The Zinc Finger Transcription Factor ZFX is Required for Maintaining the Tumorigenic Potential of Glioblastoma Stem Cells (original) (raw)
. Author manuscript; available in PMC: 2015 Mar 4.
Published in final edited form as: Stem Cells. 2014 Aug;32(8):2033–2047. doi: 10.1002/stem.1730
Abstract
Glioblastomas are highly lethal brain tumors containing tumor-propagating glioma stem cells (GSCs). The molecular mechanisms underlying the maintenance of the GSC phenotype are not fully defined. Here we demonstrate that the zinc finger and X-linked transcription factor (ZFX) maintains GSC self-renewal and tumorigenic potential by up-regulating c-Myc expression. ZFX is differentially expressed in GSCs relative to non-stem glioma cells and neural progenitor cells (NPCs). Disrupting ZFX by shRNA reduced c-Myc expression and potently inhibited GSC self-renewal and tumor growth. Ectopic expression of c-Myc to its endogenous level rescued the effects caused by ZFX disruption, supporting that ZFX controls GSC properties through c-Myc. Furthermore, ZFX binds to a specific sequence (GGGCCCCG) on the human c-Myc promoter to up-regulate c-Myc expression. These data demonstrate that ZFX functions as a critical upstream regulator of c-Myc and plays essential roles in the maintenance of the GSC phenotype. This study also supports that c-Myc is a dominant driver linking self-renewal to malignancy.
Keywords: Cancer Stem Cell, Glioblastoma, ZFX, c-Myc, Self-renewal, Tumorigenesis
Introduction
Glioblastoma (GBM) is the most prevalent and lethal type of primary brain tumor with a median survival less than 15 months [1, 2]. Despite recent therapeutic advances in the treatment of other cancers, current therapies for GBM remain largely ineffective due to therapeutic resistance and rapid tumor recurrence [3]. GBM displays remarkable cellular heterogeneity and hierarchy with self-renewing glioma stem cells (GSCs) at the apex of the hierarchical organization [4-7]. GSCs are functionally defined by their self-renewal potential, multi-lineage differentiation potency and in vivo tumorigenic capacity to propagate tumors that recapitulate the cellular hierarchy and tissue architecture of the parental tumor [8-14]. We and others have shown that GSCs actively interact with niches to promote tumor angiogenesis, cancer invasion, immune evasion, and resistance to current therapies [8, 15-19]. Recently, we found that GSCs are able to generate the majority of vascular pericytes to support vessel function and tumor growth [20]. Thus, therapeutic targeting of GSCs may suppress malignant behaviors and significantly improve GBM treatment. To target GSCs specifically and effectively, it is critical to understand better the molecular mechanisms underlying the maintenance of GSC self-renewal and tumorigenic potential.
The maintenance of stem cell properties in embryonic stem cells (ESCs), adult stem cells and cancer stem cells is controlled by similar but distinct sets of key transcription factors. An ESC-like gene expression signature has been found in poorly differentiated aggressive human tumors including GBMs that show preferential overexpression of a subset of the stem cell transcription regulators [21]. Molecular targets of c-Myc, SOX2, Nanog and Oct4 are more frequently overexpressed in poorly differentiated tumors than in well-differentiated tumors [21]. The presence of an ESC-specific signature in human malignant tumors suggests that the ESC expression signature contributes to the stem cell-like phenotype in cancers [21, 22]. Although the molecular links between stemness and neoplasia are not fully defined, it has been demonstrated that a c-Myc network actually accounts for similarities between ESC and cancer cell transcription programs [23], suggesting that the apparent similarity of cancer and ESC signatures largely reflects the pervasive nature of c-Myc regulatory networks. A genetic study also validated c-Myc as a critical regulator linking the cooperative actions of p53 and Pten in the control of normal and malignant stem cell differentiation, self-renewal and tumorigenic potential [24]. Thus, c-Myc appears to be the dominant driver connecting the stem cell phenotype to malignancy. While c-Myc is required for maintaining the proliferation and self-renewal of ESCs [25, 26], c-Myc also plays crucial roles in maintaining proliferation of cancer cells [27]. Upregulation or increased activation of c-Myc has been found in 70% of human cancers [28]. A recent study demonstrated that stabilization of c-Myc caused by mutations in the ubiquitin ligase FBXW7 drives leukemia-initiating activity of T cell acute lymphoblastic leukemia (T-ALL) [29]. Moreover, c-Myc is critically important for the maintenance of GSC self-renewal and proliferation in GBMs [24, 30]. c-Myc is preferentially expressed in GSCs relative to non-stem tumor cells in GBMs [30]. However, the transcriptional control of c-Myc expression in GSCs is poorly understood. A better understanding of the transcriptional regulation of c-Myc in GSCs may provide new insights into the molecular link between stemness and malignancy in GBM. In this study, we identified the zinc finger and X-linked transcription factor (ZFX) as a critical upstream regulator of c-Myc expression in GSCs.
ZFX is a critical transcription factor that controls the self-renewal potential of both human and murine ESCs [31-33]. ZFX and c-Myc belong to the same regulatory network of ESC self-renewal [25, 34, 35]. Mammalian ZFX protein contains several functional domains including an acidic transcriptional activation domain, a nuclear localization signal (NLS) sequence, and a DNA binding domain consisting of 13 C2H2 zinc fingers [36]. Recent studies demonstrated that ZFX also plays important roles in the nucleosome [37, 38]. Deletion of ZFX in mice impaired the self-renewal capacity of ESCs and hematopoietic stem cells (HSCs) [31]. ZFX is also required for the maintenance of the self-renewal potential of human ESCs [32]. ZFX overexpression promotes ESC self-renewal by inhibiting cell differentiation [31, 32]. In addition, loss of ZFX resulted in a significant increase of apoptotic cell death and the upregulation of stress inducible factors in ESCs [31]. These studies suggest that ZFX plays a crucial role in the maintenance of ESCs. Interestingly, ZFX has been implicated in the Myc-mediated tumorigenesis of liver cancer [39], suggesting that ZFX may also play a critical role in the pathogenesis of human cancers. However, the functional significance of ZFX in the maintenance of GSCs in GBM has not been defined, although ZFX has been shown to be associated with proliferation and survival of glioma cell lines [40, 41]. Thus, defining the potential role of ZFX in the maintenance of self-renewal and tumorigenic potential of GSCs may offer new insights into the molecular mechanisms underlying the regulation of the stem cell-like phenotype. In this study, we interrogated the potential role of ZFX in the maintenance of the GSC phenotype. We demonstrated that ZFX controls c-Myc expression to maintain the self-renewal and tumorigenic potential of GSCs. Moreover, we found that ZFX binds to a specific sequence (GGGCCCCG) on the human c-Myc promoter to mediate the upregulation of c-Myc in GSCs.
Materials and Methods
Isolation of GSCs and Non-stem Tumor Cells from GBM
De-identified GBM surgical specimens were collected from the Brain Tumor and Neuro-Oncology Center at Cleveland Clinic or University Hospitals of Case Western Reserve University in accordance with an Institutional Review Board-approved protocol. GSCs and matched non-stem tumor cells were isolated from GBM patient specimens or xenografts through cell sorting and functionally characterized as previously described [8, 20, 42, 43] with minor modification. Briefly, GBM tumors were disaggregated using the Papain Dissociation System (Worthington Biochemical) according to the manufacturer’s instructions. Isolated cells were recovered in stem cell medium (Neurobasal-A medium with B27 supplement, 10 ng/ml EGF and 10 ng/ml bFGF) for at least 6 hours to allow re-expression of surface markers, and then sorted by fluorescence-activated cell sorting (FACS) or magnetic cell sorting for GSCs using at least two surface markers (CD15/CD133). The enriched GSCs were maintained in the stem cell medium and the identity was confirmed by SOX2 and OLIG2 expression and the activation of SOX2 promoter-driven GFP expression. The cancer stem cell phenotype of GSCs was validated by functional assays of self-renewal (serial neurosphere formation at the clonal density), in vitro induction of differentiation and tumor propagation (in vivo limiting dilution assay) as previously described [42, 43]. After validation of the cancer stem cell phenotype by these functional assays, the sorted GSCs (D456, T387, T4302, T4121, T3359, T3691, T3742, CW702, CW1336 and CCF2170) were used for the in vitro and in vivo experiments.
