Cohesin gene mutations in tumorigenesis: from discovery to clinical significance (original) (raw)
Abstract
Cohesin is a multi-protein complex composed of four core subunits (SMC1A, SMC3, RAD21, and either STAG1 or STAG2) that is responsible for the cohesion of sister chromatids following DNA replication until its cleavage during mitosis thereby enabling faithful segregation of sister chromatids into two daughter cells. Recent cancer genomics analyses have discovered a high frequency of somatic mutations in the genes encoding the core cohesin subunits as well as cohesin regulatory factors (e.g. NIPBL, PDS5B, ESPL1) in a select subset of human tumors including glioblastoma, Ewing sarcoma, urothelial carcinoma, acute myeloid leukemia, and acute megakaryoblastic leukemia. Herein we review these studies including discussion of the functional significance of cohesin inactivation in tumorigenesis and potential therapeutic mechanisms to selectively target cancers harboring cohesin mutations. [BMB Reports 2014; 47(6): 299-310]
Keywords: Acute myeloid leukemia, Aneuploidy, Bladder cancer, Chromosomal instability, Cohesin, Ewing sarcoma, Glioblastoma, Mitosis, Sister chromatid cohesion, STAG2, Urothelial carcinoma
INTRODUCTION
Cohesin subunits were originally identified in yeast as mutants that displayed premature separation of sister chromatids and soon thereafter were found to form a complex required for sister chromatid cohesion in Xenopus egg extracts and mammalian cells (1,2). In vertebrates, the cohesin complex is composed of four subunits (SMC1A, SMC3, RAD21, and either STAG1 or STAG2) arranged in a ring-shaped structure that encircles chromatin (Fig. 1). Cohesin loads onto chromatin in G1 phase of the cell cycle immediately following cytokinesis, concatenates sister chromatids during DNA replication in S phase, remains chromatin bound specifically at centromeres in prophase of mitosis while the majority of cohesin along chromatid arms is released, and then the remainder of chromatin-bound cohesin is cleaved at the metaphase to anaphase transition to enable segregation of the sister chromatids into two daughter cells (3,4). Recent studies have found that cohesin containing the more abundant STAG2 subunit is essential for chromatid cohesion at centromeres and along chromosome arms, while cohesin containing the less abundant STAG1 subunit is essential for chromatid cohesion specifically at telomeres (5,6). The cohesin complex composed of SMC1A, SMC3, RAD21, and STAG1 or STAG2 is robustly expressed in all somatic cells, whereas a separate meiosis-specific cohesin complex is present in germ cells composed of SMC1B, SMC3, REC8, and STAG3 (7).
Fig. 1. Depiction of the cohesin complex present in somatic human cells.
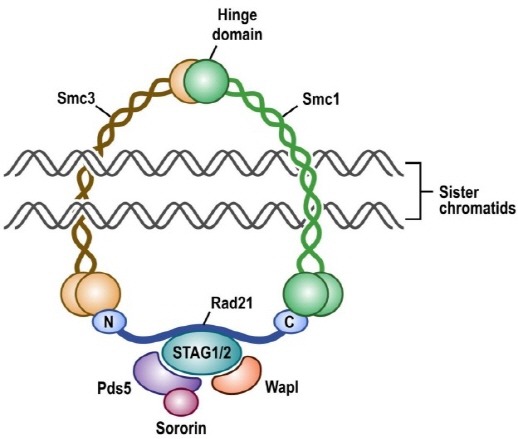
Since the initial characterization of the cohesin complex in the late 1990’s, several cohesin regulatory factors have been identified that are responsible for the loading, stability, and cleavage of cohesin during the cell cycle. Loading of cohesin onto chromatin in G1 phase is known to be dependent on the NIPBL-MAU2 heterodimer (8,9), while WAPAL, PDS5A, and PDS5B bind to chromatin-bound cohesin and promote its unloading (10,11). The establishment of sister chromatid cohesin during DNA replication in S phase requires acetylation of SMC3 by the cohesin acetyltransferases ESCO1 and ESCO2 (12,13), as well as binding of CDCA5 (Sororin) (14). At the onset of mitosis in early prophase, phosphorylation of STAG2 by PLK1 drives dissociation of the majority of cohesin along chromosome arms (15), while centromeric cohesin is protected by binding of SGOL1 (Shugoshin) (16). Activation of the anaphase promoting complex APC/C at the metaphase to anaphase transition leads to degradation of PTTG1 (Securin) and the activation of ESPL1 (Separase) (17). ESPL1 cleaves the RAD21 subunit of the remaining chromatin-bound cohesin thereby enabling segregation of the sister chromatids (18).
THE MANY CELLULAR FUNCTIONS OF COHESIN IN DEVELOPMENT AND DISEASE
Cohesin subunits are robustly expressed in all somatic cells (including terminally differentiated quiescent cells) and are now appreciated to be essential in a wide range of cellular processes including maintenance of chromatin architecture, transcriptional regulation, DNA replication, and DNA repair, in addition to their canonical role in sister chromatid cohesion and segregation (19). While studies have shown that cohesin forms a ring-like structure that encircles chromatin, no direct DNA binding motifs within the core cohesin subunits have been identified. However, emerging studies have shown that cohesin is enriched at specific chromatin loci including active transcriptional sites, pericentric heterochromatin, double-strand breaks, and stalled replication forks, suggesting cohesin localization is directed by specific DNA-binding regulatory proteins. Chromatin immunoprecipitation experiments have demonstrated that cohesin associates with the CCCTC-binding factor (CTCF), a zinc finger protein required for transcriptional insulation of gene promoters from enhancer regions, leading to enrichment of cohesin at specific genomic loci and regulation of transcription independent of its role in sister chromatid cohesion (20, 21). This cohesin-CTCF association is due to direct binding between STAG2 and CTCF, wherein CTCF is dispensable for cohesin loading onto chromatin but is required for enrichment at specific enhancer regulatory loci throughout the genome (22). Cohesin has also been identified to interact with the Mediator complex, a transcriptional co-activator which localizes to gene promoter and enhancer domains, and this interaction was found to be essential for the gene expression program in mouse embryonic stem cells (23). Most recently, cohesin was found to be enriched at pericentric heterochromatin through interaction with the histone methyltransferase SUV4-20h2 and to be essential for proper chromocenter organization (24). Cohesin has an expanding list of important functions in the maintenance of chromatin organization within the cell nucleus, which are now known to be disrupted in an array of developmental and neoplastic diseases.
In 2004 mutations in the cohesin regulatory factor NIPBL were discovered to cause Cornelia de Lange (CdL) syndrome (OMIM 122470 and 300590), a rare autosomal dominant disorder characterized by facial dysmorphism, growth delay, mental retardation, and limb abnormalities (25,26). Subsequently, mutations in cohesin core subunits SMC1A, SMC3, and RAD21 have been found in the subset of CdL patients without NIPBL mutations (27-29). Analysis of cells from CdL patients found precocious separation of sister chromatids before anaphase in mitosis, which was hypothesized to be a causative mechanism in this syndrome and a useful diagnostic assay (30). However, subsequent studies have not observed similar defects in sister chromatid cohesion (31), calling into question the functional consequence of cohesin mutations in CdL patients. Genome-wide transcriptional profiling in cells derived from CdL patients with NIPBL or SMC1A mutations versus normal subjects found a conserved pattern of transcriptional dysregulation, identifying a group of 339 genes with recurrently altered expression amongst the CdL patients and a significant correlation between the degree of transcriptional alteration and phenotypic disease severity (32). A heterozygous NIPBL knockout mouse (Nipbl+/-) has been constructed which demonstrates a developmental phenotype similar to CdL patients including small size, craniofacial anomalies, microbrachycephaly, heart defects, hearing abnormalities, delayed bone maturation, reduced body fat, behavioral disturbances, and high mortality during the first weeks of life (33). Mouse embryonic fibroblasts derived from these Nipbl+/- mice displayed normal sister chromatid cohesion but had recurrent transcriptional dysregulation in 81 genes, suggesting that NIPBL influences chromosomal regulatory interactions. More recently, a STAG1 knockout mouse was developed. Homozygous knockout of STAG1 caused embryonic lethality with significant growth delay, multi-organ hypoplasia, impaired lipid metabolism, and severe abnormalities in skeletal development, features characteristic of CdL syndrome (34). STAG1 heterozygous mice display reduced cohesin localization at CTCF-bound promoter and insulator regions and altered gene expression profiles similar to cells from CdL patients (34). Additionally, these heterozygous mice had impaired telomere replication leading to the development of aneuploidy and had increased incidence of spontaneous tumors including hepatocellular carcinomas, pancreatic intraductal papillary mucinous neoplasms, vascular neoplasms, and lymphomas (6). These studies demonstrate that cohesin inactivation both alters gene expression leading to developmental defects and sister chromatid cohesion leading to aneuploidy and increased tumorigenesis. Whether CdL patients have increased tumor incidence is not well described, but one case review of cause of death in 295 CdL patients was due to cancer in only 5 patients (three esophageal, one gastric, and one unspecified), all of whom were among the subgroup of 97 patients that survived into adulthood (35).
