E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases (original) (raw)
Abstract
E-cadherin is an essential adhesion protein as well as a tumor suppressor that is silenced in many cancers. Its adhesion-dependent regulation of signaling has not been elucidated. We report that E-cadherin can negatively regulate, in an adhesion-dependent manner, the ligand-dependent activation of divergent classes of receptor tyrosine kinases (RTKs), by inhibiting their ligand-dependent activation in association with decreases in receptor mobility and in ligand-binding affinity. E-cadherin did not regulate a constitutively active mutant RTK (Neu*) or the ligand-dependent activation of LPA receptors or muscarinic receptors, which are two classes of G protein-coupled receptors. EGFR regulation by E-cadherin was associated with complex formation between EGFR and E-cadherin that depended on the extracellular domain of E-cadherin but was independent of β-catenin binding or p120-catenin binding. Transfection of E-cadherin conferred negative RTK regulation to human melanoma and breast cancer lines with downregulated endogenous E-cadherin. Abrogation of E-cadherin regulation may contribute to the frequent ligand-dependent activation of RTK in tumors.
Keywords: cell growth, cell signaling, E-cadherin, MDCK cells, receptor tyrosine kinase
Introduction
E-cadherin, which is expressed primarily in epithelial cells, is an adhesion protein that is essential for multicellular organisms (Uemura et al, 1996;Wheelock and Johnson, 2003). It is encoded by the CDH1 gene and functions in multiple processes, including development, tissue integrity, cell migration, morphology, and polarity (Jamora and Fuchs, 2002). In non-epithelial cells such as epidermal Langerhans cells and melanocytes, E-cadherin expression may contribute to their intraepithelial location (Tang et al, 1993). E-cadherin is also a tumor suppressor whose expression is frequently reduced or silenced, and its re-expression can induce morphologic reversion (Perl et al, 1998; Hajra and Fearon, 2002). E-cadherin is a single transmembrane domain protein whose N-terminus is extracellular and C-terminus is intracellular (Hajra and Fearon, 2002; Wheelock and Johnson, 2003). Calcium is required for E-cadherin to mediate its adhesive function, and the extracellular portion of E-cadherin contains several calcium-binding sites (Boggon et al, 2002). E-cadherin molecules form homodimers on the cell surface (lateral dimerization) and carry out their adhesive function by binding, in an antiparallel fashion, to E-cadherin molecules on adjacent cells (homotypic adhesion), which may progress to extensive multimers of E-cadherin (Gumbiner, 2000; Boggon et al, 2002). E-cadherin is connected indirectly to the cytoskeleton, via non-covalent linkage of its intracellular C-terminal portion to β-catenin, which in turn is non-covalently linked to α-catenin, which binds the actin cytoskeleton. The cytoskeletal link is required for maximum homotypic activity of E-cadherin. The juxtamembrane region of the intracellular portion binds p120-catenin, which may contribute to adhesion (Thoreson et al, 2000; Ireton et al, 2002).
E-cadherin was initially considered a structural protein. More recently, it has also been recognized to regulate signaling. For example, β-catenin is a key component of the Wnt signaling pathway (Sharpe et al, 2001), in addition to its structural role in E-cadherin function. β-Catenin activity can be regulated by E-cadherin under some conditions, but this regulation may not depend on the adhesive activity of E-cadherin (Gottardi et al, 2001; Stockinger et al, 2001).
Identifying a clear relationship between the adhesive properties of E-cadherin and the putative ability of E-cadherin to regulate signaling has proven elusive. EGFR and c-Met, which are receptor protein tyrosine kinases (RTKs), have been shown to colocalize with E-cadherin to basolateral areas of polarized epithelial cells and to form multicomponent complexes that include E-cadherin, although the functional consequences of these associations have not been clearly identified (Crepaldi et al, 1994; Hoschuetzky et al, 1994; Pece and Gutkind, 2000). EGF-dependent activation of EGFR has been reported to be inhibited in an E-cadherin adhesion-dependent manner, although whether it was via a prereceptor or postreceptor mechanism was not clear (Takahashi and Suzuki, 1996). However, E-cadherin has also been found to activate transiently EGFR when cell–cell contacts were formed by switching from low-calcium conditions to high calcium (Pece and Gutkind, 2000).
These considerations have led us to examine whether a consistent signal regulating property related to the adhesive function of E-cadherin might contribute to the ability of E-cadherin to regulate cell growth negatively. In the current communication, we have tested the ability of E-cadherin to affect the response to several soluble mitogens, including ligands for RTKs of different classes and for G protein-coupled receptors (GPCRs).
Results
EGF-induced DNA synthesis in dense cells is inhibited by E-cadherin-dependent adhesion
MDCK (McRoberts et al, 1981), which is a kidney epithelial line that displays density-dependent growth inhibition, has been used extensively to characterize epithelial cell physiology. We first examined growth and intracellular signaling responses when MDCK at low and high cell density was stimulated with EGF. To determine if these activities might be regulated by E-cadherin, we incubated cells with a neutralizing monoclonal antibody (DECMA-1) that can disrupt E-cadherin adhesion complexes from MDCK and other cells (Ozawa et al, 1990) or with calcium depletion, which less-specifically disrupts cell–cell adhesions induced by E-cadherin (Gumbiner, 2000). At low density, MDCK forms islands of cells with loose junctions of E-cadherin-dependent cell–cell adhesions (Figure 1A, top left panel). At high density, the adhesions are more extensive and form tight adherens junctions (Figure 1A, lower panel; Adams et al, 1998). As expected for cells such as MDCK that display density-dependent growth inhibition, high-density cells underwent less basal DNA synthesis in low serum (0.5%) than did low-density cells, and the response to EGF was also less for high-density cells than low-density cells (Figure 1B). The lower response to EGF at high density depended on E-cadherin cell–cell adhesions; the increment in DNA synthesis induced by EGF was substantially higher in the presence of the E-cadherin neutralizing antibody than in cells incubated with control IgG. By contrast, at low density, the incremental response associated with the neutralizing antibody was only about one-third higher. Downstream intracellular signaling was assessed by determining the level of active Ras (Figure 1C, Ras·GTP), a GTPase that is activated by EGFR (Schlessinger, 2000). The EGF results paralleled those seen for DNA synthesis (Figure 1C). The differences in the responses at low versus high density were not associated with a change in the protein level of E-cadherin or Ras (Figure 1C).
Figure 1.
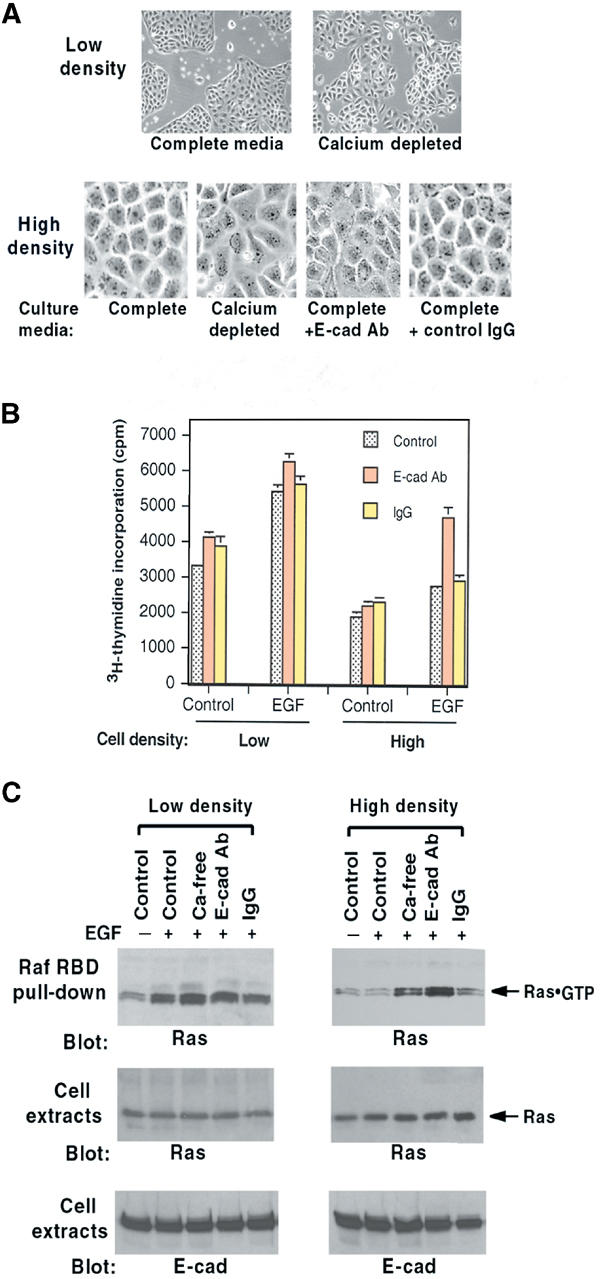
Effects of MDCK cell density on EGF responses. (A) Morphology of MDCK cells at low and high density and effects of 16 h incubation with calcium depletion, E-cadherin neutralizing antibody (E-cad Ab), or control IgG. Original magnifications were the same; images on the top panels are × 4, and those on the bottom panels × 10. (B) DNA synthesis in cells treated as in (A) and then given EGF (10 ng/ml) for 24 h. (C) Ras·GTP response of cells treated as in (A) and then stimulated with EGF (100 ng/ml) for 5 min. Ras·GTP was determined by Raf RBD pull-down in the upper panel. Total Ras and E-cadherin in cell extracts are in the middle and lower panels, respectively.
