Neat1 is a p53-inducible lincRNA essential for transformation suppression (original) (raw)
Mello et al. identify Neat1, a ncRNA constituent of paraspeckles, as a p53 target gene that plays a crucial role in suppressing transformation in response to oncogenic signals.
Keywords: p53, lincRNA, Neat1, tumor suppression, pancreatic cancer
Abstract
The p53 gene is mutated in over half of all cancers, reflecting its critical role as a tumor suppressor. Although p53 is a transcriptional activator that induces myriad target genes, those p53-inducible genes most critical for tumor suppression remain elusive. Here, we leveraged p53 ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) and RNA-seq (RNA sequencing) data sets to identify new p53 target genes, focusing on the noncoding genome. We identify Neat1, a noncoding RNA (ncRNA) constituent of paraspeckles, as a p53 target gene broadly induced by mouse and human p53 in different cell types and by diverse stress signals. Using fibroblasts derived from _Neat1_−/− mice, we examined the functional role of Neat1 in the p53 pathway. We found that Neat1 is dispensable for cell cycle arrest and apoptosis in response to genotoxic stress. In sharp contrast, Neat1 plays a crucial role in suppressing transformation in response to oncogenic signals. Neat1 deficiency enhances transformation in oncogene-expressing fibroblasts and promotes the development of premalignant pancreatic intraepithelial neoplasias (PanINs) and cystic lesions in KrasG12D-expressing mice. Neat1 loss provokes global changes in gene expression, suggesting a mechanism by which its deficiency promotes neoplasia. Collectively, these findings identify Neat1 as a p53-regulated large intergenic ncRNA (lincRNA) with a key role in suppressing transformation and cancer initiation, providing fundamental new insight into p53-mediated tumor suppression.
The role of p53 as a critical tumor suppressor is well illustrated by the fact that it is found mutated in more than half of all human cancers (Olivier et al. 2010). The highly penetrant cancer predisposition observed in Li-Fraumeni patients with germline mutations in p53 and in _p53_-null mice further highlights the importance of p53 in tumor suppression (Vousden and Prives 2009; Brady and Attardi 2010). p53 is a cellular stress sensor that responds to signals such as DNA damage and oncogene expression and triggers cell cycle arrest, senescence, or apoptosis to restrain cellular proliferation in the face of these signals. In addition, p53 also regulates other processes, including autophagy and metabolic homeostasis, which may contribute to tumor suppression (Vousden and Prives 2009; Kenzelmann Broz et al. 2013; Kruiswijk et al. 2015).p53 is a transcription factor that drives different biological responses by promoting the expression of a network of target genes. While the target genes involved in promoting cell cycle arrest or apoptosis are well characterized, the target genes responsible for mediating p53 function in tumor suppression remain enigmatic. For example, it has been shown that the target genes critical for DNA damage-induced p53-dependent cell cycle arrest or apoptosis—such as p21, Puma, and _Noxa_—are dispensable for tumor suppression (Brady et al. 2011; Li et al. 2012; Valente et al. 2013). These findings have raised the question of which p53 target genes are therefore the most essential for suppressing tumorigenesis. Recent genomic approaches, including ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) and RNA-seq (RNA sequencing), have helped to identify new p53-regulated genes whose biological functions in the p53 tumor suppression pathway could be explored (Kenzelmann Broz et al. 2013; Sánchez et al. 2014; Léveillé et al. 2015; Younger et al. 2015).
Although studies on genes in the p53 network have focused largely on protein-coding genes, in recent years, the role of p53-inducible noncoding RNAs (ncRNAs), including both microRNAs and large intergenic ncRNAs (lincRNAs), in p53 biological responses has been increasingly recognized. The first of these to be discovered, miR34a, plays an important role in restricting cellular reprogramming (as does p53) and can promote tumor suppression, by enhancing p53 activity through a positive feedback loop (Krizhanovsky and Lowe 2009; Choi et al. 2011; Okada et al. 2014). Subsequently, several p53-inducible lincRNAs, including LincRNAp21, loc285194, PANDA, and DINO, were identified (Huarte et al. 2010; Hung et al. 2011; Liu et al. 2013; Schmitt and Chang 2016; Schmitt et al. 2016). While these lincRNAs have been shown to regulate cellular proliferation and survival in vitro, cancer phenotypes using mice lacking their cognate genes have not been well explored. Analysis of such p53-regulated lincRNAs in clear genetic model systems in vivo could provide key new insight into p53-mediated tumor suppression.
To identify new ncRNAs regulated by p53 and their functional roles in the p53 pathway, we leveraged ChIP-seq and RNA-seq data sets that we generated previously in wild-type mouse embryonic fibroblasts (MEFs) treated with the DNA-damaging agent doxorubicin to activate p53 (Kenzelmann Broz et al. 2013). We identify the lincRNA Neat1 as a direct p53 target gene. NEAT1 was originally identified as an abundant nuclear ncRNA that is an essential component of paraspeckles, nuclear bodies thought to control gene expression through the nuclear retention of hyperedited RNAs (Hutchinson et al. 2007; for review, see Naganuma and Hirose 2013). Here, we interrogated the role of Neat1 in the p53 pathway using Neat1 knockout mice. Interestingly, we found that Neat1 plays an essential role in suppressing both transformation in oncogene-expressing fibroblasts and pancreatic cancer initiation by constraining mutant Kras-induced pancreatic intraepithelial neoplasias (PanINs) and cystic lesions reminiscent of intraductal papillary mucinous neoplasms (IPMNs). Additionally, we found that Neat1 deficiency in oncogene-expressing fibroblasts is associated with global changes in gene expression. Together, these findings identify Neat1 as a p53-regulated lincRNA with a fundamental role in suppressing transformation and cancer initiation, providing important new insight into p53-mediated tumor suppression pathways.
Results
Neat1 is a p53 target gene in various primary and transformed mouse and human cell types
With the goal of understanding how lincRNAs mediate downstream p53 functions, we used ChIP-seq and RNA-seq data sets that we had generated previously using primary MEFs treated with doxorubicin to identify key lincRNAs induced by p53 (Fig. 1A; Kenzelmann Broz et al. 2013). Of the 432 genes that we delineated as bound and modulated by p53, we identified two ncRNAs, Neat1 and Gm5801, although only Neat1 was significantly induced by p53, while Gm5801 was weakly repressed by p53. We found a clear p53-binding peak 1.3 kb upstream of the Neat1 transcription start site, which contained a consensus p53-binding element and to which we could confirm p53 binding by ChIP-qPCR (ChIP combined with quantitative PCR [qPCR] (Fig. 1B). To validate that Neat1 expression is activated by p53 in mouse cells, we performed qRT–PCR analysis of wild-type and _p53_−/− MEFs treated with doxorubicin and found that, indeed, Neat1 is activated by doxorubicin in a p53-dependent manner (Fig. 1C). Neat1 was also induced in a p53-dependent manner by UV-C radiation, another DNA-damaging agent that activates p53, but through a different mechanism (Fig. 1C). Finally, to determine whether p53 regulates Neat1 in other cell types, we examined Neat1 expression in doxorubicin-treated wild-type and _p53_−/− mouse embryonic stem cells (ESCs) and found largely p53-dependent induction by doxorubicin (Fig. 1D). Collectively, these findings confirm that Neat1 is bound and regulated by mouse p53 in multiple settings. These data are consistent with published reports showing that human p53 binds to the NEAT1 locus and activates its expression in human fibroblasts and cancer cell lines (Botcheva et al. 2011; Blume et al. 2015; Adriaens et al. 2016).
Figure 1.

Neat1 is a p53 target gene in mouse and human cells. (A) Experimental outline that led to the discovery of Neat1 as a p53 target gene. Wild-type and p53_−/− MEFs were either left untreated or treated with 0.2 µg/mL doxorubicin for 6 h to generate a list of doxorubicin-regulated p53-dependent RNAs using RNA-seq. Wild-type MEFs were also treated with doxorubicin for 6 h prior to ChIP-seq analysis (Kenzelmann-Broz et al. 2013). Four-hundred-thirty-two genes bound and regulated by p53 were defined, and the annotation of long ncRNAs among these pinpointed Neat1 as a novel p53-inducible target gene. (B) ChIP-qPCR testing for p53 binding at a peak identified in Neat1 by ChIP-seq analysis together with Cdkn1a as a positive control. The percentage of immunoprecipitated DNA relative to input is indicated. p53_−/− MEFs served as a negative control. (C) qRT–PCR analysis of Neat1 expression in wild-type and p53_−/− MEFs treated with 0.2 µg/mL doxorubicin (left) or 20 J/m2 UV light (right) and collected at the indicated time points, normalized to β_-actin. (D) qRT–PCR analysis of Neat1 expression in mouse ESCs treated with 0.2 µg/mL doxorubicin for the indicated times, normalized to β_-actin. (E) Human p53 ChIP-seq profiles (Younger et al. 2015) in primary human fibroblasts reveal a strong p53-binding site in the promoter of NEAT1. The top track shows the p53 ChIP sample, with the carat indicating the “called” peak as determined by DNANexus. The bottom track shows ChIP-seq input reads. The numbers in parentheses indicate the numbers of base pairs in individual half-sites matching the consensus sequence. (F) qRT–PCR analysis of NEAT1 and CDKN1A in primary human fibroblasts expressing shGFP or shp53 8 h after initiating doxorubicin treatment, normalized to β_-ACTIN. (G) Northern blot of doxorubicin-treated (for 24 h) primary human fibroblasts transfected with a scrambled siRNA (siNT), sip53, or either of two different siRNAs against NEAT1 (siN1 and siN2). The numbers below the blots correspond to the expression levels normalized to the RPLPO loading control. (H, left) NEAT1 expression levels by qRT–PCR in two different human ESCs treated with Nutlin3a for 2 d, normalized to β_-ACTIN_. (Right) NEAT1 expression levels by qRT–PCR in human ESCs expressing shGFP or shp53 and left untreated or treated with doxorubicin for the indicated times, normalized to β_-ACTIN_. (I) NEAT1 expression levels by qRT–PCR in human A549 lung cancer cells treated with Nutlin3a for 1 or 2 d, normalized to β_-ACTIN_. (J) qRT–PCR analysis of NEAT1 and CDKN1A in wild-type and p53_-null HCT116 cells treated with 0.2 µg/mL doxorubicin for different times, normalized to β_-ACTIN. (K) Northern blot of wild-type and _p53_-null HCT116 cells after doxorubicin treatment for different lengths of time. RPLPO serves as a loading control. The numbers below the blots correspond to the expression levels normalized to RPLPO. Error bars represent ±SD. (*) P ≤ 0.05; (***) P ≤ 0.001; (n.s.) nonsignificant, based on the two-tailed unpaired Student's _t_-test.