Most recently, an inactivating mutation of the meiosis-specific cohesin subunit STAG3 was found in a large consanguineous family with premature ovarian failure and was present in each of six affected family members (36). Additionally, a STAG3 knockout mouse model was generated, and female mice were sterile with oocytes arrested in early prophase I leading to oocyte depletion at one week of age (36). These findings demonstrate that the meiosis-specific cohesin complex is essential for mammalian fertility.
DISCOVERY OF COHESIN GENE MUTATIONS IN HUMAN CANCERS
Chromosome segregation defects and aneuploidy, a deviation from the expected diploid number of human chromosomes, are hallmarks of cancer that were first recognized by Theodor Boveri more than one hundred years ago, yet the genetic underpinnings of aneuploidy remain not well defined (37,38). Given its function in controlling faithful sister chromatid segregation, dysregulation of the cohesin complex was hypothesized to contribute to the development of aneuploidy during tumorigenesis since its isolation and characterization in the late 1990’s. The first report of cohesin gene alterations in human tumors was in 2008 wherein Barber et al. found somatic mutations of SMC1A, SMC3, and NIPBL in 9 out of 132 colorectal adenocarcinomas (see Table 1 for summary of cohesin gene mutations identified in human tumors), wherein they suggested that chromatid cohesion defects may underlie the chromosomal instability present in the vast majority of colorectal cancers (39). Then in 2010 an array comparative genomic hybridization study of 167 myeloid disease samples identified a chronic myelomonocytic leukemia with a deletion of RAD21 (additionally harboring an NPM1 exon 12 mutation) and a de novo acute myeloid leukemia with a deletion of STAG2 (additionally harboring an IDH1-R132C mutation) (40). These first studies provided tantalizing evidence that the cohesin complex is a frequent target of genetic alterations during tumorigenesis.
Table 1. Summary of nonsynonymous somatic mutations identified in cohesin genes in human tumor samples.
| Tumor type | Tumors with mutation in a core cohesin gene† | Tumors with mutation in core cohesin gene | Tumors with mutation in cohesin regulatory genes‡ | Technique used¶ | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| STAG2 | STAG1 | SMC3 | SMC1A | RAD21 | |||||
| Bone marrow | |||||||||
| Acute megakaryoblastic leukemia, Down syndrome | 23/49 (47%) | 9/49 (18%) | 0/49 (0%) | 1/49 (2%) | 2/49 (4%) | 11/49 (22%) | 3/49 (6%) | WES, Targeted | 49 |
| Acute megakaryoblastic leukemia, sporadic | 2/19 (11%) | 2/19 (11%) | 0/19 (0%) | 0/19 (0%) | 0/19 (0%) | 0/19 (0%) | 0/19 (0%) | WES, Targeted | 49 |
| Acute myeloid leukemia | 1/8 (13%) | 0/8 (0%) | 0/8 (0%) | 1/8 (13%) | 0/8 (0%) | 0/8 (0%) | 0/8 (0%) | WGS | 42 |
| Acute myeloid leukemia, secondary | 2/7 (29%) | 1/7 (14%) | 0/7 (0%) | 1/7 (14%) | 0/7 (0%) | 0/7 (0%) | 0/7 (0%) | WGS | 43 |
| Acute myeloid leukemia, type M1 | 7/65 (11%) | 3/65 (5%) | 0/65 (0%) | 2/65 (3%) | 2/65 (3%) | 0/65 (0%) | 1/12 (8%) | WES, Targeted | 44 |
| Acute myeloid leukemia, de novo | 26/200 (13%) | 7/200 (4%) | 0/200 (0%) | 7/200 (4%) | 7/200 (4%) | 5/200 (3%) | 4/200 (2%) | WES, WGS | 45 |
| Acute myeloid leukemia, de novo | 16/120 (13%) | 8/120 (7%) | 0/120 (0%) | 1/120 (1%) | 2/120 (2%) | 6/120 (5%) | 1/120 (1%) | WES, Targeted | 46 |
| Acute myeloid leukemia, de novo | 22/348 (6%) | 5/348 (1%) | 6/348 (2%) | 5/348 (1%) | 2/348 (1%) | 4/348 (1%) | N/A | Targeted | 47 |
| Acute myeloid leukemia, secondary | 3/37 (8%) | 2/37 (5%) | 0/37 (0%) | 0/37 (0%) | 0/37 (0%) | 1/37 (3%) | 1/37 (3%) | WES, Targeted | 46 |
| Acute myeloid leukemia, secondary | 1/41 (2%) | 0/41 (0%) | 1/41 (2%) | 0/41 (0%) | 0/41 (0%) | 0/41 (0%) | N/A | Targeted | 47 |
| Chronic myelogenous leukemia | 4/64 (6%) | 2/64 (3%) | 0/64 (0%) | 0/64 (0%) | 2/64 (3%) | 1/64 (2%) | 1/64 (2%) | WES, Targeted | 46 |
| Chronic myelomonocytic leukemia | 10/88 (11%) | 9/88 (10%) | 1/88 (0%) | 0/88 (0%) | 0/88 (0%) | 0/88 (0%) | 2/88 (2%) | WES, Targeted | 46 |
| Myelodysplastic syndrome | 19/224 (8%) | 13/224 (6%) | 1/224 (0%) | 3/224 (1%) | 0/224 (0%) | 2/224 (1%) | 2/224 (1%) | WES, Targeted | 46 |
| Myeloproliferative neoplasm | 1/77 (1%) | 1/77 (1%) | 0/77 (0%) | 0/77 (0%) | 0/77 (0%) | 0/77 (0%) | 1/77 (1%) | WES, Targeted | 46 |
| Bone/soft tissue | |||||||||
| Ewing sarcoma | 1/24 (4%) | 1/24 (4%) | N/A | N/A | N/A | N/A | N/A | Sanger | 41 |
| Brain | |||||||||
| Glioblastoma | 4/68 (6%) | 4/68 (6%) | N/A | N/A | N/A | N/A | N/A | Sanger | 41 |
| Glioblastoma | 23/291 (8%) | 12/291 (4%) | 1/291 (0%) | 4/291 (1%) | 5/291 (2%) | 1/291 (0%) | 15/291 (5%) | WES, WGS | 57 |
| Medulloblastoma | 3/125 (3%) | 1/125 (1%) | 0/125 (0%) | 1/125 (1%) | 1/125 (1%) | 0/125 (1%) | 1/125 (1%) | WES | 58 |
| Breast | |||||||||
| Adenocarcinoma | 3/100 (3%) | 1/100 (1%) | 1/100 (1%) | 0/100 (0%) | 1/100 (1%) | 0/100 (0%) | 7/100 (7%) | WES | 59 |
| Colon/Rectum | |||||||||
| Adenocarcinoma | 5/132 (4%) | 0/36 (0%) | 0/36 (0%) | 1/130 (1%) | 4/132 (3%) | 0/36 (0%) | 4/132 (3%) | Sanger | 39 |
| Pancreas | |||||||||
| Ductal adenocarcinoma | 2/50 (4%) | 2/50 (4%) | N/A | N/A | N/A | N/A | N/A | Sanger | 60 |
| Skin | |||||||||
| Melanoma | 1/48 (2%) | 1/48 (2%) | N/A | N/A | N/A | N/A | N/A | Sanger | 41 |
| Ureter/renal pelvis | |||||||||
| Urothelial carcinoma, papillary non-invasive | 2/5 (40%) | 2/5 (40%) | 0/5 (0%) | 0/5 (0%) | 0/5 (0%) | 0/5 (0%) | 1/5 (20%) | WES | 55 |
| Urothelial carcinoma, invasive | 7/21 (33%) | 6/21 (29%) | 1/21 (5%) | 0/21 (0%) | 1/21 (5%) | 0/21 (0%) | 5/21 (24%) | WES | 55 |
| Urinary bladder | |||||||||
| Urothelial carcinoma, papillary non-invasive | 9/25 (36%) | 9/25 (36%) | N/A | N/A | N/A | N/A | N/A | Sanger | 50 |
| Urothelial carcinoma, papillary non-invasive | 10/33 (30%) | 7/33 (21%) | 1/33 (3%) | 0/33 (0%) | 1/33 (3%) | 1/33 (3%) | 6/33 (18%) | WES, Targeted | 51 |
| Urothelial carcinoma, papillary non-invasive | 46/138 (33%) | 46/138 (33%) | N/A | N/A | N/A | N/A | N/A | Sanger | 53 |
| Urothelial carcinoma, superficially invasive | 6/22 (27%) | 6/22 (27%) | N/A | N/A | N/A | N/A | N/A | Sanger | 50 |
| Urothelial carcinoma, superficially invasive | 3/32 (9%) | 2/32 (6%) | 1/32 (3%) | 1/32 (3%) | 0/32 (0%) | 0/32 (0%) | 2/32 (6%) | WES, Targeted | 51 |
| Urothelial carcinoma, superficially invasive | 10/32 (31%) | 8/32 (25%) | 0/32 (0%) | 0/32 (0%) | 2/32 (6%) | 0/32 (0%) | 1/32 (3%) | WES | 52 |
| Urothelial carcinoma, superficially invasive | 16/76 (21%) | 16/76 (21%) | N/A | N/A | N/A | N/A | N/A | Sanger | 53 |
