The Listeriolysin O PEST-like Sequence Co-opts AP-2-Mediated Endocytosis to Prevent Plasma Membrane Damage during Listeria Infection (original) (raw)
. Author manuscript; available in PMC: 2019 Jun 13.
Published in final edited form as: Cell Host Microbe. 2018 Jun 13;23(6):786–795.e5. doi: 10.1016/j.chom.2018.05.006
Summary
Listeriolysin O (LLO) is a cholesterol-dependent cytolysin that mediates escape of _Listeria monocytogenes_from a phagosome, enabling growth of the bacteria in the host cell cytosol. LLO contains a PEST-like sequence that prevents it from killing infected cells, but the mechanism involved is unknown. We found that the LLO PEST-like sequence was necessary to mediate removal of LLO from the interior face of the plasma membrane where it coalesces into discrete puncta. LLO interacts with Ap2a2, an adaptor protein involved in endocytosis, via its PEST-like sequence and Ap2a2-dependent endocytosis is required to prevent LLO-induced cytotoxicity. An unrelated PEST-like sequence from a human G protein-coupled receptor (GPCR) that also interacts with Ap2a2 could functionally complement the PEST-like sequence in L. monocytogenes LLO. These data revealed that LLO co-opts the host endocytosis machinery to protect the integrity of the host plasma membrane during L. monocytogenes infection.
Keywords: LLO, membrane repair, adaptor protein complex 2, ap2a2, macrophage, pathogen, Human calcium receptor
eTOC blurb
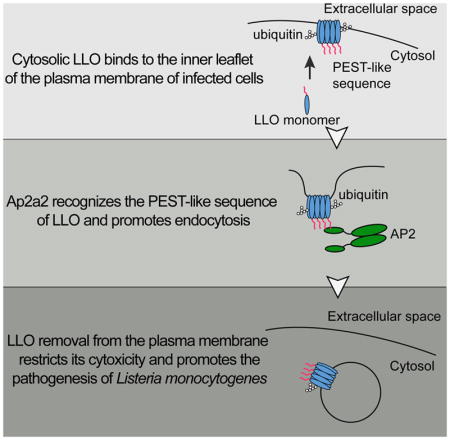
The intracellular pathogen Listeria elaborates Listeriolysin O (LLO), a cytolysin. Chen et al. discovered that the PEST-like sequence of LLO interacts with a cellular endocytosis adaptor to promote the endocytosis of plasma membrane associated LLO, thereby preventing its cytotoxicity to the infected cell. This mechanism is critical for Listeria pathogenesis.
Introduction
Intracellular bacterial pathogens represent a diverse group of microorganisms responsible for an extensive amount of worldwide morbidity and mortality. These pathogens secrete virulence factors that manipulate host cell biological processes to establish their replicative niches, which can be broadly categorized as intravacuolar or cytosolic (Mitchell, Chen, & Portnoy, 2016). Although we still have much to learn about the molecular mechanisms that govern host-remodeling processes, it is clear that establishing a replicative niche requires precise temporal and spatial regulation to restrict the activity of secreted virulence factors to specific cellular compartments (Schnupf & Portnoy, 2007).
Listeria monocytogenes is a Gram-positive food-borne facultative intracellular bacterial pathogen that can cause serious foodborne infections in immunocompromised individuals and pregnant women (Dussurget, Pizarro-Cerda, & Cossart, 2004). Subsequent to phagocytosis, L. monocytogenes escapes from a phagosomal compartment and gains access to the cytosol, where it grows rapidly within host cells (Portnoy, Auerbuch, & Glomski, 2002). Listeriolysin O (LLO) is an essential determinant of L. monocytogenes pathogenesis that mediates vacuole escape: LLO-deficient mutants fail to escape from the phagosome, do not replicate in host cells, and are completely avirulent. LLO is a member of the cholesterol-dependent cytolysins (CDCs), which is the largest family of β-barrel pore-forming toxins primarily secreted by Gram-positive bacteria (Alouf, 2001). CDCs are secreted as monomers, oligomerize (34–50 soluble monomers) upon binding to cell membranes, and mature into large transmembrane pores of ~30nm in diameter (Dunstone & Tweten, 2012;Ramachandran, Tweten, & Johnson, 2004; Shatursky et al., 1999). Whereas other CDCs are secreted by extracellular bacteria, LLO is the only CDC produced by an intracellular bacterium and can act on the interior face of the plasma membrane. Surprisingly, although LLO is a phagosome-specific cytolysin, it is also expressed throughout the entire intracellular life cycle of L. monocytogenes. Because it forms pores in host membranes, it is potentially toxic to infected host cells. However, L. monocytogenes replicates extensively within the host cytosol without killing the infected cells, suggesting the existence of mechanisms that restrict the cytosolic activity of LLO (Schnupf & Portnoy, 2007). Several potential mechanisms have been proposed to limit the cytotoxicity of LLO, including its low pH optimum and short intracellular half-life (Glomski, Gedde, Tsang, Swanson, & Portnoy, 2002; Schnupf, Portnoy, & Decatur, 2006). However, none of these proposed mechanisms had offered a satisfying answer to how LLO activity is controlled in the host cytosol, as blocking degradation or increasing its intracellular stability do not substantially increase cytotoxicity, suggesting the existence of unidentified regulatory mechanisms (Schnupf et al., 2006; Schnupf & Portnoy, 2007; Schnupf, Zhou, Varshavsky, & Portnoy, 2007).
All CDC members display a high degree of sequence similarity with a conserved core of 471 amino acids (Gonzalez, Bischofberger, Pernot, van der Goot, & Frêche, 2008). Replacing LLO with a highly related CDC, perfringolysin O (PFO), results in a L. monocytogenes strain that escapes from a phagosome, but subsequently causes rapid death of the infected cells due to loss of plasma membrane integrity (Decatur & Portnoy, 2000). LLO contains a unique N-terminal amino acid sequence that is absent in PFO, previously referred to as a PEST-like sequence, (identified by the PESTfind algorithm as rich in proline, glutamate, serine, and threonine residues). Removal of the PEST-like sequence has minor effects on LLO activity and vacuolar escape, but results in a strain that is extremely toxic to infected host cells and 10,000-fold less virulent in mice (Decatur & Portnoy, 2000). The LLO PEST-like sequence can convert an extracellular cytolysin (PFO) into a phagosome-specific lysin, which supports the intracellular replication of L. monocytogenes. These studies suggested that the PEST-like sequence contributes unique properties to LLO that make it better suited for an intracellular pathogen.
PEST-like sequences were originally identified in eukaryotic proteins with short intracellular half-lives, suggesting that they function as degradation signals (Rechsteiner & Rogers, 1996). However, other functions have been associated with PEST-like sequences from several eukaryotic cell surface proteins, such as the human calcium receptor (HCaR), the ATP-binding cassette transporter A1 (ABCA1), and the yeast α-factor receptors that are involved in the regulation of protein trafficking (Martinez, Agerholm-Larsen, Wang, Chen, & Tall, 2003; Pi et al., 2005; Roth & Davis, 2000; Roth, Sullivan, & Davis, 1998; Zhuang, Northup, & Ray, 2012). Therefore, the PEST-like sequences have alternative roles that involve interaction with other host partners. Recently, the structure of LLO was resolved and a left-handed polyproline II helix (PPII-helix) was found within the N-terminal PEST-like region. The PPII-helix is a structural motif that often acts as a platform for protein-protein interaction (Adzhubei, Sternberg, & Makarov, 2013). Indeed, several residues within the LLO PEST-like region are phosphorylated during L. monocytogenes infection, supporting the idea that the PEST-like sequence of LLO can be recognized by host components (Schnupf et al., 2006).
Determining the intrinsic features of the LLO PEST-like sequence that regulate its activity has been quite challenging because it also affects translational control of LLO. Synonymous mutations within the PEST-like sequence that alter the mRNA sequence, but not the protein sequence, increase LLO synthesis and cytotoxicity to host cells, although not to the same extent as deletion of the PEST-like sequence (Schnupf et al., 2006; 2007). These synonymous mutations result in an approximately 5-fold increase of LLO synthesis and synergize with pharmacologic agents that block the proteasome and with mutations in the N-terminal amino acid of LLO that extend its cytosolic half-life (Schnupf et al., 2006; 2007). These data suggest that a pool of LLO is degraded by the proteasome but, surprisingly, neither proteasome inhibition nor mutations that extend LLO half-life are toxic unless combined with synonymous mutations within the PEST-like region (Schnupf et al., 2006;2007). Therefore, there must be another mechanism controlling LLO toxicity.