E-cadherin reduces the mobility of EGFR in the plasma membrane
Prior to its activation, EGFR is found on the plasma membrane as inactive monomers, which dimerize and become activated in response to EGF (Schlessinger, 2000). Since EGFR can colocalize with E-cadherin in the basolateral areas of polarized epithelial cells (Hoschuetzky et al, 1994), we speculated that E-cadherin might be sequestering the EGFR monomers and interfering with their mobility, which would then inhibit their efficiency of dimerization. To address this possibility, we expressed an EGFR-CFP (cyan fluorescent protein) fusion protein in HEK293 cells (293-Ecad) that stably express E-cadherin levels similar to those in MDCK (Figure 2D) and undergo analogous morphologic changes (Li et al, 2003). At low density, the cells are rounded with weak cell–cell contacts, while at high density they are more epithelioid and have tight adherens junctions (Figure 2A). When high-density cells are incubated with the E-cadherin neutralizing antibody, they round up, partially separate from each other, and their EGFR-CFP becomes less localized to cell edges. Fluorescence recovery after photobleaching (FRAP) was used to measure the mobility of the EGFR-CFP protein in the membrane (Figure 2B). While the photobleached area recovered completely in low-density cells, recovery in high density did not return to prephotobleached values, indicating that an EGFR-CFP pool was much less mobile (left panel). The differences in EGFR-CFP mobility were not associated with a change in its surface expression (Figure 2C). Incubating high-density cells with the E-cadherin neutralizing antibody led to a recovery intermediate between the low- and high-density cells without the antibody, indicating that E-cadherin had limited the mobility of the receptors (Figure 2B, right panel). Analogous results have been reported for the mobility of E-cadherin (Adams et al, 1998).
Figure 2.
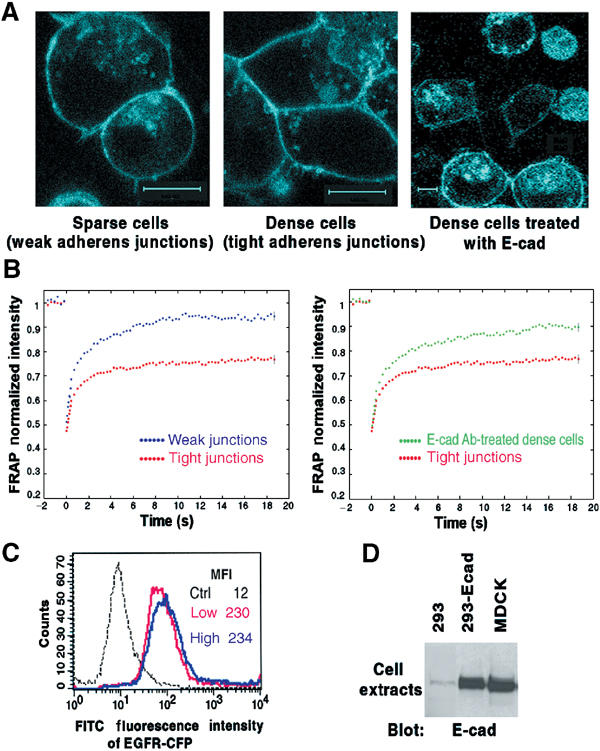
Adhesion-dependent inhibition of EGFR-CFP mobility. (A) Confocal images of HEK293 cells that stably express E-cadherin (293-Ecad) were transiently transfected with a plasmid encoding EGFR-CFP. Right panel: Cells treated with E-cadherin neutralizing antibody for 16 h. Original magnification × 100. Each bar represents 10 μm. (B) Mean FRAP values of EGFR-CFP in cells as in (A). Cell–cell junction regions from sparse, dense, or E-cad Ab-treated dense cells (as shown in panel A) were subjected to FRAP analysis. The normalized mean curves from weak versus tight adherens junctions were compared (left panel); the tight junction curve was also compared with that from the E-cad Ab-treated cells (right panel). The standard errors were calculated for each time point, but are shown only for the last time point. (C) FACS analysis of EGFR-CFP expression on the surface of cells as in (A). Cells were trypsinized and stained with anti-EGFR mAb 225 and FITC-conjugated anti-mouse IgG. MFI=mean fluorescence intensity. (D) E-cadherin expression in 293-Ecad cells. Equal amounts of cell extracts from parental HEK293, 293-Ecad, and MDCK cells were analyzed by anti-E-cadherin blotting.
E-cadherin inhibits EGF binding to EGFR and activation of EGFR/Neu
Given the decreased mobility of EGFR monomers, it was possible that the restriction to EGFR activation might be secondary to impaired receptor dimerization. An alternative explanation, which is not mutually exclusive, might be that the adhesion induced by E-cadherin had impaired the affinity of EGF binding to EGFR. This possibility was examined by Scatchard analysis of125I-EGF binding to EGFR in MDCK calls (Figure 3A). At high cell density under the growth-responsive conditions of calcium depletion or E-cadherin antibody treatment, we obtained a typical EGF binding curve (Wada et al, 1990; Rutten et al, 1996) with two distinct receptor affinity sites: 1.1 × 104 high-affinity sites per cell (Kd=0.24 nM) and 9.1 × 104 low-affinity sites (Kd=3.6 nM) for calcium depletion (middle panel), and 1.0 × 104 high-affinity sites (Kd=0.21 nM) and 9.4 × 104 low-affinity sites (Kd=6.3 nM) for E-cadherin antibody treatment (right panel). By contrast, at high cell density under the growth-inhibitory conditions of regular medium (left panel), only low-affinity receptor binding sites were present: 8.4 × 104sites (Kd=5.9 nM).
Figure 3.
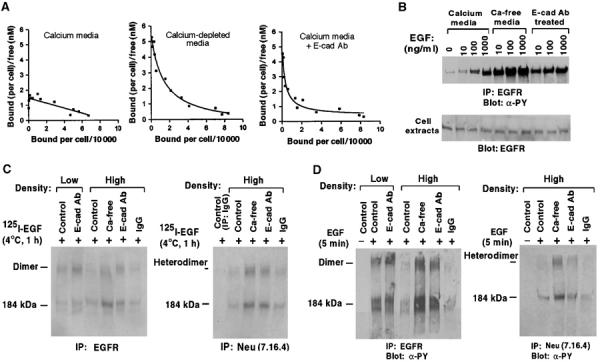
EGF binding and activation of EGFR and wild-type Neu in MDCK cells. (A) Scatchard analysis of EGF binding to MDCK-EGFR cells. Confluent MDCK-EGFR cells pretreated with media as indicated were assayed for specific EGF binding. The results were analyzed by the Scatchard method to obtain dissociation constants Kd (nM) and number of binding sites per cell (see text). The plots represent the best fit for the data. (B) High concentrations of EGF can overcome the adhesion-dependent inhibition of EGFR activation. Confluent MDCK-EGFR cells were pretreated as in Figure 1 and stimulated with the indicated concentrations of EGF for 5 min. Cell extracts were immunoprecipitated with anti-EGFR followed by antiphosphotyrosine blots (upper panel) or directly analyzed with anti-EGFR blots as loading controls (lower panel). (C) Crosslinking of125I-EGF to EGFR-EGFR homodimers and EGFR-Neu heterodimers. MDCK-EGFR cells were incubated with 0.4 nM of 125I-EGF for 1 h at 4°C followed by treatment with BS3, immunoprecipitated with EGFR antibody (left) and Neu antibody (right), and analyzed by SDS–PAGE and autoradiography. (D) EGF-dependent activation of EGFR and wild-type Neu. MDCK-EGFR cells pretreated as in Figure 1 were stimulated with EGF (100 ng/ml) for 5 min, treated with BS3, and immunoprecipitated with anti-EGFR antibody (left panels) or anti-Neu antibody (right panels) followed by immunoblotting with antiphosphotyrosine antibody.
