Properties and rates of germline mutations in humans (original) (raw)
. Author manuscript; available in PMC: 2014 Apr 1.
Published in final edited form as: Trends Genet. 2013 May 16;29(10):575–584. doi: 10.1016/j.tig.2013.04.005
Abstract
All genetic variation arises via new mutations, and therefore determining the rate and biases for different classes of mutation is essential for understanding the genetics of human disease and evolution. Decades of mutation rate analyses have focused on a relatively small number of loci because of technical limitations. However, advances in sequencing technology have allowed for empirical assessments of genome-wide rates of mutation. Recent studies have shown that 76% of new mutations originate in the paternal lineage and provide unequivocal evidence for an increase in mutation with paternal age. Although most analyses have been focused on single nucleotide variants (SNVs), studies have begun to provide insight into the mutation rate for other classes of variation, including copy number variants (CNVs), microsatellites, and mobile element insertions. Here, we review the genome-wide analyses for the mutation rate of several types of variants and suggest areas for future research.
Keywords: germline mutation rate, de novo mutation, paternal bias, paternal age, genome-wide
The fundamental process in genetics
The replication of the genome before cell division is a remarkably precise process. Nevertheless, there are some errors during DNA replication that lead to new mutations. If these errors occur in the germ cell lineage (i.e., the sperm and egg), then these mutations can be transmitted to offspring. Some of these new genetic variants will be deleterious to the organism, and a very select few will be advantageous and serve as substrates for selection. Therefore, knowledge about the rate at which new mutations appear and the properties of new mutations is critical in the study of human genetics from evolution to disease. The study of the mutation rate in humans dates further back than the discovery of the structure of DNA or the determination of DNA as the genetic material. In seminal work performed in the 1930s and 1940s, J.B.S. Haldane studied hemophilia with the assumption of a mutation/selection balance to estimate mutation rate at that locus and determine that most new mutations arose in the paternal germline[1, 2]. Until recently, most mutation rate analyses were similar to this initial work in that they extrapolated rates and properties from a handful of loci (often linked to dominant genetic disorders) (for example, see [3]). In the last few years, it has become feasible to generate large amounts of sequence data (including the genomes of parents and their offspring), and it is now possible to calculate empirically a genome-wide mutation rate. In addition, much interest has been focused on understanding the role of de novo mutations in human disease. Therefore, in this review, we seek to synthesize the recent analyses of mutation rate for multiple forms of genetic variation and discuss their implications with respect to human disease and evolution.
Single nucleotide variant (SNV) mutation rate
It is now feasible to perform whole-genome sequencing on all individuals from a nuclear family; from these data one can identify de novo mutations that “disobey” Mendelian inheritance (Box 1; Figure I). The first two papers to apply this approach were limited in scope to three families[4, 5], thus restricting the total number of de novo SNVs observed. Even with this limitation, these two analyses reported similar overall mutation rates of about 1×10−8 SNV mutation per base pair per generation, although there was considerable variation in families[4, 5]. A more recent study using whole sequence data from 78 Icelandic parent-offspring trios suggests a higher rate of 1.2×10−8 SNVs per generation from de novo mutations[6]. Another study used autozygous segments in the genomes of Hutterite trios — who were descended from a 13-generation pedigree with 64 founders — to independently calculate the same SNV mutation rate of 1.2×10−8[7]. A study of ten additional families of individuals affected with autism reported a rate of 1×10−8[8].
Box 1. Methods for discovering new mutations and estimating mutation rate.
Most of the methods developed for estimating mutation rate were developed for SNV data but can be applied more broadly to other forms of variation. The most common approach for estimating mutation rate is to use families to look for mutations carried by a child but not by either of his/her parents (Figure I). This approach has been carried out on selected loci up to whole genomes. However, it is important to note that this method can be confounded by false positives for which putative de novo variants are enriched[5]. In addition, somatic mutations in offspring of the sequenced families cannot be distinguished from germline de novo variants. The other classical approach for estimating mutation rates is to look at fixed differences between species[9, 10]. The mutation rate can then be calculated based on the estimated divergence time between the species (Figure I). Although this approach is not confounded by false positives or somatic mutations, there is uncertainty in the divergence time between humans and chimpanzees, the average generation time, and effective population sizes.
Recently, other approaches for determining mutation rate have been described. One group constructed a model of microsatellite evolution and applied this model to estimate the time to the most recent common ancestor (MRCA) for microsatellite alleles[12]. Because SNVs near the microsatellite have the same ancestry as the microsatellite, the mutation rate for SNVs could be calculated using the SNV differences between haplotypes and the time to the MRCA[12]. Another approach to estimate mutation rate involves the identification of heterozygous mutations in large regions of homozygosity by recent descent (autozygosity)[7, 120] (Figure I). Such regions are particularly abundant among founder populations providing a means for estimating mutation rate from a recent common ancestor in populations such as the Hutterites, the Amish, and the Icelandic population. Although different in many ways, these two approaches have some important similarities. Both are less susceptible to false positive and somatic mutations than analyses of de novo mutations in trios. In addition, both approaches estimate the time to the MRCA for segments of the genome in different ways but benefit by studying haplotypes with a much more recent coalescent time than humans and chimpanzees.
Figure I. Methods to discover new mutations and estimate mutation rate.
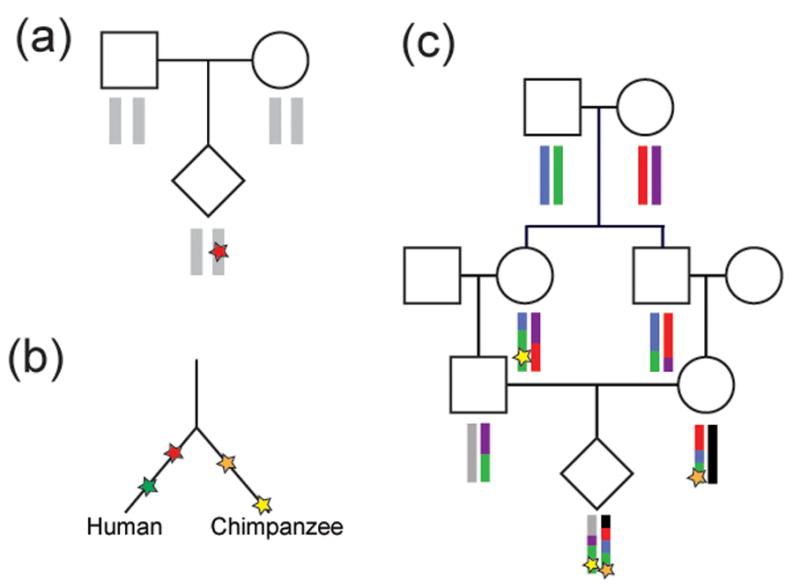
a) Sequence data from parent-offspring trios can be used to find mutations present in the child but not observed in either parent (red star). b) Fixed differences between closely related species can be identified and counted; red and green stars represent mutations occurring in the lineage leading to humans and orange and yellow stars represent mutations in the lineage leading to chimpanzees. This value, in combination with the estimated number of generations between the species, can be used to calculate mutation rate. A modification of this approach can be used within species if the coalescent time of haplotypes can be estimated[12]. c) Mutations in regions of autozygosity appear as heterozygous variants in long stretches of homozygous DNA[7, 120]. With known pedigree information, the MRCA of the autozygous haplotype can be identified and the mutation rate calculated[7].