| Urothelial carcinoma, muscle invasive | 8/64 (13%) | 8/64 (13%) | N/A | N/A | N/A | N/A | N/A | Sanger | 50 |
| Urothelial carcinoma, muscle invasive | 2/9 (22%) | 2/9 (22%) | 0/9 (0%) | 0/9 (0%) | 0/9 (0%) | 0/9 (0%) | 0/9 (0%) | WES, Targeted | 51 |
| Urothelial carcinoma, muscle invasive | 6/61 (10%) | 3/61 (5%) | 0/61 (0%) | 2/61 (3%) | 1/61 (2%) | 0/61 (0%) | 9/61 (15%) | WES | 52 |
| Urothelial carcinoma, muscle invasive | 10/80 (13%) | 10/80 (13%) | N/A | N/A | N/A | N/A | N/A | Sanger | 53 |
| Urothelial carcinoma, muscle invasive | 22/131 (17%) | 14/131 (11%) | 2/131 (2%) | 2/131 (2%) | 3/131 (2%) | 1/131 (1%) | 17/131 (13%) | WES, RNA-seq | 54 |
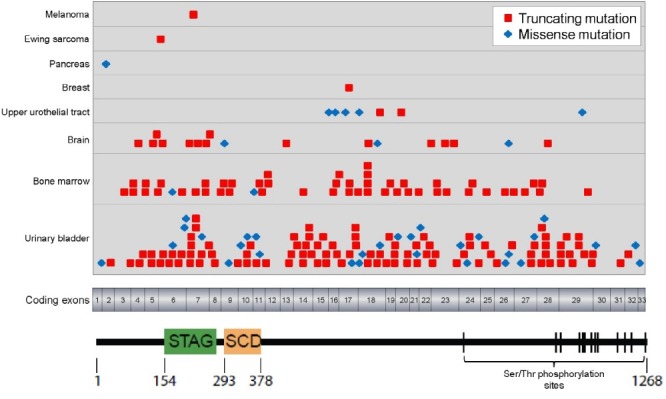
Then in 2011 Solomon et al. reported the identification of recurrent inactivating mutations of the core cohesin subunit STAG2 in a variety of human tumors and demonstrated that STAG2 inactivation is a cause of aneuploidy in human cancer cells (41). In a screen of 155 unique human cancer cell lines, complete loss of STAG2 expression was identified in 3/21 glioblastoma, 5/9 Ewing sarcoma, 1/10 melanoma, 1/6 cervical carcinoma, and 2/20 hematologic cancer cell lines and found truncating mutations or deletions in 10/12 of these samples with STAG2 loss (see Table 2 for summary of cohesin gene mutations identified in human cancer cell lines). Analysis of primary tumor samples found somatic STAG2 mutations in 4/68 glioblastoma, 1/24 Ewing sarcoma, and 1/48 melanoma samples (see Fig. 2 for diagram of STAG2 mutations reported in human tumors to date). As the STAG2 gene is on the X-chromosome, complete inactivation of STAG2 was found to only require a single mutational event due to the presence of a single X-chromosome in males and due to X-chromosome inactivation in females with the remaining wild-type allele residing on the inactivated X-chromosome. Using a monoclonal antibody directed against the C-terminus of the STAG2 protein, immunohistochemistry showed somatic loss of STAG2 expression in 6/31 glioblastomas (19%), 11/53 Ewing sarcomas (21%), 7/36 melanoma samples (19%), 1/20 medullobastomas (5%), 2/74 colorectal adenocarcinomas (3%), and 1/58 lymphomas (2%). Somatic cell gene targeting was used to correct the endogenous inactivating STAG2 mutations in two aneuploid glioblastoma cell lines (H4 and 42MGBA cells) via homologous recombination and to knockout STAG2 in a near-diploid, chromosomally stable colorectal carcinoma cell line (HCT116 cells). Repair of the STAG2 gene led to restoration of the sister chromatid cohesion defect, a decrease in abnormal mitotic figures, and a reduction in chromosomal instability in the two aneuploid glioblastoma cell lines. In contrast, STAG2 knockout in HCT116 cells led to precocious sister chromatid separation and aneuploidy including trisomies, monosomies, and de novo translocations. No significant difference in transcriptional profile was observed in these paired STAG2 mutant and wild-type glioblastoma and colorectal carcinoma cell lines, suggesting that the major tumor suppressive function of STAG2 is due to its canonical role in sister chromatid cohesin and not transcriptional regulation (41). This study provided one of the first major genetic mechanisms of aneuploidy in human tumors. However, the complete spectrum of tumors harboring cohesin mutations and the precise functional significance of cohesin mutations in the pathogenesis of specific tumor types remained undefined.
Table 2. Summary of cohesin gene mutations reported in human cancer cell lines.
| Tumor type | Cell line | Cohesin gene mutated | Mutation present | Predicted amino acid change | Reference |
|---|---|---|---|---|---|
| Acute megakaryoblastic leukemia | CMY | RAD21 | Nonsense | 3Y>Stop | 46 |
| Acute myeloid leukemia, type M2 | Kasumi-1 | RAD21 | Small insertion | 330K>frameshift | 46 |
| Acute myeloid leukemia, type M5 | P31FUJ | RAD21 | Missense | 208H>R | 46 |
| Cervical carcinoma | CaSki | STAG2 | Small deletion | 223A>truncation | 41 |
| Chronic myelogenous leukemia | MOLM-7 | SMC3 | Missense | 661R>P | 46 |
| Ewing sarcoma | A4573 | STAG2 | Small deletion | 842N>frameshift | 41 |
| Ewing sarcoma | ES-8 | STAG2 | Deletion 5 | no protein | 41 |
| Ewing sarcoma | SK-ES-1 | STAG2 | Nonsense | 735Q>Stop | 41 |
| Ewing sarcoma | TC-32 | STAG2 | Small insertion | 636Y>frameshift | 41 |
| Glioblastoma | 42MGBA | STAG2 | Nonsense | 653S>Stop | 41 |
| Glioblastoma | H4 | STAG2 | Small insertion | 357N>frameshift | 41 |
| Glioblastoma | U138MG | STAG2 | Gene deletion | no protein | 41 |
| Glioblastoma | U87MG | SMC3 | Missense | 427K>R | 41 |
| Lymphoma, immunoblastic | SR | STAG2 | Intragenic deletion | deletion 925-1092 | 41 |
| Melanoma | LOX IMVI | STAG2 | Gene deletion | no protein | 41 |
| Urothelial carcinoma | 92-1 | STAG2 | Missense | 1263L>V | 53 |
| Urothelial carcinoma | 94-10 | STAG2 | Splice site | 699N>frameshift | 50 |
| Urothelial carcinoma | HCV29 | STAG2 | Small insertion | 586T>frameshift | 53 |
| Urothelial carcinoma | UM-UC-3 | STAG2 | Complex in/del | 983K>truncation | 50 |
| Urothelial carcinoma | UM-UC-6 | STAG2 | Nonsense | 305R>Stop | 51 |
| Urothelial carcinoma | UM-UC-14 | STAG2 | Splice site | 676G>frameshift | 50 |
| Urothelial carcinoma | VM-CUB-1 | STAG2 | Small insertion | 896Y>frameshift | 50 |
| Urothelial carcinoma | VM-CUB-3 | STAG2 | Splice site | 97S>frameshift | 50 |
Fig. 2. Diagram of the STAG2 protein with the location of mutations identified in human tumor samples listed in Table 1 shown. STAG, stromal antigen domain. SCD, stromalin conserved domain.
COHESIN MUTATIONS IN ACUTE MYELOID LEUKEMIAS
Whole exome sequencing of 8 acute myeloid leukemia (AML) genomes reported in 2012 identified one AML (type M1 arising in an adult female) with an SMC3 missense mutation also harboring DNMT3A, NPM1, and FLT3 mutations (42). This AML at initial presentation had normal diploid karyotype, and upon relapse harbored a single chromosomal aberration (t(10;12) translocation) along with new ETV6 and MYO18B mutations. A subsequent whole exome sequencing study of 7 secondary AMLs arising in patients with antecedent myelodysplastic syndrome (MDS) found two AMLs with cohesin mutations, one with a truncating mutation in STAG2 and one with a truncating mutation in SMC3 (43). The STAG2 mutation was present in both the antecedent MDS and the secondary AML with additional PTPN11 and RUNX1 mutations arising de novo in the secondary AML that had normal cytogenetics. The SMC3 mutation was present at low frequency in the antecedent MDS and at high frequency in the secondary AML that also harbored NPM1 mutation and had normal cytogenetics.
