DNA Hypomethylation Perturbs the Function and Survival of CNS Neurons in Postnatal Animals (original) (raw)
Abstract
DNA methyltransferase I (Dnmt1), the maintenance enzyme for DNA cytosine methylation, is expressed at high levels in the CNS during embryogenesis and after birth. Because embryos deficient for Dnmt1 die at gastrulation, the role of Dnmt1 in the development and function of the nervous system could not be studied by using this mutation. We therefore used the cre/loxP system to produce conditional mutants that lack Dnmt1 in neuroblasts of embryonic day 12 embryos or in postmitotic neurons of the postnatal animal. Conditional deletion of the_Dnmt1_ gene resulted in rapid depletion of Dnmt1 proteins, indicating that the enzyme in postmitotic neurons turns over quickly. Dnmt1 deficiency in postmitotic neurons neither affected levels of global DNA methylation nor influenced cell survival during postnatal life. In contrast, Dnmt1 deficiency in mitotic CNS precursor cells resulted in DNA hypomethylation in daughter cells. Whereas mutant embryos carrying 95% hypomethylated cells in the brain died immediately after birth because of respiratory distress, mosaic animals with 30% hypomethylated CNS cells were viable into adulthood. However, these mutant cells were eliminated quickly from the brain within 3 weeks of postnatal life. Thus, hypomethylated CNS neurons were impaired functionally and were selected against at postnatal stages.
Keywords: DNA methylation, Dnmt1, demethylation, neural development, cre/loxP system, respiration, epigenetic mechanism, gene regulation
DNA cytosine methylation in vertebrates influences many cellular events, including gene transcription, genomic imprinting, and genome stability (Jaenisch, 1997; Jones and Gonzalgo, 1997; Robertson and Wolffe, 2000). The DNA methylation pattern in adult cells is established during gametogenesis and early embryonic development via consecutive waves of demethylation and de novo methylation (Monk et al., 1987). A family of DNA (cytosine-5) methyltransferases (Dnmts) has been identified that catalyzes the reaction of cytosine methylation in DNA (Bestor et al., 1988; Okano et al., 1998; Lyko et al., 1999; Okano et al., 1999). The first cloned family member, Dnmt1, encodes a maintenance DNA methyltransferase (Dnmt1; EC 2.1.1.37) that preferentially methylates hemi-methylated DNA that is generated after DNA replication (Bestor et al., 1988). The essential role of Dnmt1 in maintaining DNA methylation has been demonstrated by targeted mutation of the _Dnmt1_gene, which results in demethylation of the DNA in Dnmt1-deficient cells (Li et al., 1992; Lei et al., 1996). Dnmt1 mutant embryos die between embryonic day 8 (E8) and E10.5, indicating that DNA methylation is essential for embryogenesis (Li et al., 1992; Lei et al., 1996). The cause of lethality is not clear, but Dnmt1-deficient embryos exhibit numerous apoptotic cells in many tissues, including brain and liver, suggesting that DNA hypomethylation ultimately may induce an apoptotic pathway (Li et al., 1992). DNA methylation has been shown to play an essential role in the transcriptional regulation of imprinted genes (Li et al., 1993), the X-chromosome-linked Xist gene (Beard et al., 1995; Panning and Jaenisch, 1996), and retroviral intra-cisternal A particles (IAP) (Walsh et al., 1998).
The role of DNA methylation in neural development and function has not been explored. Interestingly, the mammalian brain expresses high levels of Dnmt1 both during development and in adulthood (Goto et al., 1994;Brooks et al., 1996; Trasler et al., 1996; Inano et al., 2000). The level of DNA methylation is higher in adult brain than in other tissues (Wilson et al., 1987; Tawa et al., 1990; Ono et al., 1993). Perinatally, DNA methylation levels in the brain undergo a dynamic change (Tawa et al., 1990), suggesting a role for DNA methylation in the differentiation process of the brain. Further evidence comes from a study of neuronal differentiation in PC12 cells that showed that treatment with a demethylating agent 5-azacytidine blocks neurite outgrowth and upregulates the expression of Id family transcription factors (Persengiev and Kilpatrick, 1996, 1997). Recently, DNA methylation has been associated with at least three mental retardation diseases, including the Rett, ICF, and the fragile X syndromes (for review, see Robertson and Wolffe, 2000). The neurodevelopmental disease Rett syndrome is caused by mutations in the MECP2 gene, a methylcytosine-binding protein (Amir et al., 1999). Because a major function of the MECP2 protein is to mediate methylation-induced gene suppression (Ng and Bird, 1999), the adverse effect of_MECP2_ mutations on brain development suggests that changes in DNA methylation also may affect the postnatal CNS development.Endres et al. (2000) showed that DNA methylation activity increases with transient ischemia and contributes to brain injury, suggesting that levels of DNA methylation influence adult CNS neuron survival under stress conditions.
Dnmt1-deficient embryos die around midgestation before neuronal differentiation (Li et al., 1992; Lei et al., 1996), precluding the study of DNA methylation in brain development. We therefore used the cre/loxP binary system to produce conditional knock-out mice in which the Dnmt1 gene deletion can be achieved in either brain precursor cells at E9–E12 or in postnatal CNS neurons that have exited the cell cycle. We demonstrated that, although Dnmt1 is dispensable for postmitotic neurons, Dnmt1 deficiency in brain precursor cells resulted in significant DNA hypomethylation in progeny cells, including descendant postmitotic neurons. Mutant mice carrying 95% of hypomethylated cells in the CNS died because of respiratory distress, suggesting that DNA hypomethylation perturbs vital CNS functions that are required for postnatal life. Mutant mice with 30% of hypomethylated cells survived into adulthood; however, these hypomethylated cells were eliminated rapidly from the CNS during early postnatal development.
MATERIALS AND METHODS
_Brain-specific Dnmt1 conditional mutant mice._We used the cre/loxP binary system to generate Dnmt1 conditional mutants. Details of generating the Dnmt1 conditional allele (Dnmt12lox) are reported elsewhere (Jackson-Grusby et al., 2000). Briefly, in this line of mice exons 4 and 5 of the Dnmt1 gene were flanked by loxP sites. Cre-mediated deletion of exons 4 and 5 would lead to out-of-frame splicing from exon 3 to exon 6, resulting in a null _Dnmt1_allele (Jackson-Grusby et al., 2000). To achieve Dnmt1 gene deletion in CNS precursor cells in vivo, we crossed the animals carrying the Dnmt12lox conditional allele with the nestin-cre transgenic mice. The production of nestin-cre transgenic mice has been described previously (Bates et al., 1999; Trumpp et al., 1999). For conditional gene deletion in postmitotic CNS neurons, we used the CamK-cre transgenic mice in which the cre expression is under the control of the neuronal calmodulin-kinase IIα (CamK) promoter. We also characterized the distribution of cre-mediated gene deletion by crossing the CamK-cre and nestin-cre transgenic mice with the lacZ reporter strains as described by Akagi et al. (1997) and Soriano (1999). Briefly, the brain sections from different stages of transgenic mice were processed for X-gal staining as described (Trumpp et al., 1999). The positive blue cells indicate places at which the cre-mediated loxP recombination occurred (see Fig. 3 in Results) (Akagi et al., 1997; Soriano, 1999).
Fig. 3.