Therefore, in confluent cells with active E-cadherin, the high-affinity binding sites for EGF were undetectable by Scatchard analysis in cells, but the total number of EGF binding sites was similar in the presence or absence of active E-cadherin. Consistent with this interpretation, a standard surface biotin binding protocol found that surface-accessible EGFR was not altered by E-cadherin-dependent adhesion (see Supplementary Figure 1A). It was therefore possible that EGF-dependent activation of EGFR in confluent cells might remain a dose-dependent phenomenon over a wide range of EGF concentrations. To examine this possibility, confluent cells were treated with graded doses of EGF, with tyrosine phosphorylation of EGFR serving as an indicator of receptor activation. A dose-dependent response to EGF was seen over a wide concentration range (10–1000 ng/ml; Figure 3B, calcium media). However, sensitivity to EGF was greatly reduced compared with confluent cells treated with the E-cadherin blocking antibody or calcium depletion, where saturated levels of activation were seen at 10 ng/ml EGF (Figure 3B). Under normal calcium conditions, it was only at 1000 ng/ml EGF that the level of EGFR activation approached that seen with 10 ng/ml EGF in cells treated with the blocking antibody.
These results suggested that EGF binding to EGFR should be less efficient at high cell density than under growth-permissive conditions. To test this hypothesis directly, cells at low and high density were incubated with a single concentration of 125I-EGF (0.4 nM), and the bound ligand was fixed to EGFR via a chemical crosslinker. Compared with binding to high-density cells under normal growth conditions, 125I-EGF was found to bind EGFR with greater efficiency in low-density cells or in high-density cells treated with the E-cadherin antibody or calcium depletion (Figure 3C, left panel). In addition to forming EGFR homodimers, cells that coexpress both EGFR and the EGFR family member Neu (Erb-B2) also form EGFR/Neu heterodimers in response to EGF (Wada et al, 1990). Since MDCK cells contain endogenous Neu, we analyzed the formation of EGFR/Neu heterodimers by using a Neu-specific antibody to immunoprecipitate extracts from high-density cells that had been exposed to 125I-EGF and treated with the chemical crosslinker (Figure 3C, right panel). Again, high-efficiency EGF binding was seen only in cells treated with E-cadherin antibody or calcium depletion.
To demonstrate directly that activation of EGFR homodimers and EGFR/Neu heterodimers was less efficient in confluent cells, MDCK cells in Figure 3D were exposed to EGF, treated with the chemical crosslinker, immunoprecipitated with an EGFR (left panel) or Neu (right panel) antibody, and Western blotted with an antiphosphotyrosine antibody. At high cell density under normal conditions, EGF induced low levels of activated EGFR/EGFR homodimers (left panel) and EGFR/Neu heterodimers (right panel). By contrast, incubation with the E-cadherin neutralizing antibody or calcium depletion enabled the cells to make a stronger response to EGF, while low-density cells made a robust response even without the E-cadherin neutralizing antibody.
E-cadherin inhibits activation of IGF-1R but not of two G protein-coupled receptors
To see if the ability of E-cadherin to inhibit EGFR activation might extend to other RTK classes expressed in MDCK, we examined the activation of IGF-1R by its soluble ligand IGF-1. In contrast to EGFR and other RTK classes, IGF-1R consists of preformed inactive oligomers (composed of two heterodimers) (Garrett et al, 1998; Dupont and LeRoith, 2001). Ligand-dependent activation of IGF-1R does not require further oligomerization, unlike EGFR. Therefore, the results with IGF-1R might have implications for the mechanism by which E-cadherin was inhibiting EGFR activation. If E-cadherin did not affect IGF-1R activation, it might suggest that interference with dimerization was the key mechanism underlying the effects of E-cadherin on EGFR. However, if E-cadherin did inhibit IGF-1R, it would suggest that inhibition of dimerization might not be the primary explanation for the inhibition of EGFR activation and that E-cadherin could regulate most RTKs.
The response of high-density MDCK cells to IGF-1 was monitored by DNA synthesis, MAPK activation, and Ras·GTP (Figure 4). For each parameter, the response to IGF-1 was qualitatively similar to that of EGF, the positive control, with strong stimulation seen only if cells had been treated with the E-cadherin neutralizing antibody or calcium depletion. MAPK activation by another soluble ligand, HGF, which activates a member of a third class of RTK, c-Met, also behaved similarly (Figure 4B, upper right panel). As expected, IGF-1R in low- or high-density cells were primarily preformed inactive dimers (Figure 5A, lower panels, −control). As previously seen with EGF/EGFR, a concentration of IGF-1 (50 ng/ml) that was able to induce robust IGF-1 binding and activation of IGF-1R dimers in low-density cells without treatment with the E-cadherin blocking antibody resulted in much less IGF-1R activation in high-density cells unless the cells were treated with the blocking antibody or calcium depletion (Figure 5A, upper panels).
Figure 4.
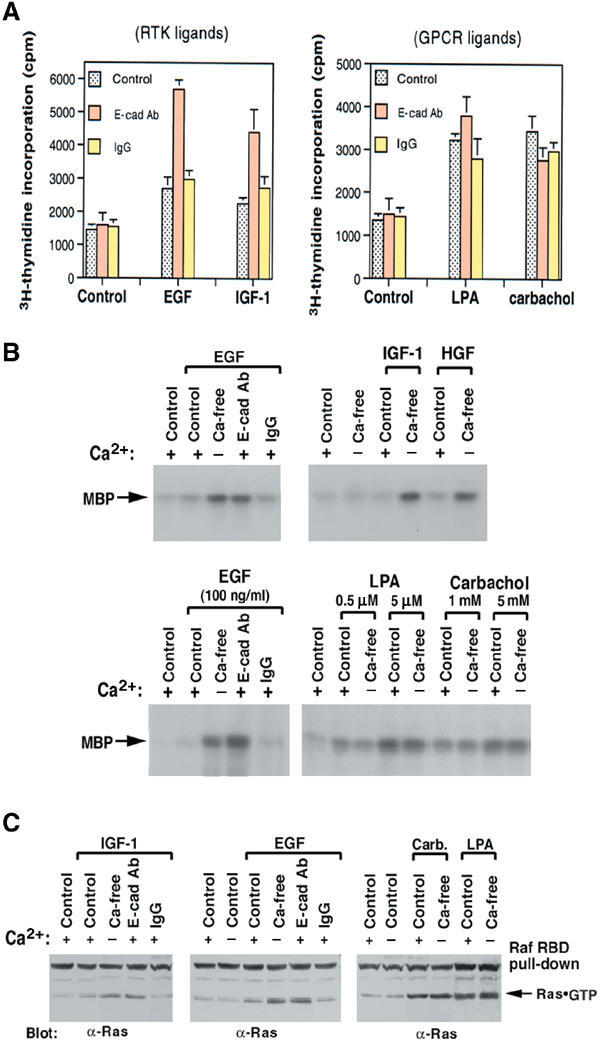
Responses of high-density cells to RTK ligands versus GPCR ligands. (A) DNA synthesis. Pretreated high-density MDCK were given EGF (10 ng/ml), IGF-1 (2 ng/ml), LPA (0.5 μM), or carbachol (0.1 mM) for 24 h, with 3H-thymidine added for the last 6 h. (B) Ligand-mediated ERK activation. Pretreated confluent MDCK cells were stimulated for 5 min with EGF (100 ng/ml), IGF-1 (20 ng/ml), HGF (20 ng/ml), or with the indicated concentrations of GPCR ligand (LPA and carbachol). Anti-ERK immunoprecipitates from cell extracts were subjected to an in vitro kinase assay using MBP as substrate. (C) Ras activation. Cells were treated as in (B) (with the high dose for LPA and carbachol), and GTP·Ras measured as in Figure 1C.