The next AML whole exome sequencing study examined the genomes of 65 type M1 AMLs that are defined by the lack of myeloid maturation and 43 type M3 AMLs that are defined by the presence of the t(15;17)(q22;21) translocation causing the PML-RARA fusion and are commonly referred to as acute promyelocytic leukemias (44). This study found cohesin mutations in 7/65 type M1 AMLs (11%) but no cohesin gene mutations in the 43 type M3 AMLs (0%). These included three M1-AMLs with STAG2 mutations, two with SMC3 mutations, and two with SMC1A mutations. All seven of these AMLs with cohesin mutations had fewer than three cytogenetic abnormalities, while 6/7 of these AMLs also harbored FLT3 mutations, 3/7 also harbored NPM1 mutations, 2/7 also harbored DNMT3A mutations, and 2/7 also harbored TET2 mutations. Given the near-diploid karyotype of all AMLs with identified cohesin mutations, the authors speculated that cohesin mutations may be selected for during leukemogenesis not due to loss of sister chromatid cohesion and induction of aneuploidy, but rather other loss of function mechanisms such as transcriptional deregulation.
In 2013 The Cancer Genome Atlas reported comprehensive genomic characterization of 200 de novo AMLs arising in adult patients with no history of myelodysplasia or myeloproliferative disorder (45). They found cohesin mutations in 26 of these AMLs (13%) including 7 with STAG2 mutations (all truncating), 7 with SMC3 mutations (5/7 missense), 7 with SMC1A mutations (6/7 missense), and 5 with RAD21 mutations (all truncating). Cohesin gene mutations were mutually exclusive in each case (e.g. no AML had multiple cohesin gene mutations) demonstrating that genetic inactivation of a single cohesin subunit is likely sufficient to disrupt the tumor suppressive function of cohesin in myeloid leukemogenesis. Rare mutations were also found in cohesin regulatory genes including ESCO2, PDS5B, and WAPAL (4/200 AMLs). Significantly co-occurring mutations with cohesin genes were NPM1, FLT3, DNMT3A, PTPN11, and PTPRT. No prognostic significance based on cohesin gene status was reported in this study.
Given the frequent mutations of cohesin genes in AML, Kon et al. chose to investigate the importance of cohesin mutations in the complete spectrum of myeloid neoplasms (46). In accordance with previous studies, cohesin mutations were identified in 16/120 de novo AMLs (13%). Additionally, cohesin mutations were found in 19/224 myelodysplastic syndromes (8%), 3/37 secondary AMLs (8%), 4/64 chronic myelogenous leukemias (6%), 10/88 chronic myelomonocytic leukemias (11%), and 1/77 myeloproliferative neoplasms (1%). No signifcant increase in cytogenetic abnormalities was observed between cohesin mutant and wild-type samples. Cohesin mutated leukemia cells had reduced levels of chromatin-bound cohesin components, and the growth of cohesin mutated leukemia cells was suppressed by forced over-expression of the respective wild-type cohesin subunit, thereby leading the authors to speculate that cohesin mutations participate in leukemogenesis through the deregulated expression of genes involved in myeloid development and differentiation (46).
In order to explore the clinical significance of cohesin mutations in AML, Thol et al. sequenced the cohesin complex subunits in a clinically annotated cohort of 348 uniformly treated adult patients with de novo AML (47). Mutations were identified in 22 patients (6%), all of which were mutually exclusive and were strongly associated with the co-occurrence of NPM1 mutations. No clear prognostic impact for survival or remission rates based on cohesin status was found.
COHESIN MUTATIONS IN ACUTE MEGAKARYOBLASTIC LEUKEMIA
Up to 10% of patients with constitutional trisomy 21 (Down syndrome) experience transient abnormal myelopoiesis (TAM) during infancy, an abnormal myeloproliferative disorder that morphologically resembles acute megakaryoblastic leukemia (AMKL) but is usually self-limiting and spontaneously resolves within 3-4 months after birth. However, 20-30% of Down syndrome patients who survive TAM during infancy will go on in the following years to develop AMKL, also known as AML type M7, that can arise sporadically outside of Down syndrome but has >500 times increased incidence in individuals with Down syndrome. It was discovered in 2002 that all TAM samples harbor truncating mutations in the 5’ coding exons of the GATA1 gene, an essential hematopoietic transcription factor, resulting in downstream re-initiation and production of a shorter GATA1 variant lacking the N-terminal activation domain (48). While the combination of GATA1 mutation and trisomy 21 are sufficient for the induction of TAM, it is unclear what the driving third genetic hit is that results in AMKL in Down syndrome patients. An exome sequencing study found that cohesin mutations were present in 23/49 (47%) of Down syndrome-associated AMKL cases (predominantly STAG2 and RAD21 mutations and occasional cases with mutations in cohesin regulatory genes, all of which were mutually exclusive) while cohesin mutations were less commonly found in sporadic AMKL (2/19 cases, 11%) and were never present in TAM (0/41 cases) (49). The majority of cohesin mutated AMKLs had two or fewer chromosomal aberrations other than trisomy 21, again supporting a non-aneuploidy driven mechanism for cohesin inactivation in myeloid leukemogenesis. This study defines a prominent role for cohesin as one of the third genetic hits responsible for the transformation of TAM into AMKL in Down syndrome patients, in addition to other identified hits including the epigenetic regulator EZH2 or the cohesin-binding insulator regulatory factor CTCF.
COHESIN MUTATIONS IN UROTHELIAL CARCINOMAS
In order to explore the complete tumor spectrum harboring STAG2 inactivation, Solomon et al. performed an immunohistochemical screen on 2,214 human tumors using a monoclonal antibody that binds at the C-terminus of the STAG2 protein (50). This screen identified somatic loss of STAG2 expression in 52/295 urothelial bladder carcinomas (18%), 3/35 Ewing sarcomas (9%), 3/48 malignant melanomas (6%), 2/99 colonic adenocarcinomas (2%), 2/112 ovarian adenocarcinomas (2%), 1/46 malignant peripheral nerve sheath tumors (2%), 1/49 gastric adenocarcinomas (2%), 1/53 uterine leiomyosarcomas (2%), 1/108 lung squamous cell carcinomas, and 1/131 gastrointestinal stromal tumors. No STAG2 loss was found in 18 bladder adenocarcinomas or 15 bladder squamous cell carcinomas, suggesting a specific role for STAG2 inactivation in urothelial carcinoma of the bladder (also referred to as transitional cell carcinoma). Sequencing of STAG2 in an independent cohort of urothelial bladder carcinomas identified mutations in 9/25 papillary non-invasive carcinomas (36%), 6/22 superficially invasive carcinomas (27%), and 8/64 muscle invasive carcinomas (13%). 21/25 of the identified mutations were truncating causing loss of STAG2 expression as seen by immunohistochemistry. Occasional urothelial carcinomas demonstrated mosaic loss of STAG2 expression and two tumors each had two independent STAG2 mutations identified with less than 50% mutant allele frequencies. These findings highlight the clonal heterogeneity of urothelial bladder carcinoma and suggest that while STAG2 inactivation occurs an early event in most bladder tumors with uniform STAG2 loss throughout, STAG2 mutations are also selected for during the clonal progression in occasional tumors. Truncating mutations of STAG2 were also identified in 5/32 urothelial carcinoma cell lines (16%). In a clinically-annotated cohort of patients with non-muscle invasive papillary urothelial bladder carcinomas treated with transurethral resection, STAG2 loss was seen in 8/34 cases (24%) and was significantly associated with improved disease-free survival. Only 1/8 of the STAG2-deficient carcinomas (12%) recurred during the follow-up period, whereas 15/26 of the STAG2-expressing carcinomas (58%) recurred including two which progressed to invasion and four which metastasized. In contrast, in a cohort of patients with muscle invasive urothelial bladder carcinomas treated with radical cystectomy, STAG2 loss was seen in 35/349 cases (10%) and was significantly associated with increased lymph node metastasis and reduced recurrence-free survival. Copy number analysis of 12 STAG2 mutant urothelial bladder carcinomas demonstrated multiple chromosomal aberrations in each of 9 tumors and no aberrations in the other 3 tumors, whereas 10/12 STAG2 wild-type urothelial bladder carcinomas contained detectable chromosomal aberrations. Re-expression of wild-type STAG2 in three urothelial carcinoma cell lines with truncating STAG2 mutations did not affect cellular proliferation in vitro, tumor growth in vivo, or mean chromosome count per cell. shRNA knockdown of STAG2 in a urothelial carcinoma cell line with wild-type STAG2 led to a modest alteration in chromosome count per cell. Overall, this study highlights that STAG2 is recurrently inactivated during urothelial carcinogenesis, at particularly high frequency in papillary non-invasive tumors, and defines a molecular subgroup of urothelial bladder carcinomas with distinct clinical outcomes (50).