In this study, we found that the PEST-like sequence of LLO interacted with Ap2a2, an adaptor protein involved in endocytosis, which promotes its removal from the host plasma membrane and protects the integrity of the host cell during L. monocytogenes infection. In addition, we discovered that a PEST-like sequence from a G protein-coupled receptor, the human calcium receptor, also interacts with Ap2a2 and can functionally replace the PEST-like sequence from LLO and promote intracellular replication of L. monocytogenes. Collectively, our results demonstrated that L. monocytogenes exploits the conserved host endocytic machinery to remove LLO from the plasma membrane, thereby preventing the premature death of infected host cells.
Results
The PEST-like sequence regulates the plasma membrane levels of LLO
The primary function of LLO is to disrupt the phagosomal membrane, allowing L. monocytogenes to reach the host cytosol. However, LLO is continually expressed by cytosolic bacteria and, as previously observed, accumulates in punctate structures around the nucleus (Figure 1A), suggesting the existence of an intrinsic feature within LLO that directs its trafficking to a specific subcellular location. In an attempt to recapitulate the fate of cytosolic LLO in a _Listeria_-free system, full-length LLO, either with or without an EGFP tag, was transiently expressed in HeLa cells. LLO expressed intracellularly also formed punctate structures resembling those observed during bacterial infection (Figure 1B and S1). Strikingly, LLO puncta also co-localized with ubiquitin and the autophagy adaptor protein p62 (Figure S2), as previously observed in infected cells (Viala, Mochegova, Meyer-Morse, & Portnoy, 2008). These data validated the use of a transfection-based assay for studying the intracellular trafficking of LLO.
Figure 1. The PEST-like sequence regulates the plasma membrane level of LLO.

BMMs were infected with EGFP expressing L. monocytogenes (green) for 6 hours. Cells were fixed and stained with nuclear dye DAPI (blue) and anti-LLO antibody (red). (B) HeLa cells were transfected with full length and truncated LLO-EGFP (green) constructs for 24 hours and stained with DAPI (blue). Image for LLOΔPEST was acquired at 15 hours post-transfection due to cytotoxicity. Scale bars are 10 μm. Images shown represent 5 independent experiments. See alsoFigure S1.
In order to map the regions of LLO responsible for its subcellular localization, several LLO truncation constructs were expressed in HeLa cells resulting in distinct cellular patterns (Figure 1B). Cells transfected with an N-terminally truncated LLO-EGFP (N100 or N200 amino acid deletions, LLO126-529-EGFP and LLO226-529-EGFP) led to enhanced localization to the plasma membrane. A C-terminal 50 amino acid truncated construct of LLO-EGFP (LLO26-480), which removed the membrane-binding domain, resulted in a diffuse pattern of expression (Figure 1B). These results suggested that, while the N-terminal sequence of LLO regulated its levels on the plasma membrane, the C-terminal membrane-binding domain was required to form cytosolic punctate structures. We further hypothesized that the PEST-like sequence was the N-terminal motif responsible for regulating the levels of LLO on the host plasma membrane. Accordingly, transient expression of LLOΔPEST in HeLa cells resulted in the accumulation of LLO on the cell surface and the detachment of cells from the coverslips (Figure 1B). These results suggested the existence of an uncharacterized role of the LLO PEST-like sequence in regulating LLO levels on the plasma membrane.
Based on the increased association of LLOΔPEST with the host plasma membrane, we reasoned that the PEST-like sequence either prevents LLO from accessing the plasma membrane or promotes its removal. In order to differentiate between these two possible mechanisms, total internal reflection fluorescence (TIRF) microscopy was utilized to monitor the dynamics of LLO association and the host plasma membrane in HeLa cells transfected with LLO-EGFP and LLOΔPEST-EGFP (Figure 2A). The LLO-EGFP signal was detected on the plasma membrane (Figure 2B), and its retention time was quantified by analyzing the kymograph, which represented the length of time that EGFP-LLO was retained on the plasma membrane (Figure 2C). More than 80% of the LLO-EGFP signal was removed from the plasma membrane within 9 minutes of observation (Figure 2D), suggesting that intracellular LLO reached the plasma membrane and was subsequently removed. The retention time of LLOΔPEST was impossible to calculate because cells transfected with constructs lacking the PEST-like sequence rounded-up and detached from with the coverslip. However, extended retention of the N-terminal truncated LLO126-529 was observed in the plasma membrane after transfection (Figure S2). Taken together, these results suggested that the PEST-like sequence of LLO does not prevent LLO binding to plasma membrane but promotes its removal from the host plasma membrane.
Figure 2. LLO can be efficiently removed from the plasma membrane in LLO-EGFP transfected cells.

(A) Schematic representation of the total internal reflection fluorescence (TIRF) imaging setup used for the experiments. (B) Frame from a TIRF acquisition time-series obtained using 100-ms exposures in every 2 seconds. The spots represent EGFP molecules. Bar, 1 μm. (C) Representative kymograph from a time-series TIRF imaging of plasma membrane-associated with LLO. Bar, 1 μm. (F) The percentage of LLO-EGFP signal removed from plasma membrane within 540 seconds of time-series obtained using 100-ms exposures (N=134 EGFP sites in 5 cells). See also Figure S2.
Host Ap2a2 interacts with the PEST-like region of LLO
We hypothesized that host proteins recognize and facilitate the internalization of membrane-associated LLO. In order to identify LLO-interacting host proteins, LLO lacking its membrane-binding domain was used as bait to screen a normalized yeast two-hybrid (Y2H) library expressing fragmented mouse cDNA. Four host proteins (Ap2a2, Filamin α, RNF25 and Snapin) were identified by yeast auxotrophic selection, and the strength of interaction was quantified by a secreted α-galactosidase quantification assay (Figure S3A and S3B). The carboxyl-terminal alpha-appendage domain derived from the adaptor-related protein complex 2 (Ap2a2), a subunit of the AP-2 complex, induced the strongest signal in the α-galactosidase quantification assay. Serial truncations of LLO were further evaluated in the Y2H assay to map the region essential for interaction with Ap2a2 (Figure 3A). Strikingly, only yeast strains expressing LLO containing the PEST-like sequence were positive interactors (Figure 3B and S3C). In contrast, PFO, a highly related CDC member that lacks the corresponding N-terminal PEST-like sequence, did not interact with Ap2a2 in the Y2H assay (Figure 3B and S3C).
Figure 3. Ap2a2 is an LLO interacting protein identified by the yeast two-hybrid assay.

(A) Schematic diagrams of different baits (LLO truncations) constructs tested in the yeast 2-hybrid assay. (B) Quantification of secreted alpha-galactosidase activity following a GAL4-based two-hybrid interaction in Y2HGold yeast colonies. The results represented the percentage of alpha-galactosidase activity of positive control (p53+ SV40 large T antigen). Bars and error represent mean ± SD of replicate measurements. N=4 biological repeats *** p<0.001 (Student’s t-test). Results shown represent at least 3 independent experiments. (C) HEK 293T and HeLa cells were co-transfected with LLO-Myc tag and Ap2a2-HA tag plasmids. At 24 hours post transfection, cells were permeabilized and stained with anti-Myc (red), anti-HA antibodies (Green), and nuclear dye DAPI (blue). Scale bars are 10 μm. (B) Co-immunoprecipitation of Ap2a2 from transfected HEK293T cells using an anti-Myc-LLO antibody. Results shown represent 3 independent experiments. See also Figure S3.
In order to confirm the interaction between LLO and Ap2a2, immunofluorescence microscopy and co-immunoprecipitation experiments were performed using HeLa and HEK293T cells co-transfected with plasmids encoding HA-tagged Ap2a2 and Myc-tagged LLO. Co-localization between full length LLO and Ap2a2 was observed in both cell types (Figure 3C). Next, the anti-Myc antibody was used to perform co-immunoprecipitation from the lysates of the transfected HEK293T cells. HA-tagged Ap2a2 co-immunoprecipitated with full-length LLO, but not with LLO lacking the PEST-like sequence (Figure 3D). These results strongly suggested that the PEST-like sequence of LLO was essential for mediating an interaction with Ap2a2, and this interaction was a unique feature of LLO among CDCs.