Figure 5.
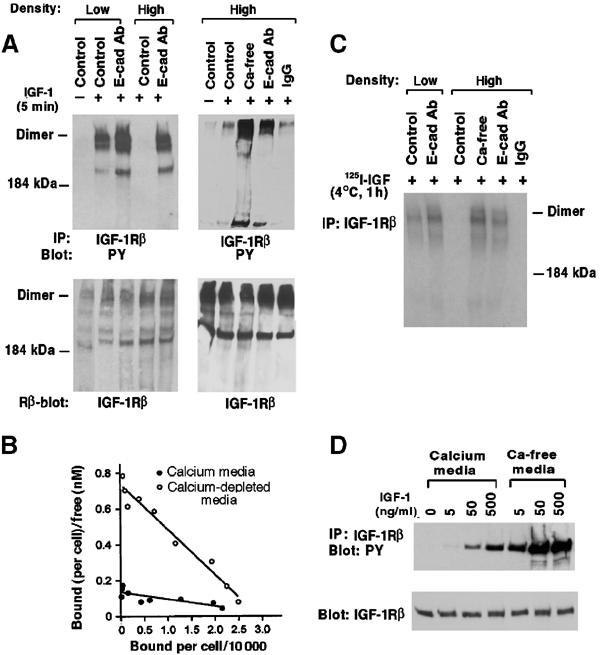
Ligand-dependent activation of IGF-1R and Scatchard analysis. (A) Activation of IGF-1R dimers. Pretreated MDCK cells were stimulated with IGF-1 (20 ng/ml) for 5 min, treated with BS3 crosslinker, and extracts were immunoprecipitated with anti-IGF-1Rβ, and immunoblotted with an antiphosphotyrosine antibody (upper panels), then stripped and reblotted with the anti-IGF-1Rβ antibody (lower panel). (B) Specific IGF-1 binding was determined for confluent MDCK cells pretreated with regular medium or calcium depletion, and the results were analyzed by the Scatchard method. See text for dissociation constants Kd (nM) and number of binding sites per cell. The plots represent the best fit for the data. (C) Crosslinking of125I-IGF-1 bound to IGF-1R. Pretreated MDCK cells were incubated with 0.2 nM125I-IGF-1 for 1 h at 4°C followed by treatment with BS3crosslinker. The anti-IGF-1Rβ immune complexes were analyzed by SDS–PAGE and autoradiography. (D) Adhesion-dependent inhibition of IGF-1R can be overcome by high concentrations of IGF-1. Confluent MDCK cells were pretreated with regular medium or calcium depletion, and stimulated with different concentrations of IGF-1. Cell extracts were immunoprecipitated with anit-IGF-1Rβ followed by antiphosphotyrosine blotting (upper panel) or anti-IGF-1Rβ blotting (lower panel).
The E-cadherin-dependent inhibition of IGF-1R activation by its ligand suggested that the binding of IGF-1 to its receptor might be reduced at high cell density. To test this possibility,125I-IGF-1 binding was analyzed by Scatchard analysis in high-density MDCK cells with regular conditions or calcium depletion, which revealed a single binding site in both cases (Figure 5B). However, the affinity with calcium depletion (3.1 × 104 sites per cell, Kd=3.9 nM) was more than seven-fold greater than for normal conditions (3.4 × 104, Kd=29 nM). The higher affinity Kd is similar to that previously reported for responsive cells (Duclos et al, 1989). Surface biotin labeling also suggested that the number of IGF-1R was independent of E-cadherin activity (see Supplementary Figure 1A).
High-density and low-density cells were also incubated with a single concentration of labeled IGF-1 and treated with the chemical crosslinker, to fix covalently the ligand bound to IGF-1R (Figure 5C). While ligand-bound receptor was readily detected in low-density cells or in high-density cells incubated with the E-cadherin neutralizing antibody or calcium depletion, ligand-bound receptor was barely detectable in cells grown under normal conditions. However, at high cell density, the cells displayed a dose-dependent increase in ligand-dependent IGF-1R activation over a wide range of IGF-1 concentrations, although their response to 500 ng/ml of IGF-1 was similar to that produced by 5 ng/ml of IGF-1 under conditions of calcium depletion (Figure 5D).
The above results indicate that E-cadherin can negatively regulate three distinct classes of RTKs: EGFR (and Neu), IGF-1R, and c-Met. To determine if this activity might extend to soluble mitogens that activate receptors other than RTKs, we exposed high-density MDCK cells to LPA or carbachol, which activate their cognate GPCRs, LPA receptors and muscarinic receptors, respectively (Rios et al, 2001). Unlike the RTKs, the two GPCRs were readily activated by their ligands at high cell density, as monitored by MAPK and RasGTP activation (Figure 4). Even with nonsaturating levels of ligand, there was no additional response following treatment with calcium depletion or the E-cadherin neutralizing antibody (Figure 4B, lower panel, and data not shown). The GPCR activation was not accompanied by EGFR activation (data not shown), in contrast to some other systems (Daub et al, 1998). Thus, E-cadherin does not regulate these GPCRs.
Mutationally activated Neu (Neu*) is not regulated by E-cadherin
If the restriction to activation of RTKs by E-cadherin is at the level of ligand–RTK interaction, an RTK that can be activated by a ligand-independent process should bypass the restriction imposed by E-cadherin. To examine this possibility, we used mutationally active Neu (Neu*), which spontaneously forms dimers and becomes constitutively activated in a ligand-independent manner (Bargmann et al, 1986). The Neu* oncoprotein was stably expressed in MDCK at low levels (to maintain the polarized cell phenotype). At high cell density, receptor dimerization, receptor activation, and downstream RasGTP signaling all occurred under normal conditions, and their levels were not further increased by the E-cadherin blocking antibody or calcium depletion (Figure 6). Thus, the ability of the Neu* oncoprotein to bypass the regulation by E-cadherin supports the inference that E-cadherin is affecting RTKs primarily by inhibiting ligand-dependent RTK activation.
Figure 6.
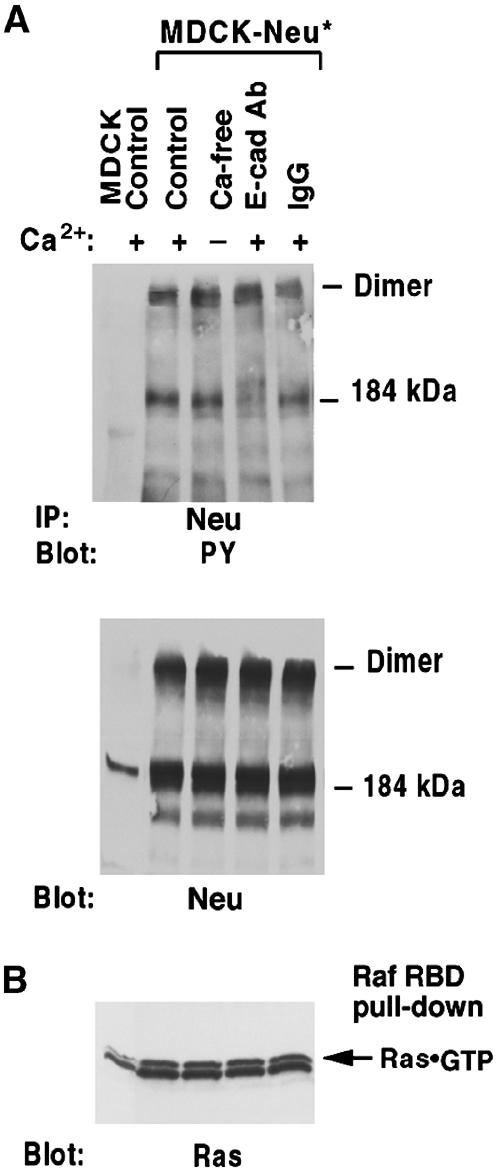
Ligand-independent activation Neu* is not regulated by E-cadherin. (A) Pretreated high-density MDCK-Neu* cells were treated with BS3crosslinker. Cell extracts were immunoprecipitated with anti-Neu followed by antiphosphotyrosine blotting, and then stripped and reblotted with anti-Neu. (B). Ras·GTP was measured as in Figure 1C.