Simultaneous to the publication of this study, two additional whole-exome sequencing studies of urothelial bladder carcinoma identifying frequent mutations of cohesin genes were also published (51,52). In the first of these other two studies, cohesin mutations were found in 10/33 papillary non-invasive tumors (30%), 3/32 superficially invasive tumors (9%), and 2/9 muscle invasive tumors (22%) (51). These were predominantly truncating mutations of STAG2, with occasional tumors harboring mutations in other core cohesin genes as well as cohesin regulatory genes. In the second of these other two studies, cohesin mutations were found in 10/32 superficially invasive tumors (31%) and 6/61 muscle invasive tumors (10%), with an additional 9 muscle invasive tumors harboring mutations in cohesin regulatory genes, predominantly ESPL1 which encodes the enzyme Separase that cleaves Rad21 at the metaphase to anaphase transition to enable sister chromatid segregation (52). In the first study, STAG2 mutation in non-muscle invasive tumors was associated with co-occurrence of FGFR3 mutations and absence of p53 overexpression. Reduced expression of STAG2 was found by immunohistochemistry in a panel of 197/671 urothelial bladder carcinomas (29%) of various grades and stages. Reduced STAG2 expression in this cohort was associated with multicentricity, increased tumor size, low stage, and low tumor grade. Additionally, reduced STAG2 expression in patients with non-muscle invasive tumors in this cohort was associated with lower risk of tumor recurrence and progression, while reduced STAG2 expression in patients with muscle invasive tumors was associated with lower risk of progression and increased overall survival. Copy number analysis of 11 low-grade papillary non-invasive urothelial carcinomas found 2 tumors with loss of one copy of chromosome 9 while the other 9 tumors had no detectable chromosomal aberrations. shRNA knockdown of STAG2 in urothelial carcinoma cell lines with wild-type STAG2 was not found to significantly alter the mean chromosome count per cell, and re-expression of wild-type STAG2 in cell lines with inactivating STAG2 mutations led to reduced colony formation in vitro (51). In the second study, invasive urothelial carcinomas with STAG2 mutations were significantly more aneuploid than invasive urothelial carcinomas without any alterations in core cohesin or cohesin regulatory genes. Additionally, patients with either superficially invasive or muscle invasive urothelial carcinomas harboring STAG2 mutations were associated with reduced overall survival compared to patients with wild-type tumors (52). An explanation for the discordant clinical outcomes based on STAG2 status in these three studies is not readily apparent, but difference in the Kaplan-Meier analysis criteria (e.g. complete loss of expression versus reduced expression by immunohistochemistry versus confirmed somatic mutation by DNA sequencing) may be a confounding factor.
In a subsequent in-depth study of STAG2 inactivation in urothelial bladder carcinoma (53), STAG2 mutations were identified in 46/138 papillary non-invasive tumors (33%), 16/76 superficially invasive tumors (21%), and 10/80 muscle invasive tumors (13%). 69/81 of the mutations (85%) identified were truncating while 12/81 were missense mutations (15%). STAG2 mutations were significantly associated with lower tumor grade, lower tumor stage, co-occurrence of FGRF3 and PIK3CA mutations, and wild-type TP53 status. No significant association of STAG2 mutation with disease recurrence in either papillary non-invasive carcinomas or invasive carcinomas was found. Immunohistochemical analysis of 120 urothelial carcinomas found 13 tumors (11%) with complete loss of STAG2 expression, all of which had truncating mutations of STAG2, and an additional 14 tumors (12%) showed mosaic loss of STAG2 expression. These findings provide further confirmation that STAG2 inactivation is both selected for early during urothelial carcinogenesis as well as during tumor progression. Copy number analysis of 220 urothelial bladder carcinomas found that STAG2 mutation was inversely related with chromosomal copy number alterations (53).
Most recently, The Cancer Genome Atlas reported comprehensive genomic analysis of 131 muscle invasive urothelial bladder carcinomas (54). In total they identified cohesin mutations in 22/131 muscle invasive carcinomas (17%), including 14 tumors (11%) with STAG2 mutations and 8 tumors with mutations in other core cohesin genes. Additionally, 17/131 tumors (13%) harbored mutations in cohesin regulatory genes including CDCA5, ESPL1, and NIPBL. Clustering of the 131 tumors based on mutations and copy number alterations identified three distinct groups, and 11/14 of the tumors with STAG2 mutations belonged to Group B enriched in tumors harboring CDKN2A deletions, FGFR3 mutations, and papillary histology. Why STAG2 mutations occur at higher frequency in particular molecular subgroups of urothelial carcinomas remains undefined (54).
Not only have cohesin gene mutations been identified in urothelial carcinomas arising in the bladder, exome sequencing of urothelial carcinomas arising in the upper urothelial tract (e.g. ureter and renal pelvis) have found cohesin mutations in 2/5 papillary non-invasive (40%) and 7/21 invasive urothelial carcinomas (33%) of the upper urothelial tract (55). In contrast, cohesin mutations have not been identified at a significant frequency in renal cell carcinoma sequencing studies (56).
OTHER TUMOR TYPES WITH COHESIN MUTATIONS
As the genomes of additional tumor types are revealed by next generation sequencing, the cohesin complex is emerging as a target of frequent of somatic alterations in a diverse range of tumors. Comprehensive genomic characterization of 291 glioblastomas by The Cancer Genome Atlas identified cohesin gene mutations in 23 tumors (8%), including 12 tumors with mutations in STAG2 (one of the most commonly mutated genes identified), as well as 15 additional tumors (5%) with mutations in cohesin regulatory genes (57). In addition to glioblastomas, whole-exome sequencing of medulloblastoma, a primitive neuroectodermal tumor arising in the cerebellum and posterior fossa of the brain of pediatric and young adults, found cohesin mutations in 3/125 cases (58). Whole-exome sequencing of 100 invasive breast adenocarcinomas identified 3 tumors with cohesin gene mutations and 7 tumors with mutations in cohesin regulatory genes (59). Whether cohesin mutations in breast carcinomas are associated with a specific histologic subtype (e.g. ductal, lobular, mucinous) or distinct clinical outcomes remains to be determined. A recent investigation into the genomes of 50 pancreatic ductal adenocarcinomas found two tumors with STAG2 alterations, one with gene deletion and one with missense mutation (60). STAG2 loss was also found in 15/344 pancreatic ductal adenocarcinomas (4%) by immunohistochemistry but was not associated with altered clinical outcome in these patients. An investigation of microsatellite-unstable gastric and colorectal adenocarcinomas found frequent frameshift mutations in the cohesin regulatory genes PDS5B and SGOL1 which harbor long mononucleotide repeats within their coding sequences (61). In addition to the frequent truncating mutations of STAG2 in Ewing sarcoma cell lines reported by Solomon et al. (41), ongoing as yet unpublished sequencing studies have also identified frequent cohesin mutations (predominantly of the STAG2 subunit) in Ewing sarcoma primary tumors samples as well. The clinical significance of cohesin mutations and the precise mechanism of tumor suppressive function in the pathogenesis of many of these specific tumor types currently remain undefined.
THERAPEUTIC TARGETING OF TUMOR CELLS HARBORING COHESIN MUTATIONS
Given the high frequency of inactivation in particular cancer types, mechanisms of selectively targeting cancer cells harboring cohesin mutations is of significant clinical interest. The RAD21 gene was originally isolated in the fission yeast S. pombe as a mutant that caused radiation sensitivity and was also found to be essential for mitosis (62). The cohesin complex is now known to be important for both DNA replication and repair, specifically at stalled replication forks and double-strand breaks (63-67). Of note, SMC1A mutant cells from a CdL patient have shown increased sensitivity to ionizing radiation and interstrand DNA crosslinking agents compared to normal controls, although this sensitivity was to a lesser extent than cells from patients with ataxia telangiectasia or Fanconi anemia syndromes (68). These findings suggest that tumors harboring cohesin mutations might have increased sensitivity to ionizing radiation and/or specific DNA damaging chemotherapeutic agents. One study has shown that knockdown of STAG2 in a pancreatic adenocarcinoma cell line led to increased sensitivity to platinum-based chemotherapy agents including cisplatin (60). Considering the potential for rational targeted therapies, inhibitors of poly(ADP-ribose) polymerase (PARP) such as olaparib have shown promise in ovarian cancers deficient in DNA repair due to mutations in BRCA1 and BRCA2. Similarly, glioblastoma cell lines harboring truncating STAG2 mutations were recently shown to have increased sensitivity to multiple unique small molecule inhibitors of PARP, leading to arrest in G2 phase of the cell cycle, more frequent micronuclei, and increased atypical mitotic figures (69). This effect of PARP inhibitors was synergistic when combined with DNA damaging chemotherapeutic agents including temozolomide or camptothecin. Together, these studies demonstrate that tumors harboring cohesin mutations might have specific sensitivities that can be rationally targeted to provide clinical benefit for the many patients with such cohesin altered cancers. Further studies are clearly warranted to continue evaluating these therapeutic strategies.
