169–181 [DOI] [PubMed] [Google Scholar]
- Bertram C, Trowern AR, Copin N, Jackson AA, Whorwood CB. 2001. The maternal diet during pregnancy programs altered expression of the glucocorticoid receptor and type 2 11beta-hydroxysteroid dehydrogenase: Potential molecular mechanisms underlying the programming of hypertension in utero. Endocrinology 142:2841–2853 [DOI] [PubMed] [Google Scholar]
- Bestor T, Laudano A, Mattaliano R, Ingram V. 1988. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells: The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol 203:971–983 [DOI] [PubMed] [Google Scholar]
- Bestor TH. 1992. Activation of mammalian DNA methyltransferase by cleavage of a Zn binding regulatory domain. EMBO J 11:2611–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocock PN, Aagaard-Tillery KM. 2009. Animal models of epigenetic inheritance. Semin Reprod Med 27:369–379 [DOI] [PubMed] [Google Scholar]
- Bogdarina I, Murphy HC, Burns SP, Clark AJ. 2004. Investigation of the role of epigenetic modification of the rat glucokinase gene in fetal programming. Life Sci 74:1407–1415 [DOI] [PubMed] [Google Scholar]
- Bogdarina I, Welham S, King PJ, Burns SP, Clark AJ. 2007. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circul Res 100:520–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, Simerly RB. 2004. Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J Neurosci 24:2797–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. 2009. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med 180:462–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton CV, Salam MT, Gilliland FD. 2011. Heritability and role for the environment in DNA methylation in AXL receptor tyrosine kinase. Epigenetics 6:895–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruin JE, Kellenberger LD, Gerstein HC, Morrison KM, Holloway AC. 2007. Fetal and neonatal nicotine exposure and postnatal glucose homeostasis: Identifying critical windows of exposure. J Endocrinol 194:171–178 [DOI] [PubMed] [Google Scholar]
- Bruin JE, Petre MA, Raha S, Morrison KM, Gerstein HC, Holloway AC. 2008. Fetal and neonatal nicotine exposure in Wistar rats causes progressive pancreatic mitochondrial damage and beta cell dysfunction. PLoS One 3:e3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdge GC, Lillycrop KA. 2010. Nutrition, epigenetics, and developmental plasticity: Implications for understanding human disease. Annu Rev Nutr 30:315–339 [DOI] [PubMed] [Google Scholar]
- Burdge GC, Lillycrop KA, Phillips ES, Slater-Jefferies JL, Jackson AA, Hanson MA. 2009. Folic acid supplementation during the juvenile-pubertal period in rats modifies the phenotype and epigenotype induced by prenatal nutrition. J Nutr 139:1054–1060 [DOI] [PubMed] [Google Scholar]
- Burdge GC, Slater-Jefferies J, Torrens C, Phillips ES, Hanson MA, Lillycrop KA. 2007. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br J Nutr 97:435–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres M, Lachuer J, Zapala MA, Redmond JC, Kudo L, Geschwind DH, Lockhart DJ, Preuss TM, Barlow C. 2003. Elevated gene expression levels distinguish human from non-human primate brains. Proc Natl Acad Sci U S A 100:13030–13035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. 2009. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 32:1076–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GQ, Gaysinskaya V, Karatayev O, Leibowitz SF. 2008. Maternal high-fat diet and fetal programming: Increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J Neurosci 28:12107–12119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Ueda Y, Dodge JE, Wang Z, Li E. 2003. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol 23:5594–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Li C, McDonald TJ, Comuzzie A, Mattern V, Nathanielsz PW. 2011. Emergence of insulin resistance in juvenile baboon offspring of mothers exposed to moderate maternal nutrient reduction. Am J Physiol Regul Integr Comp Physiol 301:R757–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravo ML, Camilo ME. 2000. Hyperhomocysteinemia in chronic alcoholism: Relations to folic acid and vitamins B(6) and B(12) status. Nutrition 16:296–302 [DOI] [PubMed] [Google Scholar]