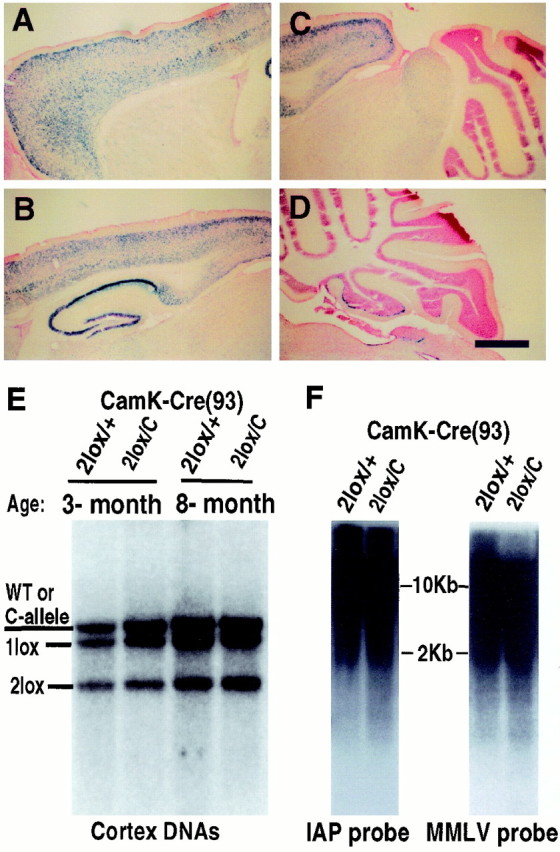
Survival of cortical neurons in the absence of Dnmt1 in vivo. A–D, X-gal staining of brain sections from a 3-week-old mouse carrying the CamK-cre transgene and a lacZ reporter gene under the control of the β-actin promoter (Akagi et al., 1997). The cells positive for β-gal enzymes (blue cells) represent those neurons having undergone cre-mediated gene recombination events. Scale bar, 675 μm.E, Southern blot analysis of cortex DNAs from conditional CamK-cre;Dnmt 2lox/+(2lox/+) heterozygous and_CamK-cre;Dnmt12lox/C_ mutant animals (2lox/C). Note that the wild-type (WT) and null C-alleles were detected at the same size in this blot. The genotypes of wild-type and C-alleles were ascertained by the absence and presence of the neomycin gene in the Dnmt1 locus. F, Methylation analysis in cortex DNAs. IAP, Intra-cisternal A particle retrovirus; MMLV, Moloney murine leukemia virus.
Some of the conditional mutants also carried the previously described_Dnmt1_ mutant N- and C-alleles (Dnmt1N or_C_), which represent _Dnmt1_hypomorphic (∼2% Dnmt1 proteins) and null alleles, respectively (Li et al., 1992; Lei et al., 1996). Southern blot analysis and PCR reactions were used for genotyping mice.
Southern and Northern blot analysis. DNA samples were extracted from brain tissues as previously described (Laird et al., 1991). RNA samples were purified with an RNAzol reagent (Tel-Test, Friendswood, TX) according to the manufacturer's procedure. DNA or RNA samples were subjected to electrophoresis and transferred to a nylon membrane (Zetabond). Hybridization of the blot was performed by the Quickhyb protocol (Stratagene, La Jolla, CA). Details of various DNA probes have been reported (Tucker et al., 1996; Jackson-Grusby et al., 2000). PhosphorImager (Fuji, Tokyo, Japan) and densitometry (Bio-Rad, Hercules, CA) analysis of the intensity of_Dnmt12lox_ and_Dnmt11lox_ alleles was used to quantify the efficiency of Dnmt1 gene deletion by the CamK-cre and nestin-cre transgenes.
Western blot analysis. Brain tissue lysates were separated by 7.5% SDS-PAGE gels, using a Bio-Rad minigel apparatus. One gel was stained with Coomassie blue to visualize whether the protein loading in each lane was even. A duplicate gel was blotted to a nylon membrane by electrotransfer for Western blotting. The procedure of Western blotting has been described (Fan and Katz, 1993). Briefly, the membrane blot was blocked in 5% milk in Tris-buffered saline and incubated with Dnmt1 primary antibodies [Dnmt1 ATG4 Ab, the rabbit anti-N-terminal Dnmt1 peptide; Dnmt1 C-term Ab, chicken anti-Dnmt1 catalytic domain peptide; see Gaudet et al. (1998) for detailed information on the antibodies], followed by peroxidase-conjugated secondary antibodies, and was visualized with enhanced ECL reagents (Amersham, Arlington Heights, IL).
Histological examination and TUNEL staining. Tissues dissected from mutant and control mice were fixed in 4% paraformaldehyde/PBS overnight and embedded in OCT for frozen sections or processed with a VIP tissue processor (Miles, Elkhart, IN) for paraffin sectioning. Serial sections were stained with hematoxylin/eosin or cresyl violet for light microscopy. For cell death analysis the TUNEL staining was performed either with a commercial in situ cell death detection kit (Boehringer Mannheim, Indianapolis, IN) or by the terminal labeling of biotinylated dNTPs with the TdT enzymes (Life Technologies, Gaithersburg, MD) as described (Ben-Sasson et al., 1995), followed with avidin–peroxidase reactions as described in the ABC kit (Vector Laboratories, Burlingame, CA).
Cortical and cerebellar cell cultures. Cortices from fetal brains (E15–E18) were dissected, treated with trypsin or papain, dissociated, and plated on coated glass coverslips, as previously described (Bonni et al., 1997). Detailed protocol of cerebellar cultures has been reported (Datta et al., 1997). At the time of plating, recombinant adenoviruses carrying the cre transgene were added into cultures at a multiplicity of infection (MOI) ratio of 10:1 (virus/cell). Cultures were harvested at different time points for Western and Southern blot analyses.
_Immunocytochemistry and whole-mount immunohistochemistry._Cultured cells were fixed with 4% paraformaldehyde and permeabilized with Triton X-100. Monoclonal β-tubulin antibody (TuJ1) and polyclonal nestin antibodies [kindly provided by Drs. A. Frankfurter (Charlottesville, VA) and R. McKay (Bethesda, MD)] were applied and visualized with fluorescein-conjugated secondary antibodies. For whole-mount staining of the diaphragm the tissues were fixed with 2% paraformaldehyde overnight, blocked in 5% goat serum with 1% DMSO overnight, and then consecutively incubated with anti-neurofilament 150 (Chemicon, Temecula, CA) and FITC-conjugated secondary antibodies. Rhodamine-conjugated α-bungarotoxin (Molecular Probes, Eugene, OR) was used to label postsynaptic AChR in the muscle. The preparations were observed on a Zeiss fluorescence scope.
Xist fluorescence in situ hybridization analysis. Cells were grown on glass coverslips, fixed in 4% paraformaldehyde, and stored in PBS. Hybridization, washing, and detection of probes have been described (Panning and Jaenisch, 1996). Briefly, coverslips were extracted with cytoskeletal buffer containing 0.5% Triton X-100, dehydrated, and incubated with the nick translation probe at 37°C overnight. Slides were washed at 39°C for three times in 2× SSC/50% formamide, three times in 2× SSC, and twice in 1× SSC for 5 min each. Biotinylated probes were detected in 2 mg/ml BSA/4× SSC at 37°C for 30 min, using FITC-avidin. Fluorescent images were captured either by a Nikon scope with Scanlytic system or on Kodak Ektachrome 1600 slide film with a Zeiss Axioskop.
Electrophysiology. Newborn mice were anesthetized with a light dose of sodium pentobarbital (Nembutal; 25–30 mg/kg, i.p.) (Paton et al., 1994) and were placed supine on a heating pad with the temperature regulated at ∼38° C (CWE C-831 Temperature Controller). Central respiratory activity was monitored by recording hypoglossal nerve discharge. A hypoglossal (XII) nerve (which innervates both the genioglossus muscle and tongue retractors) was isolated by blunt dissection with a ventral approach at the cervical level, cut distally, mounted on custom-made bipolar silver-wire electrodes, and kept in a warm mineral oil pool. Inspiratory-related efferent neuronal discharges of the hypoglossal nerve were amplified (CyberAmp 380, Axon Instruments, Foster City, CA), time-averaged with a leaky integrator (Paynter filter; time constant, 15 msec), and recorded (Thermal Array Recorder, Nihon Kohden, Tokyo, Japan). In some experiments small custom-made bipolar fish-hook electrodes were implanted into the diaphragm to record an electromyogram (EMG). Immediately after birth, small stainless steel electrodes were implanted subdermally on both sides of the chest to record an electrocardiograph (EKG). In some experiments the EKG was recorded in normal and mutant pups in utero with the mother under urethane anesthesia (1.2 gm/kg, i.p.).