CONCLUDING REMARKS
The cohesin complex is an important regulator of chromatin architecture within the cell nucleus and has expanding roles in diverse biologic processes such as DNA replication, repair, and transcription in addition to its canonical role in sister chromatid cohesin and regulation of faithful chromosome segregation during mitosis. Inherited/germline mutations in core cohesin subunits or cohesin regulatory genes lead to a spectrum of “cohesinopathies” that include Cornelia de Lange syndrome as well as premature ovarian failure. Emerging cancer genomics studies have now documented that cohesin genes are a frequent target of somatic alterations in a number of tumor types including glioblastoma, Ewing sarcoma, urothelial carcinoma, acute myeloid leukemia, and acute megakaryoblastic leukemia. Initial studies demonstrated that cohesin mutations may be a source of chromosomal instability and aneuploidy in tumors, while subsequent studies in other cancer types have found normal karyotypes in tumors harboring cohesin mutations, leading to speculation that cohesin inactivation may be causing dysregulation of other pathways during tumorigenesis, particularly altered gene expression profiles. Recent studies have begun to demonstrate that cohesin mutant tumors might have increased sensitivity to select DNA damaging agents and PARP inhibitors. Continued investigation will be necessary to define the precise mechanism(s) by which cohesin inactivation contributes to tumorigenesis and methods to effectively target cancers harboring cohesin mutations.
Acknowledgments
D.S. is supported by the UCSF Department of Pathology. J.S.K. and T.W. are supported by National Cancer Institute grants R01CA169345, R01CA115699, and R01CA159467.
References
- 1.Nasmyth K. Segregating sister genomes: The molecular biology of chromosome separation. Science. (2002);297:559–565. doi: 10.1126/science.1074757. [DOI] [PubMed] [Google Scholar]
- 2.Remeseiro S., Cuadrado A., Losada A. Cohesin in development and disease. Development. (2013);140:3715–3718. doi: 10.1242/dev.090605. [DOI] [PubMed] [Google Scholar]
- 3.Sumara I., Vorlaufer E., Gieffers C., Peters B. H., Peters J. M. Characterization of vertebrate cohesin complexes and their regulation in prophase. J. Cell Biol. (2000);151:749–762. doi: 10.1083/jcb.151.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haering C. H., Farcas A. M., Arumugam P., Metson J., Nasmyth K. The cohesin ring concatenates sister DNA molecules. Nature. (2008);454:297–301. doi: 10.1038/nature07098. [DOI] [PubMed] [Google Scholar]
- 5.Canudas S., Smith S. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J. Cell Biol. (2009);187:165–173. doi: 10.1083/jcb.200903096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Remeseiro S., Cuadrado A., Carretero M., Martinez P., Drosopoulos W. C., Canamero M., Schildkraut C. L., Blasco M. A., Losada A. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J. (2012);31:2076–2089. doi: 10.1038/emboj.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNicoll F., Stevense M., Jessberger R. Cohesin in gametogenesis. Curr. Top. Dev. Biol. (2013);102:1–34. doi: 10.1016/B978-0-12-416024-8.00001-5. [DOI] [PubMed] [Google Scholar]
- 8.Rollins R. A., Korom M., Aulner N., Martens A., Dorsett D. Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene. Mol. Cell. Biol. (2004);24:3100–3111. doi: 10.1128/MCB.24.8.3100-3111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seitan V. C., Banks P., Laval S., Majid N. A., Dorsett D., Rana A., Smith J., Bateman A., Krpic S., Hostert A., Rollins R. A., Erdjument-Bromage H., Tempst P., Benard C. Y., Hekimi S., Newbury S. F., Strachan T. Metazoan Scc4 homologs link sister chromatid cohesion to cell and axon migration guidance. PLoS Biol. (2006);4:e242. doi: 10.1371/journal.pbio.0040242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tedeschi A., Wutz G., Huet S., Jaritz M., Wuensche A., Schirghuber E., Davidson I. F., Tang W., Cisneros D. A., Bhaskara V., Nishiyama T., Vaziri A., Wutz A., Ellenberg J., Peters J. M. Wapl is an essential regulator of chromatin structure and chromosome segregation. Nature. (2013);501:564–568. doi: 10.1038/nature12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carretero M., Ruiz-Torres M., Rodriguez-Corsino M., Barthelemy I., Losada A. Pds5B is required for cohesion establishment and Aurora B accumulation at centromeres. EMBO J. (2013);32:2938–2949. doi: 10.1038/emboj.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J., Shi X., Li Y., Kim B. J., Jia J., Huang Z., Yang T., Fu X., Jung S. Y., Wang Y., Zhang P., Kim S. T., Pan X., Qin J. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol. Cell. (2008);31:143–151. doi: 10.1016/j.molcel.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Unal E., Heidinger-Pauli J. M., Kim W., Guacci V., Onn I., Gygi S. P., Koshland D. E. A molecular determinant for the establishment of sister chromatid cohesion. Science. (2008);321:566–569. doi: 10.1126/science.1157880. [DOI] [PubMed] [Google Scholar]
- 14.Rankin S., Ayad N. G., Kirschner M. W. Sororin, a substrate of the anaphase-promoting complex, is required for sister chromatid cohesion in vertebrates. Mol. Cell. (2005);18:185–200. doi: 10.1016/j.molcel.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 15.Hauf S., Roitinger E., Koch B., Dittrich C. M., Mechtler K., Peters J. M. Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol. (2005);3:e69. doi: 10.1371/journal.pbio.0030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salic A., Waters J. C., Mitchison T. J. Vertebrate shugoshin links sister centromere cohesion and kinetochore microtubule stability in mitosis. Cell. (2004);118:567–578. doi: 10.1016/j.cell.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Cohen-Fix O., Peters J. M., Kirschner M. W., Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. (1996);10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- 18.Uhlmann F., Lottspeich F., Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature. (1999);400:37–42. doi: 10.1038/21831. [DOI] [PubMed] [Google Scholar]
- 19.Mehta G. D., Kumar R., Srivastava S., Ghosh S. K. Cohesin: functions beyond sister chromatid cohesion. FEBS Lett. (2013);587:2299–2312. doi: 10.1016/j.febslet.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 20.Wendt K. S., Yoshida K., Itoh T., Bando M., Koch B., Schirghuber E., Tsutsumi S., Nagae G., Ishihara K., Mishiro T., Yahata K., Imamoto F., Aburatani H., Nakao M., Imamoto N., Maeshima K., Shirahige K., Peters J. M. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. (2008);451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 21.Parelho V., Hadjur S., Spivakov M., Leleu M., Sauer S., Gregson H. C., Jarmuz A., Canzonetta C., Webster Z., Nesterova T., Cobb B. S., Yokomori K., Dillon N., Aragon L., Fisher A. G., Merkenschlager M. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. (2008);132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 22.Xiao T., Wallace J., Felsenfeld G. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin-dependent insulation activity. Mol. Cell. Biol. (2011);31:2174–2183. doi: 10.1128/MCB.05093-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kagey M. H., Newman J. J., Bilodeau S., Zhan Y., Orlando D. A., van Berkum N. L., Ebmeier C. C., Goossens J., Rahl P. B., Levine S. S., Taatjes D. J., Dekker J., Young R. A. Mediator and cohesin connect gene expression and chromatin architecture. Nature. (2010);467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hahn M., Dambacher S., Dulev S., Kuznetsova A. Y., Eck S., Worz S., Sadic D., Schulte M., Mallm J. P., Maiser A., Debs P., von Melchner H., Leonhardt H., Schermelleh L., Rohr K., Rippe K., Storchova Z., Schotta G. Suv4-20h2 mediates chromatin compaction and is important for cohesin recruitment to heterochromatin. Genes Dev. (2013);27:859–872. doi: 10.1101/gad.210377.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krantz I. D., McCallum J., DeScipio C., Kaur M., Gillis L. A., Yaeger D., Jukofsky L., Wasserman N., Bottani A., Morris C. A., Nowaczyk M. J., Toriello H., Bamshad M. J., Carey J. C., Rappaport E., Kawauchi S., Lander A. D., Calof A. L., Li H. H., Devoto M., Jackson L. G. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. (2004);36:631–635. doi: 10.1038/ng1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tonkin E. T., Wang T. J., Lisgo S., Bamshad M. J., Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. (2004);36:636–641. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