- Crozier SR, Inskip HM, Godfrey KM, Cooper C, Harvey NC, Cole ZA, Robinson SM. Southampton Women's Survey Study Group. 2010. Weight gain in pregnancy and childhood body composition: Findings from the Southampton Women's Survey. Am J Clin Nutr 91:1745–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier SR, Robinson SM, Borland SE, Inskip HM. 2006. Dietary patterns in the Southampton Women's Survey. Eur J Clin Nutr 60:1391–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij S, Painter R, Roseboom T, Phillips D, Osmond C, Barker D, Tanck M, Michels R, Bossuyt P, Bleker O. 2006. Glucose tolerance at age 58 and the decline of glucose tolerance in comparison with age 50 in people prenatally exposed to the Dutch famine. Diabetologia 49:637–643 [DOI] [PubMed] [Google Scholar]
- Dolinoy DC, Weinhouse C, Jones TR, Rozek LS, Jirtle RL. 2010. Variable histone modifications at the Avy metastable epiallele. Epigenetics 5:637–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue SM, Rifas-Shiman SL, Gold DR, Jouni ZE, Gillman MW, Oken E. 2011. Prenatal fatty acid status and child adiposity at age 3 y: Results from a US pregnancy cohort. Am J Clin Nutr 93:780–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzielinska Z, Januszewicz A, Wiecek A, Prejbisz A, Zielinski T, Chudek J, Makowiecka-Ciesla M, Demkow M, Tyczynski P, Januszewicz M, Ruzyllo W, Naruszewicz M. 2010. Reduced kidney function estimated by cystatin C and clinical outcomes in hypertensive patients with coronary artery disease: Association with homocysteine and other cardiovascular risk factors. Kidney Blood Press R 33:139–148 [DOI] [PubMed] [Google Scholar]
- Enard W, Paabo S. 2004. Comparative primate genomics. Annu Rev Genomics Hum Genet 5:351–378 [DOI] [PubMed] [Google Scholar]
- Eskes TK. 1998. Neural tube defects vitamins and homocysteine. Eur J Pediatr 157:S139–S141 [DOI] [PubMed] [Google Scholar]
- Fança-Berthon P, Hoebler C, Mouzet E, David A, Michel C. 2010. Intrauterine growth restriction not only modifies the cecocolonic microbiota in neonatal rats but also affects its activity in young adult rats. J Pediatr Gastroenterol Nutr 51:402–413 [DOI] [PubMed] [Google Scholar]
- Farcas R, Schneider E, Frauenknecht K, Kondova I, Bontrop R, Bohl J, Navarro B, Metzler M, Zischler H, Zechner U, Daser A, Haaf T. 2009. Differences in DNA methylation patterns and expression of the CCRK gene in human and nonhuman primate cortices. Mol Biol Evol 26:1379–1389 [DOI] [PubMed] [Google Scholar]
- Feng S, Jacobsen SE, Reik W. Epigenetic reporgramming in plan and animal development Science 2010. 330:622–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferezou-Viala J, Roy AF, Serougne C, Gripois D, Parquet M, Bailleux V, Gertler A, Delplanque B, Djiane J, Riottot M, Taouis M. 2007. Long-term consequences of maternal high-fat feeding on hypothalamic leptin sensitivity and diet-induced obesity in the offspring. Am J Physiol Regul Integr Comp Physiol 293:R1056–1062 [DOI] [PubMed] [Google Scholar]
- Francis DD, Champagne FA, Liu D, Meaney MJ. 1999. Maternal care gene expression and the development of individual differences in stress reactivity. Ann N Y Acad Sci 896:66–84 [DOI] [PubMed] [Google Scholar]
- Franke RM, Park M, Belluzzi JD, Leslie FM. 2008. Prenatal nicotine exposure changes natural and drug-induced reinforcement in adolescent male rats. Eur J Neurosci 27:2952–2961 [DOI] [PubMed] [Google Scholar]
- Gama-Sosa MA, Midgett RM, Slagel VA, Githens S, Kuo KC, Gehrke CW, Ehrlich M. 1983. Tissue-specific differences in DNA methylation in various mammals. Biochimica et Biophysica Acta 740:212–219 [DOI] [PubMed] [Google Scholar]
- Garro AJ, McBeth DL, Lima V, Lieber CS. 1991. Ethanol consumption inhibits fetal DNA methylation in mice: Implications for the fetal alcohol syndrome. Alcohol Clin Exp Res 15:395–398 [DOI] [PubMed] [Google Scholar]
- Gniuli D, Calcagno A, Caristo ME, Mancuso A, Macchi V, Mingrone G, Vettor R. 2008. Effects of high-fat diet exposure during fetal life on type 2 diabetes development in the progeny. J Lipid Res 49:1936–1945 [DOI] [PubMed] [Google Scholar]