EGFR forms a complex with E-cadherin independent of _β-_catenin and p120-catenin
E-cadherin is known to form a complex with EGFR in vivo (Hoschuetzky et al, 1994). We confirmed this finding in our MDCK cells (Figure 7A), and determined in addition that Neu, Neu*, and IGF-1R also formed a complex with E-cadherin (Figure 7A, and data not shown). The complexes appeared to be less stable under calcium depletion conditions. By contrast, no complex was detected between E-cadherin and M2 or M3 muscarinic receptors (data not shown), a result that correlated with their lack of regulation by E-cadherin.
Figure 7.
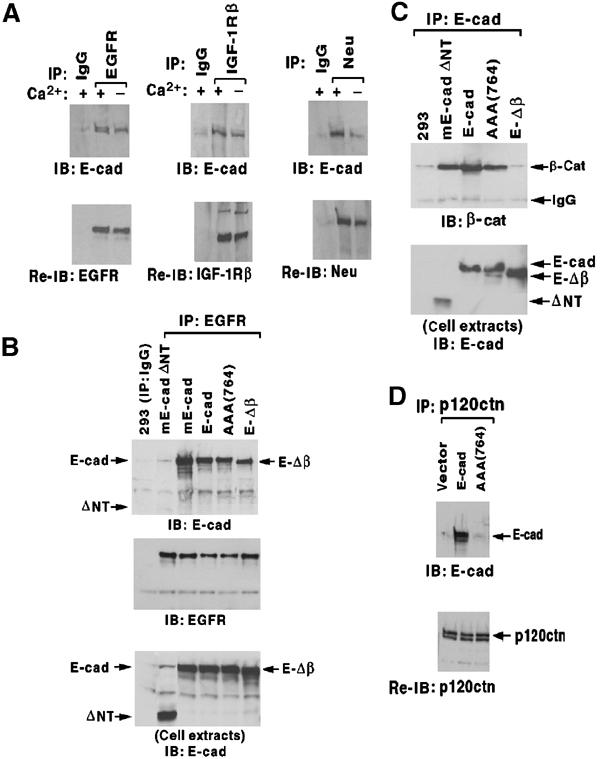
E-cadherin/RTK complex formation without β-catenin or p120-catenin. (A) E-cadherin forms complexes with RTKs in MDCK. MDCK cells expressing low levels of human EGFR (left panel) or parental MDCK cells (middle and right panels) were cultured in media with or without calcium for 16 h, and extracts were immunoprecipitated with the indicated RTK antibody (or control IgG), immunoblotted with an anti-E-cadherin antibody (upper panels), and reblotted with the respective RTK antibody (lower panels). (B–D) Clones of HEK293 cells stably expressing wild-type or mutant E-cadherin and also expressing low levels of transiently transfected EGFR (to increase the EGFR signal) are designated as in the text. (B) Complex formation with EGFR. Extracts were immunoprecipitated with anti-EGFR antibody or control IgG antibody, and immunoblotted with E-cadherin antibody (upper panel), anti-EGFR antibody (middle panel), and E-cadherin antibody (lower panel). (C) Complex formation with β-catenin. Cell extracts were immunoprecipitated with an anti-E-cadherin antibody, and immunoblotted with a β-catenin antibody (β-cat; upper panel) and the anti-E-cadherin antibody (lower panel). (D) Complex formation with p120-catenin. Extracts were immunoprecipitated with anti-p120 catenin and immunoblotted with anti-E-cadherin (upper panel), and reblotted with anti-p120 catenin (bottom panel).
To identify the E-cadherin sequences required for complex formation with EGFR, we transfected HEK293 cells with E-cadherin mutants. Although EGFR and β-catenin can bind each other (Hoschuetzky et al, 1994), we found that a premature termination mutant of E-cadherin that is deficient for β-catenin binding (E-Δβ; Gottardi et al, 2001) formed a stable complex with EGFR as efficiently as did wild-type human or mouse E-cadherin (E-cad and mE-cad, respectively), as determined by EGFR immunoprecipitation and Western blotting with an E-cadherin antibody (Figure 7B and C) or vice versa (see Supplementary Figure 2). Therefore, the interaction between EGFR and E-cadherin can occur independently of β-catenin. The site on E-cadherin that binds p120-catenin, which is the other well-described intracellular region for protein–protein interaction, was also not required, as an E-cadherin mutant deficient for p120-catenin binding (AAA(764)) (Thoreson et al, 2000) efficiently formed complexes with EGFR (Figure 7B–D andSupplementary Figure 2). However, complex formation did require the extracellular E-cadherin region, as a mutant lacking this domain (mE-cadΔNT) was deficient for binding EGFR, although it still bound β-catenin and p120-catenin (Figure 7B and C, and data not shown).
E-cadherin regulates RTKs in transformed epithelial and non-epithelial cell lines
To determine if RTK regulation resulted from forced expression of E-cadherin into an epithelial cell line with low levels of endogenous E-cadherin, wild-type and mutant E-cadherin were stably transfected into HEK293 and analyzed for their adhesive activity and their regulation of RTK activation (Figure 8). Cells expressing wild-type human (E-cad) or mouse (mE-cad) E-cadherin had strong adhesion (Figure 8A), as determined by a cell re-aggregation assay, and negatively regulated ligand-dependent activation of EGFR (Figure 8B) and IGF-1R (Figure 8C), which was abrogated by the E-cadherin blocking antibody (Figure 8B and C, right panels). By contrast, although the cells contained readily detectable levels of N-cadherin, we confirmed that EGFR did not form a complex with N-cadherin (Suyama et al, 2002) (see Supplementary Figure 2), and EGFR regulation was not affected by an N-cadherin blocking antibody (Figure 8B, right panels, and data not shown).
Figure 8.
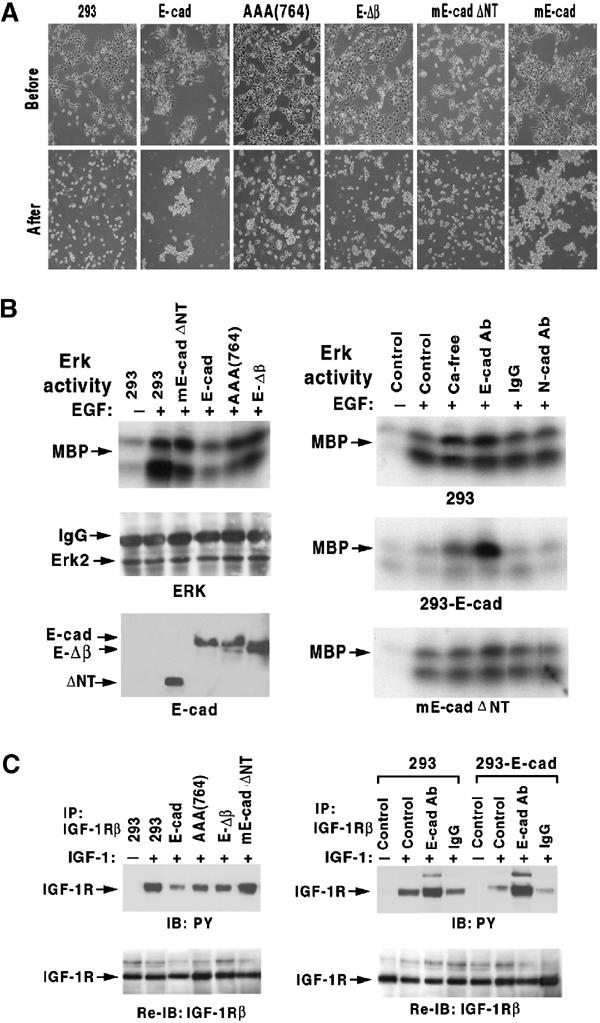
Analysis of E-cadherin mutants in HEK293 cells. Mutant lines are those designated in Figure 7. (A) Dissociation and re-aggregation assays. Photomicrographs of cell morphology before dissociation (upper panels) and after re-aggregation (lower panels). (B) ERK activation by EGF. In the left panels, confluent cells expressing the various E-cadherin mutants were stimulated for 5 min with EGF (100 ng/ml), immunoprecipitated with anti-ERK, and analyzed for ERK activity as in Figure 4B (upper panel) or for ERK protein as a loading control (middle panel); E-cadherin mutant expression was verified in the lower panel. The right panels analyze the effects of the indicated growth conditions on EGF activation of ERK in the three indicated lines. (C) IGF-1R activation by IGF-1. Confluent cells expressing the various E-cadherin mutants grown in regular medium (left panels) or in medium supplemented with E-cadherin blocking antibody (right panels) were stimulated with IGF-1 (50 ng/ml) for 5 min, and extracts were immunoblotted with antiphosphotyrosine antibody (upper panels) and reblotted with anti-IGF-1β antibody (lower panels).