RESULTS
In vitro survival of postmitotic neurons in the absence of Dnmt1
Previous studies have shown that Dnmt1 transcripts and proteins were highly expressed in the CNS during embryogenesis and in adulthood. A recent study further demonstrated that the enzyme is localized primarily in the cytoplasm of neurons (Inano et al., 2000). To determine the role of Dnmt1 in postmitotic neurons, we used the cre/loxP system to delete the Dnmt1 gene conditionally (Fig.1A). A conditional allele (referred to as Dnmt12lox), in which exons 4 and 5 of the Dnmt1 gene are flanked by loxP sites, was generated via gene targeting in embryonic stem (ES) cells. Expression of cre-recombinase causes the deletion of exons 4 and 5, which results in a null allele of Dnmt1 (designated as a_Dnmt1lox_ allele; Jackson-Grusby et al., 2000).
Fig. 1.

Conditional deletion of the Dnmt1_gene in postmitotic cerebellar neurons. A, Schematic drawing of the cre/loxP-mediated Dnmt1 gene deletion. In the Dnmt1 2lox allele exons 4 and 5 were flanked by the 34 bp loxP sequence. In the presence of cre-recombinase the two loxP sites recombined, resulting in deletion of the exons and creation of the_Dnmt11lox null allele.B, P6 _Dnmt12lox/2lox_cerebellar dissociates were infected with recombinant adenovirus carrying the cre transgene at the time of plating and then were cultured for 1 d (1d) to 14 d (14d). DNAs were extracted from the cultures and digested with Spe_I for Southern blot analysis of the efficiency of the Dnmt1 gene deletion in cerebellar cultures over time. The ratio of recombined null allele (1lox) over the sum of functional_Dnmt12lox (2lox) and 1lox alleles indicates the efficiency of gene deletion.C, Western blot analysis of Dnmt1 proteins in control and mutant neurons. Top, Levels of Dnmt1 protein were detected with Dnmt1 antibodies in cultured cerebellar neurons with or without adeno-cre infection. Bottom, A duplicate gel was stained with Coomassie blue staining to show the equal loading of the protein extracts. D, Southern blot analysis of DNA methylation in cerebellar cultures. DNAs were digested with the methyl-sensitive enzyme _Hpa_II, separated on agarose gel, transferred to the membrane, and hybridized with a centromeric minor satellite repeat probe. Small-molecular-weight fragments in Dnmt1 mutant embryonic stem cells indicate extensive demethylation of their DNA.
We first examined whether the Dnmt1 protein turns over in postmitotic cerebellar neurons. Dnmt1 protein was detected readily in dissociated cerebellar granule neurons from control_Dnmt12lox/2lox_ animals by Western blot analysis (Fig. 1C). In fact, levels of Dnmt1 in these cells were comparable with those in dividing embryonic fibroblasts (data not shown; see also Inano et al., 2000). When cerebellar cultures containing Dnmt12lox/2lox cells were infected with adenoviruses carrying the cre transgene (adeno-cre), deletion of the Dnmt1 gene occurred in 60% of cultured cells within 24 hr (Fig. 1B). Gene deletion was completed at 100% efficiency within 3 d of adenovirus infection (Fig. 1B). In parallel to the time course of gene deletion, Dnmt1 protein levels were decreased significantly within 24 hr and virtually undetectable by the end of 3 d in culture (Fig.1C). No Dnmt1 proteins were detected by Western blot analysis in adeno-cre-infected neurons after 5–7 d, using antibodies against either the N terminus (Fig. 1C) or C terminus of Dnmt1 (data not shown). This result indicates that the enzyme undergoes a rapid turnover in postmitotic cerebellar neurons.
To determine the consequence of Dnmt1 deficiency on levels of DNA methylation and neuronal survival, we examined cultured cerebellar neurons with the Dnmt1 gene deletion for a period of 2 weeks. These neurons apparently survived well in the absence of Dnmt1 during the 2 weeks of the culture period. Immunostaining with neuron-specific β-tubulin antibody (TuJ1) indicated that the neurons were healthy, with extensive neurite outgrowth (Fig.2). Southern blot analysis indicated that the depletion of Dnmt1 did not change global DNA methylation (see Fig.1D).
Fig. 2.

Survival of cerebellar neurons in the absence of Dnmt1. P6 Dnmt1 2lox/2lox cerebellar dissociates were infected with adeno-cre at the beginning and were cultured for 2 weeks. These neuron-enriched cultures were double-stained with neuron-specific β-tubulin III (TuJ1) antibodies (red) to visualize extensive neurites and with DAPI (blue) to show the healthy neuronal nuclei. Scale bar, 45 μm.
Conditional deletion of Dnmt1 in postmitotic CNS neurons in vivo
Although the above results showed that Dnmt1 is not essential for maintaining global DNA methylation and neuronal survival in vitro, it is unclear whether Dnmt1 is required for the survival of postmitotic neurons in vivo. We therefore crossed the_Dnmt12lox_ allele with a strain of CamK-cre (line 93) transgenic mice that express cre in postmitotic neurons from the perinatal stage. CamK-cre-mediated gene deletion in CNS neurons was confirmed first by a lacZ reporter gene (Akagi et al., 1997). Gene deletion was detected in a small number of forebrain neurons perinatally and reached the peak level after 3 weeks postnatally (data not shown). As shown in Figure3A–D, in 3-week-old transgenic mice CamK-cre-mediated gene deletion was widespread in the forebrain, including the cortex, hippocampus, and striatum, but virtually absent in the cerebellum (except in a few scattered Purkinje cells). The blue cells were morphologically CNS neurons, confirming the neuronal specificity of the CamK-cre transgene. The spatial and temporal distribution pattern of the_lacZ_-positive cells in the CamK-cre;lacZ reporter transgenic mice was well confirmed by Southern blot analysis of the efficiency of Dnmt1 gene deletion in various brain regions of CamK-cre;Dnmt1 conditional mutants, suggesting that CamK-cre-mediated Dnmt1 gene deletion occurs in a similar pattern as observed with the lacZ reporter gene. Conditional_Dnmt1_ mutants were recovered at the expected Mendelian ratio, indicating that Dnmt1 deficiency in postmitotic CNS neurons did not affect animal viability. Southern blot analysis showed that Dnmt1-deficient neurons were present in adult mutant brains at all of the stages that were examined, ranging from 3 to12 weeks, 8 months (Fig. 3E), and up to 17 months of age, which is the last time point that was analyzed (data not shown). The efficiency of gene deletion in the cortex of conditional mutant mice was similar to that in control conditional heterozygous mice (both at ∼50%; see Fig.3E). In addition, the percentage of Dnmt1-deficient neurons in most brain regions, except the olfactory bulb, was constant during postnatal life (Fig. 3E), suggesting that Dnmt1 deficiency in postmitotic neurons was not detrimental to long-term neuronal survival.
The long-term presence of Dnmt1-deficient neurons in_CamK-cre;Dnmt1_ conditional mutants allowed us to examine whether global DNA methylation would change in vivo over a time course of months. Southern blot analysis of DNA methylation of endogenous retroviral repeats [IAP and Moloney murine leukemia virus (MMLV)] did not show any obvious demethylation in mutant neurons that had lacked Dnmt1 for 8 months (Fig. 3F). These data indicated that Dnmt1 is not essential for maintaining global DNA methylation in postmitotic neurons in vivo.
Dnmt1 deficiency in CNS precursor cells in vivo causes global DNA hypomethylation and neonatal death of mutant animals
We next examined the consequence of the Dnmt1 gene deletion in CNS precursor cells in vivo. For this, we crossed Dnmt12lox females with male mice carrying a nestin promoter-driven cre transgene that is activated in CNS precursor cells at E9–E10 and results in almost complete gene deletion by midgestation (Bates et al., 1999; Trumpp et al., 1999).