In addition to the direct approaches in families, earlier studies employed more indirect approaches to estimate mutation rate. Using fixed differences between the human and chimpanzee genomes (Box 1; Figure I) yielded a mutation rate for SNVs of approximately 2.5×10−8 in pseudogenes where selection is not a confounding factor[9, 10], which is over twofold higher than the rates estimated from direct approaches. However, more recent comparisons of the human, chimpanzee, and gorilla genomes bring the mutation rate estimates in line with what is observed in family-based analyses[11]. Another indirect approach estimated the mutation rate for SNVs to be 1.82×10−8 using inferred ancestry of nearby microsatellites[12] (Box 1; Figure I). The difference between this mutation rate and those calculated with family information may be due to differences in filtering applied for SNVs or due to differences in sequencing methodology.
Recent genome-wide studies of the SNV mutation rate in humans have started to converge (Table 1). Studies based on whole-genome sequencing and direct estimates of de novo mutations give an average SNV mutation rate of 1.16×10−8 mutations per base pair per generation (95% confidence interval of the mean: 1.11–1.22) in 96 total families[4–8] (Table 1). However, it is important to note that all of these studies involve substantial filtering of de novo variants to remove false positives and often exclude highly repetitive regions of the genome. Given the relevance of variants in protein-coding sequence to disease, it is also important to understand the mutation rate in exonic regions. Studies from targeted sequencing of exomes or other regions have reported higher mutation rates (1.31–2.17×10−8 mutations per base pair per generation)[13–16]; this apparent increase may be due to several factors as discussed below.
Table 1.
Genome-wide estimates of SNV mutation rate.
| Type | Families | μ (×10−8) | 95% CI | % Paternal | Ref. |
|---|---|---|---|---|---|
| Whole-genome | 1 | 1.1 | 0.68–1.7 | [4] | |
| Whole-genome | 1 | 1.17 | 0.88–1.62 | 92% | [5] |
| Whole-genome | 1 | 0.97 | 0.67–1.34 | 36% | [5] |
| Whole-genome | 78 | 1.20 | 76% | [6] | |
| Whole-genome | 5 | 0.96 | 0.82–1.09 | 85% | [7] |
| Whole-genome | 10$ | 1 | 74% | [8] | |
| Targeted resequencing of 430 Mbp | 570* | 1.36 | 0.34–2.7 | [13] | |
| Whole-exome | 209† | 2.17 | 81% | [15] | |
| Whole-exome | 238‡ | 1.31 | [16] | ||
| Whole-exome | 175† | 1.5 | [14] | ||
| Indirect from microsatellites | 23# | 1.82 | 1.40–2.28 | [12] | |
| 512 Mbp of autozygosity | 5 | 1.20 | 0.89–1.43 | [7] |
Copy number variant (CNV) mutation rate
In addition to SNVs, there has been considerable effort in estimating the rates of formation of CNVs. Although CNVs are operationally defined as deletions and duplications of 50 bp or more[17], most studies have assessed de novo events only in the multi-kilobase pair range. As with SNVs, initial studies in this area focused on only a few loci. These analyses found the locus mutation rate is higher for CNVs (2.5×10−6–1×10−4 mutations per locus per generation) compared to SNVs and the rate varied by more than an order of magnitude between loci[18, 19]; data from mice suggest that the difference in rates between loci may be even larger[20]. A genome-wide analysis of large CNVs (>100 kbp) revealed a mutation rate of 1.2×10−2 CNVs per generation based on approximately 400 parent-offspring trios[21]. A significantly higher mutation rate of 3.6×10−2 mutations per generation was observed for individuals with intellectual disability likely because some of these de novo CNVs were influencing the development of the disorders observed in these individuals[22]. Using high-density microarrays and population genetic approaches, the rate of CNV formation was estimated to be 3×10−2 for variants >500 bp[23]. However, this rate is likely a lower bound since selection will remove deleterious mutations from the population and most large CNVs are estimated to be deleterious[21, 23].
Notably, when considering the total number of mutated base pairs between SNVs and CNVs, CNVs account for the vast majority. New large CNVs (>100 kbp) are relatively rare compared to SNVs: one new large CNV per 42 births (95% Poisson confidence interval: 23–97)[21] compared to an average 61 new SNVs per birth (95% confidence interval of the mean: 58–64)[5–8] (Figure 1). The average number of base pairs affected by large CNVs is 8–25 kbp per gamete (16–50 kbp per birth)[21], which is far larger than the average of 30.5 bp per gamete observed for SNVs (61 bp per birth) (Figure 1). It is important to note that the estimates for CNVs are based on microarray data that could not be used to reliably detect smaller CNVs (<100 kbp), so the mutational properties and rates of formation of these smaller variants remain unknown. Comparisons between the human and chimpanzee genomes also revealed that insertions and deletions account for close to three times the number of bases that are different compared to SNVs (3% versus 1.23%)[24]. Although caution must be exercised in the estimate of the de novo rate of CNVs, the data suggest a more than 100-fold differential between the number of base pairs affected (on average) per generation, yet only a threefold difference after 12 million years of evolution based on chimpanzee and human genome comparisons. This may reflect significant differences in the action of selection or radical rate changes since divergence for these different classes of mutations[25].
Figure 1. Comparison of the frequency and scale of different forms of genetic variation.

There is an inverse relationship between mutation size and frequency. Although SNVs occur more frequently, each mutation affects only a single base pair. In contrast, large mutations such as CNVs or chromosomal aneuploidy are rare, yet affect thousands to millions of base pairs, and even though these mutations are rare, they affect more base pairs per birth on average than SNVs. a) Average number of mutations of each type of variant per birth. b) Average number of mutated bases contributed by each type of variant per birth. (Y-axis is log10 scaled in both panels).