Ap2a2 protects host cells against LLO-induced plasma membrane damage during L. monocytogenes infection
Considering that Ap2a2 is a subunit of the AP-2 complex that is involved in clathrin-dependent endocytosis (McMahon & Boucrot, 2011), we hypothesized that the interaction between Ap2a2 and the PEST-like sequence promotes the removal of LLO from the host plasma membrane and likely represents an essential aspect of_L. monocytogenes_ pathogenesis. To evaluate the biological role of this interaction, CRISPR/Cas9-mediated gene editing was used to generate Ap2a2 knockout BMMs (Figure S4A) and to evaluate the role of Ap2a2 during the intracellular growth of L. monocytogenes. Infections were performed in the presence of gentamicin, which kills extracellular bacteria or bacteria in permeabilized cells. BMM lacking Ap2a2 supported the phagocytosis and initial growth of wild-type L. monocytogenes, but after 5 hours, the number of viable bacteria declined approximately 5-fold. In addition, an L. monocytogenes strain expressing a LLO mutant (L461T), which has higher pore forming activity at neutral pH (Glomski et al., 2002), showed a drop in CFUs greater than 10-fold at 8 hours post-infection (Figure 4A). These data showed that Ap2a2 is required for efficient replication of L. monocytogenes in macrophages.
Figure 4. Ap2a2 protects LLO-induced plasma membrane damage by recognizing the PEST-like region of LLO.

(A) The intracellular growth of the wild type 10403S strain and the L. monocytogenes_-LLO L461T strain in BMMs with the CRISPR/Cas9-mediated Ap2a2 knockout and sgRNA control BMMs. 50μg/ml gentamicin was added after 1h to kill extracellular bacteria. Results shown represent at least 3 independent experiments. (B) Schematic of_hly_fl L. monocytogenes strain. The hly and tetL (tetracycline resistance) genes are flanked by loxP sites. Cre recombinase is expressed from the cytosol-specific actA promoter. Recombination between loxP sites leads to the excision of the DNA encoding hly and_tetL. (C) Intracellular growth of the _hly_fl L. monocytogenes strain in Ap2a2 knockout and control BMMs. 50μg/ml gentamicin was added to kill extracellular L. monocytogenes. Results are representative of at least 3 independent experiments. (D) Flow cytometry analysis of SYTOX Blue staining of control and Ap2a2 knockdown BMM infected for 8 hours with an effective MOI of 1. Positively stained cells resulted from the loss of plasma membrane integrity. No gentamicin was added for plasma membrane integrity assay. ** p<0.01, *** p<0.001 (Student’s t-test). Results shown represent 2 independent experiments. See also Figure S4.
To verify that the loss of CFUs was caused by LLO produced in the cytosol, we engineered a strain of L. monocytogenes, termed hly_fl,_ that has a functional LLO, but excises the gene encoding LLO, hly, following escape of L. monocytogenes from the phagocytic vacuole (Figure 4B). Therefore, the strain initially produces LLO to facilitate escape from a phagocytic vacuole but, once in the cytosol, hly is excised resulting in an LLO-minus mutant. The excision of_hly_ was rapid and resulted in complete loss of LLO production within one and a half hours post-infection in BMMs (Figure S4B–D). Next, Ap2a2 knockout BMMs were infected with the hly_fl strain to test whether the decrease in L. monocytogenes CFUs previously observed in these cells was related to the secretion of LLO in the host cytosol. Remarkably, excision of hly from cytosolic bacteria prevented cell death and rescued L. monocytogenes growth in Ap2a2 knockout BMMs (Figure S4E), while the growth and cytotoxicity in control BMMs were not affected (Figure S4E). These results suggested that the growth defect of_L. monocytogenes in Ap2a2 knockout cells is caused by the production of LLO during the cytosolic phase of the intracellular life cycle.
We hypothesized that the drop in CFUs observed in Ap2a2 knockout cells was due to the influx of gentamicin that occurred in permeabilized cells. To test hypothesis, the integrity of the host plasma membrane was monitored by quantifying the number of infected cells positive for the membrane impermeable dsDNA dye SYTOX Blue using flow cytometry at 8 hr post- infection. As expected, the loss of CFUs directly correlated with the influx of SYTOX Blue in infected Ap2a2 knockout BMMs, while plasma membrane integrity loss was much less in BMMs infected with the _hly_fl strain (Figure 4D). These results demonstrated that LLO produced in the cytosol could cause plasma membrane damage and that Ap2a2 is necessary to protect cell integrity during L. monocytogenes cytosolic replication.
The PEST-like sequence from the human calcium receptor (HCaR-PEST) also interacts with Ap2a2 and is sufficient to rescue the cytotoxicity of an LLOΔPEST strain
The human calcium receptor (HCaR) is a G protein-coupled receptor that contains a PEST- like sequence at its C-terminus that is involved in regulating its surface expression level by promoting internalization and degradation (Zhuang et al., 2012). Similarly to LLO, the PEST- like sequence of HCaR is phosphorylated in the presence of calcium, and deletion of this sequence results in enhanced expression on the plasma membrane (Pi et al., 2005; Zhuang et al., 2012). We hypothesized that PEST-like sequences from LLO and HCaR exploited similar host mechanisms for promoting internalization. A Y2H assay was performed to test whether Ap2a2 interacts with the HCaR PEST-like sequence and a very strong interaction was detected between the PEST-like sequence from HCaR (901aa–1000aa) and the alpha-appendage domain of Ap2a2 (Figure 5Aand S5A). In addition, we confirmed that the PEST-like sequences from both LLO and HCaR were not only essential, but were sufficient to mediate an interaction with the alpha- appendage domain of Ap2a2 in the Y2H assay (Figure 5A).
Figure 5. The PEST-like sequence from human calcium receptor (HCaR-PEST) is sufficient to interact with Ap2a2 and rescue the cytotoxicity of a LLOΔPEST strain.

(A) Quantification of secreted alpha-galactosidase activity following a GAL4-based two-hybrid interaction in Y2HGold S. cerevisiae. (B) Schematic of the PEST-like and hly sequences used to complement the Δ_hly_ strain. (C) BMMs were infected with indicated bacterial mutants at an MOI of 1 without adding gentamicin. LDH release was measured at 6 hours post-infection to determine the level of cytotoxicity. N=3 biological repeats. Results shown represent at least 4 independent experiments. (D) The intracellular growth of the indicated Δ_hly_ complement strains in BMM. 50μg/ml gentamicin was added to kill extracellular L. monocytogenes. Results shown represent at least 5 independent experiments. (E) Plaque size of the indicated Δ_hly_ complement strains in murine L2 fibroblasts. 50μg/ml gentamicin was added to kill extracellular L. monocytogenes. N>20 plaques. Results shown represent at least 3 independent experiments. (F) CFUs of indicated strains from the spleen of C57BL/6 mice at 48 hours post-infection. Bars and error represent mean ± SD of replicate measurements. N=10 mice ** p<0.01, *** p<0.001 (Student’s t-test). Results shown represent at least 3 independent experiments. See also Figure S5 and S6.
A L. monocytogenes strain was constructed to test whether the PEST-like sequence from HCaR could functionally replace the PEST-like sequence of LLO (Figure 5B). The HCaR PEST chimeric strain secreted comparable amounts of LLO and had similar hemolytic activity to the LLO ΔPEST strain (Figure S6). Intracellular replication, cytotoxicity, and virulence of the chimeric strain were evaluated in the intracellular growth assay and a mouse virulence model. Although the HCaR PEST chimeric strain was slightly more toxic than wild-type L. monocytogenes, the cytotoxicity and replication defects of the LLO ΔPEST strain were largely rescued by introducing the PEST-like sequence from HCaR (Figure 6C and 6D). Next, a plaque assay was used to monitor growth, cytotoxicity, and cell-to-cell spread of L. monocytogenes. The LLO ΔPEST strain was unable to form plaques due to plasma membrane integrity loss, resulting in influx of extracellular gentamicin and killing of the intracellular bacteria. However, replacing the LLO PEST-like sequence with the HCaR PEST-like sequence rescued the plaque defect of the LLO ΔPEST strain to 76% of wild-type (6E). In addition, the transfection of the chimeric HCaR-PEST LLO also resulted in the formation of intracellular punctate structures in HeLa cells, further supporting a similar function of LLO PEST and HCaR PEST (Figure S5B). Consistent with these in vitro infection results, the virulence defect of the LLO ΔPEST strain was largely rescued by the PEST-like sequence of HCaR in a mouse model of infection (Figure 6F). These results demonstrated that the PEST-like sequence of HCaR can functionally replace much of the activity conferred by PEST-like sequence of LLO.
Figure 6. Model depicting the endocytosis and degradation of LLO synthesized in the host cytosol.