Mutant mice were obtained at the expected Mendelian frequency at all of the embryonic stages that were examined (E10.5–E19.5) but were never recovered postnatally. Careful observation of mutant mice delivered either naturally or by Cesarean section at E18.5–E19.5 revealed that neonatal mutant mice died within 1 hr of birth because of respiratory failure (see below). Southern blot analysis of E19.5 mutant embryos showed that the cre-mediated gene deletion of _Dnmt1_had occurred in ∼95% of the cells in the CNS (Fig.4A), in agreement with previous observations (Bates et al., 1999). Moderate gene deletion (<30%) was observed in muscles and kidney, and a low frequency of gene deletion was seen in lung, heart, and liver (Fig.4B). Cre-mediated Dnmt1 deletion was already detectable at E10.5 (data not shown) and was complete at E12.5 in the brain (Fig. 4B). Importantly, the proportion of brain cells carrying the Dnmt1 gene deletion remained constant throughout later stages of embryonic development (Fig.4B), suggesting that Dnmt1-deficient cells survived throughout embryogenesis. Western blot analysis showed that levels of Dnmt1 proteins were decreased greatly at E12.5 and were undetectable at E15.5 (Fig. 4C), suggesting that the enzyme turned over rapidly after gene deletion. It is worth noting that neurogenesis is the major event in the brain during embryonic development, and gliogenesis in the CNS mainly occurs postnatally (Cepko et al., 1990;Mission et al., 1991; Caviness et al., 1995). Thus, the majority of Dnmt1-deficient brain cells in conditional mutant embryos is expected to be postmitotic CNS neurons.
Fig. 4.

Deletion of the Dnmt1 gene in the brain by the paternally inherited nestin-cre transgene.A, Southern blot analysis of the efficiency of the nestin-cre-mediated Dnmt1 gene deletion in various brain regions and peripheral tissues from E19.5 conditional knock-outs. PhosphorImager analysis of mutant_Dnmt1_ 1lox (1lox) alleles and functional Dnmt12lox(2lox) alleles indicated a recombination efficiency of ∼95% in the brain. Approximately 10–30% recombination was detected in intestine, limb, cranial muscles, and kidney. Only very minor amounts (<5%) of recombination were detected in lung, heart, and liver. B, Dnmt1 gene deletion in E12.5–E18.5 brain tissues. DNA samples were collected from embryos carrying a paternally derived nestin-cre transgene and_Dnmt1_ alleles as indicated (heterozygous control,2lox/+; mutant, 2lox/2lox or_2lox/N_). PhosphorImager analysis showed that ∼95% of the Dnmt12lox allele was recombined into the null Dnmt11lox_allele in the brain from E12.5 to E18.5. C, Western blot analysis of Dnmt1 proteins in control and mutant brain tissues. Brain extracts from E12.5, E15.5, and E18.5 embryos were probed with a specific antibody against the N terminus of the Dnmt1 protein. A duplicate gel was stained with Coomassie blue as a loading control. Note that the E18.5 control sample was from a_Dnmt12lox/N embryo, which showed a reduced level of Dnmt1 proteins as compared with_Dnmt12lox/2lox_ control samples at E12.5 and E15.5. con, Control samples;mut, mutant samples.
To assess the effect of Dnmt1 deficiency on genomic methylation, we digested brain DNA with the methylation-sensitive enzyme_Hpa_II and probed the Southern blots with a centromeric minor satellite repeat probe (Fig.5A) or an IAP DNA fragment (Fig. 5B). These probes detect multiple copies of repeated DNA sequences that are highly methylated in wild-type mice. As indicated by the low-molecular-weight fragments in Figure 5,A and B, there was substantial demethylation in the brain of mutant embryos at E12.5 or older because of the deletion of the Dnmt1 gene. We also detected methylation changes in single-copy genes. As shown in Figure 5C, significant demethylation was observed at several methylation sites surrounding the coding exon of the bdnf gene. From the densitometry analysis the proportion of demethylated DNA fragments was estimated to be ∼30–40%. Although substantial DNA hypomethylation occurred in mutant brains from E12 on, CNS development seemed to proceed normally. A histological survey of mutant embryos at different embryonic stages did not show any obvious defect in the brain structure. Mutant brains at E15.5 also did not show any obvious increase in cell death as assessed by TUNEL staining (data not shown).
Fig. 5.
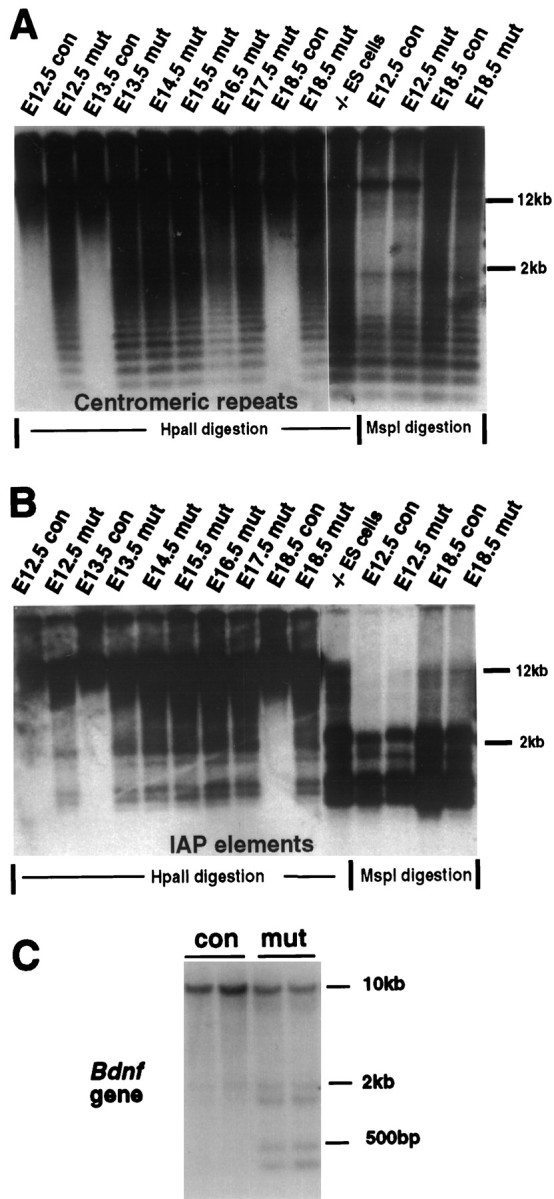
Dnmt1 deficiency results in global genomic hypomethylation. A, B, Southern blot analysis of DNA methylation in the brain. The same DNA samples in Figure1C were used for the methylation assay. DNA was digested with methyl-sensitive enzyme _Hpa_II or methyl-insensitive enzyme Msp_I (-CCGG-) and was hybridized with a centromeric minor repeat probe (A) or an IAP probe (B). The appearance of small-molecular-weight DNA fragments indicates demethylation at the CpG sites of centromeric repeats or IAP retroviruses in the genome.con,Nestin-cre;Dnmt1 +/2lox;mut, Nestin-cre;Dnmt1 2lox/2lox or_2lox/N; −/− ES cells, Demethylated DNA samples from Dnmt1 null embryonic stem cells (Lei et al., 1996). C, Brain DNA from E18.5 embryos was digested with _Sac_I and_Hpa_II enzymes and the probe with a 750 bp BDNF cDNA.
To assess the effect of DNA hypomethylation in vitro, we examined the survival and differentiation of mutant cortical neurons in dissociated cell culture at E15.5. Mutant and control cortical cells were cultured for 24 or 96 hr. After 1 d in culture ∼80% of the cells were postmitotic neurons expressing neuronal-specific β-tubulin, and the remaining 20% of the cells were precursor cells expressing the nestin intermediate filament marker that predominantly differentiated into glial-like cells in our culture conditions (Fig.6A,B). After 4 d in culture the composition of both the wild-type and mutant cultures had changed to ∼50% neurons and 50% glial-like cells because of active proliferation and differentiation of precursor cells and decreased survival of neuronal cells (Fig. 6C,D). Analysis of global DNA methylation confirmed that cultured cells remained hypomethylated throughout the culture period (Fig.6E). No difference was found in neuronal survival between the mutant and control cortical cells after either 24 or 96 hr in culture, indicating that Dnmt1 deficiency and DNA hypomethylation did not affect neuronal survival in vitro at this embryonic stage (Fig. 6A–D). Our results are consistent with the notion that genomic hypomethylation has no detectable effect on the survival of embryonic cortical neurons.