The two E-cadherin mutants that formed a stable complex with EGFR (E-Δβ and AAA(764)) retained partial adhesive activity, as expected for these mutants (Thoreson et al, 2000; Gottardi et al, 2001), and also retained some negative regulation of EGFR and IGF-1R activation (Figure 8). By contrast, the E-cadherin mutant that did not form a complex with EGFR (mE-cadΔNT) lacked both adhesive activity and RTK regulation. Thus, there was an excellent quantitative relationship between adhesive activity and RTK regulation, which also correlated qualitatively with EGFR/E-cadherin complex formation.
In tumor cell lines where E-cadherin is limiting, its forced expression is known to be growth inhibitory (Hajra and Fearon, 2002). We therefore asked whether transient transfection of wild-type E-cadherin in a human melanoma line (mel. 586) or a breast cancer line (MDA231), which respectively have low and undetectable levels of endogenous E-cadherin, would affect RTK activation (Figure 9). We also determined whether RTK regulation might be affected by the high endogenous E-cadherin levels present in another melanoma line (Figure 9, mel. 553B). The mel. 586 transfectants displayed an attenuated response to EGF and HGF, but their response to carbachol was not affected. The mel. 553B line displayed a poor HGF response that was substantially augmented by the E-cadherin blocking antibody. EGF and IGF-1 signaling was impaired in the MDA231 transfectants.
Figure 9.
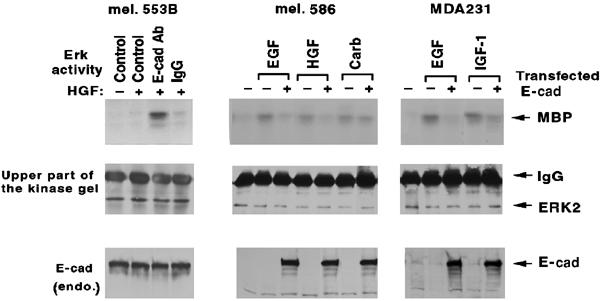
E-cadherin in human tumor cell lines regulates RTK activation. The lines were: a human melanoma cell line (mel. 553B) expressing endogenous E-cadherin (left panels), a low endogenous E-cadherin expressing human melanoma line (mel. 586) that had been transiently transfected with E-cadherin (middle panels), and a human breast cancer line (MDA231) devoid of endogenous E-cadherin that had been transiently transfected with E-cadherin (right panels). The lines were stimulated for 5 min with HGF (20 ng/ml), EGF (100 ng/ml), IGF-1 (50/ng/ml), or carbachol (5 mM), as indicated, with the mel. 553B line having first been treated with the E-cadherin blocking antibody or control IgG. Cells were analyzed for ERK activity as in Figure 4B (upper panels) and for ERK protein loading (middle panels). Extracts were also immunoblotted with an anti-E-cadherin antibody (bottom panels).
Discussion
Numerous studies indicate that diverse classes of RTKs, including EGFR/Neu, IGF-1R, and c-Met, can downregulate and inhibit E-cadherin-dependent adhesion when they induce the form of morphologic transformation known as epithelial-to-mesenchyme transition (Fujita et al, 2002; Thiery, 2002). The current results, which show that E-cadherin can inhibit the activation of EGFR/Neu, IGF-1R, and c-Met, imply that the regulation is bidirectional. By contrast, E-cadherin did not inhibit ligand-dependent activation of two classes of GPCRs, LPA receptors and muscarinic receptors. Consistent with E-cadherin having pleotropic effects and regulating RTKs, these receptors have been implicated in many normal cellular processes (Hubbard and Till, 2000).
The effects of E-cadherin on RTKs were adhesion dependent. In MDCK epithelial cells, the effects were seen at high cell density with strong adhesion. Treatments that disrupted the adhesive function of E-cadherin, such as calcium depletion or a neutralizing E-cadherin antibody, abrogated RTK regulation. The effects on RTKs were also seen in a melanoma line whose endogenous E-cadherin levels were similar to MDCK, and were reproduced by stable transfection of E-cadherin into epithelial and melanoma lines with low or undetectable levels of endogenous E-cadherin. In HEK293 cells, E-cadherin mutants that retained partial adhesion function (secondary to impairment of the β-catenin binding site or the p120-catenin binding site) also retained activity against RTKs, while an E-cadherin mutant devoid of adhesive activity (secondary to deletion of the extracellular region) did not affect RTKs.
The regulation appears to result from an E-cadherin-dependent inhibition of RTK activation by soluble ligands. In MDCK at high cell density, there was loss of high-affinity binding sites for EGF and IGF-1. However, most EGFR and IGF-1R remained available for ligand binding, since the number of ligand binding sites was similar with normal calcium or following treatment with calcium depletion or the blocking antibody, and the levels of receptor activation induced by high concentrations of ligand in normal calcium approached those seen with calcium depletion or the blocking antibody. The binding of NHS-biotin to surface-exposed EGFR and IGF-1R was also not impaired by E-cadherin. A previous report (Takahashi and Suzuki, 1996) also showed reduced EGF stimulation of DNA synthesis in confluent cultured breast cells, but its basis was not clear. These authors did not examine other ligands, did not identify cell density-dependent differences in EGF binding, did not detect efficient EGF-dependent EGFR dimerization in confluent cells pretreated with E-cadherin blocking antibody, and used cells whose level of E-cadherin at confluence was about five times higher than in sparse cells. By contrast, we found in MDCK that EGF or IGF-1 binding efficiency at high cell density was reduced compared with low cell density, affinity of ligand binding was correlated under various conditions with the efficiency of RTK activation, and E-cadherin level was independent of cell density. Furthermore, in contrast to ligand-dependent activation of wild-type RTKs, a mutant RTK that is constitutively activated without ligand, Neu*, was not regulated by E-cadherin. The ability of Neu* to override the influence of E-cadherin also supports the conclusion that ligand-dependent receptor activation is the key step regulated by E-cadherin.
Colocalization of RTKs and E-cadherin may be required for the observed negative RTK regulation. For example, ligand can induce inappropriate RTK activation and pathological cell growth in epithelial cells from polycystic kidney disease, where RTKs may be mislocated apically instead of colocalizing basolaterally with E-cadherin (Du and Wilson, 1995). E-cadherin is known to form a ligand-independent complex with RTKs (Crepaldi et al, 1994; Hoschuetzky et al, 1994; Pece and Gutkind, 2000). Although β-catenin can bind EGFR or Neu and is a substrate for these RTKs, we found here that ligand-independent complex formation between E-cadherin and EGFR did not depend on E-cadherin binding to β-catenin, a conclusion also made independently by others (Fedor-Chaiken et al, 2003), or on binding to p120-catenin. The correlation with RTK regulation suggests that complex formation between E-cadherin and EGFR may contribute to the effects of E-cadherin on EGFR regulation. The results also identify a new interaction domain on E-cadherin, as previous interactions between E-cadherin and heterologous proteins have been limited to the p120-catenin and β-catenin binding sites.
RTK regulation by two other classical cadherins, VE-cadherin and N-cadherin, has also been reported. However, these cadherins and E-cadherin have distinct effects on RTKs, via distinct mechanisms. VE-cadherin attenuates some VEGFR2 signaling while retaining other downstream effects of VEGFR2 (Carmeliet et al, 1999; Rahimi and Kazlauskas, 1999; Grazia Lampugnani et al, 2003). Attenuation is attributed to the ligand-dependent formation of a complex between VEGFR2 and VE-cadherin that requires the β-catenin binding site and is associated with β-catenin binding. Although there is no inhibition of ligand-induced dimerization of VEGFR2, the presence of VE-cadherin results in reduced phosphorylation of some VEGFR2 tyrosines, via a phosphatase-dependent mechanism. By contrast, N-cadherin positively regulates the effects of FGFR, by inhibiting the downregulation of activated receptors (Suyama et al, 2002). The effects of N-cadherin and VE-cadherin on FGFR and VEGFR2, respectively, do not extend to other classes of RTKs.