Culture of Human Neural Progenitor Cells (NPCs)
Human NPC lines derived from fetal brains (Lonza) were cultured and maintained as suspension cultures or propagated as monolayers attached on the BD stem cell Matrigel-coated dishes or coverslips in Neurobasal stem cell media supplemented with B27 and EGF/bFGF (20 ng/ml/each). These NPC lines have been validated for their potential to differentiate into neurons, astrocytes and oligodendrocytes upon induction as demonstrated in our previous studies [43, 44].
Immunoblot (IB), Immunofluorescence (IF) and Immunohistochemistry (IHC)
IB analysis of protein expression and IF staining of cells or tissue sections were performed as described [8, 20, 43]. Specific antibodies against ZFX (Cell Signaling or Sigma-Aldrich), c-Myc (Cell Signaling or Santa Cruz), SOX2 and OLIG2 (Millipore or Santa cruse), Flag and α-tubulin (Sigma-Aldrich), GFAP (Biolegend or BD Bioscience), MAP2 (Covance), TUJ1 (Covance), CD31 (Dako), Tbx3 (Abcam) and Tcl1 (Norvus) were used for IB analysis or IF staining. IHC staining on tumor and normal tissue sections was performed with an ABC kit using DAB (3,3’-Diaminobenzine) detection (Vector Lab) as previously described [15, 43]. A specific antibody against ZFX (Sigma-Aldrich) was used for IHC on paraffin embedded tumor and normal brain tissue sections including tissue microarrays (US Biomax).
Cell Differentiation Assay in Vitro
GSCs or NPCs were induced to differentiate in vitro by the addition of serum or withdrawal of growth factors as previously described [8, 43, 44]. The cells were cultured on the Matrigel-coated coverslips or dishes and then induced to differentiate in the serum-containing medium (10% FBS in α-MEM) or in Neurobasal-A medium with B27 but without EGF and bFGF. At indicated time points, cells were harvested for immunoblot analysis or fixed for immunofluorescence staining.
DNA Constructs and Lentiviral Transfection
Lentiviral clones expressing ZFX shRNA (shZFX) or NT shRNA (SHC002) were acquired from Sigma-Aldrich. Two of five shZFX clones (sh09 and sh10) displayed high efficiency of knockdown (75-85% reduction) and were used for all related experiments. A lentiviral construct expressing Flag-tagged ZFX (Flag-ZFX) or Flag-tagged c-Myc (Flag-Myc) was generated by cloning the human ZFX or c-Myc2 open reading frame (ORF) with the N-terminal Flag sequence into the pCDH-MCS-T2A-Puro-MSCV vector (System Biosciences). Viral particles were produced in 293T cells with the pACK set of helper plasmids (System Biosciences) in stem cell media. Viral stocks were concentrated by precipitation with PEG-8000 and titered according to the manufacture’s instructions.
Intracranial Tumor Formation and in Vivo Bioluminescence Imaging
Intracranial transplantation of GSCs to establish GBM xenografts was performed as previously described [8, 42, 43]. To monitor tumor growth in living animals, all GSCs used for the animal studies were transduced with firefly luciferase through lentiviral infection. GSCs expressing firefly luciferase were then transduced with NT shRNA or shZFX, and/or Flag-Myc (expression similar to endogenous level) or control vector through lentiviral infection twice at 24 hour intervals. 24 hours after the second transduction, viable cells (5×103 cells/animal or as indicated) were intracranially transplanted into athymic immunocompromised mice. For the survival experiments, animals were maintained until manifestation of neurological signs or for 180 days, whichever occurred sooner. To examine tumor growth, mouse brains implanted with T387 GSCs expressing shZFX or NT shRNA, and/or Flag-c-Myc or control vector were monitored by bioluminescence imaging or harvested simultaneously after GSC transplantation. For the imaging analysis, animals were administrated with D-luciferin intraperitoneally and anesthetized with isoflurane. The tumor luciferase images were captured by using the IVIS imaging system (Xenogen-100). All animal procedures conformed to the Cleveland Clinic IACUC approved protocol.
C-Myc Promoter Reporter Assay
To identify the activating element of c-Myc promoter in response to ZFX, the DNA fragments containing different regions of the human c-Myc promoter were cloned into a luciferase report vector pGL4.16-Luc2CP-Hygro (Promega). Mutagenesis of the potential ZFX binding site was performed using site-directed PCR mutagenesis and confirmed by sequencing. The luciferase reporter construct or the ZFX-expression construct was introduced into GSCs by using the specific transfection reagent X-tremeGENE (Roche). Two days after transfection, the luciferase activity under the control of different promoter regions was measured using a bioluminescence reader.
ZFX Promoter and the Promoter-driven GFP Expression
Human ZFX promoter (-774/+228) sequence (Switch-gear Genomics) was cloned from human genomic DNA by PCR using the following primers and then confirmed by sequencing. ZFXpro-Forward: 5′-TTC AAA ATT TTA TCG ATA AAA TCA GTT TGG CTC TGA CTG C; ZFXpro-Reverse: 5′-CCG GGC CTA ACT GCG GCC TCC. The cloned ZFX promoter was inserted into pCDH-EF1-MCS-T2A-Puro lentiviral vector to replace the internal EF1 promoter, and GFP open reading frame was cloned into MCS sites to construct the ZFX promoter-driven GFP expression.
Chromatin Immunoprecipitation (CHIP) Assay
The CHIP assay was performed using the EZ-CHIP kit (Millipore). Briefly, cell lysates were sonicated on ice to obtain DNA fragments of 200-1000 bps. The potential ZFX-bound DNA complex was immunoprecipitated with the anti-ZFX antibody (Cell Signaling) and protein G agarose beads. After the agarose beads were washed for four times, the ZFX-bound DNA fragments were eluted and then amplified by real-time quantitative PCR.
TUNEL Assay
TUNEL assays detecting apoptotic cell death on tumor sections were performed with the ApopTag Plus Peroxidase In Situ Apoptosis Kit (Millipore) according to the manufacturer’s instructions. The intensity of apoptotic cells were quantified with Image J software.
Quantitative RT-PCR
Total cellular RNA was isolated with the RNeasy Kit (Qiagen) and reverse transcribed into cDNA using the Superscript III Kit (Invitrogen). Real time PCR was performed on an Applied Biosystems 7900HT cycler using SYBR-Green Mastermix (SA Biosciences) with the following primers: c-Myc-Forward: 5’-TCA AGA GGC GAA CAC ACA AC; c-Myc-Reverse: 5’-GGC CTT TTC ATT GTT TTC CA. ZFX-Forward: 5’-GGC AGT CCA CAG CAA GAA C; ZFX-Reverse: 5’-TTG GTA TCC GAG AAA GTC AGA AG. GAPDH-Forward: 5’-GGT CTC CTC TGA CTT CAA CA; GAPDH-Reverse: 5’-GTG AGG GTC TCT CTC TTC CT. SOX2-Forward: 5’-CAA GAT GCA CAA CTC GGA GA; SOX2-Reverse: 5’-CGG GGC CGG TAT TTA TAA TC. OLIG2-Forward: 5’-GGT AAG TGC GCA ATG CTA AGC TGT; OLIG2-Reverse: 5’-TAC AAA GCC CAG TTT GCA ACG CAG. c-Myc promoter (-156/-1)-Forward: 5’-GAG CTG TGC TGC TCG CGG CCG C; c-Myc promoter (-156/-1)-Reverse: 5’-TAG ATA AAG CCC CGA AAA CCG GC. c-Myc promoter (-83/+124)-Forward: 5’-AGG GCT TCT CAG AGG CTTG; c-Myc promoter (-83/+124)-Reverse: 5’-CCT ATT CGC TCC GGA TCT C. c-Myc promoter (-1024/-874)-Forward: 5’-CCT CCC TCT CGC CCT AGC CCA G; c-Myc promoter (-1024/-874)-Reverse: 5’-GAC TGA GTC CCC CAA TTT GCT.
Statistical Analysis
All grouped data are presented as mean ± SD (standard deviation). The difference between groups was assessed by one way analysis of variance ANOVA or one way ANOVA on ranks tests. For the in vivo experiments, log rank survival analysis was performed. Sigma Stat Software (Version 3.5) was used for all statistical analyses.