- 27.Musio A., Selicorni A., Focarelli M. L., Gervasini C., Milani D., Russo S., Vezzoni P., Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. (2006);38:528–530. doi: 10.1038/ng1779. [DOI] [PubMed] [Google Scholar]
- 28.Deardorff M. A., Kaur M., Yaeger D., Rampuria A., Korolev S., Pie J., Gil-Rodriguez C., Arnedo M., Loeys B., Kline A. D., Wilson M., Lillquist K., Siu V., Ramos F. J., Musio A., Jackson L. S., Dorsett D., Krantz I. D. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. (2007);80:485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deardorff M. A., Wilde J. J., Albrecht M., Dickinson E., Tennstedt S., Braunholz D., Monnich M., Yan Y., Xu W., Gil-Rodriguez M. C., Clark D., Hakonarson H., Halbach S., Michelis L. D., Rampuria A., Rossier E., Spranger S., Van Maldergem L., Lynch S. A., Gillessen-Kaesbach G., Ludecke H. J., Ramsay R. G., McKay M. J., Krantz I. D., Xu H., Horsfield J. A., Kaiser F. J. RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. (2012);90:1014–1027. doi: 10.1016/j.ajhg.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaur M., DeScipio C., McCallum J., Yaeger D., Devoto M., Jackson L. G., Spinner N. B., Krantz I. D. Precocious sister chromatid separation (PSCS) in Cornelia de Lange syndrome. Am. J. Med. Genet. (2005);138A:27–31. doi: 10.1002/ajmg.a.30919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castronovo P., Gervasini C., Cereda A., Masciadri M., Milani D., Russo S., Selicorni A., Larizza L. Premature chromatid separation is not a useful diagnostic marker for Cornelia de Lange syndrome. Chromosome Res. (2009);17:763–771. doi: 10.1007/s10577-009-9066-6. [DOI] [PubMed] [Google Scholar]
- 32.Liu J., Zhang Z., Bando M., Itoh T., Deardorff M. A., Clark D., Kaur M., Tandy S., Kondoh T., Rappaport E., Spinner N. B., Vega H., Jackson L. G., Shirahige K., Krantz I. D. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. (2009);7:e1000119. doi: 10.1371/journal.pbio.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawauchi S., Calof A. L., Santos R., Lopez-Burks M. E., Young C. M., Hoang M. P., Chua A., Lao T., Lechner M. S., Daniel J. A., Nussenzweig A., Kitzes L., Yokomori K., Hallgrimsson B., Lander A. D. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/-) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet. (2009);5:e1000650. doi: 10.1371/journal.pgen.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remeseiro S., Cuadrado A., Gómez-López G., Pisano D. G., Losada A. A unique role of cohesin-SA1 in gene regulation and development. EMBO J. (2012);31:2090–2102. doi: 10.1038/emboj.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schrier S. A., Sherer I., Deardorff M. A., Clark D., Audette L., Gillis L., Kline A. D., Ernst L., Loomes K., Krantz I. D., Jackson L. G. Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature. Am. J. Med. Genet. (2011);155A:3007–3024. doi: 10.1002/ajmg.a.34329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caburet S., Arboleda V. A., Llano E., Overbeek P. A., Barbero J. L., Oka K., Harrison W., Vaiman D., Ben-Neriah Z., Garcia-Tunon I., Fellous M., Pendas A. M., Veitia R. A., Vilain E. Mutant cohesin in premature ovarian failure. N. Engl. J. Med. (2014);370:943–949. doi: 10.1056/NEJMoa1309635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanahan D., Weinberg R. A. Hallmarks of cancer: The next generation. Cell. (2011);144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 38.Gordon D. J., Resio B., Pellman D. Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet. (2012);13:189–203. doi: 10.1038/nrg3123. [DOI] [PubMed] [Google Scholar]
- 39.Barber T. D., McManus K., Yuen K. W., Reis M., Parmigiani G., Shen D., Barrett I., Nouhi Y., Spencer F., Markowitz S., Velculescu V. E., Kinzler K. W., Vogelstein B., Lengauer C., Hieter P. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. U. S. A. (2008);105:3443–3448. doi: 10.1073/pnas.0712384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rocquain J., Gelsi-Boyer V., Adelaide J., Murati A., Carbuccia N., Vey N., Birnbaum D., Mozziconacci M. J., Chaffanet M. Alteration of cohesin genes in myeloid diseases. Am. J. Hematol. (2010);85:717–719. doi: 10.1002/ajh.21798. [DOI] [PubMed] [Google Scholar]
- 41.Solomon D. A., Kim T., Diaz-Martinez L. A., Fair J., Elkahloun A. G., Harris B. T., Toretsky J. A., Rosenberg S. A., Shukla N., Ladanyi M., Samuels Y., James C. D., Yu H., Kim J. S., Waldman T. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. (2011);333:1039–1043. doi: 10.1126/science.1203619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding L., Ley T. J., Larson D. E., Miller C. A., Koboldt D. C., Welch J. S., Ritchey J. K., Young M. A., Lamprecht T., McLellan M. D., McMichael J. F., Wallis J. W., Lu C., Shen D., Harris C. C., Dooling D. J., Fulton R. S., Fulton L. L., Chen K., Schmidt H., Kalicki-Veizer J., Magrini V. J., Cook L., McGrath S. D., Vickery T. L., Wendl M. C., Heath S., Watson M. A., Link D. C., Tomasson M. H., Shannon W. D., Payton J. E., Kulkarni S., Westervelt P., Walter M. J., Graubert T. A., Mardis E. R., Wilson R. K., DiPersio J. F. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. (2012);481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walter M. J., Shen D., Ding L., Shao J., Koboldt D. C., Chen K., Larson D. E., McLellan M. D., Dooling D., Abbott R., Fulton R., Magrini V., Schmidt H., Kalicki-Veizer J., O M., Fan X., Grillot M., Witowski S., Heath S., Frater J. L., Eades W., Tomasson M., Westervelt P., DiPersio J. F., Link D. C., Mardis E. R., Ley T. J., Wilson R. K., Graubert T. A. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. (2012);366:1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Welch J. S., Ley T. J., Link D. C., Miller C. A., Larson D. E., Koboldt D. C., Wartman L. D., Lamprecht T. L., Liu F., Xia J., Kandoth C., Fulton R. S., McLellan M. D., Dooling D. J., Wallis J. W., Chen K., Harris C. C., Schmidt H. K., Kalicki-Veizer J. M., Lu C., Zhang Q., Lin L., O’Laughlin M. D., McMichael J. F., Delehaunty K. D., Fulton L. A., Magrini V. J., McGrath S. D., Demeter R. T., Vickery T. L., Hundal J., Cook L. L., Swift G. W., Reed J. P., Alldredge P. A., Wylie T. N., Walker J. R., Watson M. A., Heath S. E., Shannon W. D., Varghese N., Nagarajan R., Payton J. E., Baty J. D., Kulkarni S., Klco J. M., Tomasson M. H., Westervelt P., Walter M. J., Graubert T. A., DiPersio J. F., Ding L., Mardis E. R., Wilson R. K. The origin and evolution of mutations in acute myeloid leukemia. Cell. (2012);150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. (2013);368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kon A., Shih L. Y., Minamino M., Sanada M., Shiraishi Y., Nagata Y., Yoshida K., Okuno Y., Bando M., Nakato R., Ishikawa S., Sato-Otsubo A., Nagae G., Nishimoto A., Haferlach C., Nowak D., Sato Y., Alpermann T., Nagasaki M., Shimamura T., Tanaka H., Chiba K., Yamamoto R., Yamaguchi T., Otsu M., Obara N., Sakata-Yanagimoto M., Nakamaki T., Ishiyama K., Nolte F., Hofmann W. K., Miyawaki S., Chiba S., Mori H., Nakauchi H., Koeffler H. P., Aburatani H., Haferlach T., Shirahige K., Miyano S., Ogawa S. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. (2013);45:1232–1237. doi: 10.1038/ng.2731. [DOI] [PubMed] [Google Scholar]
- 47.Thol F., Bollin R., Gehlhaar M., Walter C., Dugas M., Suchanek K. J., Kirchner A., Huang L., Chaturvedi A., Wichmann M., Wiehlmann L., Shahswar R., Damm F., Gohring G., Schlegelberger B., Schlenk R., Dohner K., Dohner H., Krauter J., Ganser A., Heuser M. Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood. (2014);123:914–920. doi: 10.1182/blood-2013-07-518746. [DOI] [PubMed] [Google Scholar]
- 48.Wechsler J., Greene M., McDevitt M. A., Anastasi J., Karp J. E., Le Beau M. M., Crispino J. D. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat. Genet. (2002);32:148–152. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida K., Toki T., Okuno Y., Kanezaki R., Shiraishi Y., Sato-Otsubo A., Sanada M., Park M., Terui K., Suzuki H., Kon A., Nagata Y., Sato Y., Wang R., Shiba N., Chiba K., Tanaka H., Hama A., Muramatsu H., Hasegawa D., Nakamura K., Kanegane H., Tsukamoto K., Adachi S., Kawakami K., Kato K., Nishimura R., Izraeli S., Hayashi Y., Miyano S., Kojima S., Ito E., Ogawa S. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat. Genet. (2013);45:1293–1299. doi: 10.1038/ng.2759. [DOI] [PubMed] [Google Scholar]