- Gorog K, Pattenden S, Antova T, Niciu E, Rudnai P, Scholtens S, Splichalova A, Slotova K, Voko Z, Zlotkowska R, Houthuijs D. 2011. Maternal smoking during pregnancy and childhood obesity: Results from the CESAR Study. Matern Child Health J 15:985–992 [DOI] [PubMed] [Google Scholar]
- Grant WF, Gillingham MB, Batra AK, Fewkes NM, Comstock SM, Takahashi D, Braun TP, Grove KL, Friedman JE, Marks DL. 2011. Maternal high fat diet is associated with decreased plasma n-3 fatty acids and fetal hepatic apoptosis in nonhuman primates. PLoS One 6:e17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson BE, Allen SE, Billes SK, Williams SM, Smith MS, Grove KL. 2006. Prenatal development of hypothalamic neuropeptide systems in the nonhuman primate. Neuroscience 143:975–986 [DOI] [PubMed] [Google Scholar]
- Griffiths LJ, Hawkins SS, Cole TJ, Dezateux C. 2010. Risk factors for rapid weight gain in preschool children: Findings from a UK-wide prospective study. Int J Obes (Lond) 34:624–632 [DOI] [PubMed] [Google Scholar]
- Grove KL, Allen S, Grayson BE, Smith MS. 2003. Postnatal development of the hypothalamic neuropeptide Y system. Neuroscience 116:393–406 [DOI] [PubMed] [Google Scholar]
- Grove KL, Smith MS. 2003. Ontogeny of the hypothalamic neuropeptide Y system. Physiol Behav 79:47–63 [DOI] [PubMed] [Google Scholar]
- Gu J, Gu X. 2003. Induced gene expression in human brain after the split from chimpanzee. Trends Genet 19:63–65 [DOI] [PubMed] [Google Scholar]
- Hague WM. 2003. Homocysteine and pregnancy. Best Pract Res Clin Obstet Gynaecol 17:459–469 [DOI] [PubMed] [Google Scholar]
- Hastings JM, Jackson KS, Mavrogianis PA, Fazleabas AT. 2006. The estrogen early response gene FOS is altered in a baboon model of endometriosis. Biol Reprod 75:176–182 [DOI] [PubMed] [Google Scholar]
- Haycock PC. 2009. Fetal alcohol spectrum disorders: The epigenetic perspective Biol Reprod 81:607–617 [DOI] [PubMed] [Google Scholar]
- Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM. 2002. Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry 59:521–529 [DOI] [PubMed] [Google Scholar]
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. 2008. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A 105:17046–17049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Crabtree GR. 2010. Chromatin remodelling during development. Nature 463:474–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway AC, Cuu DQ, Morrison KM, Gerstein HC, Tarnopolsky MA. 2007. Transgenerational effects of fetal and neonatal exposure to nicotine. Endocrine 31:254–259 [DOI] [PubMed] [Google Scholar]
- Holloway AC, Lim GE, Petrik JJ, Foster WG, Morrison KM, Gerstein HC. 2005. Fetal and neonatal exposure to nicotine in Wistar rats results in increased beta cell apoptosis at birth and postnatal endocrine and metabolic changes associated with type 2 diabetes. Diabetologia 48:2661–2666 [DOI] [PubMed] [Google Scholar]
- Huang RC, Burke V, Newnham JP, Stanley FJ, Kendall GE, Landau LI, Oddy WH, Blake KV, Palmer LJ, Beilin LJ. 2007. Perinatal and childhood origins of cardiovascular disease. Int J Obes (Lond) 31:236–244 [DOI] [PubMed] [Google Scholar]
- Hult M, Tornhammar P, Ueda P, Chima C, Edstedt Bonamy A-K, Ozumba B, Norman M. 2010. Hypertension, diabetes and overweight: Looming legacies of the Biafran famine. PLoS One 5:e13582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inskip HM, Godfrey KM, Robinson SnM, Law CM, Barker DJP, Cooper CSWSSG. 2006. Cohort profile: The Southampton Women's Survey Int J Epidemiol 35:42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe RC, Ferguson-Gottschall SD, Gao W, Beam C, Fazleabas AT. 2007. Histone deacetylase inhibition and progesterone act synergistically to stimulate baboon glycodelin gene expression. J Mol Endocrinol 38:401–407 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293:1074–1080 [DOI] [PubMed] [Google Scholar]
- Jiang Y, Tsai TF, Bressler J, Beaudet AL. 1998. Imprinting in Angelman and Prader-Willi syndromes. Current Opin Genet Dev 8:334–342 [DOI] [PubMed] [Google Scholar]