We also found that the mobility of EGFR-CFP was reduced in cells at high density, as is also true of E-cadherin (Adams et al, 1998), and that abrogation of E-cadherin-dependent adhesion increased EGFR-CFP mobility, although not to the degree seen in sparse cells. This observation confirms that E-cadherin can regulate EGFR mobility in an adhesion-dependent manner. It also identifies a second level of regulation that may be relevant to RTKs whose activation is mediated by ligand-dependent oligomerization. In this situation, the reduced receptor mobility in the membrane would impair the efficiency with which inactive monomeric receptors would oligomerize. Despite its plausibility, a direct relevance of RTK mobility was not established, since activation of IGF-1R, which exists as preformed inactive oligomers, was impaired to a similar degree as EGFR. Instead, we propose that interaction of the RTK with E-cadherin in an antiparallel complex may restrict the switching of the RTK to an active conformation.
The current studies have been limited to RTK activation by soluble ligands. Although most RTK activation involves such ligands, some RTKs, such as the Eph receptors, are activated by cell-associated ligands, via a juxtacrine mechanism, and their regulation by E-cadherin may be distinct. Zantek et al (1999) have reported that in confluent cells, E-cadherin may increase the activity of Eph2 (which inhibits growth) by its ligand, rather than negatively regulating it. A possible explanation may be that at confluence, the juxtacrine activity is increased, secondary to the increased density of ligand and receptors in the basolateral area. The conditions we have studied are also distinct from those that report transient E-cadherin-dependent activation of EGFR, without ligand, which occurs following a switch from low calcium to high calcium (Pece and Gutkind, 2000).
Our findings have implications for physiologic and pathological processes associated with E-cadherin. For cells that express E-cadherin, negative regulation of RTKs may contribute to the density-dependent inhibition of cell growth, which is a multifactorial process (Nelson and Daniel, 2002). In cancer, the pathogenetic explanation given for downregulation of E-cadherin is that usually it leads to the activation of β-catenin signaling and/or to the structural loss of adhesion. Our observations suggest that downregulation of E-cadherin may also contribute to the frequent ligand-dependent activation of RTK in tumors (Blume-Jensen and Hunter, 2001).
Materials and methods
Cell culture and plasmids
MDCK cells and HEK293 cells (ATCC) were maintained in DMEM supplemented with 10% fetal bovine serum (FBS). To visualize EGFR biochemically, experiments used MDCK-EGFR, which expresses low levels of human EGFR because preliminary experiments indicated that antibodies to EGFR did not react well to canine EGFR. The HEK293 cell line stably expressing E-cadherin (293-Ecad) has been described (Li et al, 2003). Cells were cultured at 37°C in a humidified 5% CO2 atmosphere. MDCK cells were plated overnight in DMEM with 10% FBS. For low-density conditions, one-quarter as many cells were plated as for high-density conditions. Prior to each experiment, cells were then treated under the following conditions in low serum (0.5% FBS) for 16 h (24 h for DNA synthesis): (1) control, using regular DMEM (Hyclone, with calcium); (2) calcium depletion, using EMEM (BioWhittaker, without calcium); (3) E-cad Ab, using DMEM containing E-cadherin blocking antibody (DECMA-1, Sigma) 5 μl/ml (equivalent to 5 μg/ml rat IgG,); and (4) control IgG, using DMEM containing mouse or rat isotype matched IgG 5 μg/ml (Sigma). An N-cadherin blocking antibody (clone GC-4, Sigma) was also used. Dissociation and re-aggregation assays were performed as described previously with minor modifications (Li et al, 2003).
The pcDNA3-hE-cad (wild-type E-cadherin) and pcDNA3-hE-cad Δβ-catenin (mutant E-Δβ) expression constructs were provided by B Gumbiner (Gottardi et al, 2001), the EGFR-CFP expression construct with linked Neo by L Samelson (Yamazaki et al, 2002), the activated Neu*expression construct by M Greene (Weiner et al, 1989), plasmid PMS Ecad (AAA(764)) by Albert Reynolds (Thoreson et al, 2000) and was subcloned into the pcDNA3 expression vector, and plasmid IRES-mE-cad and IRES-mE-cadΔNT were by Dan Longo (Sasaki et al, 2000). Transient and stable transfections were carried out with Lipofectamine (Invitrogen) according to the manufacturer's instructions. Stable MDCK clones expressing activated Neu*were generated using Lipofectamine and selection in media containing G418 (Invitrogen).
Scatchard analysis
MDCK cells (5 × 104/well) were plated in 24-well dishes precoated with poly-D-lysine (10 μg/ml) and allowed to grow in confluence. After pretreating cells overnight with normal media with or without E-cadherin neutralization antibody, or with or without calcium-depleted media containing 0.2% FBS, cells were incubated at 4oC for 75 min with the same media containing 20 mM HEPES (pH 7.4) and 125I-IGF-1 (Perkin-Elmer) at final concentrations of 0.08–16 nM or 125I-EGF (Perkin-Elmer) at final concentrations of 0.02–25 nM. Nonspecific binding was determined with 100-fold excess cold IGF-1 or EGF, respectively. Cells were quickly washed three times with cold PBS containing 20 mM HEPES (pH 7.4) and 0.2% BSA. The cell-associated radioactivity was determined after solubilization of cells with 0.5 N NaOH. Data were analyzed on a Scatchard plot, and apparent Kd was determined by linear or nonlinear regression analysis using the Prism program.
Intracellular signaling assays
Pretreated cells were stimulated with ligand for 5 min and analyzed.
Raf-RBD pull-down assays: Using equal amounts of protein lysate (by BCA assay), the Raf-RBD pull-down assay was performed according to the manufacturer's instructions using the Ras activation Kit (Upstate Biotechnology). The pull-down pellets were washed three times with magnesium lyses buffer (Upstate Biotechnology), resuspended in Laemmli sample buffer, and separated in 15% SDS–PAGE. After transferring onto nitrocellulose membranes, the activated Ras were detected by immunoblotting using anti-pan-Ras antibody (Upstate Biotechnology).
Erk kinase assays: Cells were lysed in RIPA buffer (Qian et al, 2000). The supernatant from the cleared cell extracts was collected, and equal amounts of protein (by BCA) from extracts were immunoprecipitated with anti-ERK antibody (Santa Cruz) to precipitate ERK2. The immunopellets were processed and analyzed for in vitro kinase activity using myelin basic protein (MBP, Upstate Biotechnology) as substrate (Qian et al, 2000). Equal loading was verified by anti-ERK blotting.
For details on DNA synthesis, crosslinking of EGF and IGF-1 to receptors, FRAP analysis, co-immunoprecipitation of E-cadherin and RTKs, and surface biotinylation, see Supplementary data.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Materials
Acknowledgments
We thank Shaowei Li for the 293-Ecad cell line, Larry Samelson, Barry Gumbiner Albert Reynolds, Dan Longo, and Mark Greene for DNA constructs, Bill Vass for technical assistance, Mary Fedor-Chaiken and Robert Brackenbury for communicating prepublication information, David Salomon, Peter Nissley, Petra Lenz, and Cathy Carlin for helpful discussions, and Martha Randazzo for assistance in preparing the manuscript. We are particularly grateful to Paul Randazzo for numerous suggestions.