Cytosolically synthesized LLO is first ubiquitylated, and then the LLO monomer and aggregates are degraded by the proteasome (1). Some cytosolic LLO reaches and binds to the cholesterol-enriched plasma membrane (2). Phosphorylation and/or other post-translational modification of the PEST-like region of LLO promote its recognition and internalization of the plasma membrane associated LLO by AP-2 and prevents pore-forming activity on plasma membrane (3). Finally, LLO containing endosomes are ubiquitylated and further recognized by the autophagy machinery (4) or invaginated into MVB for degradation (5).
Discussion
Cholesterol-dependent cytolysins are secreted by many Gram-positive pathogens, but Listeriolysin O (LLO) is the only CDC produced by an intracellular pathogen. The N-terminal PEST-like sequence of LLO is unique among CDCs, and we previously reported that deletion of this sequence resulted in a strain that still escaped from a phagosome, but subsequently killed the host cell and had a 4-log virulence defect in mice (Decatur & Portnoy, 2000). Although the importance of the N-terminal region of LLO is well established as an essential component of L. monocytogenes pathogenesis (Decatur & Portnoy, 2000), its mechanism of action was unknown. The results of this study revealed that the PEST-like sequence interacts with the host protein Ap2a2, a subunit of the AP-2 complex, and triggers the removal of LLO from the plasma membrane, thereby limiting damage to the host cell. We found that Ap2a2 also interacts with the PEST-like sequence of the human GPCR HCaR, a sequence involved in regulation of protein trafficking, and we demonstrated that the PEST-like sequence of LLO was functionally replaced by the HCaR PEST-like sequence. Overall, our data demonstrated that _L. monocytogenes_exploits a conserved host mechanism of endocytosis to decrease the cytotoxicity of LLO and prevent host cell death.
One of the proposed functions of eukaryotic PEST-like sequences is to act as tags that target proteins for ubiquitination and proteasomal degradation (Rechsteiner & Rogers, 1996). However, mutations in the LLO PEST-like sequence do not affect its ubiquitination and cytosolic half-life (Schnupf et al., 2006), which suggests that the mechanism by which the PEST-like sequence controls LLO cytotoxicity is unrelated to proteasomal degradation. Interestingly, PEST-like sequences are also present in many host surface receptors, and some of these sequences have been proposed to play an important role in the regulation of protein trafficking (Tovo-Rodrigues, Roux, Hutz, Rohde, & Woods, 2014). As an example, the trafficking of the GPCR β2-adrenergic receptor depends on a proline-rich region that interacts with β-arrestin-2, which is a clathrin adaptor that binds to the Ap2b2 subunit of the AP-2 complex (Han, Kommaddi, & Shenoy, 2013; Laporte, Oakley, Holt, Barak, & Caron, 2000). Therefore, it is clear that some poly-proline or PEST-like sequences function to regulate surface expression levels.
Structural analysis of LLO revealed that the PEST-like sequence contains a poly-proline region that forms a left-handed PPII-helix, a structural feature involved in protein-protein interactions (Köster et al., 2014) and often found in binding sites of widely spread WW and SH3 domains (Macias, Wiesner, & Sudol, 2002). Interestingly, the huntingtin protein, which contains a PPII-helix, also interacts with the α-appendage domain of Ap2a2 (Darnell, Orgel, Pahl, & Meredith, 2007; Kaltenbach et al., 2007; M. W. Kim, Chelliah, Kim, Otwinowski, & Bezprozvanny, 2009), and forms intracellular aggregates reminiscent of the distribution of LLO during infection (Viala et al., 2008). Although the PEST-like sequence from HCaR lacks an apparent proline-rich region, HCaR may adopt a PPII-helix as proline is not essential for PPII-helix formation (Adzhubei et al., 2013; Eker, Griebenow, Cao, Nafie, & Schweitzer-Stenner, 2004). Further experiments are required to confirm that the PPII-helix is the structural determinant for the binding of Ap2a2 to proteins containing PEST-like sequences, including LLO and HCaR. Interestingly, the PEST-like sequences of LLO and HCaR are the target of host kinases (Pi et al., 2005; Schnupf et al., 2006), and the role of phosphorylation within the LLO PEST-like sequence is not understood. Considering that phosphorylation sites tend to be located on the interfaces of protein structural complexes and may modulate the strength of interactions (Nishi, Hashimoto, & Panchenko, 2011), it is possible that different post-translational modifications determine subsets of interaction partners that dictate function of PEST-like sequences, including the binding of Ap2a2 and the regulation of protein trafficking.
It is critical that intracellular pathogens grow inside of cells without damaging the host cytoplasmic membrane or triggering the premature death of host cells. Indeed, there exist both innate and adaptive host immune mechanisms that restrict the growth of intracellular pathogens by activating host cell death pathways (Ashida et al., 2011; Fink & Cookson, 2005; Lamkanfi & Dixit, 2010). The results of this and other studies clearly show that L. monocytogenes strains that cause premature death of infected host cells are rendered avirulent (Decatur & Portnoy, 2000; Glomski, Decatur, & Portnoy, 2003; Sauer et al., 2010). It is well appreciated that plasma membrane repair pathways play an important role in the response to extracellular exposure to pore-forming cytolysins, including CDCs. Repair of the plasma membrane is a fundamental process that allows the host cell to cope with both exogenous and endogenous stresses that could compromise its homeostasis (Blazek, Paleo, & Weisleder, 2015;Jimenez & Perez, 2017). Three models have been proposed for membrane repair in response to pore-forming toxins: (1) a patch repair model, in which Ca2+ influx-mediated actin depolymerization recruits annexins to stabilize the damaged membranes that subsequently fuse with endocytic membranes (Boye & Nylandsted, 2016; Demonbreun et al., 2016; A. K. McNeil, Rescher, Gerke, & McNeil, 2006; P. L. McNeil & Kirchhausen, 2005); (2) an endocytic model, in which lysosomal exocytosis mediates rapid internalization of the membrane-associated pore-forming toxins through caveolin-dependent endocytosis (Andrews, Almeida, & Corrotte, 2014; Corrotte et al., 2013; Idone et al., 2008); and (3) a shedding model, in which damaged membranes are sequestered into membrane blebs and then shed through microvesicles (Jimenez et al., 2014; Keyel et al., 2011; Romero et al., 2017). However, the mechanisms by which host cells deal with plasma membrane damage induced by a pore-forming toxin acting from within the host cell are largely unknown and clearly not sufficient to prevent the toxicity of PFO or PEST-minus LLO. We propose that LLO has evolved to take advantage of a conserved endocytic pathway that provides an auxiliary mechanism to limit damage to the host plasma membrane and prevent the induction of host cell death.
Pore-forming toxins like LLO are double-edged swords for intracellular pathogens. While these toxins allow pathogens to access the host cytosol, their activity must be tightly controlled and compartmentalized to avoid killing of host cells. The PEST-like sequence is the primary molecular feature that provides LLO its uniqueness and contributes to _L. monocytogenes_pathogenesis by making this toxin a phagosome-specific cytolysin with minimal cytotoxicity. We propose a model in which LLO exploits different host processes during infection to limit its cytotoxicity (Figure 6), thus providing fail-safe mechanisms to ensure completion of the L. monocytogenes intracellular life cycle. According to this model, newly secreted LLO can form cytosolic aggregates and be degraded by the proteasome in a process that does not rely on the PEST-like sequence (Schnupf et al., 2006; 2007). Next, LLO monomers, that escape proteasomal degradation, bind to the host plasma membrane and assemble into pores that are potentially lethal to the host cell. However, as revealed by this study, LLO is continuously removed from the host plasma membrane by its interaction with Ap2a2, a component of the endocytosis machinery. We further propose that LLO-containing endosomes are recognized by the autophagy machinery or invaginated into multivesicular bodies for degradation. In addition to its essential role in pathogenesis, LLO is a major antigenic determinant for the induction of an adaptive immune response during infection. Characterization of the different processes involved in LLO degradation could lead to a better understanding of antigen presentation pathways and to the development of novel vaccine strategies.
STAR Methods Text
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Daniel A. Portnoy (portnoy@berkeley.edu).
Experimental model and subject details
Immunocompetent female C57BL/6J (6–7 weeks old) and Igs2tm1.1(CAG-cas9*)Mmw/J mice (6–7 weeks old) were purchased from Jackson Laboratory. Mice were housed under specific pathogen-free biosafety level 2 conditions and maintained on a 12 hours light/dark cycle with unlimited access to water and food. All mice showed normal healthy behavior before the challenge experiments. Animal experiments were reviewed and approved by the Animal Care and Use Committee at the University of California, Berkeley.