Our own analyses bolstered the identity of NEAT1 as a p53 target gene in human cells. We also identified one strongly enriched p53 peak in our human fibroblast p53 ChIP-seq data, located 1.4 kb upstream of the transcriptional start site of NEAT1. p53 binding occurs in a position analogous to the peak found in mouse Neat1, and we found that this region harbored a site with a near-perfect match to the consensus p53-binding site (Fig. 1E; Younger et al. 2015). Moreover, we found that both the long (NEAT1_2, 23 kb) and short (NEAT1_1, 3.7 kb) isoforms of NEAT1 as well as NEAT1 paraspeckles are induced by doxorubicin in primary human fibroblasts and human ESCs in a p53-dependent manner (Fig. 1F–H; Supplemental. Fig. S1A,B). Furthermore, treatment of human ESCs and A549 lung cancer cells with Nutlin-3a, a p53 stabilizer that does not rely on DNA damage (Vassilev et al. 2004), showed that NEAT1 is induced by p53 in the absence of genotoxic stress (Fig. 1H,I). Finally, treatment of wild-type and _p5_3-null HCT116 colorectal carcinoma cells treated with doxorubicin revealed that expression of both NEAT1 isoforms is doxorubicin-inducible and that this response depends on p53 (Fig. 1J,K). Together, these findings underscore a conserved induction of NEAT1 in response to various p53-activating signals in a diversity of mouse and human cell types, raising the possibility that NEAT1 may play an important role in p53 responses.
Neat1 is dispensable for p53-dependent DNA damage responses
We next sought to elucidate the role of Neat1 in the p53 pathway. To unequivocally establish the contribution of Neat1 to p53 function, we used Neat1 knockout mice (Nakagawa et al. 2011). Notably, while MEFs derived from wild-type mice are completely proficient in forming paraspeckles in the nucleus, no paraspeckles are observed in _Neat1_−/− MEFs, as seen by costaining for Neat1 and the paraspeckle protein Sfpq (Fig. 2A). We first focused on p53 responses to acute genotoxic stress, upon which p53 directs either cell cycle arrest or apoptosis. To assess whether Neat1 is necessary for p53-dependent DNA damage responses, we exposed primary MEFs derived from wild-type, p53−/−, and _Neat1_−/− mice to 5 Gy of ionizing radiation, a classical assay to test for p53-dependent G1 cell cycle arrest. We then evaluated the cell cycle profiles 18 h after irradiation. In these assays, we observed that both wild-type and _Neat1_−/− MEFs underwent G1 cell cycle arrest to an equivalent extent, while the _p53_−/− MEFs failed to undergo G1 arrest, suggesting that Neat1 is dispensable for p53-dependent cell cycle arrest (Fig. 2B).
Figure 2.

Neat1 is dispensable for p53 acute DNA damage responses. (A, left) RNA-FISH using a Quasar 570-labeled complex probe against Neat1 to examine paraspeckles in wild-type and _Neat1_−/− primary MEFs. Nuclei were stained with DAPI. (Right) High-magnification detail of RNA-FISH against Neat1 (using a Quasar 570-labeled probe) and immunostaining of the paraspeckle protein Sfpq in wild-type and _Neat1_−/− primary MEFs. Nuclei were stained with DAPI. (B) Cell cycle arrest analysis in MEFs of different genotypes. (Left) Representative FACS analyses of 5-ethynyl-2 deoxyuridine (EdU)-incorporating and propidium iodide (PI)-stained untreated and irradiated (5 Gy) MEFs of different genotypes. (Right) Quantification of G1 arrest response in MEFs, indicated by the ratio of the S-phase fraction in irradiated cells to the S-phase fraction in untreated cells. n = 3. (C) Apoptosis analysis in MEFs of different genotypes. (Left) Representative FACS analyses of Annexin V and PI staining in E1A;HRasV12 MEFs of each genotype. (Right) Quantification of Annexin V-positive E1A;HRasV12 MEFs of different genotypes (wild type, _Neat1_−/−, and _p53_−/−) after being either left untreated (ut) or treated with 0.2 µg/mL doxorubicin for 12 or 24 h. n = 6. At least two different MEF lines were used in these experiments. Error bars represent ±SD. (*) P ≤ 0.05; (***) P ≤ 0.001; (n.s.) nonsignificant, based on the two-tailed unpaired Student's _t_-test.
The expression of the E1A and HRasV12 oncogenes sensitizes MEFs to apoptosis in response to DNA damage (Lowe et al. 1993). Thus, to test whether Neat1 plays a role in p53-dependent apoptosis, we treated _E1A;HRasV12_-expressing wild-type, _p53_−/−, and _Neat1_−/− MEFs with doxorubicin and evaluated apoptosis using Annexin V/propidium iodide (PI) staining 12 and 24 h later. Consistent with known p53 biology, _E1A;HRasV12_-expressing wild-type MEFs efficiently underwent apoptosis, while the _E1A;HRasV12_-expressing _p53_−/− MEFs remained resistant to apoptosis (Fig. 2C). The levels of apoptosis in _E1A;HRasV12_-expressing _Neat1_−/− MEFs were not statistically significantly different from those in _E1A;HRasV12_-expressing wild-type MEFs, indicating that Neat1 is dispensable for p53-dependent apoptosis. Together, these experiments suggest that, although Neat1 is induced by DNA damage, Neat1 is not necessary for the p53-dependent DNA damage responses of cell cycle arrest and apoptosis, at least in the cellular contexts examined.
Neat1 suppresses transformation in oncogene-expressing fibroblasts
Given that Neat1 is dispensable for p53 responses to genotoxic stress and that recent studies have suggested that distinct transcriptional programs are required for p53 action downstream from acute DNA damage and oncogenic signals (Brady et al. 2011; Bieging et al. 2014), we next sought to establish whether Neat1 might instead be important for p53 responses to oncogenic signals. Specifically, we examined the contribution of Neat1 to transformation suppression in a classic oncogene-expressing fibroblast model. We first investigated whether Neat1 deficiency can increase transformation by comparing _E1A;HRasV12_-expressing wild-type, _p53_−/−, and _Neat1_−/− MEFs. Importantly, Neat1 deficiency also compromises paraspeckle formation in this context, as determined by Neat1/Sfpq costaining (Fig. 3A). We confirmed next that p53 deficiency greatly enhances both clonogenic potential in low-density plating assays and anchorage-independent growth in soft agar assays relative to p53-expressing cells, as reported (Kenzelmann Broz et al. 2013). Interestingly, we found that Neat1 deficiency also significantly increases both clonogenic potential and anchorage-independent growth relative to _Neat1_-proficient cells (Fig. 3B,C), indicating that Neat1 loss promotes transformation similarly to p53 deficiency. To query whether Neat1 is the main conduit for p53-mediated transformation suppression, we attenuated p53 expression by knockdown in _E1A;HRasV12_-expressing wild-type and _Neat1_−/− MEFs. We observed an increase in anchorage-independent growth upon p53 knockdown in _E1A;HRasV12;Neat1_−/− MEFs, suggesting that Neat1 is simply one component downstream from p53 in tumor suppression (Fig. 3D). This finding is consistent with our observation that overexpression of the short isoform of Neat1 (Neat1_1) is sufficient to inhibit transformation in the absence of p53 but not as potently as p53 (Fig. 3E; Supplemental Fig. S2). Finally, to interrogate Neat1 tumor suppressor activity in vivo, we injected E1A;HRasV12;wild-type and _E1A;HRasV12;Neat1_−/− cells subcutaneously into immunocompromised mice. In concordance with our observations in vitro, Neat1 deficiency resulted in an increase in tumor mass and volume (Fig. 3F), indicating that Neat1 also has tumor suppressor activity in vivo. This finding is consistent with the p5325,26 transactivation domain 1 mutant that we generated previously (which activates only a subset of p53 target genes and yet serves as a potent tumor suppressor) being able to activate Neat1 (Fig. 3G; Brady et al. 2011). Collectively, these findings show that Neat1 plays a key role in suppressing transformation of oncogene-expressing fibroblasts, as does p53.