- 50.Solomon D. A., Kim J. S., Bondaruk J., Shariat S. F., Wang Z. F., Elkahloun A. G., Ozawa T., Gerard J., Zhuang D., Zhang S., Navai N., Siefker-Radtke A., Phillips J. J., Robinson B. D., Rubin M. A., Volkmer B., Hautmann R., Kufer R., Hogendoorn P. C. W., Netto G., Theodorescu D., James C. D., Czerniak B., Miettinen M., Waldman T. Frequent truncating mutations of STAG2 in bladder cancer. Nat. Genet. (2013);45:1428–1430. doi: 10.1038/ng.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balbas-Martinez C., Sagrera A., Carrillo-de-Santa-Pau E., Earl J., Marquez M., Vazquez M., Lapi E., Castro-Giner F., Beltran S., Bayes M., Carrato A., Cigudosa J. C., Dominguez O., Gut M., Herranz J., Juanpere N., Kogevinas M., Langa X., Lopez-Knowles E., Lorente J. A., Lloreta J., Pisano D. G., Richart L., Rico D., Salgado R. N., Tardon A., Chanock S., Heath S., Valencia A., Losada A., Gut I., Malats N., Real F. X. Recurrent inactivation of STAG2 in bladder is not associated with aneuploidy. Nat. Genet. (2013);45:1464–1469. doi: 10.1038/ng.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo G., Sun X., Chen C., Wu S., Huang P., Li Z., Dean M., Huang Y., Jia W., Zhou Q., Tang A., Yang Z., Li X., Song P., Zhao X., Ye R., Zhang S., Lin Z., Qi M., Wan S., Xie L., Fan F., Nickerson M. L., Zou X., Hu X., Xing L., Lv Z., Mei H., Gao S., Liang C., Gao Z., Lu J., Yu Y., Liu C., Li L., Fang X., Jiang Z., Yang J., Li C., Zhao X., Chen J., Zhang F., Lai Y., Lin Z., Zhou F., Chen H., Chan H. C., Tsang S., Theodorescu D., Li Y., Zhang X., Wang J., Yang H., Gui Y., Wang J., Cai Z. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. (2013);45:1459–1463. doi: 10.1038/ng.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taylor C. F., Platt F. M., Hurst C. D., Thygesen H. H., Knowles M. A. Frequent inactivating mutations of STAG2 in bladder cancer are associated with low tumour grade and stage and inversely related to chromosomal copy number changes. Hum. Mol. Genet. (2014);23:1964–1974. doi: 10.1093/hmg/ddt589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. (2014);507:515–522. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoang M. L., Chen C. H., Sidorenko V. S., He J., Dickman K. G., Yun B. H., Moriya M., Niknafs N., Douville C., Karchin R., Turesky R. J., Pu Y. S., Vogelstein B., Papadopoulos N., Grollman A. P., Kinzler K. W., Rosenquist T. A. Mutational signature of aristolochic acid exposure as revealed by whole-exome sequencing. Sci. Transl. Med. (2013);5:197ra102. doi: 10.1126/scitranslmed.3006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. (2013);499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.The Cancer Genome Atlas Research Network. Brennan C. W., Verhaak R. G., McKenna A., Campos B., Noushmehr H., Salama S. R., Zheng S., Chakravarty D., Sanborn J. Z., Berman S. H., Beroukhim R., Bernard B., Wu C. J., Genovese G., Shmulevich I., Barnholtz-Sloan J., Zou L., Vegesna R., Shukla S. A., Ciriello G., Yung W. K., Zhang W., Sougnez C., Mikkelsen T., Aldape K., Bigner D. D., Van Meir E. G., Prados M., Sloan A., Black K. L., Eschbacher J., Finocchiaro G., Friedman W., Andrews D. W., Guha A., Iacocca M., O B. P., Foltz G., Myers J., Weisenberger D. J., Penny R., Kucherlapati R., Perou C. M., Hayes D. N., Gibbs R., Marra M., Mills G. B., Lander E., Spellman P., Wilson R., Sander C., Weinstein J., Meyerson M., Gabriel S., Laird P. W., Haussler D., Getz G., Chin L. The somatic genomic landscape of glioblastoma. Cell. (2013);155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones D. T., Jager N., Kool M., Zichner T., Hutter B., Sultan M., Cho Y. J., Pugh T. J., Hovestadt V., Stutz A. M., Rausch T., Warnatz H. J., Ryzhova M., Bender S., Sturm D., Pleier S., Cin H., Pfaff E., Sieber L., Wittmann A., Remke M., Witt H., Hutter S., Tzaridis T., Weischenfeldt J., Raeder B., Avci M., Amstislavskiy V., Zapatka M., Weber U. D., Wang Q., Lasitschka B., Bartholomae C. C., Schmidt M., von Kalle C., Ast V., Lawerenz C., Eils J., Kabbe R., Benes V., van Sluis P., Koster J., Volckmann R., Shih D., Betts M. J., Russell R. B., Coco S., Tonini G. P., Schuller U., Hans V., Graf N., Kim Y. J., Monoranu C., Roggendorf W., Unterberg A., Herold-Mende C., Milde T., Kulozik A. E., von Deimling A., Witt O., Maass E., Rossler J., Ebinger M., Schuhmann M. U., Fruhwald M. C., Hasselblatt M., Jabado N., Rutkowski S., von Bueren A. O., Williamson D., Clifford S. C., McCabe M. G., Collins V. P., Wolf S., Wiemann S., Lehrach H., Brors B., Scheurlen W., Felsberg J., Reifenberger G., Northcott P. A., Taylor M. D., Meyerson M., Pomeroy S. L., Yaspo M. L., Korbel J. O., Korshunov A., Eils R., Pfister S. M., Lichter P. Dissecting the genomic complexity underlying medulloblastoma. Nature. (2012);488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oslo Breast Cancer Consortium (OSBREAC) Stephens P. J., Tarpey P. S., Davies H., Van Loo P., Greenman C., Wedge D. C., Nik-Zainal S., Martin S., Varela I., Bignell G. R., Yates L. R., Papaemmanuil E., Beare D., Butler A., Cheverton A., Gamble J., Hinton J., Jia M., Jayakumar A., Jones D., Latimer C., Lau K. W., McLaren S., McBride D. J., Menzies A., Mudie L., Raine K., Rad R., Chapman M. S., Teague J., Easton D., Langerod A., Lee M. T., Shen C. Y., Tee B. T., Huimin B. W., Broeks A., Vargas A. C., Turashvili G., Martens J., Fatima A., Miron P., Chin S. F., Thomas G., Boyault S., Mariani O., Lakhani S. R., van de Vijver M., van L., Foekens J., Desmedt C., Sotiriou C., Tutt A., Caldas C., Reis-Filho J. S, Aparicio S. A., Salomon A. V., Borresen-Dale A. L., Richardson A. L., Campbell P. J., Futreal P. A., Stratton M. R. The landscape of cancer genes and mutational processes in breast cancer. Nature. (2012);486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Evers L., Perez-Mancera P. A., Lenkiewicz E., Tang N., Aust D., Knosel T., Rummele P., Holley T., Kassner M., Aziz M., Ramanathan R. K., Von Hoff D. D., Yin H., Pilarsky C., Barrett M. T. STAG2 is a clinically relevant tumor suppressor in pancreatic ductal adenocarcinoma. Genome Med. (2014);6:9. doi: 10.1186/gm526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim M. S., An C. H., Yoo N. J., Lee S. H. Frameshift mutations of chromosome cohesion-related genes SGOL1 and PDS5B in gastric and colorectal cancers with high microsatellite instability. Hum. Pathol. (2013);44:2234–2240. doi: 10.1016/j.humpath.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 62.Birkenbihl R. P., Subramani S. Cloning and characterization of rad21 an essential gene of Schizosaccharomyces pombe involved in DNA double-strand-break repair. Nucleic Acids Res. (1992);20:6605–6611. doi: 10.1093/nar/20.24.6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sjogren C., Nasmyth K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr. Biol. (2001);11:991–995. doi: 10.1016/S0960-9822(01)00271-8. [DOI] [PubMed] [Google Scholar]
- 64.Strom L., Lindroos H. B., Shirahige K., Sjogren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell. (2004);16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 65.Strom L., Karlsson C., Lindroos H. B., Wedahl S., Katou Y., Shirahige K., Sjogren C. Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science. (2007);317:242–245. doi: 10.1126/science.1140649. [DOI] [PubMed] [Google Scholar]
- 66.Bauerschmidt C., Arrichiello C., Burdak-Rothkamm S., Woodcock M., Hill M. A., Stevens D. L., Rothkamm K. Cohesin promotes the repair of ionizing radiation-induced DNA double-strand breaks in replicated chromatin. Nucleic Acids Res. (2010);38:477–487. doi: 10.1093/nar/gkp976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tittel-Elmer M., Lengronne A., Davidson M. B., Bacal J., Francois P., Hohl M., Petrini J. H., Pasero P., Cobb J. A. Cohesin association to replication sites depends on rad50 and promotes fork restart. Mol. Cell. (2012);48:98–108. doi: 10.1016/j.molcel.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mannini L., Menga S., Tonelli A., Zanotti S., Bassi M. T., Magnani C., Musio A. SMC1A codon 496 mutations affect the cellular response to genotoxic treatments. Am. J. Med. Genet. (2012);158A:224–228. doi: 10.1002/ajmg.a.34384. [DOI] [PubMed] [Google Scholar]
- 69.Bailey M. L., O’Neil N. J., van Pel D. M., Solomon D. A., Waldman T., Hieter P. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Mol. Cancer Ther. (2014);13:724–732. doi: 10.1158/1535-7163.MCT-13-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]