- Kaminen-Ahola N, Ahola A, Maga M, Mallitt KA, Fahey P, Cox TC, Whitelaw E, Chong S. 2010. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genetics 6:e1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JL, Shi HN, Walker WA. 2011. The role of microbes in developmental immunologic programming. Pediatric Res 69:465–472 [DOI] [PubMed] [Google Scholar]
- Kelsey G. 2010. Imprinting on chromosome 20: Tissue-specific imprinting and imprinting mutations in the GNAS locus. Am J Med Genet C Semin Med Genet 154C:377–386 [DOI] [PubMed] [Google Scholar]
- Khaitovich P, Enard W, Lachmann M, Paabo S. 2006. Evolution of primate gene expression. Nat Rev Genet 7:693–702 [DOI] [PubMed] [Google Scholar]
- Kim M, Long TI, Arakawa K, Wang R, Yu MC, Laird PW. 2010. DNA methylation as a biomarker for cardiovascular disease risk. PLoS One 5:e9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnally EL, Capitanio JP, Leibel R, Deng L, LeDuc C, Haghighi F, Mann JJ. 2010. Epigenetic regulation of serotonin transporter expression and behavior in infant rhesus macaques. Genes Brain Behav 9:575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk SL, Samuelsson AM, Argenton M, Dhonye H, Kalamatianos T, Poston L, Taylor PD, Coen CW. 2009. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS One 4:e5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiser C, Schaffrath Rosario A, Mensink GB, Prinz-Langenohl R, Kurth BM. 2009. Potential determinants of obesity among children and adolescents in Germany: Results from the cross-sectional KiGGS Study. BMC Public Health 9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutcherov Y, Mai JK, Ashwell KW, Paxinos G. 2002. Organization of human hypothalamus in fetal development. J Comp Neurol 446:301–324 [DOI] [PubMed] [Google Scholar]
- Kwok MK, Schooling CM, Lam TH, Leung GM. 2010. Paternal smoking and childhood overweight: evidence from the Hong Kong “Children of 1997.” Pediatrics 126:e46–56 [DOI] [PubMed] [Google Scholar]
- Langley SC, Jackson AA. 1994. Increased systolic blood pressure in adult rats induced by fetal exposure to maternal low protein diets. Clin Sci 86:217–222; discussion 121 [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Phillips GJ, Benediktsson R, Gardner DS, Edwards CR, Jackson AA, Seckl JR. 1996. Protein intake in pregnancy placental glucocorticoid metabolism and the programming of hypertension in the rat. Placenta 17:169–172 [DOI] [PubMed] [Google Scholar]
- Lawrence J, Chen M, Xiong F, Xiao D, Zhang H, Buchholz JN, Zhang L. 2011. Foetal nicotine exposure causes PKCepsilon gene repression by promoter methylation in rat hearts. Cardiovasc Res 89:89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary SD, Smith GD, Rogers IS, Reilly JJ, Wells JC, Ness AR. 2006. Smoking during pregnancy and offspring fat and lean mass in childhood. Obesity (Silver Spring) 14:2284–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Latham KE, VandeVoort CA. 2008. Effects of in vitro maturation on gene expression in rhesus monkey oocytes. Physiol Genomics 35:145–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legler J, Hamers T, van Eck van der Sluijs-van de Bor M, Schoeters G, van der Ven L, Eggesbo M, Koppe J, Feinberg M, Trnovec T. 2011. The OBELIX project: Early life exposure to endocrine disruptors and obesity. Am J Clin Nutr 94:19335–19385 [DOI] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. 2005. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 102:11070–11075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. 2008. Evolution of mammals and their gut microbes. Science 320:1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. 2008. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat Rev Micro 6:776–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI. 2006. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124:837–848 [DOI] [PubMed] [Google Scholar]
- Li C, Schlabritz-Loutsevitch NE, Hubbard GB, Han V, Nygard K, Cox LA, McDonald TJ, Nathanielsz PW. 2009. Effects of maternal global nutrient restriction on fetal baboon hepatic insulin-like growth factor system genes and gene products. Endocrinology 150:4634–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YF, Langholz B, Salam MT, Gilliland FD. 2005. Maternal and grandmaternal smoking patterns are associated with early childhood asthma. Chest 127:1232–1241 [DOI] [PubMed] [Google Scholar]
- Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. 2005. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr 135:1382–1386 [DOI] [PubMed] [Google Scholar]
- Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. 2007. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Brit J Nutr 97:1064–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linglart A, Bastepe M, Jüppner H. 2007. Similar clinical and laboratory findings in patients with symptomatic autosomal dominant and sporadic pseudohypoparathyroidism type Ib despite different epigenetic changes at the GNAS locus. Clin Endocrinol 67:822–831 [DOI] [PubMed] [Google Scholar]
- Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, Sharma S, Pearson D, Plotsky PM, Meaney MJ. 1997. Maternal care hippocampal glucocorticoid receptors and hypothalamic-pituitary-adrenal responses to stress. Science 277:1659–1662 [DOI] [PubMed] [Google Scholar]
- Liu J, Litman D, Rosenberg MJ, Yu S, Biesecker LG, Weinstein LS. 2000. A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J Clin Invest 106:1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Balaraman Y, Wang G, Nephew KP, Zhou FC. 2009. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 4:500–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. 2008. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci 28:10576–10586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8[thinsp]A resolution. Nature 389:251–260 [DOI] [PubMed] [Google Scholar]
- Lumey L, Stein AD, Kahn HS, Romijn J. 2009. Lipid profiles in middle-aged men and women after famine exposure during gestation: The Dutch Hunger Winter Families Study. Am J Clin Nutr 89:1737–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumey L, Stein AD, Kahn HS, van der Pal-de Bruin KM, Blauw G, Zybert PA, Susser ES. 2007. Cohort profile: The Dutch Hunger Winter Families Study. Int J Epidemiol 36:1196–1204 [DOI] [PubMed] [Google Scholar]
- Maccani MA, Avissar-Whiting M, Banister CE, McGonnigal B, Padbury JF, Marsit CJ. 2010. Maternal cigarette smoking during pregnancy is associated with downregulation of miR-16, miR-21, and miR-146a in the placenta. Epigenetics 5:583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltepe E, Bakardjiev AI, Fisher SJ. 2010. The placenta: Transcriptional, epigenetic, and physiological integration during development. J Clin Invest 120:1016–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangrio E, Lindstrom M, Rosvall M. 2010. Early life factors and being overweight at 4 years of age among children in Malmo Sweden. BMC Public Health 10:764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JF, Johnston CS, Han CT, Benyshek DC. 2000. Nutritional origins of insulin resistance: A rat model for diabetes-prone human populations. J Nutr 130:741–744 [DOI] [PubMed] [Google Scholar]
- Maze I, Nestler EJ. 2011. The epigenetic landscape of addiction. Ann N Y Acad Sci 1216:99–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, Grove KL. 2009. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 119:323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, Meaney MJ, Szyf M. 2008. Diet and the epigenetic (re)programming of phenotypic differences in behavior. Brain Res 1237:12–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ER, 3rd, Juraschek S, Pastor-Barriuso R, Bazzano LA, Appel LJ, Guallar E. 2010. Meta-analysis of folic acid supplementation trials on risk of cardiovascular disease and risk interaction with baseline homocysteine levels. Am J Cardiology 106:517–527 [DOI] [PubMed] [Google Scholar]
- Mills JL, McPartlin JM, Kirke PN, Lee YJ, Conley MR, Weir DG, Scott JM. 1995. Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet 345:149–151 [DOI] [PubMed] [Google Scholar]