Method Details
Construction of L. monocytogenes strains
The L. monocytogenes strains used in this study are listed in the reagent or resource table. All mutant strains were derived from Δ_hly_ (DP-L2161) (Jones & Portnoy, 1994). Mutant alleles of the LLO gene (hly) were cloned into _Listeria_integration plasmid pPL2 under the hly promoter (Lauer, Chow, Loessner, Portnoy, & Calendar, 2002) and introduced into the L. monocytogenes_Δ_hly background. All resulting mutations were confirmed by sequencing. All L. monocytogenes strains were grown in brain heart infusion (BHI) at 30°C overnight prior to each experiment unless otherwise specified.
To generate hly_fl strains, loxP_ sites were inserted into the L. monocytogenes genome flanking hly, and Cre recombinase, which mediates DNA recombination between_loxP_ sites, was inserted into the genome under the control of the _L. monocytogenes_cytosol-specific actA promoter, as previously described (Reniere et al., 2015).
Hemolytic activity screen and quantification
The hemolytic activity assay was performed in a 96-well V-bottom styrene plate with hemolysis buffer (35 mM sodium phosphate, 125 mM sodium chloride, 10mM cysteine hydrochloride, adjusting pH to 5.5 or 7.0). Supernatant fluid from of logarithmic phase culture grown in BHI were serially diluted two-fold in 100μl assay buffer, and then 100μl of 5% sheep red blood cells (HemoStat Laboratories) were added to each well. The plates were incubated shaking at 37°C, and then pelleted in the V-bottom. Supernatant was transferred from the V-bottom plate into corresponding locations in a flexible polyvinyl chloride flat bottom 96-well plate, and the absorbance at 450 nm was measured for each well (Spectramax340) and analyzed with SoftMax Pro v1.2 software (Molecular Devices Corp.). Hemolytic activity was presented as the percentage of sheep red blood cells that had been lysed completely by 1% Triton X-100.
To quantify the excision efficiency of hly_fl inside BMMs,_ BMMs were infected with _hly_fl and lysed in water at different time post infection. Lysates were then spread on blood agar plate and both hemolytic and ahemolytic colonies were enumerated as the excision of hly results in loss of hemolytic activity.
Yeast Two Hybrid
The Matchmaker Gold Yeast Two-Hybrid System (Clontech) was used to identify the interaction between LLO and host proteins. LLO, without its membrane-binding domain (26–480aa), was inserted between the BamHI and PstI sites of pGBKT7 vector (Clontech) to generate pGBKT7-LLO as the bait, then transformed into Y2HGold Competent Cells. Universal Mouse Mate & Plate™ Normalized Library (Clontech 630482) was used as the prey library to screen the LLO interacting host proteins. The appendage domain of mouse Ap2a2 cDNA was inserted between the BamHI and PstI sites of the pGADT7 vector (Clontech) to generate pGADT7-Ap2a2 as the prey to confirm the interaction. _S. cerevisiae_containing pGBKT7-53 and pGADT7-T plasmids was used as a positive control, and S. cerevisiae containing pGBKT7-lam + pGADT7-T plasmids was used as a negative control. Plasmids were transformed into Y2HGold Competent Cells according to the small-scale transformation and mating procedure described in the Matchmaker Gold Yeast Two-Hybrid System user manual (Clontech). The Y2HGold bait strain and Y187 pray strain were mated in 2xYPDA and plated on SD/-Leu/-Trp/ (DDO) or SD/-Ade/-His/-Leu/-Trp/X-a-Gal/AbA (QDO/X/A) agar plate. The activity of the secreted α-galactosidase enzyme was measured according to manufacturer’s instructions (Clontech). Briefly, diploid S. cerevisiae strains were cultured in DDO medium and supernatants were collected from the overnight culture (20 hours post inoculation) and mixed with PNP-α-Gal Solution (Clontech). The reaction was terminated after 3 hours of incubation and optical density at 410nm was measured by SPECTRAmax (Molecular Devices).
Intracellular Growth Curves
Bone marrow derived macrophages (BMMs) were isolated from 6- to 8-wk-old female mice and cultured as described (Mitchell et al., 2015). BMMs were infected with WT or mutant L. monocytogenes at a MOI of 0.2 (~1 bacterium per 5 macrophages, which results in the infection of 5–10% of the BMMs) as previously described (Mitchell et al., 2015). Thirty minutes after addition of bacteria, macrophage monolayers were washed three times with PBS, and fresh medium was added. At 1 hour post-infection, gentamicin was added at 50 μg/ml to kill extracellular bacteria. Replication was quantified by enumerating intracellular colony forming units (CFU). Three biological repeat samples were taken and analyzed from each time point (N=3).
Plaque Assay
Plaque formation in mouse L2 fibroblasts was carried out as previously described (Sun, Camilli, & Portnoy, 1990), with modifications to the methods of measurement (Skoble, Portnoy, & Welch, 2000). Briefly, L2 fibroblasts, maintained in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific) containing 10 % fetal bovine serum (VWR Life Science) and grown to confluence in 6-well tissue culture plates, were infected with 2×106 of washed overnight cultures of_L. monocytogenes_. At 1-hour post-infection, cell monolayers were washed three times with PBS and the medium was replaced with DMEM agar containing 10μg/ml gentamicin. The infected cells were then incubated for an additional 3 days. The plaques were visualized 8 hours after addition of another DMEM agar overlay containing 10μg/ml gentamicin and 2.5% neutral red (GIBCO/Invitrogen). The relative plaque size was measured by ImageJ software (NIH) and reported as a percentage of the wild-type plaque size.
Lactate Dehydrogenase (LDH) Release Assay
The LDH release assay was performed as previously described (Decker & Lohmann-Matthes, 1988). Twenty-four well plates were seeded with 5×105 BMM per well and each well was infected with 2×106 of washed overnight cultures of L. monocytogenes (or mock-infected) for 30 mins. Macrophages were subsequently washed three times with PBS and then BMM media without gentamicin was added. At 6 hours post-infection, 60 μl supernatant from infected BMMs was added to 60 μl LDH detection reagent in 96-well plate and the absorbance of each well was measured on a SpectraMax 340 spectrophotometer (Molecular Devices) at wavelength 490 nm. The lysis values were calculated as a percentage of cells lysed completely by 1% Triton X-100. Percent maximal LDH release was determined using the following formula: % Maximal LDH release = [(experimental LDH release) − (spontaneous LDH release)]/[(maximum LDH release) − (spontaneous LDH release)].
SYTOX Blue Nucleic Acid Stain and Flow Cytometry Analysis
SYTOX Blue was used to evaluate the plasma membrane integrity of L. monocytogenes infected BMMs. Briefly, 1×106 BMMs were seeded into each well of non-TC treated 6 well plates and were infected with 2×106 of washed overnight cultures of L. monocytogenes (or mock-infected) for 30 min. BMMs were subsequently washed three times with PBS and then cultured with media containing 1μM SYTOX Blue. At 8 hours post-infection, BMMs were washed two times with PBS and then detached from plate by incubating at 4°C for 10mins with cold PBS. Detached BMMs were then analyzed by LSR Fortessa using 405nm excitation with a 450/50 nm bandpass filter.
Cell transfection and co-immunoprecipitation
HeLa and HEK293T cells (preserved in our laboratory) were maintained in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific) containing 10% fetal bovine serum (VWR Life Science), 100 units/ml penicillin, and 100 μg/ml streptomycin. One day before transfection, 1×105 cells were seeded into each well of a 24-well plate containing one microscope coverslips (Thermo Fisher Scientific). When the cells reached ~80 % confluence, the LLO-pcDNA3.0 or LLO-pEGFP-N1 plasmid was transfected into the cells using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. For co-immunoprecipitation, HEK293T cells were co-transfected using Ap2a2-pCMV-HA together with LLO26-529-pCMV-Myc or LLOΔPEST56-529-pCMV-Myc vector. All cells were cultured at 37°C with 5 % CO2.
Whole cell extracts of transfected cells were prepared at 24 hours post-transient transfection in a mild lysis buffer consisting of 20 mM Tris, 150 mM NaCl, 2 % glycerol, 1.6 mM EDTA, 0.5 % Triton X-100 and a protease inhibitor cocktail (Cell Signaling Technology). The supernatant was collected and incubated overnight at 4 °C with a mouse monoclonal anti-c-Myc antibody (Clontech) and Protein G Dynabeads (Thermo Fisher Scientific) at 4 °C. The beads were washed with 1 ml washing buffer A (20 mM Tris, 150 mM NaCl) four times and the precipitates were eluted with 300 μl elution buffer (0.1 M glycine at pH 3). Finally, the eluted proteins were prepared for loading on SDS-PAGE and immunoblotting using rabbit polyclonal anti-HA-Tag antibody and mouse monoclonal anti-c-Myc antibody (Clontech).