Results
ZFX Is Preferentially Expressed by GSCs in Human GBMs
To determine expression pattern of ZFX in GSC populations and non-stem glioma cells in human GBMs, we performed co-immunofluorescence staining of ZFX with several GSC markers on GBM tumor sections. ZFX was preferentially expressed in the cancer cells co-expressing the GSC markers (SOX2, OLIG2 and CD133) in several human primary GBMs (Fig. 1A-1C and Supplemental Fig. S1A) and GBM xenografts (Supplemental Fig. S1B and S1C). In addition, ZFX-expressing cells were often found proximal to blood vessels marked by CD31 staining in primary GBMs (Fig. 1D), consistent with the fact that GSCs are mainly localized in perivascular niches [16, 45]. To determine whether ZFX is commonly expressed in human GBM tumors, we examined ZFX expression in GBM tissue microarrays containing 74 human primary GBMs. ZFX was expressed in a subpopulation of cancer cells in the majority (69 of 74 cases, 93.2%) of GBM samples (Fig. 1E and Supplemental Table S1). In contrast, ZFX was not detected in human normal brain tissues adjacent to GBM tumors (Fig. 1E) or normal mouse brains (Supplemental Fig. S1D). These data demonstrate that ZFX is preferentially expressed in GSCs in the majority of human GBMs.
Figure 1.
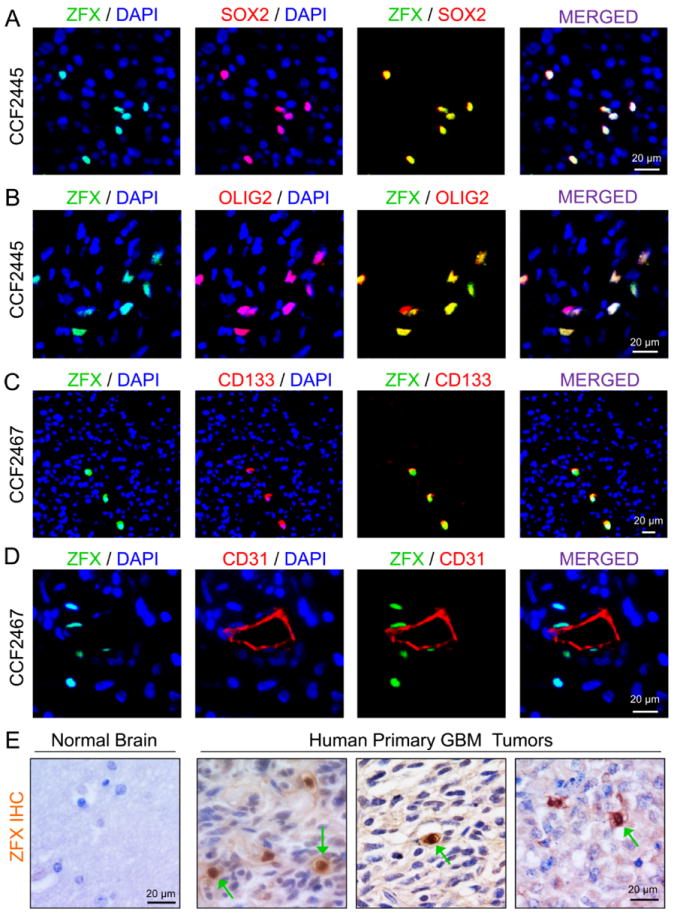
ZFX is preferentially expressed in a fraction of cancer cells expressing GSC markers in primary GBMs.
(A-C): Immunofluorescence (IF) staining of ZFX and GSC markers in primary GBMs. Frozen sections of GBM tumors (CCF2445 and CCF2467) were co-immunostained with specific antibodies against ZFX (in green) and a GSC marker (SOX2, OLIG2 or CD133, in red) and then counterstained with DAPI to show nuclei (in blue). ZFX is co-expressed in the cancer cells expressing the GSC markers.
(D): IF staining of ZFX (in green) and the endothelial cell marker CD31 (in red) in a primary GBM. Sections were counterstained with DAPI (in blue). ZFX-expressing cells are localized in the perivascular niche.
(E): Immunohistochemical (IHC) staining of ZFX in human brain tissue and primary GBMs. Tissue sections were counterstained with hematoxylin to mark nuclei. The positive cells (indicated by green arrows) are shown in brown color. Scale bars represent 20 μm.
To further confirm the specific expression of ZFX in GSCs, we examined ZFX protein levels in matched GSCs and non-stem tumor cells isolated from six primary GBMs or xenografts. GSCs were isolated from GBM tumors through dual labeled fluorescence-activated cell sorting (FACS) using at least two putative GSC surface markers (CD15+/CD133+), and validated by expression of GSC transcription factors (SOX2 and OLIG2) and the ability to activate the SOX2 promoter-driven GFP expression (data not shown). The enrichment for GSCs was further confirmed through three functional assays: (1) self-renewal potential measured by tumorsphere formation at clonal (single cell) density; (2) multipotency tested by cell differentiation induction in vitro; and (3) tumor formation capacity evaluated by limiting dilution assay [8, 15, 42, 43, 46]. Immunoblot analysis showed that ZFX protein levels were commonly higher in GSCs than matched non-stem tumor cells (Fig. 2A). This result was validated by immunofluorescence co-staining of ZFX and several GSC markers (SOX2, OLIG2 and c-Myc) in matched T387 GSCs and non-stem glioma cells (Fig. 2B, 2C and Supplemental Fig. S2A). ZFX was also co-expressed with GSC markers such as OLIG2 in T4121 GSC tumorspheres (Fig. 2D). As GSCs share similar properties with neural progenitor cells (NPCs), we examined ZFX expression in human NPC lines. Immunoblot analysis indicated that ZFX and c-Myc levels in NPCs were much lower than that in GSCs, although both GSCs and NPCs expressed similar levels of SOX2 (Fig. S2B). This result was validated by immunofluorescence co-staining of ZFX, c-Myc and SOX2 in NPCs (Supplemental Fig. S2C and S2D). Consistently, the subventricular zone (SVZ) or dentate gyrus (DG) region that is enriched with NPCs in mouse brain displayed abundant SOX2 expression but low ZFX expression (Supplemental Fig. S2E). Collectively, these data demonstrate that ZFX is preferentially expressed in GSCs relative to non-stem tumor cells and NPCs.
Figure 2.
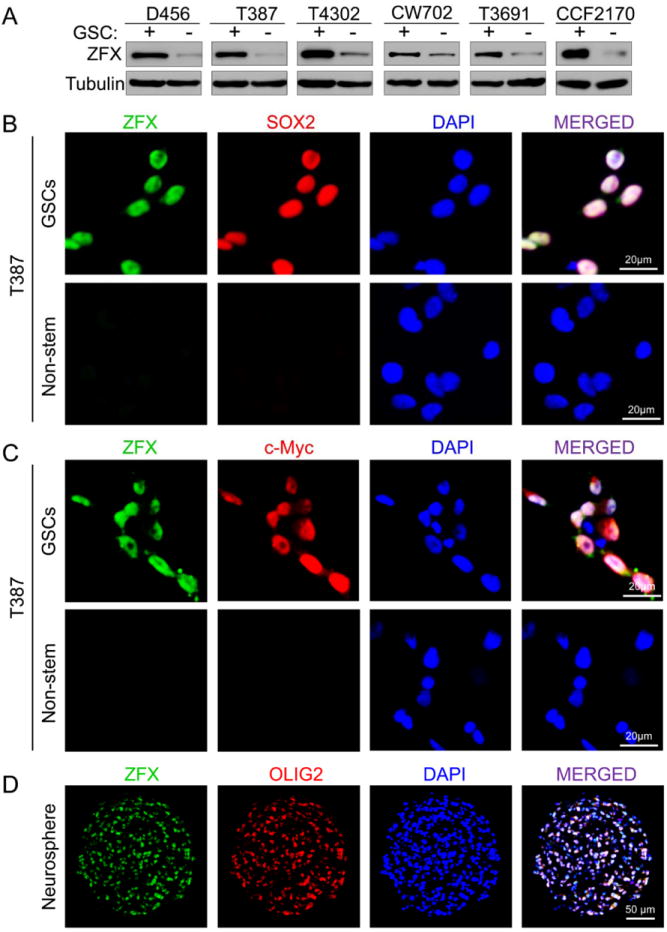
ZFX is differentially expressed in GSCs relative to non-stem tumor cells.
(A): Immunoblot analysis of ZFX protein levels in matched GSCs (+) and non-stem tumor cells (-) isolated from six primary GBM tumors or GBM xenografts.
(B and C): IF staining of ZFX and the GSC markers in matched T387 GSCs and non-stem tumor cells. The sorted GSCs and non-stem tumor cells from a GBM tumor (T387) were co-immunostained with specific antibodies against ZFX (in green) and a GSC marker (SOX2 or c-Myc, in red) and then counterstained with DAPI to show nuclei (in blue).