- Miranda RC, Pietrzykowski AZ, Tang Y, Sathyan P, Mayfield D, Keshavarzian A, Sampson W, Hereld D. 2010. MicroRNAs: Master regulators of ethanol abuse and toxicity? Alc Clin Exp Res 34:575–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Alvers KM, Crump EM, Rowland NE. 2009. Effect of high-fat diet during gestation lactation, or postweaning on physiological and behavioral indexes in borderline hypertensive rats. Am J Physiol Regul Integr Comp Physiol 296:R20–R28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani T, Suzuki K, Kondo N, Yamagata Z. 2007. Association of maternal lifestyles including smoking during pregnancy with childhood obesity. Obesity (Silver Spring) 15:3133–3139 [DOI] [PubMed] [Google Scholar]
- Monk D, Arnaud P, Apostolidou S, Hills FA, Kelsey G, Stanier P, Feil R, Moore GE. 2006. Limited evolutionary conservation of imprinting in the human placenta. Proc Natl Acad Sci U S A 103:6623–6628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SM, Ekbom A. 2002. Smoking during pregnancy and diabetes mellitus in a British longitudinal birth cohort. BMJ 324:26–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BC, O’Reilly RL, Singh SM. 2008. DNA methylation and mRNA expression of SYN III a candidate gene for schizophrenia. BMC Med Genet 9:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso G, Gambino R, Cassader M. 2010. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care 33:2277–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro HA, Seidler FJ, Schwartz RD, Baker FE, Dobbins SS, Slotkin TA. 1989. Prenatal exposure to nicotine impairs nervous system development at a dose which does not affect viability or growth. Brain Res Bull 23:187–192 [DOI] [PubMed] [Google Scholar]
- Ng HK, Novakovic B, Hiendleder S, Craig JM, Roberts CT, Saffery R. 2010. Distinct patterns of gene-specific methylation in mammalian placentas: Implications for placental evolution and function. Placenta 31:259–268 [DOI] [PubMed] [Google Scholar]
- Nijland MJ, Mitsuya K, Li C, Ford S, McDonald TJ, Nathanielsz PW, Cox LA. 2010. Epigenetic modification of fetal baboon hepatic phosphoenolpyruvate carboxykinase following exposure to moderately reduced nutrient availability. J Physiol 588:1349–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. 2008. Prenatal exposure to maternal depression neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 3:97–106 [DOI] [PubMed] [Google Scholar]
- Ouko LA, Shantikumar K, Knezovich J, Haycock P, Schnugh DJ, Ramsay M. 2009. Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes: Implications for fetal alcohol spectrum disorders. Alcohol Clin Exp Res 33:1615–1627 [DOI] [PubMed] [Google Scholar]
- Pai AA, Bell JT, Marioni JC, Pritchard JK, Gilad Y. 2011. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet 7:e1001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. 2008. Brain chromatin remodeling: A novel mechanism of alcoholism. J Neurosci 28:3729–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaum RL, Sterner KN, Noviello CM, Stewart C-B, Disotell TR. 2005. Catarrhine primate divergence dates estimated from complete mitochondrial genomes: Concordance with fossil and nuclear DNA evidence. J Human Evol 48:237–257 [DOI] [PubMed] [Google Scholar]
- Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, Bleker OP. 1998. Glucose tolerance in adults after prenatal exposure to famine. Lancet 351:173–177 [DOI] [PubMed] [Google Scholar]
- Ravelli ACJ, van Der Meulen JHP, Osmond C, Barker DJP, Bleker OP. 1999. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr 70:811–816 [DOI] [PubMed] [Google Scholar]
- Refsum H, Ueland PM, Nygard O, Vollset SE. 1998. Homocysteine and cardiovascular disease. Annu Rev Med 49:31–62 [DOI] [PubMed] [Google Scholar]
- Reik W. 2007. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447:425–432 [DOI] [PubMed] [Google Scholar]
- Riviere G, Lienhard D, Andrieu T, Vieau D, Frey BM, Frey FJ. 2011. Epigenetic regulation of somatic angiotensin-converting enzyme by DNA methylation and histone acetylation. Epigenetics 6:478–489 [DOI] [PubMed] [Google Scholar]
- Rodriguez JS, Zürcher NR, Keenan KE, Bartlett TQ, Nathanielsz PW, Nijland MJ. 2011. Prenatal betamethasone exposure has sex specific effects in reversal learning and attention in juvenile baboons. Am J Obstet Gynecol 204:545.e541–545.e510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseboom TJ, Van der Meulen JHP, Osmond C, Barker DJP, Ravelli ACJ, Bleker OP. 2000a. Plasma lipid profiles in adults after prenatal exposure to the Dutch famine. Am J Clin Nutr 72:1101–1106 [DOI] [PubMed] [Google Scholar]