Figure 3.

Neat1 suppresses transformation in oncogene-expressing MEFs. (A) RNA-FISH against Neat1 (using a Quasar 570-labeled probe) and immunostaining of the paraspeckle protein Sfpq in E1A;HRasV12 and _E1A;HRasV12;Neat1_−/− MEFs. Nuclei were stained with DAPI. (B) Clonogenic potential of E1A;HRasV12 MEFs of different genotypes (wild type, _Neat1_−/−, and _p53_−/−) assayed using a low-density plating assay. Colonies were stained with crystal violet. (Left) Representative wells from clonogenic assays are shown. (Right) Dots represent average colony numbers from triplicate samples, integrating results from three different experiments using two to four different MEF lines per genotype. (C) Anchorage-independent growth of E1A;HRasV12 MEFs of different genotypes (wild type, _Neat1_−/−, and _p53_−/−) in a soft agar colony assay. Colonies were stained with Giemsa. (Left) Representative wells are shown. (Right) Dots represent average colony numbers from triplicate samples. Two independent experiments using two to four MEF lines per genotype were performed. (D) Anchorage-independent growth of E1A;HRasV12 MEFs of different genotypes (wild type and _Neat1_−/−) upon the introduction of control shRNA or shRNA against p53. Colonies were stained with Giemsa. (Left) Representative wells are shown. (Right) Dots represent average colony numbers for triplicate samples from two independent experiments using two MEF lines per genotype. (E) Clonogenic potential of _E1A;HRasV12; p53_−/− MEFs after Neat1 or p53 overexpression. pLex-empty served as a negative control. (Left) Representative wells from clonogenic assays are shown. (Right) Dots represent average colony number for triplicate samples from two independent experiments. Independent experiments for B_–_E were performed with both different MEF lines and some MEF lines multiple times to ensure both repetition and representation by multiple MEF lines. (F, left) Average tumor volumes as a function of time in Scid mice injected with _E1A;HRasV12_-expressing wild-type and _Neat1_−/− MEFs. The dots represent the average of tumors from the left and right flanks of a given animal. Two different E1A;HRasV12;Neat1+/+ and three different E1A;HRasV12;Neat1_−/− MEF lines were used, totaling four and six tumors of each genotype, respectively. (Middle) Images of the tumors at the end of the experiment, 22 d after injection. (Right) Tumor weight at day 22. (G) Neat1 expression levels by qRT–PCR in homozygous p53 LSL-wt and p53 LSL-25,26 primary MEFs upon adeno-Cre-induced reactivation of p53, normalized to β_-actin. Error bars represent ±SD. (*) P ≤ 0.05; (***) P ≤ 0.001, based on the two-tailed unpaired Student's _t_-test.
Neat1 suppresses transformation in pancreatic cancer cells
As cancers derived from epithelia or carcinomas represent the majority of human cancers, we next sought to examine the role for Neat1 in a carcinoma model. We focused specifically on pancreatic ductal adenocarcinoma (PDAC), which can be modeled in mice by _Pdx1_-Cre-mediated expression of activated KrasG12D in the pancreas, as we discovered that Neat1 expression is also p53-dependent in KrasG12D-expressing premalignant pancreatic epithelium in an RNA-seq data set that we generated (Fig. 4A; SS Mello, LJ Valente, N Raj, JA Seoane, BM Flowers, J McClendon, KT Bieging-Rolett, J Lee, D Ivanochko, MM Kozak, et al., in prep.). Based on this observation, we hypothesized that Neat1 could be involved in p53-dependent PDAC suppression. To test this idea, we assessed whether Neat1 overexpression could also reduce the tumorigenicity of _p53_-null pancreatic cancer cells using a clonogenic potential assay. Indeed, we found that, as with p53, overexpression of Neat1 in a _p53_−/− pancreatic cancer cell line decreased clonogenic potential, and this was associated with an increase in paraspeckles (Fig. 4B,C; Supplemental Fig. S3). These data indicate that ectopic Neat1 expression can suppress transformation in different cell types.
Figure 4.
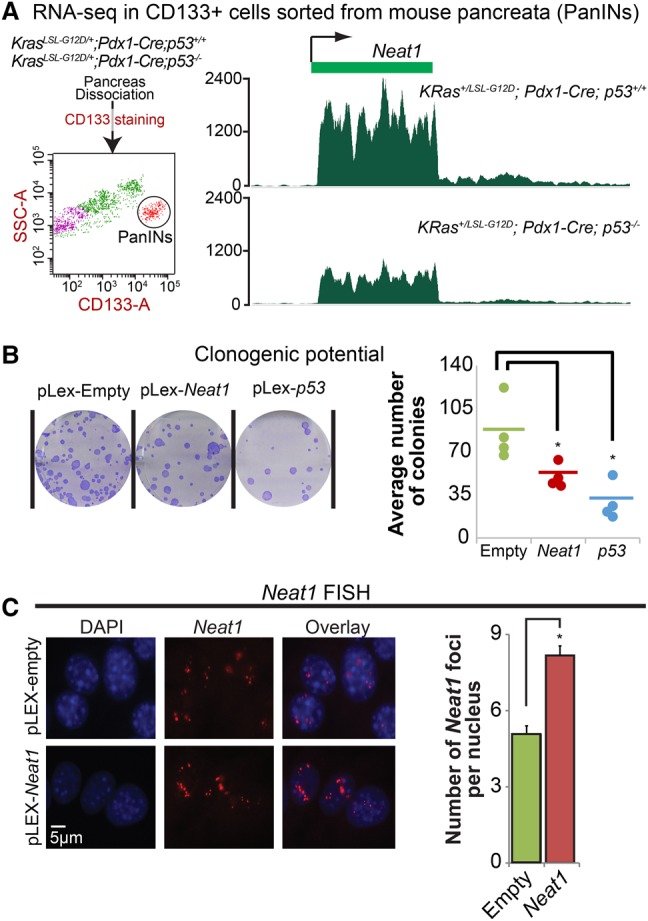
Neat1 is associated with suppression of pancreatic cancer cell growth. (A) Tracks representing Neat1 expression levels in RNA-seq data from CD133+ FACS-sorted mouse pancreata from KRas+/LSL-G12D;Pdx1-Cre;p53+/+ mice (top) and KRas+/LSL-G12D;_Pdx1-Cre;p53_−/− mice (bottom). (B) Clonogenic potential of a PDAC cell line derived from KRas G12D;Pdx1-Cre;p53 fl/fl mice after Neat1 or p53 overexpression. pLex-empty served as a negative control. (Left) Representative wells from clonogenic assays are shown. (Right) Dots indicate average colony number of triplicates from each independent experiment. n = 4. (*) P < 0.05, based on the two-tailed unpaired Student's _t_-test. (C) RNA-FISH for Neat1 in _p53_-null PDAC cells after empty vector or Neat1 transduction. (Left) Representative RNA-FISH images for Neat1. Nuclei were stained with DAPI. (Right) Average number of Neat1 foci ± SEM per nucleus. (*) P ≤ 0.05, based on the two-tailed unpaired Student's _t_-test.
Neat1 deficiency promotes pancreatic cancer initiation
p53 is known to serve as a barrier to PDAC development in KRas+/LSL-G12D mice expressing Cre recombinase under the control of a pancreatic-specific promoter. Given our initial data highlighting the capacity of Neat1 to suppress transformation of pancreatic cancer cells, we sought to determine whether Neat1 can restrain pancreatic cancer initiation in vivo. As RNA-FISH in pancreas tissue showed that the numbers of Neat1 paraspeckles in KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/− mice are significantly lower than in KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/+ mice (Fig. 5A), we used both Neat1 heterozygous and _Neat1-_null mice to determine the role of Neat1 in suppression of pancreatic cancer initiation. Pancreatic cancer can arise through the dedifferentiation of pancreatic acinar cells into ductal-like cells, a process known as acinar-to-ductal metaplasia (ADM), leading to premalignant lesions known as PanINs, which ultimately progress to PDAC (Guerra et al. 2007; Zhu et al. 2007; Morris et al. 2010; Kopp et al. 2012). PDAC has also been reported to arise from cystic lesions known as IPMNs, which are thought to originate from ductal cells (Matthaei et al. 2011; von Figura et al. 2014).
Figure 5.