Fig. 6.

Survival of hypomethylated cortical neurons_in vitro_. A–D, E15.5 cortical cells from control animals (con,Dnmt 2lox/2lox in A and_C_) and_Nestin-cre;Dnmt12lox/2lox_ mutants (mut, in B and D) after 24 or 96 hr in culture. Cultured cells were double-stained with a monoclonal TuJ1 antibody against neuronal-specific β-tubulin III (green) and a polyclonal antibody against nestin intermediate filaments for precursor cells (red). Note that in 24 hr cultures ∼80% of cells were postmitotic neurons. In 96 hr cultures the neurons and glial-like cells (nestin-positive) were ∼50% each, and no difference in these ratios was observed between control and mutant cultures. Scale bar, 60 μm. E, DNA hypomethylation in cultured E15.5 cortical cells. DNAs from cortical cells cultured for 24 or 96 hr were assayed for global demethylation by probing with IAP retro-elements. Demethylated IAP DNA fragments were detected readily in the mutant cultures. con, Control_Dnmt2lox/2lox_ cultures;mut, conditional knock-out cultures.
We also examined DNA hypomethylation at individual cell levels by using dissociated cortical cultures. It has been shown that the_Xist_ gene on the active X chromosome is silenced by methylation at the time of X inactivation but is activated by demethylation, leading to ectopic X inactivation (Beard et al., 1995;Panning and Jaenisch, 1996). In female cells Xist mRNA is expressed from and associated with the inactive X in every cell. Fluorescence in situ hybridization (FISH) analysis showed that a strong Xist mRNA signal was detected in all of the neurons explanted from the brain of E15.5 control females, but not from control male embryos (Fig. 7). In contrast, a strong Xist mRNA signal, similar to the one seen in the control female cells, was seen in a portion of neurons explanted from mutant male embryos (Fig. 7, third panel). Quantification of Xist_-expressing cells showed that ectopic_Xist activation occurred in 4–8% of mutant cells explanted at E15.5 and E19.5 that had been cultured for 1–7 d (Table1).
Fig. 7.

Expression of Xist mRNA in neuronal and glial cells in culture. Xist FISH analysis was performed as described in Materials and Methods. _Xist_RNA expression (red dots in DAPI-stained blue nuclei) was detected in a portion (4–8%) of male mutant cells in 1-d-old E15.5 cortical neuronal cultures (also see Table 1). A few mutant cells also express the Xist transcripts in distributed granules within the cells (third panel), characteristic of cells in the early G1 phase of cell cycle (Clemson et al., 1996).con, Control embryos (Dnmt1 2lox/2lox); mut, conditional mutant embryos. Scale bar, 7 μm.
Table 1.
Number of _Xist_-expressing cells in male mutant cortical cultures
| Days in culture | Xist+ | DAPI nuclei | Xist/DAPI (%) | |
|---|---|---|---|---|
| E15.5 mutants | 1 | 32 | 832 | 3.85 |
| 39 | 818 | 4.77 | ||
| 29 | 358 | 8.10 | ||
| 4 | 44 | 855 | 5.15 | |
| 7 | 26 | 834 | 3.12 | |
| E19.5 mutants | 1 | 48 | 710 | 6.76 |
Dnmt1 deficiency and DNA hypomethylation result in defects in neuronal respiratory control
To assess the possible cause of lethality in neonatal mutant mice that lack Dnmt1 in the brain, we examined E18.5 and E19.5 mutant mice delivered by Cesarean section. In contrast to the control mice that survived after the Cesarean section, all mutant mice died within 1 hr of delivery. Although mutant mice occasionally gasped, they did not initiate coordinated rhythmic breathing. Postmortem examination showed that their lungs failed to inflate, confirming respiratory failure (data not shown).
Multiple causes could lead to respiratory failure, including a defect in neural rhythmogenesis or motor output of the respiratory pattern generator, occlusion of the respiratory tract, delay in lung development, or abnormalities secondary to the failure of the cardiovascular or other vital systems. We first examined the behavior of mutant fetuses within the uterus. Mutant mice _in utero_had normal body movement, either spontaneous or in response to mechanical stimuli such as a pinch, suggesting that they had a normal sensorimotor reflex. We also monitored EKGs of the heart in utero and found a normal heart rate in mutant mice (control, 356/min; mutant, 346–364/min). This suggests that respiratory failure in mutant animals was not caused by malfunction of the cardiovascular system. At the light microscope level the morphology of various lung cells in conditional mutant animals appeared normal (data not shown). This is consistent with the observation that little of _Dnmt1_gene deletion was observed in the lung (see Fig. 4A). Similarly, histological examination did not show any obvious abnormalities in respiratory muscle groups, including the intercostal and diaphragm muscles. In addition, diaphragm muscles were innervated normally by the respiratory phrenic nerve originating from the cervical spinal cord (data not shown). These observations suggest that the respiratory failure in the mutant was not caused by abnormalities of the peripheral respiratory system.
To investigate whether the respiratory failure in the Dnmt1 mutants was caused by a defect in the neural control of respiration in the CNS, we monitored the neuronal activity of the 12th cranial hypoglossal nerve and performed a diaphragmatic electromyogram in E18.5 and E19.5 mice delivered by Cesarean section. It is known that the majority of hypoglossal neuronal discharges is inspiratory, with a bursting period similar to that of phrenic nerve activity. Indeed, many hypoglossal motor neurons receive innervations from premotor inspiratory neurons located within the respiratory pattern generator in the ventrolateral medulla (Withington-Wray et al., 1988; Ono et al., 1994) (for review, see St. John, 1998). Figure8A shows that the hypoglossal nerve activity in normal mice displayed a rhythmic but gasping-like pattern ∼5 min after birth (frequency, 12/min), which was converted to an eupneic pattern with increased respiratory frequency (50/min) and heart rate (260/min) within 10–20 min. In contrast, mutant mice at birth produced only sporadic gasping activity, which never developed into an eupneic pattern (Fig.8A). Although occasionally showing arrhythmia, the initial heartbeat rate in mutant mice was close to that of control animals immediately after birth. Some 20–30 min after delivery, however, the heartbeat of mutant mice became increasingly irregular and sporadic (∼30/min), and the animals died shortly thereafter.
Fig. 8.

Lack of respiratory drive in the 12th cranial nerve. A, Electrophysiological recordings of descending respiratory discharge from hypoglossal nerve. Control (con, Dnmt1 2lox/2lox) and conditional mutant mice were dissected from the uterus at E18.5 and E19.5 and immediately prepared for 12th nerve recording as described in the Materials and Methods. Only occasional spontaneous gasping was observed in mutant mice. The gasping discharge also could be induced occasionally by tail pinching of mutant mice when an initial recording did not show any spontaneous signals. Similar results were obtained with six control and five mutant mice. Insets for each trace are EKG recordings obtained with subcutaneous electrodes (the EKG in the second control trace was derived from the diaphragmatic EMG recording). B, Normal morphology of hypoglossal motor neurons in the brainstem. E18.5 control (CON) and mutant embryos were fixed with 10% formalin and processed for paraffin histology. Brain sections were stained with cresyl violet. No obvious morphological difference was observed between control and mutant hypoglossal motor nuclei. Scale bar, 37.5 μm.
The ability of the mutant mice to produce gasping activity in the hypoglossal nerve and diaphragm suggests that their respiratory motor efferent pathway was functional. Histological examination showed that the hypoglossal motor nucleus in the brainstem of mutant mice was intact (Fig. 8B). It is possible that a defect in the neural control of respiration may result from disrupted respiratory rhythmogenesis or neurotransmission in the Dnmt1-deficient brain.
DNA hypomethylation results in rapid cell death in the postnatal brain
The observations described so far suggest that Dnmt1_deletion and subsequent hypomethylation did not affect the survival of neuronal cells in the prenatal brain, although mutant animals died immediately after delivery. We were interested in investigating the fate of brain cells in postnatal mutant animals. In the experiments described above, Dnmt1 had been deleted in ∼95% of brain cells (see Fig. 4A,B) by expression of the cre recombinase from a paternally transmitted nestin-cre transgene crossed with females carrying the Dnmt12lox target allele. However, when the nestin-cre transgene was transmitted maternally and the Dnmt12lox target allele was derived paternally, the recombination frequency was only 30% instead of 95% in the adult brain, indicating that the cre transgene was imprinted and the expression level was dependent on parental origin (compare Figs. 9A and4A,B). The fraction of brain cells that performed the cre-mediated recombination was already ∼30% at E12 with maternal nestin-cre transmission, and this fraction did not increase during later development. This was shown by crossing females carrying the nestin-cre transgene with males carrying a lacZ reporter gene (the Rosa26 reporter; Soriano, 1999), which resulted in ∼30% lacZ_-positive cells in the brains of E12, newborn, and adult animals (data not shown). To determine the fraction of_Dnmt1 mutant cells in animals carrying a maternally derived nestin-cre transgene and a paternally derived_Dnmt12lox allele, we performed Southern blot analyses with E12 and newborn brain tissues. Loop-out frequency was ∼30% at E12 (Fig. 9B, lane 1) and at birth (Fig. 9A,B), indicating that the fraction of mutant cells did not change during embryonic development. Thus, mutant mice carrying the maternally derived nestin-cre transgene had a mosaic brain in which Dnmt1-deficient (Dnmt11lox) and Dnmt1-proficient (Dnmt12lox) cells coexisted and _Dnmt1_-deficient cells were not selected against during the embryonic stages of development.
Fig. 9.

Postnatal loss of Dnmt1-deficient brain cells in mutant mice with maternal inheritance of the nestin-cre transgene.A, Southern blot analysis of the Dnmt1_gene deletion in DNA from different brain regions of mutant mice after maternal inheritance of the nestin-cre transgene. At the newborn stage ∼30–35% of Dnmt1 gene deletion was detected in both mutant and heterozygous brains. FB, Forebrain;Col, colliculus; CB, cerebellum;BS, brainstem; SP, spinal cord.B, Dnmt1-deficient brain cells are eliminated during postnatal development. Deletion of the Dnmt1 gene was maximal by E12.5 (Br, whole brain) and remained constant in the brain throughout the late stage of embryogenesis and at the postnatal day 1 (P1; also see A). However, only a very small number of cells (4–6%) carrying the_Dnmt1 deletion was detected in the cortex (CX) and cerebellum (CB) in 2-week-old (P14) mutant mice. By P21, Dnmt1-deficient cells were not detectable by Southern blot analysis in the cortex, cerebellum, or other regions of the brain (data not shown).mutant, Mutant mice with the_Nestin-cre;Dnmt1_ 2lox/N genotype.con, Control samples from_Nestin-cre;Dnmt1+/2lox_ mice, which showed constant levels of the Dnmt1 deletion at P1 and P21.
Because newborn mutants with a maternally transmitted cre transgene were born alive and normal, we examined whether_Dnmt1_-deficient neurons survived postnatally. To assess the survival of Dnmt1-deficient cells, we performed Southern blot analyses to determine the percentage of mutant cells in brain tissues at different postnatal stages. Figure 9B shows that the fraction of brain cells with cre-mediated Dnmt1 deletion in heterozygous mice (Dnmt1+/2lox carrying the maternally derived nestin-cre transgene) remained constant during the first 3 weeks of postnatal development. In contrast, the frequency of cre-mediated Dnmt1 deletion in mutant mice was initially 30% at the newborn stage but decreased significantly by P14. At 3 weeks of age Dnmt1-deficient cells were not detectable by Southern blot analysis in either the cerebellum or cortex (Fig. 9B) or other brain regions (data not shown). These results indicate that Dnmt1-deficient neurons were eliminated from the postnatal brain, in contrast to the prenatal brain in which hypomethylation has no apparent effect on cell survival.
DISCUSSION
Previous work had established that Dnmt1 is highly expressed in embryonic and postnatal neurons of the brain (Goto et al., 1994; Brooks et al., 1996; Trasler et al., 1996; Inano et al., 2000). This finding was surprising, given that the known function of Dnmt1 is to maintain the parental methylation pattern of the daughter DNA strands in mitotic cells. Dnmt1 is, therefore, highly expressed during S-phase in mitotic cells but is downregulated in resting cells (Szyf et al., 1991). Because deletion of Dnmt1 from the germline causes apoptosis and early embryonic lethality (Li et al., 1992), this mutant line was not useful for studying the possible function of DNA methylation in brain development. As a first approach to assessing the role of methylation during brain development and postnatal life, we generated a_Dnmt1_ conditional allele that can be deleted at postmitotic CNS neurons or E12 CNS precursor cells. Although Dnmt1 is not essential for maintaining global DNA methylation in postmitotic neurons, the enzyme is required for the methylation of mitotic precursor cells and their daughter cells. Hypomethylated CNS cells survived through the late stages of embryogenesis but died postnatally. In addition, hypomethylation in the brain leads to abnormal neural control of respiration at birth. These findings indicated that DNA methylation is crucial for the function and survival of postnatal CNS neurons.
When Dnmt1 gene deletion is mediated by the nestin-cre transgene, Dnmt1 deficiency occurred in the brain at E12.5, a stage when neurogenesis is still actively ongoing (Austin and Cepko, 1990;Cepko et al., 1990). This suggests that precursor cells in the embryonic mutant brain are able to generate mature neurons (and non-neuronal cells) in the absence of Dnmt1. The mutant cells and their mitotic descendants survive for at least another 7 d until birth, as supported by both in vivo and _in vitro_evidence. Indeed, the fraction of Dnmt1-deficient cells, either 95% or 30%, depending on the parental origin of the nestin-cre transgene, remained constant in the mutant brain from E12.5 to the newborn stage. Histological examination of mutant fetal brains did not show any obvious increase in the number of pyknotic nuclei and TUNEL-positive cells, suggesting that DNA hypomethylation did not increase the rate of neuronal cell death in vivo. These observations argue that DNA hypomethylation did not confer a selective disadvantage on the survival of the hypomethylated embryonic neurons in vivo. Likewise, the in vitro survival of embryonic cortical neurons explanted from mutant brains was indistinguishable from that of controls. Nevertheless, we cannot exclude the possibility that, in the mutant brain, a small fraction of Dnmt1-deficient cells may undergo rapid turnover that is below our detection level.
In contrast to the significant demethylation observed in the CNS neurons of Nestin-cre;Dnmt1 mutants, Dnmt1-deficient neurons in CamK-cre;Dnmt1 conditional mutants were long-lived and did not show any significant demethylation in the highly methylated DNA repeats. The difference in DNA methylation between_Nestin-cre;Dnmt1_ and CamK-cre;Dnmt1 mutant cells could be attributable simply to the fact that only Dnmt1-deficient cells in Nestin-cre;Dnmt1 conditional mutants undergo mitosis that results in passive DNA demethylation. The stable DNA methylation pattern in Dnmt1-deficient neurons of_CamK-cre;Dnmt1_ conditional mutants is not surprising, because there is no direct evidence that DNA methylation undergoes any dynamic turnover in normal adult neuronal cells.