Other classes of genetic variation
In addition to CNVs and SNVs, there are many other forms of genetic variation that arise by completely different mutational processes and consequently have distinct biases. The largest, of course, are aneuploidies — the duplication or deletion of an entire chromosome. Due to the severity of these mutations (the most well-studied aneuploidy is Down syndrome), most aneuploidies are lethal in utero. Studies of spontaneous abortions and embryos created with in vitro fertilization suggest that 30%–60% of embryos and 0.3% of newborns have a chromosomal aneuploidy (reviewed in [26]) (Figure 1). Interestingly, there are substantial differences between chromosomes in the incidence of aneuploidy; trisomies of chromosomes 16, 18, 21, and the sex chromosomes are most prevalent[27]. Chromosomal aneuploidies are thought to primarily arise during meiosis I through several mechanisms. Most simply, homologous chromosomes can fail to pair or stay paired in meiosis potentially due to lack of recombination events[28]. However, trisomies can also arise if sister chromatids improperly segregate during meiosis I[29] (Figure 1), and it appears as though different chromosomes may be primarily affected by different mechanisms[26].
Other forms of genetic variation have been less well characterized, often due to methodological biases in their discovery leading to reduced sensitivity. The rate of small insertions and deletions or “indels” has been reported as approximately 0.20 ×10−9 per site per generation for insertions and 0.53×10−9–0.58×10−9 per site per generation for deletions; this corresponds to roughly 6% of the SNV mutation rate[3, 30] (Figure 1). Whole-genome sequence data from the 1000 Genomes Project suggested that each individual carries about one-tenth the number of indels compared to SNVs[31], but comparison of two Sanger-sequenced human genomes suggested a ratio closer to one-fifth[32]. The estimates from short-read sequencing must be considered conservative, since repetitive and low complexity regions of the genome have been difficult to assay because short reads harboring indels are difficult to map, especially in low complexity regions of the genome where this type of variation is enriched.
In addition to indels, several recent studies have focused on the rate of mobile element insertions (MEIs). The MEI rate has been estimated at about 2.5×10−2 per genome per generation or 1 in 20 births (for the active retrotransposons: Alu, L1, and SVA)[33] (Figure 1). It should be noted that comparative analyses of great ape genomes have suggested that this rate has varied radically in different lineages over the last 15 million years of human/great ape evolution. Unlike SNVs, the rate of MEIs has been far less clocklike over the course of evolution[34]. Within the human lineage, the insertions of Alus constitute the majority of MEI events with a rate of 2–4.6×10−2 per genome per generation or approximately 1 in 20 births[33, 35], whereas LI and SVA insertions are much rarer occurring at 3–4×10−3 per genome per generation (1 per about 100–150 births)[33, 36] and 6.5×10−4 per genome per generation (1 per 770 births)[33], respectively. However, these rates were primarily calculated indirectly using assumptions of the SNV mutation rate, so additional studies based on direct estimates from families are warranted. Given the low frequency of such occurrences and biases in terms of their integration into AT-rich and repetitive DNA, such analyses will require very large sample sizes and deeply sequenced genomes with preferably long reads in order to provide a reliable estimate.
Several loci in the genome are especially prone to mutation, including microsatellites[37], rDNA gene clusters[38], and segmental duplications (SDs)[39, 40]. A recent genome-wide analysis of over two thousand known microsatellites in over 24 thousand Icelandic trios revealed a mutation rate of 2.73×10−4 mutations per locus per generation for dinucleotide repeats and ~10×10−4 mutations per locus per generation for tetranucleotide repeats[12], which is similar to original projections based on population genotype data and Mendelian inconsistencies in families[37, 41]. It is important to note that this rate is several orders of magnitude greater than the rate for SNVs (base for base), underscoring that microsatellites are an extraordinary reservoir of new mutation. In addition, the mutation rate of individual microsatellites increases with average allele length and repeat uniformity likely because it is easier for DNA polymerase to slip on longer, purer repeats[12, 37, 42, 43] (reviewed in [44]) (Figure 2). Interestingly, there are length constraints on di- and tetranucleotide repeats where very long alleles tend to mutate to short ones and vice versa[12]; in contrast, studies of loci associated with trinucleotide repeat disorders indicate a polarity toward increasing length where mutability depends on the length and purity of the repeat tract length (reviewed in [45]). This property, where the increasing repeat length increases the probability of new mutation, has been described as dynamic mutation in contrast to the bulk of static mutations in the human genome[46].
Figure 2. Common mechanisms leading to biases in mutation.

a) CpG dinucleotides are the sites of cytosine methylation and frequent mutation. 5-methyl-cytosine can be deaminated to thymine (red). This mutation can either be repaired by mismatch repair pathways (reviewed in [121]) or be replicated to yield a cytosine to thymine mutation. b) Indels can occur by polymerase slippage during replication if these events are not repaired by mismatch repair (reviewed in [121]), especially in regions of low complexity such as microsatellites. Replication slippage is shown (red) on the newly synthesized strand leading to an insertion. c) Regions flanked by highly identical SDs (black boxes) are prone to NAHR. Recombination between homologous chromosomes (blue and magenta) occurs in paralogous regions leading to duplication of genes ABC in one of the recombined chromosomes and deletion on the other. d) Replicated homologous chromosomes are shown in black and gray. Premature loss of cohesion between sister chromatids can lead to separation of chromatids in meiosis I (black) leading to cells with only one chromatid or three chromatids. Trisomy results after meiosis II when one gamete ends up with an extra chromatid (red).
Although generated by a different mechanism involving non-allelic homologous recombination (NAHR) (Figure 2), clusters of ribosomal RNA genes (rDNA), centromeric satellites, and SDs also show extraordinary rates of mutation. The mutation rate for rDNA is estimated to be 0.11 per gene cluster per generation leading to an incredible diversity of rDNA alleles[38]. Centromeric satellites are also large regions of highly duplicated DNA where unequal crossover is rampant[47, 48]. The mutability of these regions gives rise to large differences in chromosomal length among individuals[49]; however, the repetitive nature of these regions has made them historically difficult to study other than by Southern blot and pulsed-field gel electrophoresis[50]. There is emerging data that SDs similarly are highly dynamic regions of the genome and prone to recurrent mutation. Copy number polymorphisms (CNPs), for example, are significantly enriched in regions of SDs[51, 52]; 90% of CNP genes map to SDs[53, 54]. Similar to satellites and rDNA, this bias is due, in large part, to the propensity for these segments to undergo NAHR[55–57]. As a result, CNPs in SDs are less likely to be in linkage disequilibrium with nearby SNPs[58, 59]. In addition, significant overlap between CNV loci in humans and nonhuman primates is likely due to recurrent mutation rather than ancestral polymorphism[60, 61].