(D): IF staining of ZFX (in green) and the GSC marker OLIG2 (in red) on frozen sections of T4121 GSC tumorspheres. Sections were counterstained with DAPI to mark nuclei (in blue). ZFX and OLIG2 were co-expressed in the majority of cells in the GSC tumorspheres.
Scale bars represent 20 μm (B and C) and 50 μm (D).
ZFX Is Required for Self-renewal and Proliferation of GSCs
To define the functional significance of the preferential expression of ZFX in GSCs, we examined the effect of ZFX downregulation by shRNA on the maintenance of GSCs. Targeting ZFX in T387 GSCs by lentiviral-mediated shRNA (sh09 or sh10) reduced ZFX protein by 75-85% (Fig. 3A). Disrupting ZFX by the shRNA markedly reduced tumorsphere formation of the GSCs as the sphere size and number dramatically decreased after ZFX knockdown (Fig. 3B-3D). In addition, ZFX knockdown significantly reduced proliferation of the GSCs (Fig. 3E) but showed little effect on non-stem glioma cells and NPCs (Supplemental Fig. S3A and S3B). These results were validated in four GSC populations and matched non-stem tumor cells derived from different GBM tumors (data not shown). To further confirm the functional link between ZFX expression and GSC maintenance, we examined the change in ZFX expression as GSCs differentiate. ZFX and other GSC markers including c-Myc and SOX2 gradually decreased during GSC differentiation, while the expression of the astrocyte marker GFAP and the neuronal marker MAP2 increased (Fig. 3F). Immunofluorescence confirmed that ZFX expression was abolished on day 6 after induction of GSC differentiation (Fig. 3G). To further trace ZFX promoter activity in GSCs and the differentiated cells, we cloned the human ZFX promoter and constructed a ZFX promoter-driven GFP expression (ZFXpro-GFP) in a lentiviral vector (Supplemental Fig. S3C). Most cells in the tumorspheres derived from the GSCs transduced with the ZFXpro-GFP expressed high levels of GFP (Supplemental Fig. S3D), while the differentiated cells derived from the ZFXpro-GFP-GSCs largely lost GFP expression but increased the fraction of cells expressing GFAP or TUJ1 (a neuronal marker) after induction of differentiation (Supplemental Fig. S3E and S3F), suggesting that the ZFX promoter is activated in GSCs but silenced in the differentiated cells. These data demonstrate that ZFX is required for maintaining the self-renewal and proliferation potential of GSCs.
Figure 3.
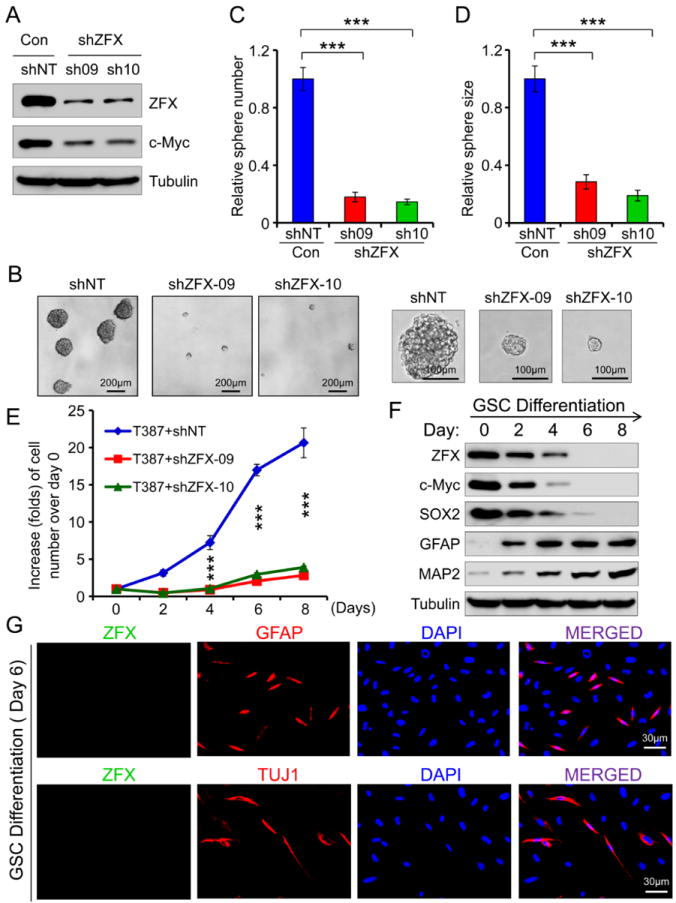
ZFX is required for the maintenance of GSC self-renewal and growth in vitro.
(A): Immunoblot analysis of ZFX and c-Myc in the GSCs transduced with ZFX shRNA (sh09 and sh10) or non-targeting (NT) shRNA (control). ZFX knockdown by shRNA reduced c-Myc expression in the GSCs.
(B-D): Tumorsphere formation of T387 GSCs expressing shZFX (sh09 or sh10) or NT shRNA. Disrupting ZFX impaired GSC tumorsphere formation (B). Quantifications show that ZFX knockdown significantly reduced the GSC tumorsphere number (C) and size (D). ***, p<0.001.
(E): Growth curves of GSCs expressing shZFX or NT shRNA (control). GSCs were transduced with shZFX (sh09 or sh10) or NT shRNA and then measured for cell growth over a time course. Disrupting ZFX significantly inhibited GSC growth. ***, p<0.001.
(F): Immunoblot analysis of ZFX, the GSC markers (c-Myc and SOX2) and the differentiation markers (GFAP and MAP2) during differentiation of GSCs. ZFX, c-Myc and SOX2 gradually decreased while the differentiation markers GFAP (for astrocytes) and MAP2 (for neuronal lineages) increased during the differentiation.
(G): IF staining of ZFX (in green) and GFAP (in red) or TUJ1 (a neuronal marker, in red) in differentiated cells (day 6) derived from GSCs (CCF2170). Nuclei were stained with DAPI (in blue). The differentiated cells lost ZFX expression. Scale bars represent 30 μm (G). Data are means ± SD.
Targeting ZFX Disrupted GBM Tumor Growth and Increased the Survival of Mice Bearing GSC-derived Xenografts
One of defining functional phenotypes of cancer stem cells is their potent tumor propagation ability. To address the functional requirement for ZFX in the maintenance of the tumorigenic potential of GSCs, we examined the effect of ZFX disruption by shRNA on tumor formation of GSCs. The sorted T387 GSCs were transduced with firefly luciferase and ZFX shRNA (shZFX-09 or shZFX-10) or non-targeting shRNA and then transplanted into the brains of immunocompromised mice. Bioluminescence imaging analysis monitoring tumor development indicated that animals bearing the GSCs expressing shZFX markedly delayed the tumor progression relative to the control group bearing the GSCs expressing NT shRNA (Fig. 4A). Necropsy of animals sacrificed simultaneously 21 days after implantation revealed that GSCs transduced with shZFX (sh-09 or sh-10) frequently failed to generate tumors or had reduced tumor size relative to the GSCs expressing NT shRNA (Fig. 4B). Moreover, mice intracranially implanted with the GSCs transduced with shZFX survived significantly longer than the control mice implanted with the GSCs transduced with NT shRNA (Fig. 4C). TUNEL assay demonstrated a significant increase in apoptotic cell death occurred in the infrequent GBM tumors derived from the GSCs expressing shZFX (Fig. 4D and 4E). Taken together, these data demonstrate that ZFX is required for maintaining the tumorigenic capacity of GSCs in vivo, suggesting that ZFX might be a potential target that may be disrupted to inhibit the tumorigenic potential of GSCs.
Figure 4.