The survival of hypomethylated embryonic CNS neurons in_Nestin-cre;Dnmt1_ conditional knock-outs is in apparent contrast to the extensive apoptosis observed in the brain of the_Dnmt1_ mutant embryos just after gastrulation (Li et al., 1992). We consider the following mutually nonexclusive possibilities: (1) DNA hypomethylation in the Dnmt1 conditional neuronal cells may be less extensive than in the rapidly dividing non-neuronal cells of the gastrulating mutant embryos. For example, in_Dnmt1_ homozygous mutant male embryos ectopic Xist_gene activation was seen in ∼15% of the cells (Panning and Jaenisch, 1996), whereas only 4–8% of brain cells showed ectopic_Xist transcription after cre-mediated Dnmt1_deletion, perhaps reflecting a less severe genomic demethylation in the neurons as compared with somatic cells of the postgastrulation_Dnmt1 null embryo. To reach a critical level of hypomethylation, cells have to undergo several rounds of DNA replication after the deletion of Dnmt. It is possible that the number of cell divisions that separate postmitotic neurons from their precursor cells (where the cre-mediated Dnmt1 deletion occurs at E9.5–E12) is limited, resulting in less pronounced genomic demethylation in Dnmt1-deficient neurons than in the cells of_Dnmt1_−/−embryos at the postgastrulation stage. This may lead to less ectopic_Xist_ activation and less pronounced apoptosis. (2)Dnmt3a and 3b are known to be expressed in the developing brain (Okano et al., 1998), and these enzymes may compensate for the loss of Dnmt1 function in mutant neurons. (3) Finally, it is possible that prenatal neurons are intrinsically more resistant to the consequences of genomic DNA demethylation than the cells of the postgastrulation embryo.
As an epigenetic factor, DNA methylation patterns may be subject to active regulation in the nervous system in response to particular stimuli. Endres et al. (2000) recently demonstrated that levels of DNA methylation activity in the brain actually are increased with ischemic injury, and this increase is partly dependent on Dnmt1 activity. Blocking Dnmt1 activities, either genetically or pharmacologically, is protective to the injured neurons, suggesting that a balance of DNA methylation levels is important for neuronal survival (Endres et al., 2000). Brooks et al. (1996) suggested that Dnmt1 expression in postmitotic neurons may serve to maintain DNA methylation after base-excision repair of the G:T mismatch that can occur with deamination of the methylated cytosine. Recently, Inano et al. (2000)demonstrated that Dnmt1 protein in adult CNS neurons is localized primarily in the cytoplasm, raising the possibility that Dnmt1 may play a novel function other than DNA methylation in these cells. Fuks et al. (2000) demonstrated that Dnmt1 itself contains a transcription repression domain that directly recruits histone deacetylases, suggesting an active role for Dnmt1 in chromatin remodeling. Furthermore, Dnmt1 is an active component of several repressive transcriptional complexes that can directly target transcriptional silencing to particular classes of genes or during the S-phase of the cell cycle (Robertson et al., 2000; Rountree et al., 2000). These findings certainly provide us with the clues to look further into the molecular and cellular changes in Dnmt1-deficient neurons in_Nestin-cre;Dnmt1_ and CamK-cre;Dnmt1 conditional mutants.
The lethality of Nestin-cre;Dnmt1 mutant animals appears to be caused by respiratory failure. These mutant animals never initiated breathing and showed highly impaired spontaneous neuronal activity recorded from the 12th cranial nerve. The neural mechanism underlying this defect is still unclear. It is possible that genomic hypomethylation alters the expression pattern of genes that are involved either directly or indirectly in respiratory control and thus interferes with the initiation of normal breathing. It is of interest to note that mutations of a methyl-binding protein MECP2 have been associated with the neurodevelopmental disease Rett syndrome (Amir et al., 1999) in which the patients often exhibit respiration irregularities in addition to behavioral defects and mental retardation (Kerr, 1992; Naidu, 1997; Armstrong et al., 1999). Because a major function of the MECP2 protein is to mediate methylation-induced gene silencing, it is possible that the phenotype of DNA hypomethylation in the postnatal brain may overlap partially with what is observed in MECP2 deficiency. Understanding the role of DNA methylation in brain development and function at the molecular level also may shed light on the disease mechanisms in Rett syndrome.
Footnotes
This work was supported by National Institutes of Health Grants R35 CA44339 (to R.J.), HL52925 and HL60064 (to C.S.P.), HD18184 (to C.B.W.), and CA44338 (to J. M. Bishop). Financial support also comes from a Medical Foundation postdoctoral fellowship (G.F.), from a National Institutes of Health postdoctoral fellowship (R.Z.C.), and from the Deutsche Forschungsgemeinschaft (A.T.). We thank Jessie Dausman, Ruth Flannery, and Jeanne Reis for technical support.
Correspondence should be addressed to Dr. Rudolf Jaenisch, Whitehead Institute for Biomedical Research, Nine Cambridge Center, Cambridge, MA 02142. E-mail: jaenisch@wi.mit.edu.
Dr. Bates's present address: Genetics Institute, Cambridge, MA 02140.
Dr. Lee's present address: Department of Biology, University of California, San Diego, La Jolla, CA 92093.
Dr. Kühn's present address: Artemis Pharmaceuticals, Cologne, Germany.
Dr. Trumpp's present address: Swiss Institute for Experimental Cancer Research, CH-1066 Epalinges, Switzerland.
REFERENCES
- 1.Akagi K, Sandig V, Vooijs M, Van der Valk M, Giovannini M, Strauss M, Berns A. Cre-mediated somatic site-specific recombination in mice. Nucleic Acids Res. 1997;25:1766–1773. doi: 10.1093/nar/25.9.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amir RE, Van Den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong DD, Dunn JK, Schultz RJ, Herbert DA, Glaze DG, Motil KJ. Organ growth in Rett syndrome: a postmortem examination analysis. Pediatr Neurol. 1999;20:125–129. doi: 10.1016/s0887-8994(98)00124-6. [DOI] [PubMed] [Google Scholar]
- 4.Austin CP, Cepko CL. Cellular migration patterns in the developing mouse cerebral cortex. Development. 1990;110:713–732. doi: 10.1242/dev.110.3.713. [DOI] [PubMed] [Google Scholar]
- 5.Bates B, Rios M, Trumpp A, Chen C, Fan G, Bishop JM, Jaenisch R. Neurotrophin-3 is required for proper cerebellar development. Nat Neurosci. 1999;2:115–117. doi: 10.1038/5669. [DOI] [PubMed] [Google Scholar]
- 6.Beard C, Li E, Jaenisch R. Loss of methylation activates Xist in somatic but not in embryonic cells. Genes Dev. 1995;9:2325–2334. doi: 10.1101/gad.9.19.2325. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Sasson SA, Sherman Y, Gavrieli Y. Identification of dying cells—in situ staining. In: Schwarz L, Osborne BA, editors. Cell death. Academic; San Diego: 1995. pp. 29–39. [PubMed] [Google Scholar]
- 8.Bestor T, Laudano A, Mattaliano R, Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol. 1988;203:971–983. doi: 10.1016/0022-2836(88)90122-2. [DOI] [PubMed] [Google Scholar]
- 9.Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- 10.Brooks PJ, Marietta C, Goldman D. DNA mismatch repair and DNA methylation in adult brain neurons. J Neurosci. 1996;16:939–945. doi: 10.1523/JNEUROSCI.16-03-00939.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time, and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- 12.Cepko CL, Austin CP, Walsh C, Ryder EF, Halliday A, Fields-Berry S. Studies of cortical development using retrovirus vectors. Cold Spring Harb Symp Quant Biol. 1990;55:265–278. doi: 10.1101/sqb.1990.055.01.029. [DOI] [PubMed] [Google Scholar]