Non-random distribution of new mutations
Given the tendency for certain types of loci to mutate, it is not surprising that new SNV and CNV mutations are not random. Several reported and predicted properties of new SNVs have been confirmed in recent genome-wide analyses. First, transitions outnumber transversions by twofold for de novo SNVs[4, 5, 30]. The rate of mutation at CpG dinucleotides has been observed to be 10- to 18-fold the rate of non-CpG dinucleotides[3, 6, 7, 30]. CpG dinucleotides are predicted to be more mutagenic because these are preferential sites of cytosine methylation, and spontaneous deamination of 5-methylcytosine yields thymine and thus creates a cytosine to thymine mutation (Figure 2). Considering that most estimates of de novo mutation rate have been based on sequencing technology that biases against particularly GC-rich DNA[31, 62], our estimates almost certainly represent a lower bound.
A number of different properties besides GC content have been associated with variation in mutation rate, including nucleosome occupancy and DNaseI hypersensitivity, replication timing, recombination rate, transcription, and repeat content[8, 63–68]. The higher mutation rates reported in or near protein-coding regions may be explained in part by the higher GC-content of these regions[13, 15, 16] in combination with the effects of transcription associated mutations[67]. Interestingly, a recent study of human RNA-seq data and human-macaque divergence found that an increase of twofold in gene expression leads to a 15% increase in mutation due to transcription associated mutagenesis (TAM)[67]. In addition, there is a strand asymmetry in mutations in transcribed regions of the genome where mutations induced from DNA damage (C to T, A to G, G to T, and A to T) are increased on the non-transcribed strand likely due to exposure of single-stranded DNA during transcription[66, 67, 69]. The transcribed strand, in contrast, is subject to RNA polymerase stalling leading to the recruitment of transcription coupled repair (TCR) machinery, which corrects some mutations (reviewed by [70]). The opposing forces of TAM and TCR lead to a bias towards G and T bases on the coding strand[67, 69].
Recent whole-genome sequencing studies have confirmed the non-randomness of mutations, which have been reported as an enrichment for clustered de novo SNVs. It was recently reported that 2%–3% of de novo SNVs are part of multinucleotide mutations, or mutations within 20 bp of another de novo SNV[71]. Similarly, a recent study reported an enrichment of SNVs (2% of de novo variants) within 10 kbp that could not be fully explained by GC content or multinucleotide mutations[7]. Finally, other recent work[8] confirmed previous reports of large deviations in the distribution of de novo SNVs compared to what would be expected under a model of random mutation[66, 72]. These studies suggest that a model of random SNV mutation is inaccurate at many different levels. With additional genome-wide mutation rate data, it should also be possible to assign local SNV mutation rates across the genome. Such biases are critical to assessing the significance of new mutations at a locus-specific level with respect to disease[73], especially as the community begins to explore the noncoding landscape.
Similar to SNVs, new CNVs are non-randomly distributed. Long stretches of highly paralogous sequences (SDs or low copy repeats) in direct orientation predispose to NAHR, which leads to deletions and duplications of the intervening sequence[39, 40] (Figure 2). The process of NAHR is involved in a greater fraction of large CNVs, although it does not contribute much to the formation of smaller (<50 kbp) CNVs[23, 74], which are thought to arise as a result of errors in replication or microhomology-mediated mutation[75–78]. Loci flanked by paralogous sequences have significantly higher rates of CNV mutation compared to loci outside of these regions[51, 79], and many of the CNVs in these regions have been strongly associated with diseases, including developmental delay, autism, and epilepsy (reviewed in [80]). Within loci flanked by SDs, there are differences in the rates of CNV formation. These differences are largely due to the presence of directly oriented SDs and the size and level of sequence identity of the flanking duplications. Thus, larger and more identical duplications provide better substrates for NAHR leading to higher rates of CNV formation[81, 82] (Figure 2). Moreover, as the size of CNVs increased so did the probability that the variants occurred de novo reflecting the effect of strong selection against such large variants[82] (Figure 3). Interestingly, NAHR “hotspots” often show structural variation in the flanking SDs that mediate the NAHR events. These structural variants lead to haplotypes that are prone to and protected from recurrent deletion because of differences in their genomic architecture and content of the flanking SDs[79, 83–86]. Interestingly, many of these “structural” haplotypes occur at very different frequencies among human populations leading to differences in ethnic predilection to recurrent copy number variation and disease[86, 87].
Figure 3. Larger CNVs are more likely to be de novo.
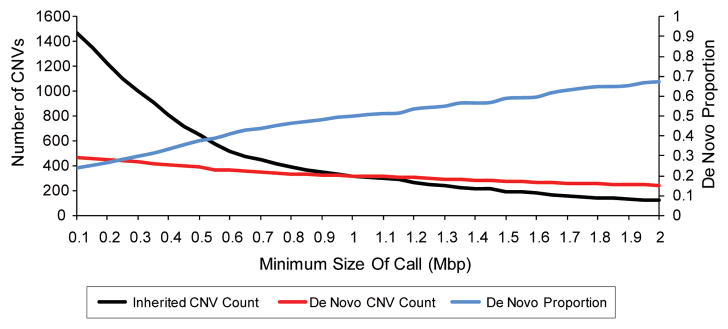
Size distributions of CNVs from over 15,000 children with developmental delay are plotted. Inherited CNVs are in black and de novo CNVs are in red with the number of CNVs on the left-hand y-axis. The proportion of CNVs that are de novo is plotted in blue with the de novo proportion on the right-hand y-axis. Reproduced from [82].
Parental bias and paternal age effects
It has long been hypothesized and observed that more mutations arise on the paternal germline[2, 88], and this difference is thought to be due to the larger number and continuous nature of cell divisions in spermatogenesis. Female eggs arise from a finite number of 22–33 cell divisions whereas male sperm monotonically increase every 15–16 days as a result of mitotic maintenance of the spermatogonial pool (reviewed in [89]). The dependence of SNV mutation on replication dictates an increase in mutations with advancing paternal age[88]. Whole-genome and whole-exome sequencing studies have confirmed the paternal bias for SNVs. The combined studies report that 76% (95% binomial CI = 73%–80%) of new mutations arise in the paternal germline based on 497 new mutations where the parental origin has been ascertained[6–8, 15].