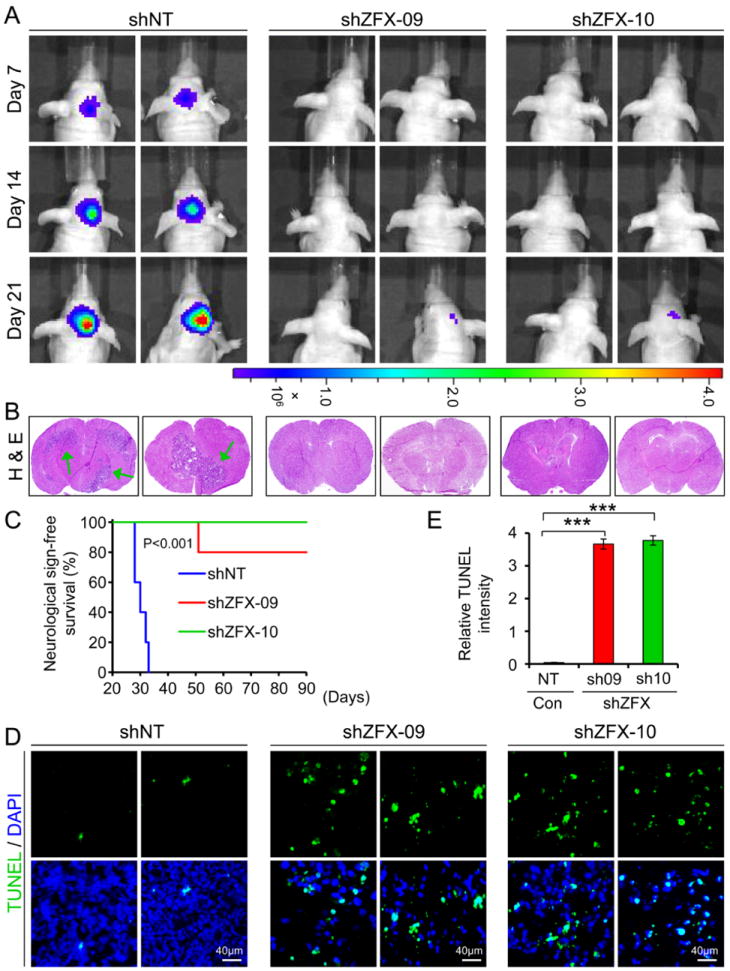
Disrupting ZFX potently inhibited GSC tumor growth and significantly increased survival of animals bearing the xenografts.
(A): In vivo bioluminescent imaging of GBM xenografts derived from luciferase-labeled GSCs expressing shZFX (sh09 or sh10) or NT shRNA. GSCs (T387) were transduced with firefly luciferase and shZFX or NT shRNA through lentiviral infection, and then transplanted into brains immunocompromised mice. Mice bearing the intracranial xenografts were monitored after the GSC transplantation. Representative images at indicated days post-injection are shown.
(B): Representative images of cross sections (hematoxylin and eosin stained) of mouse brains harvested on day 21 post-transplantation of the GSCs expressing shZFX or NT shRNA. Arrows indicated tumors in brains. No tumor or only small tumor was found in brains implanted with the GSCs expressing shZFX.
(C): Kaplan-Meier survival curves of mice implanted with GSCs expressing shZFX (sh09 or sh10) or NT shRNA. Disrupting ZFX significantly increased survival of animals bearing the GSC-derived xenografts. P<0.001.
(D and E): TUNEL assay detecting apoptosis (in green) in GBM tumors derived from the GSCs expressing shZFX or NT shRNA. Nuclei were stained with DAPI (in blue). Quantification (E) shows that a significant increase of apoptotic cell death was found in the xenografts derived from the GSCs expressing shZFX (sh09 or sh10). ***, p<0.001. Scale bars represent 40 μm (D). Data are means ± SD.
Ectopic Expression of ZFX Augmented GSC Proliferation and Tumorigenic Potential
Because ZFX is critical for maintaining GSC proliferation and tumorigenic potential, we examined whether ectopic expression of ZFX impacts GSC proliferation, tumorsphere formation and tumor formation in vivo. Forced expression of ZFX (Flag-ZFX) in T387 GSCs increased c-Myc expression as demonstrated by immunoblot analysis and immunofluorescence (Fig. 5A and 5B). As expected, ectopic expression of ZFX enhanced GSC tumorsphere formation (Fig. 5C-5E) and cell growth in vitro (Fig. 5F). To examine whether forced expression of ZFX enhances the tumorigenic potential of GSCs, we monitored the tumor growth of GBM xenografts derived from the luciferase-labeled GSCs that were transduced with Flag-ZFX or vector control. Bioluminescence imaging of the intracranial tumors indicated that ectopic expression of ZFX significantly augmented tumor growth of the GSC-derived xenografts (Fig. 5G and 5H). As a consequence, ectopic expression of ZFX in the GSCs significantly reduced the survival of mice bearing the GSC-derived xenografts (Fig. 5I). Collectively, these data demonstrate that forced expression of ZFX enhanced GSC proliferation and tumorigenic potential, further supporting that ZFX plays a critical role in maintaining GSC malignant properties.
Figure 5.
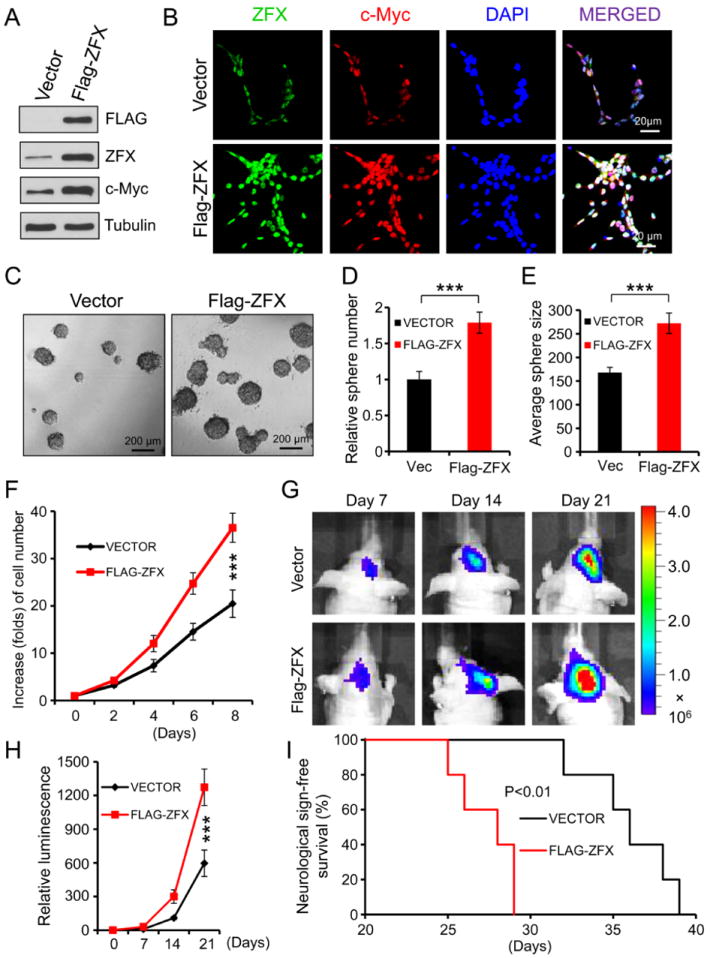
Ectopic expression of ZFX augmented GSC tumorsphere formation and tumor progression.
(A): Immunoblot analysis of ZFX and c-Myc protein levels in the GSCs transduced with Flag-ZFX or vector control. Ectopic expression of ZFX (Flag-ZFX) up-regulates c-Myc expression.
(B): IF staining of ZFX (in green) and c-Myc (in red) in T387 GSCs transduced with Flag-ZFX or vector control. Nuclei were counterstained with DAPI (in blue). Forced expression of ZFX (Flag-ZFX) increased c-Myc levels.
(C-E): Tumorsphere formation of GSCs expressing Flag-ZFX or vector control. Representative images of tumorspheres derived from the GSCs transduced with Flag-ZFX or vector control are shown (C). Quantifications indicate that ectopic expression of ZFX (Flag-ZFX) significantly increased the GSC tumorsphere number (D) and size (E). ***, p<0.004.
(F): Cell growth curves of GSCs expressing Flag-ZFX or vector control. GSCs transduced with Flag-ZFX or vector control were measured for cell growth over a time course (day 0 to day 8). Forced expression of ZFX (Flag-ZFX) significantly enhanced the GSC growth. ***, p<0.001.
(G and H): Bioluminescence imaging of GBM xenografts derived from GSCs transduced with Flag-ZFX or vector control. Sorted GSCs were transduced with firefly luciferase and Flag-ZFX or vector control through lentiviral infection, and then transplanted into brains of immunocompromised mice. Mice bearing the intracranial xenografts were monitored at indicated days after GSC transplantation. Representative images are shown (G). Luminescent quantification (H) indicated that ectopic expression of ZFX significantly augmented the GSC tumor growth in mouse brains. **, p<0.01.
(I): Kaplan-Meier survival curves of mice intracranially implanted with GSCs expressing Flag-ZFX or vector control. Forced expression of ZFX (Flag-ZFX) in the GSCs significantly reduced survival of animals bearing the GSC-derived tumors. *, p<0.01.
Scale bars represent 20 μm (B) 200 μm (C). Data are means ± SD.