- 13.Clemson CM, McNeil JA, Willard HF, Lawrence JB. Xist RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J Cell Biol. 1996;132:259–275. doi: 10.1083/jcb.132.3.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 15.Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, Lipski A, Jaenisch R, Moskowitz MA, Dirnagl U. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. 2000;20:3175–3181. doi: 10.1523/JNEUROSCI.20-09-03175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan G, Katz DM. Non-neuronal cells inhibit catecholaminergic differentiation of primary sensory neurons: role of leukemia inhibitory factor. Development. 1993;118:83–93. doi: 10.1242/dev.118.1.83. [DOI] [PubMed] [Google Scholar]
- 17.Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet. 2000;24:88–91. doi: 10.1038/71750. [DOI] [PubMed] [Google Scholar]
- 18.Gaudet F, Talbot D, Leonhardt H, Jaenisch R. A short DNA methyltransferase isoform restores methylation in vivo. J Biol Chem. 1998;273:32725–32729. doi: 10.1074/jbc.273.49.32725. [DOI] [PubMed] [Google Scholar]
- 19.Goto K, Numata M, Komura JI, Ono T, Bestor TH, Kondo H. Expression of DNA methyltransferase gene in mature and immature neurons as well as proliferating cells in mice. Differentiation. 1994;56:39–44. doi: 10.1046/j.1432-0436.1994.56120039.x. [DOI] [PubMed] [Google Scholar]
- 20.Inano K, Suetake I, Ueda T, Miyake Y, Nakamura M, Okada M, Tajima S. Maintenance-type DNA methyltransferase is highly expressed in postmitotic neurons and localized in the cytoplasmic compartment. J Biochem. 2000;128:315–321. doi: 10.1093/oxfordjournals.jbchem.a022755. [DOI] [PubMed] [Google Scholar]
- 21.Jackson-Grusby L, Beard C, Possemato R, Fambrough D, Csankovszki G, Dausman J, Lee P, Wilson CB, Lander E, Jaenisch R (2000) Loss of genomic methylation cause p53-dependent apoptosis and epigenetic deregulation. Nat Genet, in press. [DOI] [PubMed]
- 22.Jaenisch R. DNA methylation and imprinting: why bother? Trends Genet. 1997;13:323–329. doi: 10.1016/s0168-9525(97)01180-3. [DOI] [PubMed] [Google Scholar]
- 23.Jones P, Gonzalgo M. Altered DNA methylation and genome instability: a new pathway to cancer? Proc Natl Acad Sci USA. 1997;94:2103–2105. doi: 10.1073/pnas.94.6.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerr AM. A review of the respiratory disorder in the Rett syndrome. Brain Dev. 1992;[Suppl] 14:43–45. [PubMed] [Google Scholar]
- 25.Laird P, Zijderveld A, Linders K, Rudnicki M, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei H, Oh S, Okano M, Juttermann R, Goss K, Jaenisch R, Li E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 27.Li E, Bestor T, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 28.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 29.Lyko F, Ramsahoye BH, Kashevsky H, Tudor M, Mastrangelo MA, Orr-Weaver TL, Jaenisch R. Mammalian (cytosine-5) methyltransferases cause genomic DNA methylation and lethality in Drosophila. Nat Genet. 1999;23:363–366. doi: 10.1038/15551. [DOI] [PubMed] [Google Scholar]
- 30.Mission JP, Takahashi T, Caviness VS., Jr Ontogeny of radial and other astroglial cells in murine cerebral cortex. Glia. 1991;4:138–148. doi: 10.1002/glia.440040205. [DOI] [PubMed] [Google Scholar]
- 31.Monk M, Boubelik M, Lehnert S. Temporal and regional changes in DNA methylation in the embryonic, extra-embryonic, and germ cell lineages during mouse embryo development. Development. 1987;99:371–382. doi: 10.1242/dev.99.3.371. [DOI] [PubMed] [Google Scholar]
- 32.Naidu S. Rett syndrome: a disorder affecting early brain growth. Ann Neurol. 1997;42:3–10. doi: 10.1002/ana.410420104. [DOI] [PubMed] [Google Scholar]
- 33.Ng HH, Bird A. DNA methylation and chromatin modification. Curr Opin Genet Dev. 1999;9:158–163. doi: 10.1016/s0959-437x(99)80024-0. [DOI] [PubMed] [Google Scholar]
- 34.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 35.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 36.Ono T, Uehara Y, Kurishita A, Tawa R, Sakurai H. Biological significance of DNA methylation in the ageing process. Age Ageing. 1993;22:S34–S43. doi: 10.1093/ageing/22.suppl_1.s34. [DOI] [PubMed] [Google Scholar]
- 37.Ono T, Ishiwata Y, Inaba N, Kuroda T, Nakamura Y. Hypoglossal premotor neurons with rhythmical inspiratory-related activity in the cat: localization and projection to the phrenic nucleus. Exp Brain Res. 1994;98:1–12. doi: 10.1007/BF00229103. [DOI] [PubMed] [Google Scholar]
- 38.Panning B, Jaenisch R. DNA hypomethylation can activate Xist expression and silence X-linked genes. Genes Dev. 1996;10:1991–2002. doi: 10.1101/gad.10.16.1991. [DOI] [PubMed] [Google Scholar]
- 39.Paton JF, Ramirez JM, Richter DW. Functionally intact in vitro preparation generating respiratory activity in neonatal and mature mammals. Pfl∫gers Arch. 1994;428:250–260. doi: 10.1007/BF00724504. [DOI] [PubMed] [Google Scholar]
- 40.Persengiev SP, Kilpatrick DL. Nerve growth factor-induced differentiation of neuronal cells requires gene methylation. NeuroReport. 1996;8:227–231. doi: 10.1097/00001756-199612200-00046. [DOI] [PubMed] [Google Scholar]
- 41.Persengiev SP, Kilpatrick DL. The DNA methyltransferase inhibitor 5-azacytidine specifically alters the expression of helix-loop-helix proteins Id1, Id2, and Id3 during neuronal differentiation. NeuroReport. 1997;8:2091–2095. doi: 10.1097/00001756-199707070-00001. [DOI] [PubMed] [Google Scholar]
- 42.Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1:11–19. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- 43.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. Dnmt1 forms a complex with Rb, E2F1, and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 44.Rountree MR, Bachman KE, Baylin SB. Dnmt1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269–277. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- 45.St. John W. Neurogenesis of patterns of automatic ventilatory activity. Prog Neurobiol. 1998;56:97–117. doi: 10.1016/s0301-0082(98)00031-8. [DOI] [PubMed] [Google Scholar]
- 46.Soriano P. Generalized lacZ expression with the ROSA26 cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 47.Szyf M, Bozovic V, Tanigawa G. Growth regulation of mouse DNA methyltransferase gene expression. J Biol Chem. 1991;266:10027–10030. [PubMed] [Google Scholar]
- 48.Tawa R, Ono T, Kurishita A, Okada S, Hirose S. Changes of DNA methylation level during pre- and postnatal periods in mice. Differentiation. 1990;45:44–48. doi: 10.1111/j.1432-0436.1990.tb00455.x. [DOI] [PubMed] [Google Scholar]
- 49.Trasler JM, Trasler DG, Bestor TH, Li E, Ghibu F. DNA methyltransferase in normal and Dnmtn/Dnmtn mouse embryos. Dev Dyn. 1996;206:239–247. doi: 10.1002/(SICI)1097-0177(199607)206:3<239::AID-AJA2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 50.Trumpp A, Depew MJ, Rubenstein JLR, Bishop JM, Martin GR. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev. 1999;13:3136–3148. doi: 10.1101/gad.13.23.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tucker KL, Beard C, Dausmann J, Jackson-Grusby L, Laird, Lei H, Li E, Jaenisch R. Germ-line passage is required for establishment of methylation and expression patterns of imprinted but not of nonimprinted genes. Genes Dev. 1996;10:1008–1020. doi: 10.1101/gad.10.8.1008. [DOI] [PubMed] [Google Scholar]
- 52.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 53.Wilson VL, Smith RA, Ma S, Cutler RG. Genomic 5-methyldeoxycytidine decreases with age. J Biol Chem. 1987;262:9948–9951. [PubMed] [Google Scholar]
- 54.Withington-Wray DJ, Mifflin SW, Spyer KM. Intracellular analysis of respiratory-modulated hypoglossal motoneurons in the cat. Neuroscience. 1988;25:1041–1051. doi: 10.1016/0306-4522(88)90057-7. [DOI] [PubMed] [Google Scholar]