Immune Fluorescent Microscopy and Total Internal Reflection Microscopy
Microscope coverslips containing the infected or transfected cells were washed twice with phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde (PFA) for 15 min. Then, coverslips were incubated for 30 min in permeabilization/blocking buffer (PBS containing 2% bovine serum albumin and 0.1% saponin) at room temperature. Primary antibodies that recognize LLO (gift from Pascale Cossart), p62 (Fitzgerald, Acton, MA, USA; no. 20R-PP001; 1:200 dilution), ubiquitylated proteins (Enzo Life Sciences), HA tag (Clontech), or Myc tag (Clontech) were diluted in PB buffer containing 0.1% saponin, added to coverslips and incubated for 60 min. The coverslips were washed 6 times with 0.1% saponin in PBS. The secondary antibodies AlexaFluor-488 goat anti-mouse IgG (Thermo Fisher Scientific), rhodamine red-X goat anti-rabbit IgG (Thermo Fisher Scientific), phalloidin AlexaFluor-647 (Thermo Fisher Scientific) were diluted in PB buffer, added to coverslips, and incubated for 1 hour. The coverslips were washed 3 times in 0.1% saponin in PBS and mounted on a drop of ProLong Gold antifade reagent containing 4′6,-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific). Imaging was acquired on an Olympus IX81 epifluorescence microscope or a KEYENCE BZ-X710 fluorescent microscope using a 100× objective.
TIRF microscopy images were captured using MetaMorph software on an Olympus IX81 epifluorescence microscope using an APON 60×/1.49 NA TIRF oil objective. The system was maintained at 37°C using a weather station chamber and temperature controller. At 16–24 hours before imaging, cells were seeded onto uncoated coverslips and transfected with LLO-pEGFP N1 plasmids. During imaging, cells were maintained in DMEM without phenol red (Life Technologies) that was supplemented with 5% FBS and 10 mM HEPES (Thermo Fisher Scientific). A 488-nm solid-state laser (Melles Griot) and a 561-nm diode-pumped solid-state laser (Melles Griot) were used to excite GFP and RFP fluorophores, respectively. For lifetime analysis, images were acquired every 2s for 3–6 min, with an exposure time of 500 ms. Kymographs was generated by ImageJ (National Institutes of Health) from images acquired from one representative experiment.
Ap2a2 Knockdown in Cas9+ BMM
Lentiviruses were use to deliver guide RNAs into bone marrow cells that overexpress Streptococcus pyogenes CRISPR associated protein 9 (Cas9) endonuclease to generate Ap2a2 knockout BMMs. Lentivirus plasmid pLX-sgRNA was acquired from Addgene (Addgene plasmid # 50662). Two independent mouse Ap2a2-specific sgRNAs were predicted using the CRISPR design web portal from the Broad Institute. The sgRNA sequences were amplified by PCR and cloned into pLX-sgRNA vector. One microgram of pLX-sgRNA plasmid was co-transfected with 100ng VSV-G and 900ng psPAX2 plasmids into 3.5×106 HEK293T cells in a 6-well plate using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. Media was changed 24 hours after transfection. The virus-containing supernatant was collected 48 and 72 hours after transfection and passed through a 0.45 μm filter. Bone marrow cells derived from the femurs of mice expressing Cas9 (The Jackson Laboratory) were infected with pooled lenti-virus carrying both sgRNAs in media containing 8 μg/mL of polybrene. One day after infection, virus particles were removed and fresh BMM media was added to differentiate the bone-marrow cells into macrophages. Three days later, cells were selected with 5μg/mL blasticidine for another 3 days. Cell lysates were analyzed using immunoblotting and a mouse monoclonal anti-Ap2a2 antibody (Abcam) in order to evaluate knockout efficiency. Mouse monoclonal Anti-β-actin antibody (Abcam) was used to detect β-actin as a loading control.
In Vivo Infections
Eight-week old female C57BL/6J mice (The Jackson Laboratory) were infected intravenously with 1 × 105 CFU of logarithmic phase L. monocytogenes sub-cultured in BHI at 37 °C. Forty-eight hours post-infection, livers and spleens mice were harvested from infected, homogenized in 0.1% NP-40 and plated on BHI agar to enumerate CFU.
Quantification and Statistical Analysis
Statistical analysis was performed using GraphPad Prism software (v.6.02). Data was judged to be statistically significant when p < 0.05 by two-tailed Student’s t test. Student’s t test for the analysis of each experiment was specified in figure legends. (*, p < 0.05; **, p < 0.01;***, p < 0.001). Detailed information on the n of biological samples and animals used can be found in figure legends and STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-LLO antibody | Gift from Pascale Cossart | Clone B3-19 |
| Mouse monoclonal anti-Ubiquitin antibody, clone FK2 | Enzo Life Sciences | Cat# PW8805 |
| Mouse monoclonal anti-p62 antibody | Enzo Life Sciences | Cat# 20R-PP001 |
| Rabbit polyclonal anti-HA-Tag Antibody | Clontech | Cat# 631207 |
| Mouse monoclonal anti-c-Myc Antibody | Clontech | Cat# 631206 |
| Mouse monoclonal Anti-Ap2a2 antibody | Abcam | Cat# ab220065 |
| Mouse monoclonal Anti-β-actin antibody | Abcam | Cat# ab8226 |
| AlexaFluor-488 goat anti-mouse IgG | Thermo Fisher Scientific | Cat# A11029 |
| Rhodamine goat anti-rabbit IgG | Thermo Fisher Scientific | Cat# R-6394 |
| Phalloidin AlexaFluor-647 | Thermo Fisher Scientific | Cat# A12379 |
| Bacterial and Virus Strains | ||
| Listeria monocytogenes 10403s::pHyper-GFP-pPL2 | Lab strain | DP-L6643 |
| Listeria monocytogenes 10403s Δ_hly::hly_-pPL2 | This study | DP-L6644 |
| Listeria monocytogenes 10403s Δ_hly_:: ΔPEST _hly_-pPL2 | This study | DP-L6645 |
| Listeria monocytogenes 10403s Δ_hly_:: HCaR PEST_hly_-pPL2 | This study | DP-L6646 |
| Listeria monocytogenes 10403s hlyfl | This study | DP-L6647 |
| Listeria monocytogenes 10403s hlyfl:: pactA-Cre-pPL2e | This study | DP-L6648 |
| Listeria monocytogenes 10403s Δ_hly_ | Lab strain | DP-L2161 |
| Listeria monocytogenes 10403s L461T | Lab strain | DP-L4017 |
| Biological Samples | ||
| Defibrinated sheep blood | Hemostat | Cat# DSB100 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SYTOX™ Blue Dead Cell Stain, for flow cytometry | Thermo Fisher Scientific | Cat# ab220065 |
| Dropout Supplement –Ade/–His/–Leu/–Trp | Clontech | Cat# 630428 |
| Dropout Supplement –Leu/–Trp | Clontech | Cat# 630417 |
| Dropout Supplement –Leu | Clontech | Cat# 630414 |
| Dropout Supplement –Trp | Clontech | Cat# 630413 |
| X-alpha-Gal | Clontech | Cat# 630462 |
| Aureobasidin A | Clontech | Cat# 630499 |
| Dulbecco’s Modified Eagle Medium | Thermo Fisher Scientific | Cat# 11965-084 |
| Premium Grade Fetal Bovine Serum (FBS) | VWR Life Science | Cat# 97068-085 |
| L-glutamine | Thermo Fisher Scientific | Cat# 25030024 |
| Neutral Red Solution | Sigma-Aldrich | Cat# N2889 |
| Cell Lysis Buffer | Cell Signaling Technology | Cat# 9803s |
| HEPES | Thermo Fisher Scientific | Cat# 15630106 |
| Critical Commercial Assays | ||
| Matchmaker Gold Yeast Two-Hybrid System | Clontech | Cat# 630489 |
| Mate & Plate™ Library - Universal Mouse (Normalized) | Clontech | Cat# 630482 |
| QIAprep Spin Miniprep | Qiagen | Cat# 27115 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat# 11668019 |
| CloneAmp HiFi PCR Premix | Clontech | Cat# 639298 |
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat# 10004D |
| pLX-sgRNA | Addgene | Cat# 631604 |
| pCMV-Myc & pCMV-HA Vector Set | Clontech | Cat# 631604 |
| Experimental Models: Cell Lines | ||
| Human: HeLa S3 cell line | Lab stock | RRID:CVCL_0058 |
| Human: HEK293T cell line | Lab stock | https://www.atcc.org/Products/All/CRL-11268.aspx |
| L2 | Lab stock | https://www.atcc.org/products/all/CCL-149.aspx |
| Experimental Models: Organisms/Strains | ||
| Saccharomyces cerevisiae: Y2HGold Yeast Strain | Clontech | Cat# 630498 |
| Saccharomyces cerevisiae: Y187 Yeast Strain | Clontech | Cat# 630457 |