ZFX Controls c-Myc Expression in GSCs by Binding to a Specific Sequence on the c-Myc Promoter
To determine the molecular mechanisms underlying ZFX-mediated GSC maintenance, we screened for key regulators of GSCs temporally related to ZFX down-regulation or the downstream targets immediately affected by ZFX knockdown. Surprisingly, Tbx3 and Tcl1, two well-known ZFX downstream targets in ESCs [31], were not affected by ZFX knockdown in T387 GSCs (Supplemental Fig. S4). Instead, we identified c-Myc as an early downstream effector of ZFX in GSCs (Fig. 3A and S4). Real time PCR analysis confirmed that ZFX disruption caused a rapid and significant down-regulation of c-Myc expression in the GSCs before the expression change of other GSC markers (SOX2 and OLIG2) (Fig. 6A). Immunofluorescence validated that ZFX disruption in the GSCs resulted in a rapid decrease of c-Myc protein levels (Fig. 6B). Consistently, forced expression of ZFX (Flag-ZFX) increased expression of c-Myc as demonstrated by both immunoblot analysis and immunofluorescence (Fig. 6C and 6D). To determine whether ZFX directly controls c-Myc expression in GSCs, we cloned different regions of the human c-Myc promoter [47] into a luciferase reporter construct and tested promoter activity in response to ectopic expression of ZFX (Fig. 6E). Forced expression of ZFX significantly increased the reporter activity driven by all promoter fragments containing the P2 region (-147 to +51) but not the promoter fragment lacking the P2 region (Fig. 6E), suggesting that the P2 region of the c-Myc promoter contains a ZFX response element. Sequence analysis indicated the P2 region of the c-Myc promoter contains a putative ZFX-binding sequence (GGGCCCCG) [31, 48]. A mutation of the putative ZFX-binding site (GGGCCCCG → GGATCCTA) on the P2 region abolished the promoter activity in response to ZFX ectopic expression (Fig. 6F and 6G). To determine whether ZFX binds to the P2 region of c-Myc promoter, we performed chromatin immunoprecipitation (ChIP) assay with the anti-ZFX specific antibody. PCR analysis confirmed that the anti-ZFX antibody specifically pulled down the c-Myc promoter fragment containing the P2 region but not the upstream fragment containing the P1 region and lacking the P2 region (Fig. 6H). These data demonstrate that ZFX up-regulates c-Myc expression by binding to a specific sequence (GGGCCCCG) on the P2 region of the c-Myc promoter. As c-Myc is an important factor required for the maintenance of self-renewal and proliferation potential of embryonic stem cells (ESCs) and GSCs, our data suggest that ZFX may control the maintenance of the GSC phenotype through regulation of c-Myc expression.
Figure 6.
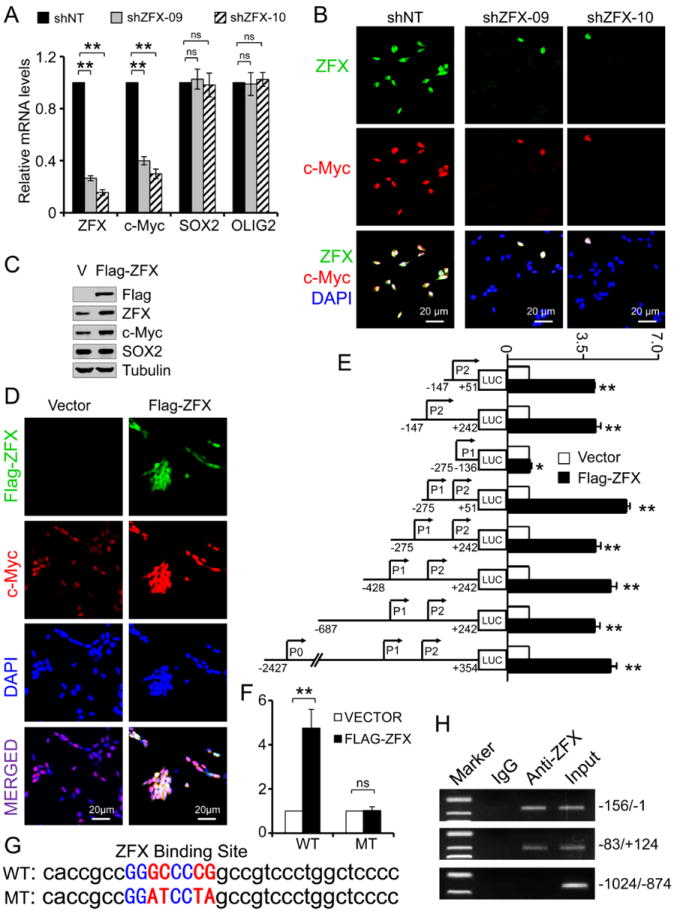
ZFX controls c-Myc expression by binding to a specific sequence on the human c-Myc promoter.
(A): RT-PCR analysis of ZFX, c-Myc, SOX2 and OLIG2 mRNA levels in GSCs expressing shZFX (sh09 or sh10) or NT shRNA. ZFX knockdown rapidly decreased expression of c-Myc but not SOX2 and OLIG2 in the GSCs. **, p<0.01; ns, p>0.05.
(B): IF staining of ZFX (in green) and c-Myc (in red) in GSCs transduced with NT shRNA or shZFX (shZFX-09 or shZFX-10). Nuclei were stained with DAPI (in blue). ZFX disruption by shRNA attenuated c-Myc expression in T387 GSCs.
(C): Immunoblot analysis of c-Myc and SOX2 expression after ectopic expression of ZFX (Flag-ZFX) in the GSCs. Forced expression of ZFX up-regulates c-Myc but not SOX2.
(D): IF staining of Flag-ZFX (in green) and c-Myc (in red) in GSCs transduced with Flag-ZFX or vector control. Nuclei were counterstained with DAPI (in blue). Ectopic expression of Flag-ZFX increased c-Myc levels in the GSCs.
(E): Analyses of c-Myc promoter activity using luciferase reporter assay in GSCs in response to ZFX ectopic expression. DNA fragments containing indicated regions of human c-Myc promoter were cloned into the luciferase reporter system. GSCs were transduced with the luciferase report system and Flag-ZFX or vector control. The relative luciferase activity were measured and quantified. The c-Myc promoter fragment lacking the P2 region lost the response to ZFX ectopic expression. **, p<0.001; *, _p_>0.05.
(F and G): Luciferase reporter assay showing mutations in the conserved ZFX binding site (G) on P2 region of c-Myc promoter abolished the promoter activity in response to ZFX overexpression. WT: wild type; MT: mutated. **, p<0.001; ns, _p_>0.05.
(H): Chromatin immunoprecipitation (ChIP) assay and PCR analysis of ZFX-binding fragments of c-Myc promoter in GSCs. CHIP assay was performed with an anti-ZFX specific antibody or IgG control. DNA fragments of the c-Myc promoter regions bound to ZFX were amplified by PCR and then confirmed by sequencing. Scale bars represent 20 μm (B and D). Data are means ± SD.
Ectopic Expression of c-Myc to Its Endogenous Level Rescued the Phenotype Caused by ZFX Disruption
To confirm c-Myc upregulation as a critical molecular mechanism mediating the effects of ZFX on the maintenance of GSC properties and tumorigenic potential, we examined whether ectopic expression of c-Myc to its endogenous level could rescue the effects caused by ZFX disruption in vitro and in vivo. GSCs derived from GBM tumors (CCF2170 or T387) were transduced with Flag-tagged human c-Myc (Flag-c-Myc) or vector control and shZFX (sh09 or sh10) or NT shRNA. Immunoblot analysis confirmed that the level of ectopic expression of Flag-c-Myc was similar to its endogenous level in GSCs (Supplemental Fig. S5A). The ectopic expression of c-Myc was not affected by ZFX knockdown, while the endogenous c-Myc was markedly reduced by ZFX disruption. Ectopic expression of c-Myc to its endogenous level in the GSCs was able to rescue the impaired tumorsphere formation caused by ZFX knockdown (Supplemental Fig. S5B-S5D) and restored the cell growth affected by ZFX disruption (Supplemental Fig. S5E). Because ZFX disruption in the GSCs significantly inhibited GBM tumor growth and increased animal survival, we examined whether ectopic expression of c-Myc could restore the GSC tumor growth. Expression of c-Myc to its endogenous level indeed rescued GSC tumor growth impaired by ZFX downregulation as demonstrated by in vivo bioluminescence imaging (Fig. 7A and 7B). Consistently, ectopic expression of c-Myc also attenuated the increased survival of mice bearing the GSC-derived tumors expressing shZFX (Fig. 7C). Furthermore, TUNEL assay indicated that expression of c-Myc attenuated the increased apoptosis caused by ZFX knockdown in GSC-derived GBM tumors (Fig. 7D and 7E). These data demonstrate that ectopic expression of c-Myc to its endogenous level in GSCs largely attenuated the effects of ZFX disruption on GSC growth in vitro and tumor progression in vivo, indicating that ZFX controls c-Myc expression to maintain the GSC phenotype and tumorigenic potential.