- Roseboom TJ, Van der Meulen JHP, Osmond C, Barker DJP, Ravelli ACJ, Schroeder-Tanka JM, Van Montfrans GA, Michels RPJ, Bleker OP. 2000b. Coronary heart disease after prenatal exposure to the Dutch famine 1944-45. Heart 84:595–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseboom TJ, Van Der Meulen JHP, Ravelli ACJ, Osmond C, Barker DJP, Bleker OP. 2000c. Plasma fibrinogen and factor VII concentrations in adults after prenatal exposure to famine. Brit J Haematol 111:112–117 [DOI] [PubMed] [Google Scholar]
- Roth TL, Lubin FD, Sodhi M, Kleinman JE. 2009. Epigenetic mechanisms in schizophrenia. Biochimica et Biophysica Acta 1790:869–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, Piersma AH, Ozanne SE, Twinn DF, Remacle C, Rowlerson A, Poston L, Taylor PD. 2008. Diet-induced obesity in female mice leads to offspring hyperphagia adiposity, hypertension, and insulin resistance: A novel murine model of developmental programming. Hypertension 51:383–392 [DOI] [PubMed] [Google Scholar]
- Schaible TD, Harris RA, Dowd SE, Smith CW, Kellermayer R. 2011. Maternal methyl-donor supplementation induces prolonged murine offspring colitis susceptibility in association with mucosal epigenetic and microbiomic changes. Human Mol Genet 20:1687–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJ, Badger TM. 2008. Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol 294:R528–538 [DOI] [PubMed] [Google Scholar]
- Sharma RP, Grayson DR, Gavin DP. 2008. Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: Analysis of the National Brain Databank microarray collection. Schizophrenia Res 98:111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan U, Misra D, Marazita ML, Foxman B. 2009. Vaginal and oral microbes host genotype and preterm birth. Med Hypotheses 73:963–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegers-Theunissen RP, Boers GH, Trijbels FJ, Eskes TK. 1991. Neural-tube defects and derangement of homocysteine metabolism. New Engl J Med 324:199–200 [DOI] [PubMed] [Google Scholar]
- Stoger R. 2008. Epigenetics and obesity. Pharmacogenomics 9:1851–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susser ES, Lin SP. 1992. Schizophrenia after prenatal exposure to the Dutch Hunger Winter of 1944–1945. Arch Gen Psychiatry 49:983–988 [DOI] [PubMed] [Google Scholar]
- Suter M, Abramovici A, Showalter L, Hu M, Shope CD, Varner M, Aagaard-Tillery K.2010. In utero tobacco exposure epigenetically modifies placental CYP1A1 expression. Metabolism 59:1481–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter M, Bocock P, Showalter L, Hu M, Shope C, McKnight R, Grove K, Lane R, Aagaard-Tillery K. 2011. Epigenomics: Maternal high-fat diet exposure in utero disrupts peripheral circadian gene expression in nonhuman primates FASEB J 25:714–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter MA, Aagaard-Tillery KM. 2009. Environmental influences on epigenetic profiles. Semin Reprod Med 27:380–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice JA. 2010. The vital amines: Too much of a good thing?: Comment on “Effects of lowering homocysteine levels with B vitamins on cardiovascular disease cancer, and cause-specific mortality.” Arch Intern Med 170:1631–1633 [DOI] [PubMed] [Google Scholar]
- Toledo-Rodriguez M, Lotfipour S, Leonard G, Perron M, Richer L, Veillette S, Pausova Z, Paus T. 2010. Maternal smoking during pregnancy is associated with epigenetic modifications of the brain-derived neurotrophic factor-6 exon in adolescent offspring. Am J Med Genet. 153B:1350–1354 [DOI] [PubMed] [Google Scholar]
- Torrens C, Brawley L, Anthony FW, Dance CS, Dunn R, Jackson AA, Poston L, Hanson MA. 2006. Folate supplementation during pregnancy improves offspring cardiovascular dysfunction induced by protein restriction. Hypertension 47:982–987 [DOI] [PubMed] [Google Scholar]
- Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J. 2005. Obesity and metabolic syndrome in circadian clock mutant mice. Science 308:1043–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Gordon JI. 2009. The core gut microbiome energy balance and obesity. J Physiol 587:4153–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. 2009. A core gut microbiome in obese and lean twins. Nature 457:480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. 2007. The human microbiome project. Nature 449:804–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vael C, Desager K. 2009. The importance of the development of the intestinal microbiota in infancy. Curr Opin Pediatr 21:794–800 [DOI] [PubMed] [Google Scholar]
- Vassallo MF, Walker WA. 2008. Neonatal microbial flora and disease outcome. Nestle Nutr Workshop Ser Pediatr Program 61:211–224 [DOI] [PubMed] [Google Scholar]
- Villeneuve LM, Reddy MA, Natarajan R. 2011. Epigenetics: Deciphering its role in diabetes and its chronic complications. Clin Exp Pharmacol Physiol 38:401–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F. 2006. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439:871–874 [DOI] [PubMed] [Google Scholar]
- von Kries R, Bolte G, Baghi L, Toschke AM. 2008. Parental smoking and childhood obesity—Is maternal smoking in pregnancy the critical exposure? Int J Epidemiol 37:210–216 [DOI] [PubMed] [Google Scholar]
- Vucetic Z, Kimmel J, Totoki K, Hollenbeck E, Reyes TM. 2010. Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology 151:4756–4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gomutputra P, Wolgemuth DJ, Baxi LV. 2010. Acute alcohol exposure induces apoptosis and increases histone H3K9/18 acetylation in the mid-gestation mouse lung. Reprod Sci 17:384–390 [DOI] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL. 2003. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23:5293–5300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterland RA, Travisano M, Tahiliani KG, Rached MT, Mirza S. 2008. Methyl donor supplementation prevents transgenerational amplification of obesity. Int J Obes (Lond) 32:1373–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. 2004a. Epigenetic programming by maternal behavior. Nat Neurosci 7:847–854 [DOI] [PubMed] [Google Scholar]
- Weaver IC, Diorio J, Seckl JR, Szyf M, Meaney MJ. 2004b. Early environmental regulation of hippocampal glucocorticoid receptor gene expression: Characterization of intracellular mediators and potential genomic target sites. Ann N Y Acad Sci 1024:182–212 [DOI] [PubMed] [Google Scholar]
- Wildfang E, Radabaugh TR, Vasken Aposhian H. 2001. Enzymatic methylation of arsenic compounds. IX. Liver arsenite methyltransferase and arsenate reductase activities in primates. Toxicology 168:213–221 [DOI] [PubMed] [Google Scholar]
- Xu J, Gordon JI. 2003. Honor thy symbionts. Proc Natl Acad Sci U S A 100:10452–10459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano E, Martinez-Samayoa PM, Bautista CJ, Deas M, Guillen L, Rodriguez-Gonzalez GL, Guzman C, Larrea F, Nathanielsz PW. 2005. Sex differences in transgenerational alterations of growth and metabolism in progeny (F2) of female offspring (F1) of rats fed a low protein diet during pregnancy and lactation. J Physiol 566:225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang F, Didelot X, Bruce KD, Cagampang FR, Vatish M, Hanson M, Lehnert H, Ceriello A, Byrne CD. 2009. Maternal high fat diet during pregnancy and lactation alters hepatic expression of insulin like growth factor-2 and key microRNAs in the adult offspring. BMC Genomics 10:478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Rollet M, Pan YX. 2011. Maternal protein restriction during pregnancy induces CCAAT/enhancer-binding protein (C/EBPbeta) expression through the regulation of histone modification at its promoter region in female offspring rat skeletal muscle. Epigenetics 6:161–170 [DOI] [PubMed] [Google Scholar]
- Zhou FC, Balaraman Y, Teng M, Liu Y, Singh RP, Nephew KP. 2011. Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol Clin Exp Res 35:735–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvonic S, Ptitsyn AA, Conrad SA, Scott LK, Floyd ZE, Kilroy G, Wu X, Goh BC, Mynatt RL, Gimble JM. 2006. Characterization of peripheral circadian clocks in adipose tissues. Diabetes 55:962–970 [DOI] [PubMed] [Google Scholar]