References
- Adams CL, Chen YT, Smith SJ, Nelson WJ (1998) Mechanisms of epithelial cell–cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin-green fluorescent protein. J Cell Biol 142: 1105–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI, Hung MC, Weinberg RA (1986) The neu oncogene encodes an epidermal growth factor receptor-related protein. Nature 319: 226–230 [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411: 355–365 [DOI] [PubMed] [Google Scholar]
- Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L (2002) C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science 296: 1308–1313 [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oostuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E (1999) Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98: 147–157 [DOI] [PubMed] [Google Scholar]
- Crepaldi T, Pollack AL, Prat M, Zborek A, Mostov K, Comoglio PM (1994) Targeting of the SF/HGF receptor to the basolateral domain of polarized epithelial cells. J Cell Biol 125: 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub H, Wallasch C, Laukenau A, Herrlich A, Ullrich A (1998) Signal characteristics of G protein-transactivated EGF receptor. EMBO J 1997: 7032–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Wilson PD (1995) Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am J Physiol 269: C487–495 [DOI] [PubMed] [Google Scholar]
- Duclos M, Houdebine LM, Djiane J (1989) Comparison of insulin-like growth factor 1 and insulin effects on prolactin-induced lactogenesis in the rabbit mammary gland in vitro. Mol Cell Endocrinol 65: 129–134 [DOI] [PubMed] [Google Scholar]
- Dupont J, LeRoith D (2001) Insulin and insulin-like growth factor I receptors: similarities and differences in signal transduction. Horm Res 55 (Suppl 2): 22–26 [DOI] [PubMed] [Google Scholar]
- Fedor-Chaiken M, Hein PW, Stewart JC, Brackenbury R, Kinch MS (2003) E-cadherin binding modulates EGF receptor activation. Cell Commun Adhes 10: 105–118 [PubMed] [Google Scholar]
- Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W (2002) Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 4: 222–231 [DOI] [PubMed] [Google Scholar]
- Garrett TP, McKern NM, Lou M, Frenkel MJ, Bentley JD, Lovrecz GO, Elleman TC, Cosgrove LJ, Ward CW (1998) Crystal structure of the first three domains of the type-1 insulin-like growth factor receptor. Nature 394: 395–399 [DOI] [PubMed] [Google Scholar]
- Gottardi CJ, Wong E, Gumbiner BM (2001) E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol 153: 1049–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grazia Lampugnani M, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, Orsenigo F, Cattelino A, Kemler R, Daniel TO, Dejana E (2003) Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J Cell Biol 161: 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner BM (2000) Regulation of cadherin adhesive activity. J Cell Biol 148: 399–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajra KM, Fearon ER (2002) Cadherin and catenin alterations in human cancer. Genes Chromosomes Cancer 34: 255–268 [DOI] [PubMed] [Google Scholar]
- Hoschuetzky H, Aberle H, Kemler R (1994) Beta-catenin mediates the interaction of the cadherin–catenin complex with epidermal growth factor receptor. J Cell Biol 127: 1375–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SR, Till JH (2000) Protein tyrosine kinase structure and function. Annu Rev Biochem 69: 373–398 [DOI] [PubMed] [Google Scholar]
- Ireton RC, Davis MA, van Hengel J, Mariner DJ, Barnes K, Thoreson MA, Anastasiadis PZ, Matrisian L, Bundy LM, Sealy L, Gilbert B, van Roy F, Reynolds AB (2002) A novel role for p120 catenin in E-cadherin function. J Cell Biol 159: 465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamora C, Fuchs E (2002) Intercellular adhesion, signalling and the cytoskeleton. Nat Cell Biol 4: E101–E108 [DOI] [PubMed] [Google Scholar]
- Li S, Braverman R, Li H, Vass WC, Lowy DR, DeClue JE (2003) Regulation of cell morphology and adhesion by the tuberous sclerosis complex (TSC1/2) gene products in human kidney epithelial cells through increased E-cadherin/beta-catenin activity. Mol Carcinogen 37: 98–109 [DOI] [PubMed] [Google Scholar]
- McRoberts JA, Taub M, Saier MH (1981) The Madin Darby canine kidney (MDCK) cell line. In Sato G (ed) Functionally Differentiated Cell Lines, New York: Alan R Liss, Inc. p. 117–139 [Google Scholar]
- Nelson PJ, Daniel TO (2002) Emerging targets: molecular mechanisms of cell contact-mediated growth control. Kidney Int Suppl 61 (Suppl 1): 99–105 [DOI] [PubMed] [Google Scholar]
- Ozawa M, Hoschutzky H, Herrenknecht K, Kemler R (1990) A possible new adhesive site in the cell-adhesion molecule uvomorulin. Mech Dev 33: 49–56 [DOI] [PubMed] [Google Scholar]
- Pece S, Gutkind JS (2000) Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell–cell contact formation. J Biol Chem 275: 41227–41233 [DOI] [PubMed] [Google Scholar]
- Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G (1998) A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392: 190–193 [DOI] [PubMed] [Google Scholar]
- Qian X, Esteban L, Vass WC, Upadhyaya C, Papageorge AG, Yienger K, Ward JM, Lowy DR, Santos E (2000) The Sos1 and Sos2 Ras-specific exchange factors: differences in placental expression and signaling properties. EMBO J 19: 642–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi N, Kazlauskas A (1999) A role for cadherin-5 in regulation of vascular endothelial growth factor receptor 2 activity in endothelial cells. Mol Biol Cell 10: 3401–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios CD, Jordan BA, Gomes I, Devi LA (2001) G-protein-coupled receptor dimerization: modulation of receptor function. Pharmacol Ther 92: 71–87 [DOI] [PubMed] [Google Scholar]
- Rutten MJ, Dempsey PJ, Luttropp CA, Hawkey MA, Sheppard BC, Crass RA, Deveney CW, Coffey RJ Jr (1996) Identification of an EGF/TGF-alpha receptor in primary cultures of guinea pig gastric mucous epithelial cells. Am J Physiol 270: G604–G612 [DOI] [PubMed] [Google Scholar]
- Sasaki CY, Lin H, Morin PJ, Longo DL (2000) Truncation of the extracellular region abrogates cell contact but retains the growth-suppressive activity of E-cadherin. Cancer Res 60: 7057–7065 [PubMed] [Google Scholar]
- Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103: 211–225 [DOI] [PubMed] [Google Scholar]
- Sharpe C, Lawrence N, Martinez Arias A (2001) Wnt signalling: a theme with nuclear variations. BioEssays 23: 311–318 [DOI] [PubMed] [Google Scholar]
- Stockinger A, Eger A, Wolf J, Beug H, Foisner R (2001) E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J Cell Biol 154: 1185–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama K, Shapiro I, Guttman M, Hazan RB (2002) A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell 2: 301–314 [DOI] [PubMed] [Google Scholar]
- Takahashi K, Suzuki K (1996) Density-dependent inhibition of growth involves prevention of EGF receptor activation by E-cadherin-mediated cell–cell adhesion. Exp Cell Res 226: 214–222 [DOI] [PubMed] [Google Scholar]
- Tang A, Amagai M, Granger LG, Stanley JR, Udey MC (1993) Adhesion of epidermal Langerhans cells to keratinocytes mediated by E-cadherin. Nature 361: 82–85 [DOI] [PubMed] [Google Scholar]
- Thiery JP (2002) Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2: 442–454 [DOI] [PubMed] [Google Scholar]
- Thoreson MA, Anastasiadis PZ, Daniel JM, Ireton RC, Wheelock MJ, Johnson KR, Hummingbird DK, Reynolds AB (2000) Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J Cell Biol 148: 189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Oda H, Kraut R, Hayashi S, Kotaoka Y, Takeichi M (1996) Zygotic Drosophila E-cadherin expression is required for processes of dynamic epithelial cell rearrangement in the_Drosophila_ embryo. Genes Dev 10: 659–671 [DOI] [PubMed] [Google Scholar]
- Wada T, Qian XL, Greene MI (1990) Intermolecular association of the P185Neu protein and EGF receptor modulates EGF receptor function. Cell 61: 1339–1347 [DOI] [PubMed] [Google Scholar]
- Weiner DB, Liu J, Cohen JA, Williams V, Greene MI (1989) A point mutation in the neu oncogene mimics ligand induction of receptor aggregation. Nature 339: 230–231 [DOI] [PubMed] [Google Scholar]
- Wheelock MJ, Johnson KR (2003) Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol 19: 207–235 [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Zaal K, Hailey D, Presley J, Lippincott-Schwartz J, Samelson LE (2002) Role of Grb2 in EGF-stimulated EGFR internalization. J Cell Sci 115: 1791–1802 [DOI] [PubMed] [Google Scholar]
- Zantek ND, Azimi M, Fedor-Chaiken M, Wang B, Brackenbury R, Kinch MS (1999) E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ 10: 629–638 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Materials