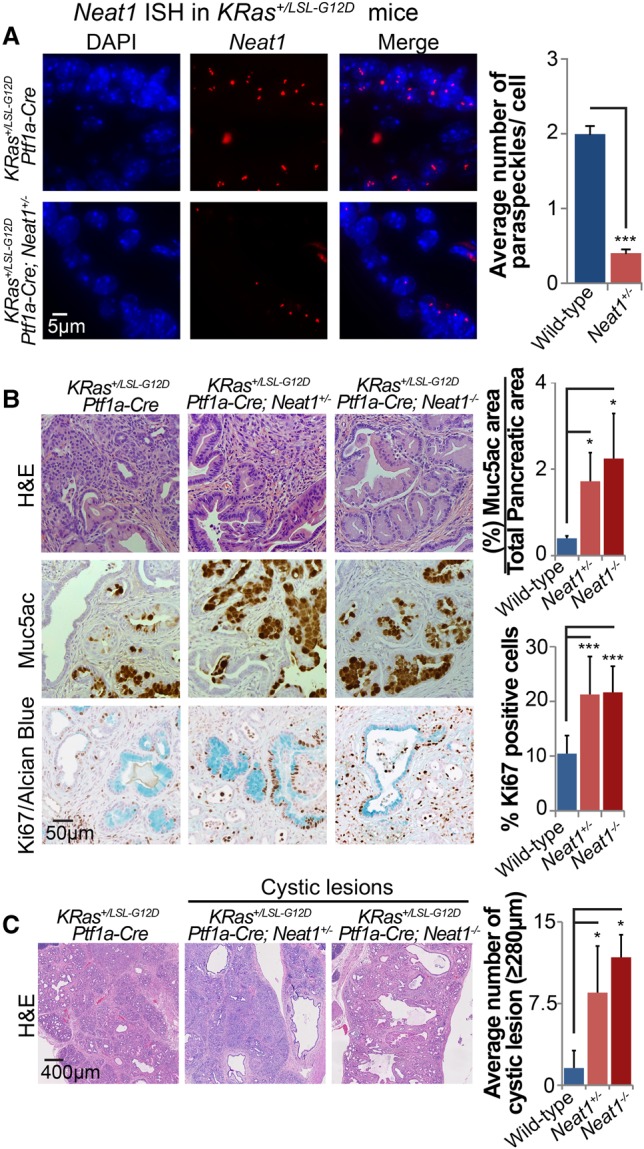
Neat1 suppresses pancreatic cancer initiation in vivo upon pancreatitis. (A) RNA-FISH for Neat1 in pancreas sections of cerulein-treated KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/+ and KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/− mice. (Left) Representative RNA-FISH images for Neat1. Nuclei were stained with DAPI. (Right) Average Neat1 foci ± SD per nucleus (B) Pancreas histology of KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/+ (n = 13), Kras+/LSL-G12D;Ptf1a-Cre;Neat1+/− (n = 6), and Kras+/LSL-G12D;_Ptf1a-Cre;Neat1_−/− (n = 4) mice with acute pancreatitis 7 d after cerulein treatment. (Left) Representative hematoxylin and eosin (H&E), Muc5ac (PanIN marker), and Ki67/Alcian blue costaining (markers of proliferation and PanINs, respectively) of pancreata from cerulein-treated cohorts. (Top right) Average PanIN area ± SD as a percentage of total pancreas area, as determined by Muc5ac quantification. (Bottom right) Average percentage ± SD of proliferating PanIN cells per mouse pancreas, as determined by the counting of at least 1000 Alcian blue cells. (C, left) Representative low-magnification H&E staining of pancreata from cerulein-treated cohorts, evidencing large cystic lesions reminiscent of human IPMN lesions. (Right) Average number of cystic lesions per mouse ± SD. Cystic lesions are defined by size criteria (diameter ≥280 µm). (*) P ≤ 0.05; (***) P ≤ 0.001, based on the two-tailed unpaired Student's _t_-test.
We first examined the consequences of Neat1 deficiency for ADM and PanIN formation in KRas+/LSL-G12D;Ptf1a-Cre mice treated with cerulein, an inducer of pancreatitis that enhances pancreatic cancer initiation by triggering ADM and PanIN formation (Guerra et al. 2011). Interestingly, we found that both KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/− and KRas+/LSL-G12D;_Ptf1a-Cre;Neat1_−/− mouse pancreata displayed dramatic ADM (with a great increase in PanIN burden marked by Muc5ac or Alcian blue positivity) relative to KRas+/LSL-G12D;Ptf1a-Cre; wild-type mouse pancreata (Fig. 5B). These findings underscore a critical function for Neat1 in suppressing pancreatic cancer initiation. The increased propensity for ADM observed with Neat1 deficiency was further supported by an ex vivo ADM transdifferentiation assay using organoid cultures of acini from Kras LSL-G12D;Neat1+/+ and Kras LSL-G12D;_Neat1_−/− mice (Supplemental Fig. S4). In addition to increasing ADM, Neat1 deficiency also increased PanIN burden by enhancing the percentage of Ki67-positive cells in PanINs relative to _Neat1_-expressing PanINs, suggesting that Neat1 also restricts proliferation in PanINs (Fig. 5B). Interestingly, we also observed that both heterozygous and homozygous loss of Neat1 led to the increased formation of cystic lesions lined by mucinous epithelium, some of which were low-grade, while others had clear papillae, histologically similar to human IPMNs presenting gastric-type differentiation (Fig. 5C; Supplemental Figs. S5, S7). Together, these findings suggest that Neat1 loss enhances the formation of multiple types of preneoplastic lesions in the context of Kras activation and pancreatitis.
To analyze the role of Neat1 in maintaining pancreatic homeostasis in aging mice, we next explored whether Neat1 deficiency promotes spontaneous ADM and PanIN formation or the generation of IPMN-like lesions in the absence of pancreatitis. We aged KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/+, KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/−, and KRas+/LSL-G12D;_Ptf1a-Cre;Neat1_−/− mice for 5 mo and found that _Neat1_-deficient mice presented a dramatic loss of normal acinar parenchyma and an accumulation of PanIN lesions relative to KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/+ mice, as indicated by hematoxylin and eosin (H&E) staining and diminished expression of the acinar marker amylase accompanied by positivity for the ductal marker Ck19 (Fig. 6A). Similar to what we observed in cerulein-treated mice at early time points, we found that the loss of Neat1 also increased the percentage of Ki67-positive cells in Ck19-positive lesions of aging mice (Fig. 6B). Interestingly, we again observed a significant increase in mucinous cystic lesions in aging KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/− and KRas+/LSL-G12D;_Ptf1a-Cre;Neat1_−/− mice compared with KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/+ mice (Fig. 6C; Supplemental Figs. S6, S7). While Neat1 deficiency enhanced the formation of both PanIN and IPMN-like preneoplastic lesions (Fig 6), there was no increased cancer predisposition at this time point, suggesting that Neat1 loss promotes efficient ADM and cystic lesion/IPMN formation but that it may take more time or potentially cooperating genetic lesions to fully develop cancer. Together, these findings suggest that Neat1 suppresses the development of pancreatic neoplasias in vivo and does so by restricting ADM, limiting PanIN proliferation, and suppressing the development of cystic lesions.
Figure 6.
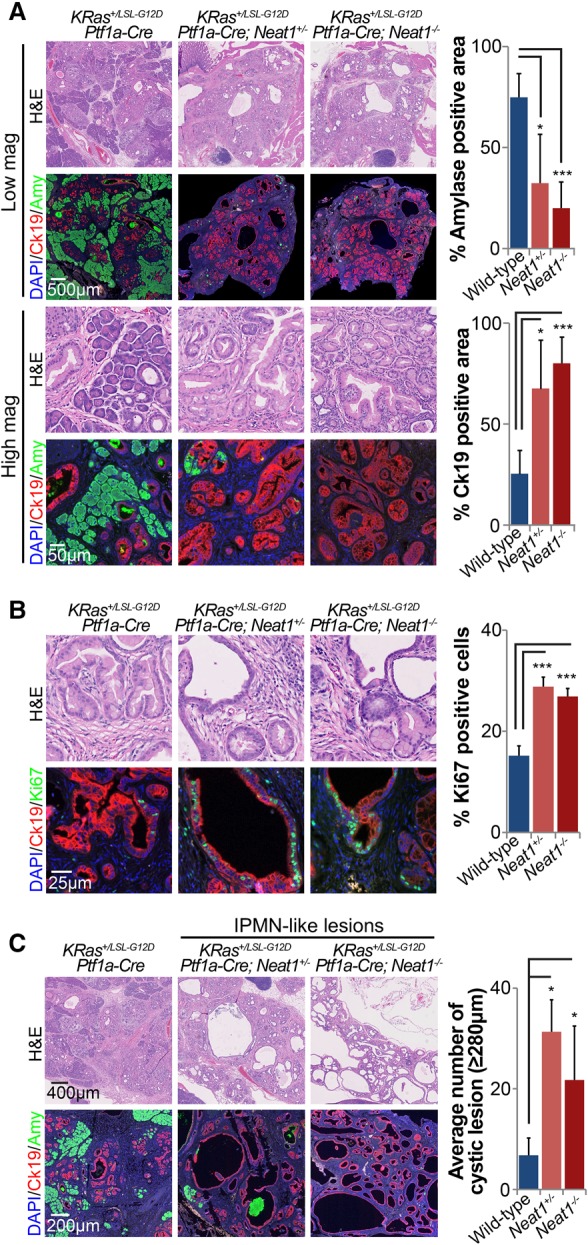
Neat1 suppresses pancreatic cancer initiation in vivo in aging mice. (A) Pancreas histology of 5-mo-old KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/+ (n = 5), Kras+/LSL-G12D;Ptf1a-Cre;Neat1+/− (n = 3), and Kras+/LSL-G12D;_Ptf1a-Cre;Neat1_−/− (n = 4) mice. (Left) Representative H&E, amylase + Ck19 double-immunofluorescence staining (specific markers of acinar cells and epithelial cells of ductal origin, respectively) of pancreata from aged cohorts. (Right) Average amylase and Ck19-positive areas ± SD as a percentage of total pancreas area. (B, left) Representative H&E and Ck19 + Ki67 staining (markers of epithelial lesions and proliferation, respectively) of pancreata from aged cohorts. (Right) Percentage ± SD of proliferating Ck19-positive epithelial cells, as determined by counting at least 1000 cells. (C, left) Representative low-magnification H&E staining and amylase + Ck19 double-immunofluorescence staining of pancreata from aged cohorts, evidencing large cystic lesions reminiscent of human IPMN lesions. (Right) Average number of cystic lesions per mouse ± SD. Cystic lesions are defined by size criteria (diameter ≥280 µm) ± SD. (*) P ≤ 0.05; (***) P ≤ 0.001, based on the two-tailed unpaired Student's _t_-test.
Neat1 deficiency induces global gene expression profile changes
To gain insight into how Neat1 loss might promote both transformation and the development of preneoplastic lesions, we leveraged our tractable in vitro E1A;HRasV12 cell model to analyze genome-wide expression profiles in the presence and absence of Neat1 by RNA-seq. Comparison of gene expression profiles in four E1A;HRasV12;Neat1+/+ and four _E1A;HRasV12;Neat1_−/− MEF lines revealed that loss of Neat1 results in significant expression level changes in ∼1300 genes (_q_-value 0.005) (Fig. 7A). Analysis using Enrichr (Chen et al. 2013; Kuleshov et al. 2016) revealed that most up-regulated genes were related to mechanisms of protein synthesis (data not shown), a process that is commonly enhanced during tumorigenesis (Truitt and Ruggero 2016). Enrichr also identified gene expression programs down-regulated upon Neat1 loss in _E1A;HRasV12_-expressing cells, including nervous system development and function and axon guidance programs, which have been associated previously with cancer development (Fig. 7B,C; Dallol et al. 2003; Chedotal et al. 2005; Biankin et al. 2012; Mi Je et al. 2013; Göhrig et al. 2014). We confirmed the differential expression of various genes in these categories by qRT–PCR analysis in E1A;HRasV12;wild-type and _E1A;HRasV12;Neat1_−/− MEF lines, focusing on genes such as Srgap3, Dll1, Reln, and Plxna4, which have been shown previously to display tumor suppressor activity (Sato et al. 2006; Balakrishnan et al. 2009; Zhang et al. 2011; Castellano et al. 2016). Moreover, given that deficiency in SWI/SNF complex function leads to the formation of IPMNs in mouse models for PDAC (von Figura et al. 2014; Roy et al. 2015), we also queried the status of chromatin remodeling gene expression in _E1A;HRasV12;Neat1_−/− MEFs using the RNA-seq data. Interestingly, we found that expression of various SWI/SNF components, such as Smarca1 and Smarcc2, was reduced in _Neat1_-deficient cells relative to controls (Fig. 7C,D). Importantly, the SWI/SNF complex, including the Smarca1 and Smarcc2 subunits, has been shown to have an extensive role in tumor suppression (Weissman and Knudsen 2009; Wilson and Roberts 2011; Amankwah et al. 2013; Takeshima et al. 2015). Additionally, we found that Ogt, a chromatin modifier involved in histone GlcNAcylation, is down-regulated in _Neat1_−/− cells. To determine whether this compromise in gene expression could also explain the pancreatic phenotypes that we observed, we examined the expression of some of these genes in _Neat1_-deficient pancreata. Interestingly, many, but not all, of these genes display diminished expression with Neat1 loss in the pancreas, suggesting that these gene expression changes could underlie the phenotypes seen in _Neat1-_deficient pancreata (Fig. 7E).
Figure 7.

Neat1 deficiency triggers global gene expression program changes. (A) Scatter plot of the log2 (fold-change) versus the mean reads count per gene, generated using RNA-seq expression profiling data from E1A;HRasV12;wild-type and _E1A;HRasV12;Neat1_−/− MEFs. The dots represent differentially expressed genes according to DEseq2 analysis. (B) Table with Reactome categories found down-regulated in _E1A;HRasV12;Neat1_−/− MEFs. (C) Heat map representing the expression of the top differentially expressed genes in E1A;HRasV12;wild-type and _E1A;HRasV12;Neat1_−/− MEFs. (D) qRT–PCR analysis of genes involved in axon guidance, GABA receptor activation, and chromatin remodeling in E1A;HRasV12;wild-type and _E1A;HRasV12;Neat1_−/− MEFs, normalized to Gapdh. n = 6. (E) qRT–PCR analysis of expression of genes involved in axon guidance, GABA receptor activation, and chromatin remodeling in pancreata of KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/+ and KRas+/LSL-G12D;_Ptf1a-Cre;Neat1_−/− mice 7 d after cerulein treatment, normalized to Gapdh. n = 2. (F) Gene set enrichment analysis heat map of genes contributing to enrichment (left) and enrichment plot of pancreas development genes differentially expressed in E1A;HRasV12;Neat1+/+ and _E1A;HRasV12;Neat1_−/− MEFs (right). False discovery rate is 0.052. (G) qRT–PCR analysis of pancreas development genes found differentially expressed in E1A;HRasV12 MEFs in pancreata of KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/+ and KRas+/LSL-G12D;_Ptf1a-Cre;Neat1_−/− mice 7 d after cerulein treatment, normalized to Gapdh. (*) P ≤ 0.05, based on the one-tailed unpaired Student's _t_-test.
The enhanced formation of ADMs, PanINs, and mucinous cystic lesions in KRasG12D-expressing _Neat1-_deficient mice suggests that these mice are more susceptible to the dedifferentiation process that precedes the generation of preneoplastic lesions. To investigate whether Neat1 has a role in inhibiting dedifferentiation, we used gene set enrichment analysis (GSEA) to analyze our MEF RNA-seq data and test whether Neat1 deficiency impacts the expression of genes involved in pancreas development. We generated a GSEA signature gene set based on a previously published compilation of gene regulatory networks involved in pancreas development and differentiation (Arda et al. 2013). We found that the expression of various pancreas development genes, such as Dll1, Gata6, Bhlha15, Mist1, Foxa1, and Neurod1, is decreased in E1A;HRasV12;Neat1_−/− cells relative to E1A;HRasV12;Neat1+/+ MEFs (Fig. 7F). Furthermore, we found that a subset of these genes—_Bhlha15 and _Sox9_—is also down-regulated in _Neat1_-deficient pancreata (Fig. 7G). Interestingly, Bhlha15 has a key role in pancreas development, as it is necessary for acinar cell differentiation and maintenance of the exocrine pancreas (Shi et al. 2009; Direnzo et al. 2012; Martinelli et al. 2013). Moreover, Sox9, known as a master regulator of the pancreatic developmental program, is responsible for maintaining the embryonic and adult ductal state (Seymour 2014), and a decrease in Sox9-positive cells was associated previously with mucinous cystic lesions (Tanaka et al. 2013). These findings suggest that Neat1 deficiency impacts the regulatory networks involved in the differentiation and maintenance of pancreatic acinar and ductal cells, processes that could directly explain the susceptibility of KRasG12D-expressing _Neat1-_deficient mice to the formation of different types of pancreatic preneoplastic lesions. Collectively, these findings suggest that the ability of Neat1 to globally regulate gene expression—with effects on diverse transcriptional programs—provides a potential mechanism for how Neat1 acts to suppress transformation and tumor initiation.
Discussion
Here, we leveraged ChIP-seq and RNA-seq data from p53 wild-type and _p53_−/− MEFs to identify p53-regulated ncRNAs that might help in understanding downstream components in p53 biological responses. We demonstrate that Neat1/NEAT1 is a bona fide direct p53 target gene in diverse mouse and human cell types and that it is induced in response to different stress signals. Moreover, we queried its biological function downstream from p53 and provided the first evidence that Neat1 is dispensable for p53-dependent cell cycle arrest or apoptosis responses to DNA damage. In dramatic contrast, however, we showed that Neat1 does play a critical role in p53-dependent tumor suppression in oncogene-expressing fibroblast tumors and in suppression of pancreatic cancer initiation. Thus, our findings provide key genetic evidence that Neat1 is a novel component of the p53 tumor suppression program.
Previous studies on NEAT1 in cancer have focused primarily on NEAT1 expression levels during the development of various types of human cancers. In some studies, NEAT1 levels were found to increase during tumorigenesis, and high levels of NEAT1 expression were associated with worse prognosis (Li et al. 2015; He et al. 2016; Ma et al. 2016; Wang et al. 2016). NEAT1 can also promote cell survival and/or proliferation of human cancer cell lines, and Neat1 acts as an oncogene in mice subjected to the DMBA-TPA skin carcinogenesis protocol (Adriaens et al. 2016). However, beyond the suggestion that NEAT1 is an oncogene, other studies have suggested that NEAT1 acts as a tumor suppressor in certain contexts. NEAT1 is down-regulated in some cancers relative to normal tissue, augmented NEAT1_2 levels were shown to predict better overall survival in colorectal cancer patients, and increased NEAT1 levels were associated with enhanced apoptosis in irradiated chronic lymphocytic leukemia cells (Gibb et al. 2011; Blume et al. 2015; Wu et al. 2015). Moreover, NEAT1_2 was found to inhibit cellular proliferation (Wu et al. 2015). These seemingly contradictory results may reflect cell type-specific roles for NEAT1 in tumorigenesis. Importantly, these piecemeal and contradictory findings underscore the critical need for analysis of Neat1 function in a genetically tractable animal model, such as Neat1 knockout mice, to derive an unequivocal understanding of the role of Neat1 in tumorigenesis.
Several molecular functions have been proposed for NEAT1 that could relate to how it serves as a tumor suppressor. First, NEAT1 is essential for the formation and maintenance of paraspeckles, (Clemson et al. 2009; Sasaki et al. 2009; Sunwoo et al. 2009), which have been suggested as sites of nuclear retention for adenosine-to-inosine (A-to-I) edited RNAs, thereby exerting an effect on gene expression at a post-transcriptional level (Prasanth et al. 2005; Chen and Carmichael 2009; Fox and Lamond 2010). Recent reports have also suggested that NEAT1 may act at the post-transcriptional level by interacting with splicing factors and RNA 3′ end processing factors to modulate the proper maturation of precursor mRNAs (West et al. 2014). NEAT1 can also regulate genes at the transcriptional level by sequestering transcriptional regulators into paraspeckles (Hirose et al. 2014) or binding the DNA of active genes (West et al. 2014) to increase active chromatin marks such as histone H3K4 trimethylation and histone H3K9 acetylation in these genes (Chakravarty et al. 2014), suggesting yet another pleotropic mechanism by which it could regulate the gene expression. Consistent with these transcriptional effects, the enhanced transformation and pancreatic neoplasia that we observed in the absence of Neat1 are associated with altered gene expression programs in specific functional categories such as axon guidance, GABA A receptor activation, and chromatin remodeling. Interestingly, mutations in axon guidance genes were associated previously with pancreatic cancer in a study profiling the mutational landscape of this disease (Biankin et al. 2012). Indeed, some of the axon guidance genes with diminished expression upon Neat1 deficiency have activities consistent with tumor suppression: Srgap3, which is involved in the inhibition of anchorage-independent growth; Dll1, which can reduce tumor growth and vascularization; Reln, which can restrain Ras and PI3K; and Plxna4, which is involved in the inhibition of bFGF and VEGF-induced cell proliferation (Kigel et al. 2011; Zhang et al. 2011; Lahoz and Hall 2013; Castellano et al. 2016). It will be interesting to reveal the transcriptional programs most critical for NEAT1 activity in tumor suppression in more detail in future studies.
The mechanisms underlying Neat1/NEAT1 function described above may also provide key insights into how it might modulate cellular responses during tumor suppression. Neat1/NEAT1 has been shown previously to control several aspects of cell behavior by enhancing or decreasing cell division, inhibiting cell death, and increasing migration (Chakravarty et al. 2014; Chen et al. 2015; Choudhry et al. 2015; Wu et al. 2015; Ma et al. 2016; Wang et al. 2016). Our findings in the pancreatic cancer model support the notion that Neat1 inhibits cell proliferation. In addition, the observation that Neat1/NEAT1 levels increase during differentiation of a variety of cell types, including ESCs, muscle cells, neuronal cells, and glial cells, suggests that NEAT1 may play a role in cellular differentiation (Lehnert et al. 2007; Chen and Carmichael 2009; Sunwoo et al. 2009; Mercer et al. 2010; Zeng et al. 2014). Moreover, studies of _Neat1_-null mice have shown that Neat1 is required for proper corpus luteum differentiation and mammary gland development (Nakagawa et al. 2014; Standaert et al. 2014). Consistent with a role in regulating differentiation is our observation that Neat1 deficiency in Kras _G12D_-expressing acini triggers increased ADM, a dedifferentiation event through which terminally differentiated acinar cells reprogram into ductal cells and then PanINs, leading ultimately to PDAC development (Kopp et al. 2012). In addition, we found that Kras _G12D_-expressing _Neat1_-deficient mice are prone to develop cystic lesions, suggesting that Neat1 could also be acting in more than one way to limit pancreatic neoplasia. Ductal cells undergo a dedifferentiation process to become IPMN lesions (Roy et al. 2015), and our results thus suggest that Neat1 is also involved in the maintenance of terminally differentiated ductal cells in the context of oncogenic Kras. The decreased expression of pancreatic differentiation genes observed upon Neat1 loss support an important role for Neat1 in differentiation in the pancreas. Interestingly, _Neat1_−/− mice in the context of wild-type KRas are not reported to develop cancer, and we similarly did not detect any noticeable developmental defects or pancreas abnormalities of _Neat1_−/− mice (Supplemental Fig. S8), reinforcing the idea that Neat1 tumor suppressor activities are triggered only upon oncogenic stress. Collectively, our findings suggest that Neat1 function in differentiation could be the basis for its activity as a suppressor of transformation and pancreatic neoplasia.
Despite the unequivocal importance of p53 in tumor suppression, the mechanisms through which it suppresses cancer development remain elusive. Recent studies have suggested that the best-characterized p53 functions— inducing cell cycle arrest or apoptosis in response to genotoxic stresses—as well as the well-studied p53 target genes involved in these responses (p21, Noxa, and Puma) are dispensable for tumor suppression (Brady et al. 2011; Li et al. 2012; Valente et al. 2013). New strategies are therefore needed to elucidate the molecular underpinnings of p53 tumor suppressor function. Here, the use of genomic approaches such as ChIP-seq and RNA-seq has helped to greatly expand the repertoire of genes known to be directly regulated by p53. Our discovery that Neat1/NEAT1 is a conserved p53-inducible lincRNA with a critical role in p53-dependent transformation suppression provides a key piece to the p53 tumor suppression puzzle. Understanding the cellular and molecular basis for how NEAT1 acts as a tumor suppressor will ultimately greatly expand our understanding of p53-mediated tumor suppression.
Materials and methods
Cell culture experiments
MEFs, colorectal cancer cells (HCT116), and pancreatic cancer cells were cultured in DMEM containing 10% FBS. Primary human fibroblasts are originally from Coriell Cell Repository and were cultured in DMEM containing 15% FBS. Human ESCs H9 (Wicell) and LSJ2 (Stanford) were maintained as described (Conklin et al. 2012). Mouse ESCs were cultured together with irradiated feeders in DMEM supplemented with 20% stem cell certified FBS, 10% NEAA, LIF, and β-mercaptoethanol. Doxorubicin (Sigma) treatment was at 0.2 μg/mL, and Nutlin-3a (Sigma-Aldrich) was at 10 µM. UV-C treatment was at 20 J/m2, and ionizing radiation dose was 5 Gy of γ radiation. Lentiviral infections for gene silencing or overexpression were performed as described (Brady et al. 2011). siRNA transfection using sequences against p53 (Dharmacon, M-003329-03) or NEAT1 were performed using Dharmafect 4 (Dharmacon) according to the manufacture's protocol, and siGENOME nontargeting siRNA pools (Dharmacon, D-001206-14) were used as a control. For clonogenic assays in which Neat1 or p53 was overexpressed, _E1A;HRasV12;p53_−/− MEFs were transduced with pLEX MCS-empty (negative control), pLEX MCS-p53 (positive control), or pLEX MCS-Neat1.
Cell cycle arrest and apoptosis assays
MEFs were irradiated, 5-ethynyl-2′-deoxyuridine (EdU)-pulsed after 14 h for 4 h, and processed using the Click-iT EdU Alexa fluor 488 imaging kit (ThermoFisher Scientific) according to the manufacturer's protocol. For apoptosis experiments, E1A;HRasV12 MEFs were treated with doxorubicin for 12 or 24 h and stained with Annexin V FITC (Invitrogen) and PI, following the manufacturer's protocol. Both cell cycle and apoptosis experiments were assessed by flow cytometry.
Clonogenic assays, anchorage-independent growth assays, and subcutaneous tumor studies
For the clonogenic assays, E1A;HRasV12 MEFs were plated in triplicate on six-well plates at 150 cells per well and left to grow for ∼12 d. Cells were fixed with 10% formalin and stained with 0.1% crystal violet. Anchorage-independent growth assays were performed as described previously (Kenzelmann Broz et al. 2013). Plates were scanned, and colony number quantification was performed manually for clonogenic assays, while OpenCFU (Geissmann 2013) was used to quantify the scanned images of wells from the anchorage-independent growth assays. Subcutaneous tumor studies were performed as described (Brady et al. 2011).
RNA-seq and data sets
Total RNA was extracted from wild-type and Neat1−/− E1A;HRasV12 MEFs using the Qiagen RNeasy mini extraction kit (Qiagen) according to the manufacturer's protocol. RNA-seq libraries were generated using the Illumina TruSeq kit (version 2) following the manufacturer's instructions. The libraries were sequenced on a HiSeq 4000 system (Illumina), and the RNA-seq reads were analyzed with Basespace's RNA Express pipeline (RNA Express Legacy version: 1.0.0), which encompasses alignment using the STAR aligner (Dobin et al. 2013) and differential expression analysis using DESEQ2 (Love et al. 2014). Differentially regulated genes were also analyzed using Enrichr (Chen et al. 2013; Kuleshov et al. 2016) to detect which biological pathways are being altered upon Neat1 loss. GSEA was used to test whether Neat1 deficiency impacts the expression of genes involved in pancreas development (Subramanian et al. 2005). A pancreas development signature was generated based on a previously published study (Arda et al. 2013). The RNA-seq data generated by this work is available in the Gene Expression Omnibus (GEO) database under accession number GSE100098. For the identification of NEAT1/Neat1 as a p53 target gene, previously generated mouse ChIP-seq and RNA-seq data sets (GSE46240) as well as a human p53 ChIP-seq data set (GSE55727) were used.
qRT–PCR, RNA-FISH, and Northern blot analysis
RNA was isolated using Trizol (Invitrogen) and reverse-transcribed using MMLV reverse transcriptase (Invitrogen) and random primers. qPCR was performed with Power SYBR Green PCR master mix (Thermo Fisher) and a 7900HT Fast real-time PCR machine (Applied Biosystems). A standard curve was used to quantify the samples. ChIP-qPCR was performed as described (Kenzelmann Broz et al. 2013). Primer sequences for qRT–PCR and ChIP-qPCR are listed in Supplemental Table S1. RNA-FISH was performed using Stellaris RNA-FISH complex probe sets (Biosearch Technologies) according to the manufacturer's protocol. The mouse Neat1 probe set was Quasar 570-labeled, while the human NEAT1 probe set was FAM (6-carboxyfluorescein)-labeled. Double staining with Sfpq was performed using a rabbit polyclonal anti-Sfpq antibody (1:200; Bethyl Laboratories). Northern blotting was performed as described (Johnson et al. 2005).
Mouse models for pancreatic cancer
Pancreatitis was triggered by treating 8-wk-old KRas+/LSL-G12D;Ptf1a-Cre, KRas+/LSL-G12D;Ptf1a-Cre;Neat1+/−, and KRas+/LSL-G12D;_Ptf1a-Cre;Neat1_−/− mice with eight hourly intraperitoneal injections of cerulein (100 μg per kilogram of body weight; Sigma-Aldrich) over 2 d, as described previously (Jensen et al. 2005). Mice were sacrificed 7 d after cerulein treatment, and the pancreata were analyzed by different histological parameters. Spontaneous transformation was also assessed in mice aged for 5 mo. Both groups were evaluated by a trained pathologist specializing in pancreatic cancer. Mice were on a 129/Sv and C57BL/6 mixed background.
Histology and immunohistochemistry
Tissue specimen processing, sectioning, and H&E staining were performed using standard protocols. Immunohistochemistry was performed using the VectaStain Elite ABC kit (Vector Laboratories) according to the manufacturer's protocol. The antibodies used were mouse anti-MUC5AC (1:500; ThermoFisher), mouse anti-Ki67 (1:100; BD Pharmingen), rat anti-Ck19 (1:750; University of Iowa), and goat anti-amylase (1:100; Santa Cruz Biotechnology). The sections were counterstained with hematoxylin or Alcian blue/nuclear fast red using the NovaUltra Alcian blue stain kit (IHC World) according to the manufacturer's instructions. For Ck19 and amylase staining, the sections were stained using anti-goat Alexa 488 (1:200; Invitrogen) and anti-rat Alexa 594 (1:200; Invitrogen) and counterstained with DAPI. Pictures were taken using a Leica microscope and/or with a NanoZoomer 2.0-RS slide scanner (Hamamatsu). Analysis of the PanIN and mucinous cystic lesion areas and Ki67 staining was performed using ImageJ. To simulate the size criterion used to diagnose IPMNs in humans, we also used a size criterion (diameter ≥280) to call cystic lesions/IPMNs. This size was based on the ability to distinguish large cystic lesions from PanIN lesions. Further classification of these lesions was performed based on their lining using H&E and Muc5ac staining.
Supplementary Material
Supplemental Material
Acknowledgments
We thank J.C. Marine, B.M. Flowers, and A. Kaiser for critical discussions and reading of the manuscript. We thank P. Chu of the Stanford Comparative Medicine Histology Research Core Laboratory for technical assistance with tissue processing, sectioning, and staining; A. Fox and G. Pierron for plasmids; N. Bardeesy for p53-deficient pancreatic cancer cells; J.C. Marine for Neat1 knockout MEFs; and E. Majunder for technical assistance. This work was supported by funding from the National Institutes of Health (R01 ES020260 to J.R. and L.D.A., and R35 CA197591 to L.D.A.) and the Lustgarten Foundation (J.S.).
Footnotes
Supplemental material is available for this article.
References
- Adriaens C, Standaert L, Barra J, Latil M, Verfaillie A, Kalev P, Boeckx B, Wijnhoven PWG, Radaelli E, Vermi W, et al. 2016. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat Med 22: 861–868. [DOI] [PubMed] [Google Scholar]
- Amankwah EK, Thompson RC, Nabors LB, Olson JJ, Browning JE, Madden MH, Egan KM. 2013. SWI/SNF gene variants and glioma risk and outcome. Cancer Epidemiol 37: 162–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arda HE, Benitez CM, Kim SK. 2013. Gene regulatory networks governing pancreas development. Dev Cell 25: 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan A, Penachioni JY, Lamba S, Bleeker FE, Zanon C, Rodolfo M, Vallacchi V, Scarpa A, Felicioni L, Buck M, et al. 2009. Molecular profiling of the ‘plexinome’ in melanoma and pancreatic cancer. Hum Mutat 30: 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras M-C, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch A-M, Wu J, et al. 2012. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491: 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging KT, Mello SS, Attardi LD. 2014. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 14: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume CJ, Hotz-Wagenblatt A, Hullein J, Sellner L, Jethwa A, Stolz T, Slabicki M, Lee K, Sharathchandra A, Benner A, et al. 2015. p53-dependent non-coding RNA networks in chronic lymphocytic leukemia. Leukemia 29: 2015–2023. [DOI] [PubMed] [Google Scholar]
- Botcheva K, McCorkle SR, McCombie WR, Dunn JJ, Anderson CW. 2011. Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle 10: 4237–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Attardi LD. 2010. p53 at a glance. J Cell Sci 123: 2527–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Broz DK, Basak S, Park EJ, McLaughlin ME, et al. 2011. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145: 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano E, Molina-Arcas M, Krygowska AA, East P, Warne P, Nicol A, Downward J. 2016. RAS signalling through PI3-kinase controls cell migration via modulation of Reelin expression. Nat Commun 7: 11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Sboner A, Nair SS, Giannopoulou E, Li R, Hennig S, Mosquera JM, Pauwels J, Park K, Kossai M, et al. 2014. The oestrogen receptor α-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat Commun 5: 5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chedotal A, Kerjan G, Moreau-Fauvarque C. 2005. The brain within the tumor: new roles for axon guidance molecules in cancers. Cell Death Differ 12: 1044–1056. [DOI] [PubMed] [Google Scholar]
- Chen L-L, Carmichael GG. 2009. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol Cell 35: 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Kong J, Ma Z, Gao S, Feng X. 2015. Up regulation of the long non-coding RNA NEAT1 promotes esophageal squamous cell carcinoma cell progression and correlates with poor prognosis. Am J Cancer Res 5: 2808–2815. [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Lin C-P, Ho JJ, He X, Okada N, Bu P, Zhong Y, Kim SY, Bennett MJ, Chen C, et al. 2011. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol 13: 1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry H, Albukhari A, Morotti M, Haider S, Moralli D, Smythies J, Schödel J, Green CM, Camps C, Buffa F, et al. 2015. Tumor hypoxia induces nuclear paraspeckle formation through HIF-2α dependent transcriptional activation of NEAT1 leading to cancer cell survival. Oncogene 34: 4482–4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A, Lawrence JB. 2009. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Molecular Cell 33: 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin JF, Baker J, Sage J. 2012. The RB family is required for the self-renewal and survival of human embryonic stem cells. Nat Commun 3: 1244. [DOI] [PubMed] [Google Scholar]
- Dallol A, Morton D, Maher ER, Latif F. 2003. SLIT2 axon guidance molecule is frequently inactivated in colorectal cancer and suppresses growth of colorectal carcinoma cells. Cancer Res 63: 1054–1058. [PubMed] [Google Scholar]
- Direnzo D, Hess DA, Damsz B, Hallett JE, Marshall B, Goswami C, Liu Y, Deering T, Macdonald RJ, Konieczny SF. 2012. Induced Mist1 expression promotes remodeling of mouse pancreatic acinar cells. Gastroenterology 143: 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AH, Lamond AI. 2010. Paraspeckles. Cold Spring Harb Perspect Biol 2: a000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann Q. 2013. OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS One 8: e54072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb EA, Vucic EA, Enfield KSS, Stewart GL, Lonergan KM, Kennett JY, Becker-Santos DD, MacAulay CE, Lam S, Brown CJ, et al. 2011. Human cancer long non-coding RNA transcriptomes. PLoS One 6: e25915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göhrig A, Detjen KM, Hilfenhaus G, Körner JL, Welzel M, Arsenic R, Schmuck R, Bahra M, Wu JY, Wiedenmann B. 2014. Axon guidance factor SLIT2 inhibits neural invasion and metastasis in pancreatic cancer. Cancer Res 74: 1529–1540. [DOI] [PubMed] [Google Scholar]
- Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, Barbacid M. 2007. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11: 291–302. [DOI] [PubMed] [Google Scholar]
- Guerra C, Collado M, Navas C, Schuhmacher Alberto J, Hernández-Porras I, Cañamero M, Rodriguez-Justo M, Serrano M, Barbacid M. 2011. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 19: 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Jiang B, Ma J, Li Q. 2016. Aberrant NEAT1 expression is associated with clinical outcome in high grade glioma patients. APMIS 124: 169–174. [DOI] [PubMed] [Google Scholar]
- Hirose T, Virnicchi G, Tanigawa A, Naganuma T, Li R, Kimura H, Yokoi T, Nakagawa S, Bénard M, Fox AH, et al. 2014. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol Biol Cell 25: 169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al. 2010. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142: 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, et al. 2011. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet 43: 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A. 2007. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JN, Cameron E, Garay MVR, Starkey TW, Gianani R, Jensen J. 2005. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology 128: 728–741. [DOI] [PubMed] [Google Scholar]
- Johnson TM, Hammond EM, Giaccia A, Attardi LD. 2005. The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat Genet 37: 145–152. [DOI] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. 2013. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 27: 1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigel B, Rabinowicz N, Varshavsky A, Kessler O, Neufeld G. 2011. Plexin-A4 promotes tumor progression and tumor angiogenesis by enhancement of VEGF and bFGF signaling. Blood 118: 4285–4296. [DOI] [PubMed] [Google Scholar]
- Kopp Janel L, von Figura G, Mayes E, Liu F-F, Dubois Claire L, Morris Iv John P, Pan Fong C, Akiyama H, Wright Christopher VE, Jensen K, et al. 2012. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22: 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Lowe SW. 2009. Stem cells: the promises and perils of p53. Nature 460: 1085–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruiswijk F, Labuschagne CF, Vousden KH. 2015. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 16: 393–405. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. 2016. Enrichr: a comprehensive gene set enrichment analysis Web server 2016 update. Nucleic Acids Res 44: W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahoz A, Hall A. 2013. A tumor suppressor role for srGAP3 in mammary epithelial cells. Oncogene 32: 4854–4860. [DOI] [PubMed] [Google Scholar]
- Lehnert SA, Reverter A, Byrne KA, Wang Y, Nattrass GS, Hudson NJ, Greenwood PL. 2007. Gene expression studies of developing bovine longissimusmuscle from two different beef cattle breeds. BMC Dev Biol 7: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léveillé N, Melo CA, Rooijers K, Díaz-Lagares A, Melo SA, Korkmaz G, Lopes R, Moqadam FA, Maia AR, Wijchers PJ, et al. 2015. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat Commun 6: 6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. 2012. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149: 1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li Y, Chen W, He F, Tan Z, Zheng J, Wang W, Zhao Q, Li J. 2015. NEAT expression is associated with tumor recurrence and unfavorable prognosis in colorectal cancer. Oncotarget 6: 27641–27650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Huang J, Zhou N, Zhang Z, Zhang A, Lu Z, Wu F, Mo Y-Y. 2013. LncRNA loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res 41: 4976–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. 1993. p53 is required for radiation induced apoptosis in mouse thymocytes. Nature 362: 847–849. [DOI] [PubMed] [Google Scholar]
- Ma Y, Liu L, Yan F, Wei W, Deng J, Sun J. 2016. Enhanced expression of long non-coding RNA NEAT1 is associated with the progression of gastric adenocarcinomas. World J Surg Oncol 14: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli P, Canamero M, del Pozo N, Madriles F, Zapata A, Real FX. 2013. Gata6 is required for complete acinar differentiation and maintenance of the exocrine pancreas in adult mice. Gut 62: 1481–1488. [DOI] [PubMed] [Google Scholar]
- Matthaei H, Schulick RD, Hruban RH, Maitra A. 2011. Cystic precursors to invasive pancreatic cancer. Nat Rev Gastroenterol Hepatol 8: 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Qureshi IA, Gokhan S, Dinger ME, Li G, Mattick JS, Mehler MF. 2010. Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci 11: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi Je E, Gwak M, Oh H, Ryoung Choi M, Jin Choi Y, Lee SH, Jin Yoo N. 2013. Frameshift mutations of axon guidance genes ROBO1 and ROBO2 in gastric and colorectal cancers with microsatellite instability. Pathology 45: 645–650. [DOI] [PubMed] [Google Scholar]
- Morris JP, Cano DA, Sekine S, Wang SC, Hebrok M. 2010. β-Catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest 120: 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma T, Hirose T. 2013. Paraspeckle formation during the biogenesis of long non-coding RNAs. RNA Biol 10: 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Naganuma T, Shioi G, Hirose T. 2011. Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J Cell Biol 193: 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Shimada M, Yanaka K, Mito M, Arai T, Takahashi E, Fujita Y, Fujimori T, Standaert L, Marine J-C, et al. 2014. The lncRNA Neat1 is required for corpus luteum formation and the establishment of pregnancy in a subpopulation of mice. Development 141: 4618–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada N, Lin C-P, Ribeiro MC, Biton A, Lai G, He X, Bu P, Vogel H, Jablons DM, Keller AC, et al. 2014. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev 28: 438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, Hainaut P. 2010. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2: a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, Zhang MQ, Spector DL. 2005. Regulating gene expression through RNA nuclear retention. Cell 123: 249–263. [DOI] [PubMed] [Google Scholar]
- Roy N, Malik S, Villanueva KE, Urano A, Lu X, Von Figura G, Seeley ES, Dawson DW, Collisson EA, Hebrok M. 2015. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev 29: 658–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez Y, Segura V, Marín-Béjar O, Athie A, Marchese FP, González J, Bujanda L, Guo S, Matheu A, Huarte M. 2014. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat Commun 5: 5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki YTF, Ideue T, Sano M, Mituyama T, Hirose T. 2009. MENε/β noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc Natl Acad Sci 106: 2525–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Fukushima N, Chang R, Matsubayashi H, Goggins M. 2006. Differential and epigenetic gene expression profiling identifies frequent disruption of the RELN pathway in pancreatic cancers. Gastroenterology 130: 548–565. [DOI] [PubMed] [Google Scholar]
- Schmitt AM, Chang HY. 2016. Long noncoding RNAs in cancer pathways. Cancer Cell 29: 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AM, Garcia JT, Hung T, Flynn RA, Shen Y, Qu K, Payumo AY, Peres-da-Silva A, Broz DK, Baum R, et al. 2016. An inducible long noncoding RNA amplifies DNA damage signaling. Nat Genet 48: 1370–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour PA. 2014. Sox9: a master regulator of the pancreatic program. Rev Diabet Stud 11: 51–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, Zhu L, Sun Y, Bettencourt R, Damsz B, Hruban RH, Konieczny SF. 2009. Loss of the acinar-restricted transcription factor Mist1 accelerates Kras-induced pancreatic intraepithelial neoplasia. Gastroenterology 136: 1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standaert L, Adriaens C, Radaelli E, Keymeulen A, Blanpain C, Hirose T. 2014. The long noncoding RNA Neat1 is required for mammary gland development and lactation. RNA 20: 1844–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunwoo H, Dinger ME, Wilusz JE, Amaral PP, Mattick JS, Spector DL. 2009. MENε/β nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res 19: 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima H, Niwa T, Takahashi T, Wakabayashi M, Yamashita S, Ando T, Inagawa Y, Taniguchi H, Katai H, Sugiyama T, et al. 2015. Frequent involvement of chromatin remodeler alterations in gastric field cancerization. Cancer Lett 357: 328–338. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kuroki T, Adachi T, Ono S, Hirabaru M, Soyama A, Kitasato A, Takatsuki M, Hayashi T, Eguchi S. 2013. Evaluation of SOX9 expression in pancreatic ductal adenocarcinoma and intraductal papillary mucinous neoplasm. Pancreas 42: 488–493. [DOI] [PubMed] [Google Scholar]
- Truitt ML, Ruggero D. 2016. New frontiers in translational control of the cancer genome. Nat Rev Cancer 16: 288–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente Liz J, Gray Daniel HD, Michalak Ewa M, Pinon-Hofbauer J, Egle A, Scott Clare L, Janic A, Strasser A. 2013. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep 3: 1339–1345. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303: 844–848. [DOI] [PubMed] [Google Scholar]
- von Figura G, Fukuda A, Roy N, Liku ME, Morris Iv JP, Kim GE, Russ HA, Firpo MA, Mulvihill SJ, Dawson DW, et al. 2014. The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat Cell Biol 16: 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137: 413–431. [DOI] [PubMed] [Google Scholar]
- Wang P, Wu T, Zhou H, Jin Q, He G, Yu H, Xuan L, Wang X, Tian L, Sun Y, et al. 2016. Long noncoding RNA NEAT1 promotes laryngeal squamous cell cancer through regulating miR-107/CDK6 pathway. J Exp Clin Cancer Res 35: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman B, Knudsen KE. 2009. Hijacking the chromatin remodeling machinery: impact of SWI/SNF perturbations in cancer. Cancer Res 69: 8223–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Davis CP, Sunwoo H, Simon MD, Sadreyev RI, Wang PI, Tolstorukov MY, Kingston RE. 2014. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell 55: 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BG, Roberts CWM. 2011. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11: 481–492. [DOI] [PubMed] [Google Scholar]
- Wu Y, Yang L, Zhao J, Li C, Nie J, Liu F, Zhuo C, Zheng Y, Li B, Wang Z, et al. 2015. Nuclear-enriched abundant transcript 1 as a diagnostic and prognostic biomarker in colorectal cancer. Mol Cancer 14: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger ST, Kenzelmann-Broz D, Jung H, Attardi LD, Rinn JL. 2015. Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res 43: 4447–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, Xu Y, Xu L, Yu X, Cheng J, Yang L, Chen S, Li Y. 2014. Inhibition of long non-coding RNA NEAT1 impairs myeloid differentiation in acute promyelocytic leukemia cells. BMC Cancer 14: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JP, Qin HY, Wang L, Liang L, Zhao XC, Cai WX, Wei YN, Wang CM, Han H. 2011. Overexpression of Notch ligand Dll1 in B16 melanoma cells leads to reduced tumor growth due to attenuated vascularization. Cancer Lett 309: 220–227. [DOI] [PubMed] [Google Scholar]
- Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. 2007. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol 171: 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material