Multiple studies have confirmed that the number of de novo mutations increases with the age of the father[6, 8, 15]. Yet, the data remain conflicted on the magnitude and model of this effect (Figure 4). In one study of the whole-genome sequences of two parent-offspring trios, for example, a paternal bias was observed in one trio and a maternal bias in the other[5]. If the increase in de novo mutations was solely due to the increased number of cell divisions in sperm production as a man aged, then it would be expected that there should be a linear relationship between paternal age and number of mutations. The data from these recent publications are not inconsistent with a linear model that estimates that the number of mutations increases by 1–2 mutations per year of the father’s life[6, 8]. However, others have suggested that an exponential increase of ~3% per year may be a slightly better fit for this data[6]. Further studies with larger ranges of paternal ages (especially older fathers) are needed to resolve this issue.
Figure 4. Relationship between paternal age and de novo mutations.

Current fitted models are shown of the increase in SNV mutations with paternal age from whole-exome and whole-genome sequencing of parent-offspring trios. There is some difference between the studies in regards to the magnitude of this effect, but sample sizes were relatively low and more studies, especially with elderly fathers, are needed to achieve a more precise estimate. The paternal age is on the x-axis, the left-hand y-axis shows the number of mutations per genome per birth and the right-hand y-axis shows the number of mutations per exome per birth. Exome data from 189 trios yielded an increase of 0.04 exonic mutations per year of paternal age (dashed green line)[15]; the smaller number of mutations compared to the whole-genome studies is consistent with the smaller target (protein-coding exons). Whole-genome data from 78 trios yielded an increase of 2.01 mutations per year (blue)[6]. Whole-genome data from 10 families yielded an increase of 1.02 mutations per year (red)[8].
An important consideration in paternal bias and age effects is the selective potential of de novo mutations on spermatogonial cells. Recent analysis has revealed that mutations in several genes (e.g., FGFR2, FGFR3, HRAS, and PTPN11) likely confer growth advantages to spermatogonial cells leading to further proliferation of sperm carrying those mutations — even though mutations in these genes lead to autosomal dominant disorders at the organismal level, including Apert syndrome (FGFR2) and achondroplasia (FGFR3)[90, 91]. A strong “paternal age effect” has been observed for these disorders[92, 93] with mutations in these genes at a rate exceeding linear expectation[94, 95]. Mutations associated with these disorders are almost exclusively paternal (95%–100%), gain-of-function missense mutations. These observations are consistent with a model of selfish spermatogonial selection where mutations confer growth advantages to spermatogonial cells leading to a clonal proliferation in the testis which, in turn, contributes disproportionately to the number of mutant sperm as a man ages[90, 91]. These genes are likely the reason that previous studies focused on select autosomal dominant loci estimated a faster than linear increase of mutations with paternal age[94, 95]. With the exception of a few loci such as these, the available data are consistent with a linear increase of mutations with advancing paternal age[6, 8], primarily as a result of increased cell division and replication errors.
In addition to SNVs, other forms of genetic variation have been assessed for parental origin and association with increased parental age. Similar to SNVs, a strong paternal bias has also been reported for mutations at microsatellites with a paternal to maternal ratio of 3.3:1. Once again, the number of microsatellite mutations increases linearly with paternal age[12]. Parental origin has also been assessed for structural variation, albeit limited to children with developmental delay where parental data were available. A paternal bias has been observed for large chromosomal rearrangements visible by microscopy, including deletions, duplications, and translocations[96]. Similarly, CNVs (>150 kbp) also have been reported to have a paternal bias with 90 out of 118 of all de novo CNVs arising on the paternal haplotype (76%; binomial 95% CI = 69%–84%)[22]. This result is driven primarily by mechanisms other than NAHR where no significant difference is found in the number of events between paternal and maternal origin. Similar to SNVs, the number of non-NAHR CNVs increased with paternal age[22]. So far, the only exception to the rule of paternal origin for new mutations and increase with paternal age is chromosomal aneuploidy, including Down syndrome (trisomy of chromosome 21), where the vast majority of mutations originate in the maternal germline and the risk of aneuploidy increases exponentially with maternal age (reviewed extensively in [26, 27]).
New mutations, selection, and human disease
There has been much recent interest in identifying de novo mutations that play a role in the development of human disease; knowledge of the patterns of human mutation is critical to the interpretation of these studies. Some broad themes are beginning to emerge. First, it is clear that deleterious de novo mutations contribute significantly to human disease and probably have played a more important role in all diseases than previously anticipated as a result of the super exponential increase in the human population over the last 5,000 years[97–99]. Exome sequencing revealed an increase in the number of de novo loss-of-function SNVs in individuals with autism[15, 16, 100] and schizophrenia[101]. The story is similar for CNVs, where individuals with neurocognitive diseases show an increase in de novo CNVs[79, 102–104]. Interestingly, individuals with autism in families with multiple affected individuals also show an increased number of de novo CNVs compared to their siblings even though the multiplex nature of these families would suggest a primarily inherited model of disease[21].
Given the data that de novo SNVs contribute to disease in combination with an increase in mutation rate with paternal age, there has been considerable discussion regarding the effect of paternal age on disease[105]. However, it is important to consider the potential magnitude of this effect, which is likely to be modest. Even if there are two new mutations per year of paternal age or a doubling of mutations every 16.5 years[6], most of these new mutations will be neutral and not contribute to disease. These data are consistent with epidemiological data that suggest a modest, albeit significant, increase in prevalence of disease in children from elderly fathers — there is a twofold increase in relative risk of a child developing autism from a father over 55 years of age when compared to a father less than 29 years of age[106]. The notable exceptions are diseases caused by mutations in spermatogonial selection genes, where the effect of paternal age increases much more significantly[91].
Inferring dates of human evolution
The increasing number of direct analyses in human families has led to discussion aimed at resolving these new rate estimates with our knowledge of important dates in human evolution. This stems from the fact that the mutation rates calculated directly in human families are about half of that calculated based on sequence divergence and fossil record[107, 108]. As a result of these updated mutation rates, generation times in the great ape lineages may be longer than previously thought[107]. Taken together, this pushes divergence times further back, and these dates are more in line with the fossil record in some cases but seem ridiculous in others (see [107, 108] for a detailed discussion). However, if mutation rates calculated from whole-genome sequencing of human families represent a lower bound as discussed above, then rates from direct and indirect approaches would be more concordant and the lengthening of divergence times would be overestimated. Moreover, there is also considerable uncertainty in terms of the effect of paternal age with respect to ancestral populations, and this may account for some of the difference between direct and indirect estimates of mutation rate. Adding to the complexity, there is good evidence that mutation rates have not remained constant over evolutionary time with a slowdown in hominids — likely a consequence of generational time[9, 109]. Outside of humans, there is little genome-wide data on the extent of this slowdown even among closely related species.
Concluding remarks
In the last few years, genomic technologies have made it possible to obtain direct knowledge concerning rates of human mutation. Recent studies are converging on similar SNV mutation rates, quantifying the male mutation bias and its relationship with paternal age. The current rate estimate for SNVs likely represents a lower bound because of biases in next-generation sequencing technology[31, 62] and the stringent filtering required to remove false positive calls. In addition, we have gained new insight into the mutational properties of large CNVs, their regional biases within the genome, and their genomic impact. However, our understanding of the properties of human mutation is far from complete. Many studies have focused on identifying de novo mutations in individuals with disease, and this may introduce biases in our understanding of the natural processes of mutation. Large studies of individuals from relatively healthy families will provide valuable insight into the general patterns of mutation. It also remains unclear how mutation rate increases with paternal age and the number of genes subject to spermatogonial selection. Many of the recent de novo mutations associated with autism have been found in genes potentially important in cell growth and chromatin modification — it is possible that mutations in these also confer growth advantage in the testis. One approach may be to sequence more families with many children or children born from particularly old fathers. It will also be important to sequence DNA from multi-generation families in order to understand what fraction of new mutations discovered specifically in the blood are transmitted to the next generation. In light of the critical importance of new mutations in understanding evolution, efforts to sequence genomes from nonhuman primate families should be a high priority in order to understand how the rate has changed in different lineages. Although discussed briefly, we are still lacking reliable estimates of the mutation rate and the complexity of short indels and smaller CNVs, especially those mapping within SDs. One promising approach would be to use sequencing of large-insert clones to fully phase long haplotypes[110], which would allow parental origin to be determined for all de novo mutations and enable better interpretation of indels. Understanding the mutation rate of SDs and centromeric satellite sequences will likely require single molecular sequencing with very long reads (>50 kbp)[111, 112] and accurate de novo assembly.
Although we are beginning to understand the pattern of germline mutation, somatic mutation processes are largely unknown outside of cancer studies. Somatic mutations, however, have the potential to contribute to diseases other than cancer and may be subjected to very different mutational biases as a result of differences in repair and replication between meiotic and mitotic tissues (reviewed in [113, 114]). Such mutations can be identified as genetic differences either between tissues from the same donor or differences between monozygotic twins. Given the proportion of the somatic mutation compared to the germline alleles in a population of cells or a tissue sample and with some assumptions, one can currently estimate approximately where in development the mutation occurred[114, 115]. There is compelling evidence that somatic structural variants accumulate with age likely as a result of an increasing number of replication copy errors[116]. The continued development of single-cell whole-genome sequencing technologies will revolutionize this area of research. It has already allowed for analysis of somatic mutations in tumor samples[117], embryos[118], and haplotype phasing of individual cells[119]. Its application to sperm and egg will allow for the calculation of the true germline mutation rate and provide data on effects of positive and negative selection of mutations within germ cells. Such technologies coupled with advances in genome sequencing will ultimately allow scientists to generate ontogenic maps of mutation tracking the origin and fate of somatic mutations during the development of organisms.
Highlights.
- We describe insights into mutation rate from high-throughput genome sequencing of families.
- A paternal bias and age-effect in mutation has been quantified at the genome-wide level.
- Copy number variants arise less frequently than point mutations but affect more bases.
- Future research will yield insight into the mutation rate of other forms of variation.
Acknowledgments
We thank Santhosh Girirajan and Bradley Coe for sharing data and figures. We are grateful to Andrew Wilkie, Anne Goriely, and Peter Sudmantfor helpful discussions and to T onia Brown for assistance with manuscript preparation. We would like to thank Jacob Michaelson and Jonathan Sebat for sharing a prepublication version of their manuscript. C.D.C. was supported by a Ruth L. Kirschstein National Research Service Award (NRSA) (F32HG006070). E.E.E. is an Investigator of the Howard Hughes Medical Institute.
GLOSSARY BOX
Autozygosity
Large regions of homozygous sequence inherited from a recent ancestor. They are also referred to as homozygosity by recent descent
De novo mutation
A mutation observed in a child but not in the parents. Such mutations are assumed to have occurred in one of the parental germlines
Haplotype phase
Determination of which alleles segregate on the same physical chromosomes. For example, which alleles of nearby variants in a child occur on the chromosome inherited from the father
Microsatellite
A locus comprised of a simple repeat of DNA bases. The repeating unit is most often comprised of two, three, or four bases
rDNA
The regions of the genome encoding for ribosomal RNA. These are comprised of repeating units of either 2.2 kbp located on chromosome 1 or 43 kbp located on the acrocentric chromosomes
Retrotransposon
A DNA sequence that copies itself through an mRNA intermediate and reinserts the copied sequence through reverse transcription into a new location in the genome
Segmental duplication (SD)
A segment (>1 kbp) of high sequence identity (>90%) that exists at two or more locations in a genome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haldane JBS. The rate of spontaneous muation of a human gene. Journal of Genetics. 1935;31:317–326. doi: 10.1007/BF02717892. [DOI] [PubMed] [Google Scholar]
- 2.Haldane JB. The mutation rate of the gene for haemophilia, and its segregation ratios in males and females. Ann Eugen. 1947;13:262–271. doi: 10.1111/j.1469-1809.1946.tb02367.x. [DOI] [PubMed] [Google Scholar]
- 3.Kondrashov AS. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat. 2003;21:12–27. doi: 10.1002/humu.10147. [DOI] [PubMed] [Google Scholar]
- 4.Roach JC, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conrad DF, et al. Variation in genome-wide mutation rates within and between human families. Nat Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong A, et al. Rate of de novo mutations and the importance of father/’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell CD, et al. Estimating the human mutation rate using autozygosity in a founder population. Nature genetics. 2012;44:1277–1281. doi: 10.1038/ng.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michaelson Jacob J, et al. Whole-Genome Sequencing in Autism Identifies Hot Spots for De Novo Germline Mutation. Cell. 2012;151:1431–1442. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li WH, Tanimura M. The molecular clock runs more slowly in man than in apes and monkeys. Nature. 1987;326:93–96. doi: 10.1038/326093a0. [DOI] [PubMed] [Google Scholar]
- 10.Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scally A, et al. Insights into hominid evolution from the gorilla genome sequence. Nature. 2012;483:169–175. doi: 10.1038/nature10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun JX, et al. A direct characterization of human mutation based on microsatellites. Nat Genet. 2012 doi: 10.1038/ng.2398. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Awadalla P, et al. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am J Hum Genet. 2010;87:316–324. doi: 10.1016/j.ajhg.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scherer SW, et al. Challenges and standards in integrating surveys of structural variation. Nat Genet. 2007;39:S7–15. doi: 10.1038/ng2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39:S43–47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 19.Turner DJ, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egan CM, et al. Recurrent DNA copy number variation in the laboratory mouse. Nat Genet. 2007;39:1384–1389. doi: 10.1038/ng.2007.19. [DOI] [PubMed] [Google Scholar]
- 21.Itsara A, et al. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hehir-Kwa JY, et al. De novo copy number variants associated with intellectual disability have a paternal origin and age bias. Journal of medical genetics. 2011;48:776–778. doi: 10.1136/jmedgenet-2011-100147. [DOI] [PubMed] [Google Scholar]
- 23.Conrad DF, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 25.Marques-Bonet T, et al. A burst of segmental duplications in the genome of the African great ape ancestor. Nature. 2009;457:877–881. doi: 10.1038/nature07744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagaoka SI, et al. Human aneuploidy: mechanisms and new insights into an age-old problem. Nature reviews Genetics. 2012;13:493–504. doi: 10.1038/nrg3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nature reviews Genetics. 2001;2:280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- 28.Henderson SA, Edwards RG. Chiasma frequency and maternal age in mammals. Nature. 1968;218:22–28. doi: 10.1038/218022a0. [DOI] [PubMed] [Google Scholar]
- 29.Angell RR. Predivision in human oocytes at meiosis I: a mechanism for trisomy formation in man. Human genetics. 1991;86:383–387. doi: 10.1007/BF00201839. [DOI] [PubMed] [Google Scholar]
- 30.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen JQ, et al. Variation in the ratio of nucleotide substitution and indel rates across genomes in mammals and bacteria. Molecular biology and evolution. 2009;26:1523–1531. doi: 10.1093/molbev/msp063. [DOI] [PubMed] [Google Scholar]
- 33.Stewart C, et al. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS genetics. 2011;7:e1002236. doi: 10.1371/journal.pgen.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Locke DP, et al. Comparative and demographic analysis of orang-utan genomes. Nature. 2011;469:529–533. doi: 10.1038/nature09687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cordaux R, et al. Estimating the retrotransposition rate of human Alu elements. Gene. 2006;373:134–137. doi: 10.1016/j.gene.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 36.Ray DA, Batzer MA. Reading TE leaves: new approaches to the identification of transposable element insertions. Genome research. 2011;21:813–820. doi: 10.1101/gr.110528.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weber JL, Wong C. Mutation of human short tandem repeats. Human molecular genetics. 1993;2:1123–1128. doi: 10.1093/hmg/2.8.1123. [DOI] [PubMed] [Google Scholar]
- 38.Stults DM, et al. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome research. 2008;18:13–18. doi: 10.1101/gr.6858507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends in genetics: TIG. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 40.Bailey JA, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 41.Whittaker JC, et al. Likelihood-based estimation of microsatellite mutation rates. Genetics. 2003;164:781–787. doi: 10.1093/genetics/164.2.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eichler EE, et al. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nature genetics. 1994;8:88–94. doi: 10.1038/ng0994-88. [DOI] [PubMed] [Google Scholar]
- 43.Ballantyne KN, et al. Mutability of Y-chromosomal microsatellites: rates, characteristics, molecular bases, and forensic implications. American journal of human genetics. 2010;87:341–353. doi: 10.1016/j.ajhg.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellegren H. Microsatellites: simple sequences with complex evolution. Nature reviews Genetics. 2004;5:435–445. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 45.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nature reviews Genetics. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richards RI, Sutherland GR. Dynamic mutation: possible mechanisms and significance in human disease. Trends Biochem Sci. 1997;22:432–436. doi: 10.1016/s0968-0004(97)01108-0. [DOI] [PubMed] [Google Scholar]
- 47.Waye JS, Willard HF. Structure, organization, and sequence of alpha satellite DNA from human chromosome 17: evidence for evolution by unequal crossing-over and an ancestral pentamer repeat shared with the human X chromosome. Mol Cell Biol. 1986;6:3156–3165. doi: 10.1128/mcb.6.9.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alkan C, et al. The role of unequal crossover in alpha-satellite DNA evolution: a computational analysis. Journal of computational biology: a journal of computational molecular cell biology. 2004;11:933–944. doi: 10.1089/cmb.2004.11.933. [DOI] [PubMed] [Google Scholar]
- 49.Mahtani MM, Willard HF. Pulsed-field gel analysis of alpha-satellite DNA at the human X chromosome centromere: high-frequency polymorphisms and array size estimate. Genomics. 1990;7:607–613. doi: 10.1016/0888-7543(90)90206-a. [DOI] [PubMed] [Google Scholar]
- 50.Warburton PE, Willard HF. Genomic analysis of sequence variation in tandemly repeated DNA. Evidence for localized homogeneous sequence domains within arrays of alpha-satellite DNA. Journal of molecular biology. 1990;216:3–16. doi: 10.1016/s0022-2836(05)80056-7. [DOI] [PubMed] [Google Scholar]
- 51.Sharp AJ, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nature reviews Genetics. 2006;7:552–564. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- 54.Bailey JA, et al. Human copy number polymorphic genes. Cytogenet Genome Res. 2008;123:234–243. doi: 10.1159/000184713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Conrad DF, et al. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet. 2010;42:385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kidd JM, et al. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell. 2010;143:837–847. doi: 10.1016/j.cell.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mills RE, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Locke DP, et al. Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am J Hum Genet. 2006;79:275–290. doi: 10.1086/505653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campbell CD, et al. Population-genetic properties of differentiated human copy-number polymorphisms. Am J Hum Genet. 2011;88:317–332. doi: 10.1016/j.ajhg.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perry GH, et al. Hotspots for copy number variation in chimpanzees and humans. Proc Natl Acad Sci U S A. 2006;103:8006–8011. doi: 10.1073/pnas.0602318103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee AS, et al. Analysis of copy number variation in the rhesus macaque genome identifies candidate loci for evolutionary and human disease studies. Hum Mol Genet. 2008;17:1127–1136. doi: 10.1093/hmg/ddn002. [DOI] [PubMed] [Google Scholar]
- 62.Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stamatoyannopoulos JA, et al. Human mutation rate associated with DNA replication timing. Nature genetics. 2009;41:393–395. doi: 10.1038/ng.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ying H, et al. Evidence that localized variation in primate sequence divergence arises from an influence of nucleosome placement on DNA repair. Molecular biology and evolution. 2010;27:637–649. doi: 10.1093/molbev/msp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen CL, et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome research. 2010;20:447–457. doi: 10.1101/gr.098947.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hodgkinson A, Eyre-Walker A. Variation in the mutation rate across mammalian genomes. Nature reviews Genetics. 2011;12:756–766. doi: 10.1038/nrg3098. [DOI] [PubMed] [Google Scholar]