| Mouse: C57BL/6J strain female | The Jackson Laboratory | Cat# 000664; RRID:IMSR_JAX:000664 |
| Mouse: Igs2tm1.1(CAG-cas9*)Mmw/J female | The Jackson Laboratory | Cat# 027650 |
| Oligonucleotides | ||
| Ap2a2 CRISPR guide sgRNA 1 | AGGGCGAGACCCATAAACGT | Broad institute GPP Web Portal https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| Ap2a2 CRISPR guide sgRNA 2 | TCACCTGACCTAGTTCCCAT | Broad institute GPP Web Portal https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| Recombinant DNA | ||
| LLO-pcDNA3 | This study | DP-E6649 |
| LLO26-529-pEGFP-N1 | This study | DP-E6650 |
| LLO126-529-pEGFP-N1 | This study | DP-E6651 |
| LLO226-529-pEGFP-N1 | This study | DP-E6652 |
| LLO26-480-pEGFP-N1 | This study | DP-E6653 |
| LLO-N26-180-pGBKT7 | This study | DP-E6654 |
| LLO-M170-320-pGBKT7 | This study | DP-E6655 |
| LLO-C310-480-pGBKT7 | This study | DP-E6656 |
| LLOΔPEST26-480-pGBKT7 | This study | DP-E6657 |
| PFO37-455-pGBKT7 | This study | DP-E6658 |
| LLO-PEST-pGBKT7 | This study | DP-E6659 |
| HCaR-PEST-pGBKT7 | This study | DP-E6660 |
| Ap2a2(appendage domain)-pGADT7 | This study | DP-E6661 |
| Filamin α-pGADT7 | This study | DP-E6662 |
| RNF25-pGADT7 | This study | DP-E6663 |
| Snapin-pGADT7 | This study | DP-E6664 |
| LLO26-529-pCMV-Myc | This study | DP-E6665 |
| LLOΔPEST 56-529-pCMV-Myc | This study | DP-E6666 |
| Ap2a2(appendage domain)-pCMV-HA | This study | DP-E6667 |
| pLX-Ap2a2 sgRNA 1 | This study | DP-E6668 |
| pLX-Ap2a2 sgRNA 2 | This study | DP-E6669 |
| pactA-Cre-pPL2e | This study | DP-E6670 |
| _hly_-pPL2 | This study | DP-E6671 |
| ΔPEST _hly_-pPL2 | This study | DP-E6672 |
| HCaR PEST _hly_-pPL2 | This study | DP-E6673 |
| Software and Algorithms | ||
| Prism | GraphPad | ver 6.02; RRID: SCR_007370 |
| ImageJ | NIH | ver 1.49a; RRID:SCR_003070 |
| FlowJo | FlowJo | Ver 8.7; RRID:SCR_001155 |
| SoftMax Pro | Molecular Devices | Ver 1.2 |
| Other | ||
| Spectramax340 | Molecular Devices | NA |
| Olympus IX81 epifluorescence microscope | Olympus | NA |
| BZ-X710 KEYENCE fluorescent microscope | Keyence | NA |
| LSR Fortessa | BD Biosciences | NA |
Supplementary Material
supplement
Highlights.
- The PEST-like sequence of Listeriolysin O (LLO) regulates its plasma membrane levels
- LLO interacts with the endocytosis adaptor Ap2a2 via its PEST-like sequence
- Ap2a2 is required to prevent LLO-induced plasma membrane damage and cytotoxicity
- A PEST-like sequence of a cellular GPCR can rescue the cytotoxicity of a LLOΔPEST strain
Acknowledgments
We thank Sarah Stanley (UC Berkeley) for generously providing us with mouse femurs and David Drubin and his lab (UC Berkeley) for assistance on TIRF microscopy. This work was supported by National Institutes of Health grants 1P01 AI063302 (D.A.P.) and 1R01 AI027655 (D.A.P.), and by the DTRA funding grant HDTRA1-13-1-0003 (C.C. & D.A.P.). G.M. was supported by fellowships from Fonds de recherche du Québec - Nature et technologies (FRQNT), Fonds de recherche du Québec - Santé (FRQS), and the Natural Sciences and Engineering Research Council of Canada (NSERC). B.N.N. was supported by the National Science Foundation Graduate Research Fellowship under grant no. DGE 1106400.
Footnotes
Author Contributions
C.C. and D.A.P. designed and supervised this study. C.C., B.N.N., G.M., S.R.M, and D.M. conducted all experiments. C.C. and D.A.P. wrote the paper. G.M. and B.N.N. edited the paper.
Declaration of Interests
Daniel A. Portnoy has a consulting relationship with and a financial interest in Aduro Biotech. Both he and the company stand to benefit from the commercialization of the results of this research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adzhubei AA, Sternberg MJE, Makarov AA. Polyproline-II helix in proteins: structure and function. Journal of Molecular Biology. 2013;425(12):2100–2132. doi: 10.1016/j.jmb.2013.03.018. [DOI] [PubMed] [Google Scholar]
- Alouf JE. Pore-forming bacterial protein toxins: an overview. Current Topics in Microbiology and Immunology. 2001;257:1–14. [PubMed] [Google Scholar]
- Andrews NW, Almeida PE, Corrotte M. Damage control: cellular mechanisms of plasma membrane repair. Trends in Cell Biology. 2014;24(12):734–742. doi: 10.1016/j.tcb.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, Sasakawa C. Cell death and infection: a double-edged sword for host and pathogen survival. The Journal of Cell Biology. 2011;195(6):931–942. doi: 10.1083/jcb.201108081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazek AD, Paleo BJ, Weisleder N. Plasma Membrane Repair: A Central Process for Maintaining Cellular Homeostasis. Physiology (Bethesda, Md) 2015;30(6):438–448. doi: 10.1152/physiol.00019.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye TL, Nylandsted J. Annexins in plasma membrane repair. Biological Chemistry. 2016;397(10):961–969. doi: 10.1515/hsz-2016-0171. [DOI] [PubMed] [Google Scholar]
- Corrotte M, Almeida PE, Tam C, Castro-Gomes T, Fernandes MC, Millis BA, et al. Caveolae internalization repairs wounded cells and muscle fibers. eLife. 2013;2:e00926. doi: 10.7554/eLife.00926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell G, Orgel JPRO, Pahl R, Meredith SC. Flanking polyproline sequences inhibit beta-sheet structure in polyglutamine segments by inducing PPII-like helix structure. Journal of Molecular Biology. 2007;374(3):688–704. doi: 10.1016/j.jmb.2007.09.023. [DOI] [PubMed] [Google Scholar]
- Decatur AL, Portnoy DA. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science. 2000;290(5493):992–995. doi: 10.1126/science.290.5493.992. [DOI] [PubMed] [Google Scholar]
- Decker T, Lohmann-Matthes ML. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. Journal of Immunological Methods. 1988;115(1):61–69. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- Demonbreun AR, Quattrocelli M, Barefield DY, Allen MV, Swanson KE, McNally EM. An actin-dependent annexin complex mediates plasma membrane repair in muscle. The Journal of Cell Biology. 2016;213(6):705–718. doi: 10.1083/jcb.201512022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunstone MA, Tweten RK. Packing a punch: the mechanism of pore formation by cholesterol dependent cytolysins and membrane attack complex/perforin-like proteins. Current Opinion in Structural Biology. 2012;22(3):342–349. doi: 10.1016/j.sbi.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussurget O, Pizarro-Cerda J, Cossart P. Molecular determinants of Listeria monocytogenes virulence. Annual Review of Microbiology. 2004;58(1):587–610. doi: 10.1146/annurev.micro.57.030502.090934. [DOI] [PubMed] [Google Scholar]
- Eker F, Griebenow K, Cao X, Nafie LA, Schweitzer-Stenner R. Tripeptides with ionizable side chains adopt a perturbed polyproline II structure in water. Biochemistry. 2004;43(3):613–621. doi: 10.1021/bi035740. [DOI] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glomski IJ, Decatur AL, Portnoy DA. Listeria monocytogenes mutants that fail to compartmentalize listerolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect Immun. 2003;71(12):6754–6765. doi: 10.1128/IAI.71.12.6754-6765.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glomski IJ, Gedde MM, Tsang AW, Swanson JA, Portnoy DA. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. The Journal of Cell Biology. 2002;156(6):1029–1038. doi: 10.1083/jcb.200201081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MR, Bischofberger M, Pernot L, van der Goot FG, Frêche B. Bacterial pore-forming toxins: the (w)hole story? Cellular and Molecular Life Sciences: CMLS. 2008;65(3):493–507. doi: 10.1007/s00018-007-7434-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SO, Kommaddi RP, Shenoy SK. Distinct roles for β-arrestin2 and arrestin-domain-containing proteins in β2 adrenergic receptor trafficking. EMBO Reports. 2013;14(2):164–171. doi: 10.1038/embor.2012.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. The Journal of Cell Biology. 2008;180(5):905–914. doi: 10.1083/jcb.200708010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez AJ, Perez F. Plasma membrane repair: the adaptable cell life-insurance. Current Opinion in Cell Biology. 2017;47:99–107. doi: 10.1016/j.ceb.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. ESCRT machinery is required for plasma membrane repair. Science. 2014;343(6174):1247136. doi: 10.1126/science.1247136. [DOI] [PubMed] [Google Scholar]