Figure 7.
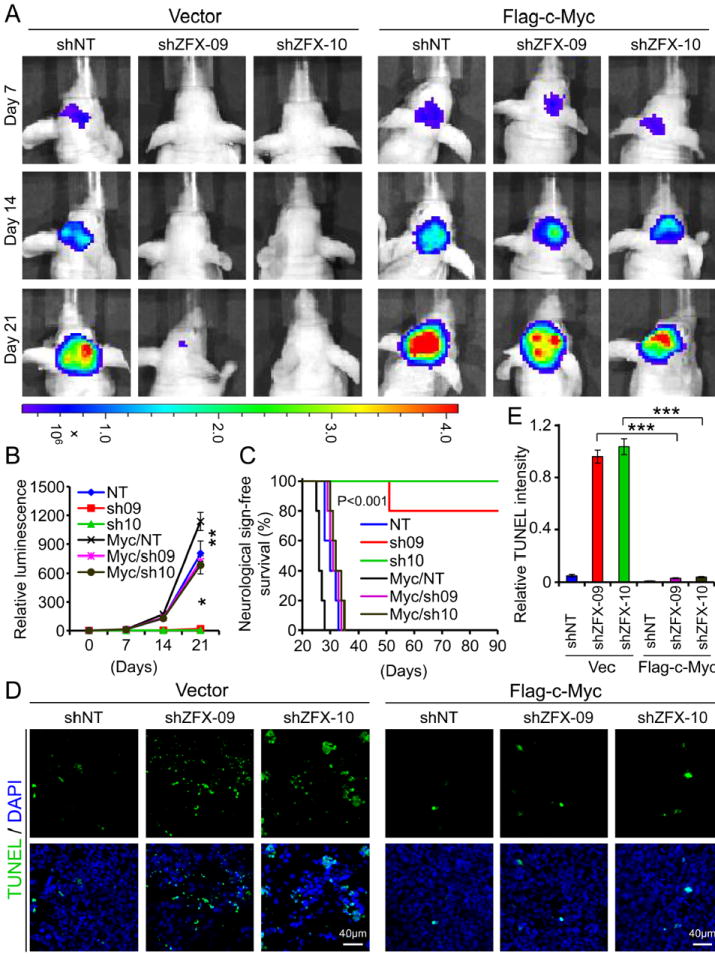
Ectopic expression of c-Myc to its endogenous level was able to rescue growth inhibition of GSC tumors caused by ZFX disruption.
(A and B): In vivo bioluminescent imaging of GBM xenografts derived from luciferase-labeled GSCs transduced with Flag-c-Myc or vector control in combination with shZFX (sh09 or sh10) or NT shRNA. Luciferase-labeled GSCs were transduced with Flag-c-Myc or vector control and shZFX or NT shRNA through lentiviral infection, and then transplanted into brains of immunocompromised mice. Mice bearing the intracranial xenografts were monitored after the GSC transplantation. Representative images at indicated days post-transplantation are shown (A). Quantification of luminescence indicates that ectopic expression of c-Myc restored GSC tumorigenic potential impaired by ZFX disruption (B). *, p<0.001; **, p>0.05.
(C): Kaplan-Meier survival curves of mice implanted with GSCs transduced with Flag-ZFX or vector control in combination with shZFX (sh09 or sh10) or NT shRNA. Mice implanted with the GSCs were maintained until the development of neurological signs. Ectopic expression of c-Myc in the GSCs significantly attenuated the increased survival of mice caused by ZFX disruption in the GSC-derived tumors.
(D and E): TUNEL assay detecting apoptosis (in green) in GBM tumors derived from GSCs expressing Flag-ZFX or vector in combination with shZFX or NT shRNA. Quantification (E) of TUNEL intensity shows that ectopic expression of c-Myc abolished the increased apoptotic cell death caused by ZFX disruption in the GSC-derived xenografts. ***, p<0.001.
Scale bars represent 40 μm (D). Data are means ± SD.
Discussion
The zinc finger transcription factor ZFX plays a critical role in modulating the self-renewal of both murine and human embryonic stem cells (ESCs) [31, 32]. ZFX inactivation in mice resulted in small animal size and reduced number of germ cells in both male and female animals [49]. Reduced ZFX expression led to a loss of self-renewal of human ESCs while ZFX overexpression enhanced the clonogenicity and decreased spontaneous differentiation of hESCs [32]. These studies suggest that ZFX is an essential self-renewal regulator that has a conserved role in the maintenance of human and murine ESCs. Although cancer stem cells (CSCs) are distinct from ESCs, it is possible that CSCs and ESCs share a similar molecular program for maintaining the self-renewal potential [21-23]. Thus, it is important to determine the functional significance of ZFX in the maintenance of the stem cell-like properties in GBMs. As disrupting ZFX in GSCs markedly inhibited GSC proliferation and survival and potently induced apoptotic cell death in the GSC-derived GBM tumor, ZFX also has a role in the maintenance of GSC survival. However, in ESCs, ZFX maintains stem cells by suppressing differentiation and thus targeting ZFX in ESCs induced cell differentiation [31, 32]. It seems that disrupting ZFX in ESCs and GSCs resulted in similar but distinct phenotypes, suggesting that ZFX may mediate through different mechanisms to control the stem cell phenotype in ESCs and GSCs. It is likely that ZFX differentially regulates downstream targets in ESCs and GSCs. ZFX has been shown to regulate expression of a network of downstream targets involved in the self-renewal and proliferation of ESCs [33, 34]. Two of downstream direct targets of ZFX in ESCs are Tbx3 and Tcl1 [31]. However, ZFX knockdown or overexpression in GSCs did not shown an effect on expression of both Tbx3 and Tcl1 (Figure S4). Instead of Tbx3 and Tcl1, c-Myc is the main downstream target of ZFX in GSCs. Our rescue experiments demonstrated that ZFX controls c-Myc up-regulation to maintain the GSC phenotype and tumorigenic potential, as ectopic expression of c-Myc to its endogenous level in GSCs was able to rescue the effects caused by ZFX disruption in vitro and in vivo. Thus, ZFX may signal through distinct downstream targets to exert its differential roles in the maintenance of ESCs and GSCs.
c-Myc has been studied extensively for its instrumental role in proliferation and growth of normal and neoplastic cells. Abnormal expression or regulation of c-Myc has been found in diverse human tumors and often correlates with poor prognosis and advanced stage of malignancies [28, 50]. c-Myc is critically required for the maintenance of pluripotent stem cells [25, 26]. Introduction of c-Myc with other key transcription factors (SOX2/Oct4/Klf4) generates induced pluripotent stem cells (iPSCs) [51, 52]. Moreover, several studies have demonstrated that c-Myc also functions as an oncogenic protein and plays crucial roles in maintaining the self-renewal, proliferation and tumorigenic potential of cancer stem cells including GSCs [24, 30, 53]. A recent study demonstrated that abnormal upregulation of c-Myc caused by the inactivation of its negative regulator FBXW7 (a c-Myc ubiquitin ligase) led to proliferation of leukemia stem cells in T cell acute lymphoblastic leukemia (T-ALL) [29]. Thus, c-Myc serves as a functional link connecting stemness and malignancy. It has been shown that the proliferation, growth and survival of GSCs critically depend on c-Myc expression [24, 30]. Targeting c-Myc impaired proliferation and survival of GSCs and significantly suppressed GSC tumor growth [30]. Consistently, c-Myc is significantly up-regulated in GSCs relative to matched non-stem tumor cells. However, the molecular mechanisms associated with the upregulation of c-Myc in GSCs were not fully understood. Our study revealed that c-Myc expression in GSCs is controlled by ZFX that is preferentially expressed in GSCs. In addition, we found that ZFX binds to a specific sequence on the c-Myc promoter to regulate c-Myc expression. ZFX-regulated c-Myc expression is required for maintaining the GSC phenotype and tumorigenic potential. A recent study demonstrated that ZFX controls leukemia stem cell self-renewal and propagation and prevents differentiation of acute myeloid leukemia (AML) and T-lymphoblastic leukemia (T-ALL) [54]. This study also showed that ZFX contributes to gene induction and transformation by Myc overexpression in myeloid progenitors [54], suggesting a molecular link between ZFX and Myc in different cancer stem or progenitor cells. Interestingly, under hypoxic conditions HIF-2α has been shown to bind to c-Myc promoter and enhance c-Myc expression to promote tumor progression [55, 56]. Our previous study showed that HIF-2α is differentially expressed in GSCs and is preferentially stabilized in GSCs in response to hypoxia [42]. Thus, c-Myc is a critical regulatory node that may mediate different signals for the maintenance of self-renewal, proliferation, survival and tumorigenic potential in varied conditions. As c-Myc is universally important to survival of both normal and neoplastic cells, therapeutic targeting of c-Myc is not practical. However, manipulation of its upstream regulator ZFX may effectively eliminate GSCs with fewer side effects. As disrupting ZFX in vivo potently induced apoptosis, suppressed tumor growth and increased the survival of animals bearing the GBM xenografts, therapeutic targeting of ZFX may significantly improve patient survival.
Conclusion
Our data demonstrate that the zinc finger transcription factor ZFX controls c-Myc expression to maintain the self-renewal and tumorigenic potential of GSCs. ZFX is preferentially expressed in GSCs. Silencing ZFX by shRNA reduced c-Myc expression and potently inhibited GSC self-renewal and tumor growth, indicating that ZFX is required for the maintenance of the GSC phenotype. Ectopic expression of c-Myc to its endogenous level was able to rescue the phenotypes caused by ZFX disruption, suggesting that ZFX maintains GSC properties through c-Myc upregulation. We also found that ZFX binds to a specific sequence (GGGCCCCG) on the human c-Myc promoter to regulate c-Myc expression in GSCs. Our studies reveal that ZFX-mediated c-Myc regulation plays a critical role in connecting stemness to neoplasia. Thus, ZFX is a potential molecular target of GSCs for future development of anti-GBM therapeutics.
Supplementary Material
supplemental figures and legends
Acknowledgments
We thank the Brain Tumor and Neuro-Oncology Centers at Cleveland Clinic and University Hospitals of Case Western Reserve University for providing GBM surgical specimens for this study. We are grateful to members in Dr. Rich’s laboratory for the scientific discussion. We also thank Cathy Shemo and Sage O’Bryant of the Flow Cytometry Core, Judith Drazba of the Imaging Core at Cleveland Clinic Lerner Research Institute for their assistance. This work was supported by the Cleveland Clinic Foundation and a NIH R01 grant (NS070315) to S.B.
Footnotes
Author Contributions
X.F. and S.B.: designed the experiments, analyzed the data and wrote the manuscript; X.F., Z.H., W.Z., and Q.W.: performed the experiments. A.E.S. and R.E.M. :provided GBM surgical specimens; R.E.M.: performed pathological analyses; J.S.Y., G.O. and J.N.R.: provided scientific inputs and helped to edit the manuscript.
Disclosure Of Potential Conflicts Of Interests
The authors declare no potential conflicts of interests.
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1002/stem.1730
References
- 1.Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 4.Bonavia R, Inda MM, Cavenee WK, et al. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 2011;71:4055–4060. doi: 10.1158/0008-5472.CAN-11-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–296. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venere M, Fine HA, Dirks PB, et al. Cancer stem cells in gliomas: identifying and understanding the apex cell in cancer’s hierarchy. Glia. 2011;59:1148–1154. doi: 10.1002/glia.21185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 8.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 9.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 10.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ignatova TN, Kukekov VG, Laywell ED, et al. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 12.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 13.Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 14.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 15.Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 16.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 17.Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei J, Barr J, Kong LY, et al. Glioma-associated cancer-initiating cells induce immunosuppression. Clin Cancer Res. 2010;16:461–473. doi: 10.1158/1078-0432.CCR-09-1983. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Cheng L, Huang Z, Zhou W, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139–152. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, Orkin SH. Embryonic stem cell-specific signatures in cancer: insights into genomic regulatory networks and implications for medicine. Genome Med. 2011;3:75. doi: 10.1186/gm291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Woo AJ, Chu J, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313–324. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng H, Ying H, Yan H, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–1133. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Xu H, Yuan P, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Chu J, Shen X, et al. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 29.King B, Trimarchi T, Reavie L, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153:1552–1566. doi: 10.1016/j.cell.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Wang H, Li Z, et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One. 2008;3:e3769. doi: 10.1371/journal.pone.0003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galan-Caridad JM, Harel S, Arenzana TL, et al. Zfx controls the self-renewal of embryonic and hematopoietic stem cells. Cell. 2007;129:345–357. doi: 10.1016/j.cell.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harel S, Tu EY, Weisberg S, et al. ZFX controls the self-renewal of human embryonic stem cells. PLoS One. 2012;7:e42302. doi: 10.1371/journal.pone.0042302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu G, Kim J, Xu Q, et al. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009;23:837–848. doi: 10.1101/gad.1769609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ouyang Z, Zhou Q, Wong WH. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc Natl Acad Sci U S A. 2009;106:21521–21526. doi: 10.1073/pnas.0904863106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.North M, Sargent C, O’Brien J, et al. Comparison of ZFY and ZFX gene structure and analysis of alternative 3’ untranslated regions of ZFY. Nucleic Acids Res. 1991;19:2579–2586. doi: 10.1093/nar/19.10.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teif VB, Vainshtein Y, Caudron-Herger M, et al. Genome-wide nucleosome positioning during embryonic stem cell development. Nat Struct Mol Biol. 2012;19:1185–1192. doi: 10.1038/nsmb.2419. [DOI] [PubMed] [Google Scholar]
- 38.Gokhman D, Livyatan I, Sailaja BS, et al. Multilayered chromatin analysis reveals E2f, Smad and Zfx as transcriptional regulators of histones. Nat Struct Mol Biol. 2013;20:119–126. doi: 10.1038/nsmb.2448. [DOI] [PubMed] [Google Scholar]
- 39.O’Donnell KA, Keng VW, York B, et al. A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc Natl Acad Sci U S A. 2012;109:E1377–1386. doi: 10.1073/pnas.1115433109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Y, Su Z, Huang Y, et al. The Zfx gene is expressed in human gliomas and is important in the proliferation and apoptosis of the human malignant glioma cell line U251. J Exp Clin Cancer Res. 2011;30:114. doi: 10.1186/1756-9966-30-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu Z, Li K, Xu D, et al. ZFX regulates glioma cell proliferation and survival in vitro and in vivo. J Neurooncol. 2013;112:17–25. doi: 10.1007/s11060-012-1032-z. [DOI] [PubMed] [Google Scholar]
- 42.Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guryanova OA, Wu Q, Cheng L, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011;19:498–511. doi: 10.1016/j.ccr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Z, Wu Q, Guryanova OA, et al. Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat Cell Biol. 2011;13:142–152. doi: 10.1038/ncb2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hjelmeland AB, Lathia JD, Sathornsumetee S, et al. Twisted tango: brain tumor neurovascular interactions. Nat Neurosci. 2011;14:1375–1381. doi: 10.1038/nn.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bao S, Wu Q, Li Z, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68:6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 48.L’Haridon M, Paul P, Xerri JG, et al. Transcriptional regulation of the MHC class I HLA-A11 promoter by the zinc finger protein ZFX. Nucleic Acids Res. 1996;24:1928–1935. doi: 10.1093/nar/24.10.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luoh SW, Bain PA, Polakiewicz RD, et al. Zfx mutation results in small animal size and reduced germ cell number in male and female mice. Development. 1997;124:2275–2284. doi: 10.1242/dev.124.11.2275. [DOI] [PubMed] [Google Scholar]
- 50.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006;16:318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 51.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 53.Radke J, Bortolussi G, Pagenstecher A. Akt and c-Myc induce stem-cell markers in mature primary p53(-)/(-) astrocytes and render these cells gliomagenic in the brain of immunocompetent mice. PLoS One. 2013;8:e56691. doi: 10.1371/journal.pone.0056691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weisberg SP, Smith-Raska MR, Esquilin JM, et al. ZFX Controls Propagation and Prevents Differentiation of Acute T-Lymphoblastic and Myeloid Leukemia. Cell Rep. 2014;6:528–540. doi: 10.1016/j.celrep.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gordan JD, Bertout JA, Hu CJ, et al. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–113. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplemental figures and legends