- 67.Park C, et al. Genomic evidence for elevated mutation rates in highly expressed genes. EMBO Rep. 2012;13:1123–1129. doi: 10.1038/embor.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koren A, et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. American journal of human genetics. 2012;91:1033–1040. doi: 10.1016/j.ajhg.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Green P, et al. Transcription-associated mutational asymmetry in mammalian evolution. Nature genetics. 2003;33:514–517. doi: 10.1038/ng1103. [DOI] [PubMed] [Google Scholar]
- 70.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 71.Schrider DR, et al. Pervasive multinucleotide mutational events in eukaryotes. Curr Biol. 2011;21:1051–1054. doi: 10.1016/j.cub.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matassi G, et al. Chromosomal location effects on gene sequence evolution in mammals. Current biology: CB. 1999;9:786–791. doi: 10.1016/s0960-9822(99)80361-3. [DOI] [PubMed] [Google Scholar]
- 73.O’Roak BJ, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kidd JM, et al. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat Meth. 2010;7:365–371. doi: 10.1038/nmeth.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith CE, et al. Template switching during break-induced replication. Nature. 2007;447:102–105. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- 76.Lee JA, et al. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 77.Payen C, et al. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS genetics. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hastings PJ, et al. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sharp AJ, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 80.Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev. 2009;19:196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu P, et al. Frequency of nonallelic homologous recombination is correlated with length of homology: evidence that ectopic synapsis precedes ectopic crossing-over. American journal of human genetics. 2011;89:580–588. doi: 10.1016/j.ajhg.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cooper GM, et al. A copy number variation morbidity map of developmental delay. Nature genetics. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Osborne LR, et al. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29:321–325. doi: 10.1038/ng753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koolen DA, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 85.Zody MC, et al. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet. 2008 doi: 10.1038/ng.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Antonacci F, et al. A large, complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat Genet. 2010 doi: 10.1038/ng.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steinberg KM, et al. Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nature genetics. 2012;44:872–880. doi: 10.1038/ng.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Crow JF. The origins, patterns and implications of human spontaneous mutation. Nature reviews Genetics. 2000;1:40–47. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- 89.Hurst LD, Ellegren H. Sex biases in the mutation rate. Trends in genetics: TIG. 1998;14:446–452. doi: 10.1016/s0168-9525(98)01577-7. [DOI] [PubMed] [Google Scholar]
- 90.Goriely A, et al. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science. 2003;301:643–646. doi: 10.1126/science.1085710. [DOI] [PubMed] [Google Scholar]
- 91.Goriely A, Wilkie AO. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. American journal of human genetics. 2012;90:175–200. doi: 10.1016/j.ajhg.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cohen MM, Jr, et al. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992;42:655–659. doi: 10.1002/ajmg.1320420505. [DOI] [PubMed] [Google Scholar]
- 93.Orioli IM, et al. Effect of paternal age in achondroplasia, thanatophoric dysplasia, and osteogenesis imperfecta. Am J Med Genet. 1995;59:209–217. doi: 10.1002/ajmg.1320590218. [DOI] [PubMed] [Google Scholar]
- 94.Risch N, et al. Spontaneous mutation and parental age in humans. American journal of human genetics. 1987;41:218–248. [PMC free article] [PubMed] [Google Scholar]
- 95.Crow JF. The high spontaneous mutation rate: is it a health risk? Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8380–8386. doi: 10.1073/pnas.94.16.8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thomas NS, et al. Parental and chromosomal origin of unbalanced de novo structural chromosome abnormalities in man. Human genetics. 2006;119:444–450. doi: 10.1007/s00439-006-0157-6. [DOI] [PubMed] [Google Scholar]
- 97.Nelson MR, et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Keinan A, Clark AG. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science. 2012;336:740–743. doi: 10.1126/science.1217283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tennessen JA, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu B, et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nature genetics. 2011;43:864–868. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de Vries BB, et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Walsh T, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 105.Kondrashov A. Genetics: The rate of human mutation. Nature. 2012;488:467–468. doi: 10.1038/488467a. [DOI] [PubMed] [Google Scholar]
- 106.Hultman CM, et al. Advancing paternal age and risk of autism: new evidence from a population-based study and a meta-analysis of epidemiological studies. Molecular psychiatry. 2011;16:1203–1212. doi: 10.1038/mp.2010.121. [DOI] [PubMed] [Google Scholar]
- 107.Langergraber KE, et al. Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:15716–15721. doi: 10.1073/pnas.1211740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Scally A, Durbin R. Revising the human mutation rate: implications for understanding human evolution. Nature reviews Genetics. 2012;13:745–753. doi: 10.1038/nrg3295. [DOI] [PubMed] [Google Scholar]
- 109.Elango N, et al. Variable molecular clocks in hominoids. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1370–1375. doi: 10.1073/pnas.0510716103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kitzman JO, et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol. 2011;29:59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Branton D, et al. The potential and challenges of nanopore sequencing. Nature biotechnology. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eid J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 113.Erickson RP. Somatic gene mutation and human disease other than cancer: an update. Mutat Res. 2010;705:96–106. doi: 10.1016/j.mrrev.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 114.Frank SA. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(Suppl 1):1725–1730. doi: 10.1073/pnas.0909343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Abyzov A, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012 doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Forsberg LA, et al. Age-related somatic structural changes in the nuclear genome of human blood cells. American journal of human genetics. 2012;90:217–228. doi: 10.1016/j.ajhg.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Voet T, et al. Breakage-fusion-bridge cycles leading to inv dup del occur in human cleavage stage embryos. Human mutation. 2011;32:783–793. doi: 10.1002/humu.21502. [DOI] [PubMed] [Google Scholar]
- 119.Fan HC, et al. Whole-genome molecular haplotyping of single cells. Nature biotechnology. 2011;29:51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Alkuraya FS. Autozygome decoded. Genetics in medicine: official journal of the American College of Medical Genetics. 2010;12:765–771. doi: 10.1097/GIM.0b013e3181fbfcc4. [DOI] [PubMed] [Google Scholar]
- 121.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]