- Jones S, Portnoy DA. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect Immun. 1994;62(12):5608–5613. doi: 10.1128/iai.62.12.5608-5613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genetics. 2007;3(5):e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyel PA, Loultcheva L, Roth R, Salter RD, Watkins SC, Yokoyama WM, Heuser JE. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. Journal of Cell Science. 2011;124(Pt 14):2414–2423. doi: 10.1242/jcs.076182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary structure of Huntingtin amino-terminal region. Structure (London, England: 1993) 2009;17(9):1205–1212. doi: 10.1016/j.str.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster S, van Pee K, Hudel M, Leustik M, Rhinow D, Kühlbrandt W, et al. Crystal structure of listeriolysin O reveals molecular details of oligomerization and pore formation. Nature Communications. 2014;5:3690. doi: 10.1038/ncomms4690. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host & Microbe. 2010;8(1):44–54. doi: 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Holt JA, Barak LS, Caron MG. The interaction of beta-arrestin with the AP-2 adaptor is required for the clustering of beta 2-adrenergic receptor into clathrin-coated pits. The Journal of Biological Chemistry. 2000;275(30):23120–23126. doi: 10.1074/jbc.M002581200. [DOI] [PubMed] [Google Scholar]
- Lauer P, Chow MYN, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. Journal of Bacteriology. 2002;184(15):4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias MJ, Wiesner S, Sudol M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Letters. 2002;513(1):30–37. doi: 10.1016/s0014-5793(01)03290-2. [DOI] [PubMed] [Google Scholar]
- Martinez LO, Agerholm-Larsen B, Wang N, Chen W, Tall AR. Phosphorylation of a pest sequence in ABCA1 promotes calpain degradation and is reversed by ApoA-I. The Journal of Biological Chemistry. 2003;278(39):37368–37374. doi: 10.1074/jbc.M307161200. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nature Publishing Group. 2011;12(8):517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- McNeil AK, Rescher U, Gerke V, McNeil PL. Requirement for annexin A1 in plasma membrane repair. The Journal of Biological Chemistry. 2006;281(46):35202–35207. doi: 10.1074/jbc.M606406200. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Kirchhausen T. An emergency response team for membrane repair. Nature Reviews. Molecular Cell Biology. 2005;6(6):499–505. doi: 10.1038/nrm1665. [DOI] [PubMed] [Google Scholar]
- Mitchell G, Chen C, Portnoy DA. Strategies Used by Bacteria to Grow in Macrophages. Microbiology Spectrum. 2016;4(3) doi: 10.1128/microbiolspec.MCHD-0012-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell G, Ge L, Huang Q, Chen C, Kianian S, Roberts MF, et al. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infection and Immunity. 2015:IAI.00110–15. doi: 10.1128/IAI.00110-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi H, Hashimoto K, Panchenko AR. Phosphorylation in protein-protein binding: effect on stability and function. Structure (London, England: 1993) 2011;19(12):1807–1815. doi: 10.1016/j.str.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi M, Oakley RH, Gesty-Palmer D, Cruickshank RD, Spurney RF, Luttrell LM, Quarles LD. Beta-arrestin- and G protein receptor kinase-mediated calcium-sensing receptor desensitization. Molecular Endocrinology (Baltimore, Md) 2005;19(4):1078–1087. doi: 10.1210/me.2004-0450. [DOI] [PubMed] [Google Scholar]
- Portnoy DA, Auerbuch V, Glomski IJ. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. The Journal of Cell Biology. 2002;158(3):409–414. doi: 10.1083/jcb.200205009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Tweten RK, Johnson AE. Membrane-dependent conformational changes initiate cholesterol-dependent cytolysin oligomerization and intersubunit beta-strand alignment. Nature Structural & Molecular Biology. 2004;11(8):697–705. doi: 10.1038/nsmb793. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends in Biochemical Sciences. 1996;21(7):267–271. [PubMed] [Google Scholar]
- Reniere ML, Whiteley AT, Hamilton KL, John SM, Lauer P, Brennan RG, Portnoy DA. Glutathione activates virulence gene expression of an intracellular pathogen. Nature. 2015;517(7533):170–173. doi: 10.1038/nature14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero M, Keyel M, Shi G, Bhattacharjee P, Roth R, Heuser JE, Keyel PA. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death and Differentiation. 2017;24(5):798–808. doi: 10.1038/cdd.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Davis NG. Ubiquitination of the PEST-like endocytosis signal of the yeast a-factor receptor. The Journal of Biological Chemistry. 2000;275(11):8143–8153. doi: 10.1074/jbc.275.11.8143. [DOI] [PubMed] [Google Scholar]
- Roth AF, Sullivan DM, Davis NG. A large PEST-like sequence directs the ubiquitination, endocytosis, and vacuolar degradation of the yeast a-factor receptor. The Journal of Cell Biology. 1998;142(4):949–961. doi: 10.1083/jcb.142.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host & Microbe. 2010;7(5):412–419. doi: 10.1016/j.chom.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnupf P, Portnoy DA. Listeriolysin O: a phagosome-specific lysin. Microbes and Infection/Institut Pasteur. 2007;9(10):1176–1187. doi: 10.1016/j.micinf.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Schnupf P, Portnoy DA, Decatur AL. Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: role of the PEST3like sequence. Cellular Microbiology. 2006;8(2):353–364. doi: 10.1111/j.1462-5822.2005.00631.x. [DOI] [PubMed] [Google Scholar]
- Schnupf P, Zhou J, Varshavsky A, Portnoy DA. Listeriolysin O secreted by Listeria monocytogenes into the host cell cytosol is degraded by the N-end rule pathway. Infect Immun. 2007;75(11):5135–5147. doi: 10.1128/IAI.00164-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatursky O, Heuck AP, Shepard LA, Rossjohn J, Parker MW, Johnson AE, Tweten RK. The mechanism of membrane insertion for a cholesterol-dependent cytolysin: a novel paradigm for pore-forming toxins. Cell. 1999;99(3):293–299. doi: 10.1016/s0092-8674(00)81660-8. [DOI] [PubMed] [Google Scholar]
- Skoble J, Portnoy DA, Welch MD. Three regions within ActA promote Arp2/3 complex-mediated actin nucleation and Listeria monocytogenes motility. The Journal of Cell Biology. 2000;150(3):527–538. doi: 10.1083/jcb.150.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun AN, Camilli A, Portnoy DA. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun. 1990;58(11):3770–3778. doi: 10.1128/iai.58.11.3770-3778.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovo-Rodrigues L, Roux A, Hutz MH, Rohde LA, Woods AS. Functional characterization of G-protein-coupled receptors: A bioinformatics approach. Neuroscience. 2014;277:764–779. doi: 10.1016/j.neuroscience.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viala JPM, Mochegova SN, Meyer-Morse N, Portnoy DA. A bacterial pore-forming toxin forms aggregates in cells that resemble those associated with neurodegenerative diseases. Cellular Microbiology. 2008;10(4):985–993. doi: 10.1111/j.1462-5822.2007.01100.x. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Northup JK, Ray K. Large putative PEST-like sequence motif at the carboxyl tail of human calcium receptor directs lysosomal degradation and regulates cell surface receptor level. The Journal of Biological Chemistry. 2012;287(6):4165–4176. doi: 10.1074/jbc.M111.271528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplement