RNA base-pairing complexity in living cells visualized by correlated chemical probing (original) (raw)
Significance
How do we determine the structure of an RNA, particularly in cells? Chemical probing is a broadly applicable strategy for mapping RNA structure. However, chemical probing experiments do not observe base pairs directly and hence are unable to unambiguously define global RNA structure or resolve structural dynamics. We describe an approach, PAIR-MaP, that harnesses correlations in how nucleotides are chemically modified to simultaneously map local RNA structure and directly detect base pairs. PAIR-MaP visualizes long-range helices and pseudoknots in complex RNAs with high specificity and resolution, including in living cells, and further reveals alternative base-pairing states. PAIR-MaP makes it possible to determine RNA structure and dynamics in cells with high resolution, confidence, and throughput.
Keywords: RNA structure modeling, duplex detection, RNA dynamics, single molecule, RING-MaP
Abstract
RNA structure and dynamics are critical to biological function. However, strategies for determining RNA structure in vivo are limited, with established chemical probing and newer duplex detection methods each having deficiencies. Here we convert the common reagent dimethyl sulfate into a useful probe of all 4 RNA nucleotides. Building on this advance, we introduce PAIR-MaP, which uses single-molecule correlated chemical probing to directly detect base-pairing interactions in cells. PAIR-MaP has superior resolution compared to alternative experiments, can resolve multiple sets of pairing interactions for structurally dynamic RNAs, and enables highly accurate structure modeling, including of RNAs containing multiple pseudoknots and extensively bound by proteins. Application of PAIR-MaP to human RNase MRP and 2 bacterial messenger RNA 5′ untranslated regions reveals functionally important and complex structures undetected by prior analyses. PAIR-MaP is a powerful, experimentally concise, and broadly applicable strategy for directly visualizing RNA base pairs and dynamics in cells.
RNA molecules are strongly driven to fold back on themselves into base-paired secondary structures. These structures play central roles in RNA biology, from mediating complex functions such as RNA catalysis and specific ligand recognition, to more broadly tuning RNA sequence accessibility to regulate processes such as translation initiation (1, 2). Furthermore, many RNAs fold into multiple structures, enabling molecular switching functions (3). Accurately resolving RNA structure and its potential dynamic complexity is therefore essential for understanding RNA function.
Chemical probing experiments are among the most broadly useful classes of experiments for characterizing RNA structure (4–6). SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) reagents, dimethyl sulfate (DMS), or other chemical probes are used to selectively modify conformationally flexible nucleotides and reactivity is measured using sequencing approaches such as mutational profiling (MaP) (7). These reactivity data provide powerful insight into local RNA structure and can be used to guide accurate RNA structure modeling (7–9). Nevertheless, chemical probing experiments are limited in that they do not detect RNA base pairing interactions directly—structure can only be inferred based on compatibility with reactivity data. In some cases, the reactivity data may be equally compatible with multiple structures. Even if the structure inference problem is uniquely defined, follow-up mutational analysis is often desired to obtain direct evidence of pairing interactions. Chemical probing data are also poorly suited for resolving alternative structural states of dynamic RNAs. Finally, conventional chemical probing data are difficult to interpret for RNAs bound by proteins or in cells.
To address the limitations of chemical probing experiments, new strategies have been developed that use scanning mutagenesis and chemical probing (mutate-and-map) to identify interacting nucleotides (10) or detect RNA duplexes by cross-linking and proximity ligation (11, 12). However, both of these classes of experiments are laborious, with the former limited to in vitro settings and the latter having low resolution (10 to 20 nucleotides [nt]) and insufficient information to rank and define complete RNA structures (13). We recently introduced a third strategy that uses single-molecule chemical probing experiments (14) to detect correlated modifications between paired nucleotides (15), but this approach was also limited to in vitro settings and the underlying mechanism has been questioned (10). Thus, current duplex detection strategies retain substantial limitations, being restricted either to in vitro contexts or lacking the desired quantitative accuracy and experimental concision.
Here, we introduce a strategy that converts the classic reagent DMS into a probe of all 4 RNA nucleotides. We combine this advance with new analysis algorithms to demonstrate that single-molecule correlated chemical probing sensitively and specifically detects RNA duplexes in cells, comprising a strategy we term PAIR-MaP (pairing ascertained from interacting RNA strands measured by mutational profiling). PAIR-MaP permits simultaneous measurement of local chemical probing data and duplex interactions via one straightforward chemical probing experiment, enabling accurate RNA structure modeling and revealing alternative RNA structural states. Application of PAIR-MaP to human RNase MRP and the Escherichia coli S2- and S4-binding autoregulatory elements reveals previously undetected, functionally important structural features for these RNAs, highlighting the broad potential of PAIR-MaP for understanding RNA biology.
Results
DMS Probes Structure of All 4 Nucleotides.
DMS is among the most commonly used RNA chemical probes, favored for its cell permeability and ability to heavily modify RNA molecules during correlated chemical probing experiments. However, a major limitation is that DMS does not typically react with the base pairing face of guanosine (G) and uridine (U) nucleotides due to protonation of the respective N1 and N3 positions at neutral pH (pKa ≈ 9.2; Fig. 1_A_) (4, 16). We discovered that DMS can be converted into a useful probe of all 4 nucleotides by performing modification at pH 8, which promotes transient deprotonation of G and U and reaction with DMS. Optimized buffer conditions consisting of 200 mM bicine at pH 8.0 were found to maintain a well-controlled pH without quenching the DMS reaction (SI Appendix, Supporting Methods). These optimized conditions were used to perform multiple-hit DMS probing of natively extracted (termed “cell-free”) total E. coli RNA, and DMS methylation sites were detected using the single-molecule MaP strategy (14). Analysis of the 16S and 23S ribosomal RNAs (rRNAs) reveals that U and G nucleotides are consistently modified in a structure-specific manner: Single-stranded U and G positions are modified at average rates of 1.3% and 0.7%, respectively, whereas paired positions are protected and have ∼4-fold lower modification rates (Fig. 1 B and C). The modification rate for U and G residues is ∼10-fold lower than for A and C (Fig. 1_B_) but is sufficiently high for quantification by the MaP strategy (14).
Fig. 1.

DMS probes all 4 RNA nucleotides. (A) Deprotonation equilibrium of G and U nucleotides (16). (B) DMS modification rates measured by MaP for E. coli 16S and 23S rRNAs probed under cell-free conditions in buffered bicine (pH 8.0). Histograms are shown for single-stranded (red) and paired (blue) nucleotides based on the accepted folded structures. (C) Normalized DMS reactivities for a representative region of the 16S rRNA (nucleotides 693 to 718). U and G nucleotides are highlighted (blue). Red, orange, and black denote high, moderate, and low reactivity, respectively. The secondary structure is indicated by arcs (bottom). (D) Receiver operating characteristic (ROC) curves for DMS-MaP reactivity profiles calculated for different RNAs. E. coli ncRNAs (RNase P and tmRNA) are shown in orange, E. coli rRNAs (5S, 16S, and 23S rRNAs) are shown in red, and human ncRNAs (U1 snRNA and RMRP) are shown in blue. Cell-free 1M7 SHAPE data from the E. coli 16S and 23S rRNA (17) is provided as a reference. Area under the ROC curve (AUC) values are provided in SI Appendix, Table S1.
Benchmarking across a diverse panel of RNAs with known structures confirmed that DMS reactivity at U and G residues provides an informative measurement of nucleotide pairing status. In addition to the 16S and 23S rRNAs, we used MaP to quantify DMS modification of 5S rRNA, RNase P, and transfer-messenger RNA (tmRNA) in cell-free E. coli RNA. We also performed DMS probing experiments on cell-free total RNA from human Jurkat cells and quantified modifications of U1 small nuclear RNA and RNase MRP (RMRP). Remarkably, in these cell-free experiments, DMS reactivity discriminates single-stranded versus paired U residues with accuracy comparable to that for A and C nucleotides and also performs comparably to SHAPE reactivity (Fig. 1_D_). DMS reactivity is less discriminative for G nucleotides but is still informative (Fig. 1_D_; area under the receiver operating characteristic curve [AUC] = 0.6 to 0.7; SI Appendix, Table S1). The decreased specificity observed for G modifications is most likely attributable to nonspecific DMS modification at the N7 position of G (4) that is partially detected by MaP.
We also assessed whether DMS is an effective probe of G and U nucleotides in cells. Living E. coli or human Jurkat cultures were supplemented with bicine probing buffer and treated with DMS. As expected, DMS is less effective at discriminating single-stranded versus paired nucleotides in cells due to protection by proteins, particularly for the E. coli rRNAs (Fig. 1 C and D). Nonetheless, DMS still measures structure-specific modification of U nucleotides in cells in all RNAs, again with similar discriminatory power as for A and C nucleotides (Fig. 1 C and D). DMS reactivity at G nucleotides is weakly informative for E. coli noncoding RNA (ncRNA) structure (AUC = 0.6) but is uninformative for human ncRNAs and E. coli rRNAs in cells (AUC = 0.48 to 0.55; SI Appendix, Table S1).
Combined, our data clearly show that DMS is an effective probe of all 4 RNA nucleotides at pH 8.0, including in living bacterial and human cells. Separately, our data also demonstrate that the MaP strategy, in conjunction with the ShapeMapper bioinformatics pipeline (17), detects DMS modifications with a high degree of structural specificity without need for specialized enzymes or separate counting of termination events (18, 19).
PAIR-MaP Enables Direct Visualization of RNA Base-Pairing Complexity.
The ability to probe all 4 nucleotides with DMS is an important experimental innovation but does not address the core limitation of conventional RNA structure probing analysis: Structures are not visualized directly but only inferred based on consistency with a 1-dimensional reactivity profile. A unique advantage of MaP compared to alternative “seq” readout strategies is that MaP allows measurement of multiple, correlated DMS modifications within a single RNA molecule (14). We previously showed that we could use correlated chemical probing to detect correlated modifications that occur between A–U and G–C base pairs in model in vitro transcripts (15). However, we were unable to detect base pairs in endogenous RNAs due to low DMS reactivity at G and U positions. We now exploit PAIR-MaP to directly detect pairing interactions in endogenous RNAs, including in living cells, at high resolution and specificity.
PAIR-MaP is predicated on detecting correlated DMS modifications on opposing strands of paired duplexes (Fig. 2_A_). While paired nucleotides are normally protected, equilibrium fluctuations transiently expose paired bases, mediating low but detectable rates of DMS modification. Chance modification of one base will permanently destabilize the base pair, increasing the probability of subsequent DMS modification at either the directly opposing base or neighboring bases (Fig. 2_A_). We detect these characteristic correlated modification signals by performing correlation analysis over 3-nt windows, which amplifies the weak modification signals of paired nucleotides by summing over nearest neighbors. Paired duplexes can then be specifically identified as lowly reactive, complementary 3-nt windows that are modified in a correlated manner (Fig. 2_B_). Significantly, PAIR-MaP detects duplexes formed in the predominant structure of an RNA as well as duplexes formed in lesser but appreciably populated alternative or misfolded structures. We therefore classify PAIR-MaP correlations into 2 classes (Fig. 2_B_ and SI Appendix, Fig. S1). “Principal” correlations are defined to occur between lowly reactive positions and are unambiguously the strongest correlation for each set of interacting nucleotides, providing high-confidence indicators of the predominant structure. “Minor” correlations represent weaker correlations or occur between moderately reactive nucleotides and report on unstable and alternative RNA duplexes. An abridged description of the PAIR-MaP algorithm is provided in Materials and Methods, and full details are provided in SI Appendix, Supporting Methods and Fig. S1.
Fig. 2.

PAIR-MaP enables direct detection of principal and alternative base-pairing interactions. (A) Correlated modification mechanism. (B) Strategy for identifying base-pairing interactions from correlated probing data. (C and D) In vitro PAIR-MaP data collected on the adenine riboswitch without (C) adenine and (D) in the presence of 100 μM adenine. Per-nucleotide DMS reactivities (black, yellow, and red) are shown in the middle. PAIR-MaP correlations are shown at the bottom, with principal correlations (dark blue) and minor correlations (light blue) colored with varying intensity based on Z-score significance (Materials and Methods). In C, the known (20) apoA (gray) and apoB (purple) secondary structures are shown at the top. In D, the ligand-bound crystallographic structure (43) (black) is shown with long-range tertiary interactions indicated by dashed lines.
As an initial validation of our strategy, we used PAIR-MaP to probe an in vitro transcript of the Vibrio vulnificus add adenine riboswitch, an established model system known to adopt multiple structures (Fig. 2_C_) (20, 21). PAIR-MaP immediately reveals the complex structural landscape of the riboswitch. In the absence of adenine ligand, PAIR-MaP reports a superposition of multiple pairing interactions recapitulating the ligand-free aptamer (apoA) and alternative structure (apoB) equilibrium (Fig. 2_C_). The relative strengths of the P1, P1B, and P2 helix correlations are also consistent with reported stabilities of these helices (populations between 20 and 50%) (20, 21). Upon addition of the adenine ligand, the PAIR-MaP correlation network markedly consolidates. All _apoB_-specific correlations disappear, consistent with the expected depopulation of the apoB state (expected population <25%), while P1 correlations significantly strengthen (expected population >75%) (20, 21). We also observe several minor correlations arising from tertiary interactions and indirect cooperative folding interactions, representing false-positive base pairs (but true tertiary interactions). Thus, other types of structural correlations can occasionally pass through the PAIR-MaP filtering algorithm. Combined, these data validate that PAIR-MaP directly visualizes RNA base pairing and structural complexity.
Direct Visualization of RNA Base Pairing in Cells.
We next benchmarked PAIR-MaP using endogenous E. coli and human RNAs, probed in the cell-free state. PAIR-MaP again provides a detailed visualization of the architectures of these diverse RNAs. For the 16S and 23S rRNAs, extensive correlations clearly define individual domains, including numerous duplexes spanning >350 nt (Fig. 3 and SI Appendix, Figs. S2–S4). PAIR-MaP correlations also clearly define pseudoknots (PKs) in tmRNA and RNase P (Fig. 3_E_ and SI Appendix, Fig. S2).
Fig. 3.

Detection of long-range base pairs, PKs, and misfolding in endogenous RNAs. (A) PAIR-MaP data collected on the E. coli 16S rRNA under cell-free conditions. The accepted secondary structure is shown at the top, with duplexes lacking sufficient read depth or exceeding PAIR-MaP read-length limitations indicated in gray. Normalized DMS data and PAIR-MaP correlations are shown at the middle and bottom, respectively, following the same scheme as in Fig. 2. Misfolded regions implied by high DMS reactivity and nonnative PAIR-MaP signals are highlighted in gold. (B) Detail of misfolding in the 136 to 227 region of the 16S rRNA. At the left, misfolded base pairs observed in the cell-free DMS+PAIR-MaP structure model (purple), with corroborating PAIR-MaP correlations shown at the bottom (arrow). In the middle, misfolded base pairs predicted by prior SHAPE-MaP analysis of the cell-free structure (green; ref. 17). At the right, structure observed by crystallography for the intact ribosome. (C_–_F) Cell-free and in-cell PAIR-MaP data for human U1 (C and D) and E. coli tmRNA (E and F). Accepted secondary structures are shown at the top, normalized DMS reactivities in the middle, and PAIR-MaP data at the bottom. Known duplexes that overlap primer-binding sites and thus lack PAIR-MaP data are drawn in gray. tmRNA PKs are labeled.
Under cell-free conditions, principal PAIR-MaP correlations are strongly predictive of the known secondary structure with an average positive predictive value (ppv) of 88%. Furthermore, many of the “false-positive” correlations (corresponding to correlations that do not match the known secondary structure) are consistent with misfolding of the deproteinized RNAs and, indeed, provide direct evidence of such misfolding. Of particular note, we observe strong PAIR-MaP signals supporting misfolding of the 136 to 227 region of the 16S rRNA (Fig. 3_B_); prior SHAPE probing studies suggested that this region significantly populates an alternative conformation in the absence of proteins, but the validity of this misfolding event had remained controversial (8, 22, 23). We also observe PAIR-MaP signals supporting previously suggested misfolding events elsewhere in the 16S rRNA, 23S rRNA, and human RMRP (Fig. 3_A_ and SI Appendix, Figs. S3 and S4) (8, 24, 25). When these regions with clear alternative folds are excluded, the average ppv of principal correlations increases to 92% (Table 1). The remaining false positives are likely a mixture of indirect interactions reflective of cooperative folding events and “real” misfolding interactions that we cannot confidently assess.
Table 1.
Accuracy of PAIR-MaP analysis and DMS+PAIR-MaP guided structure modeling
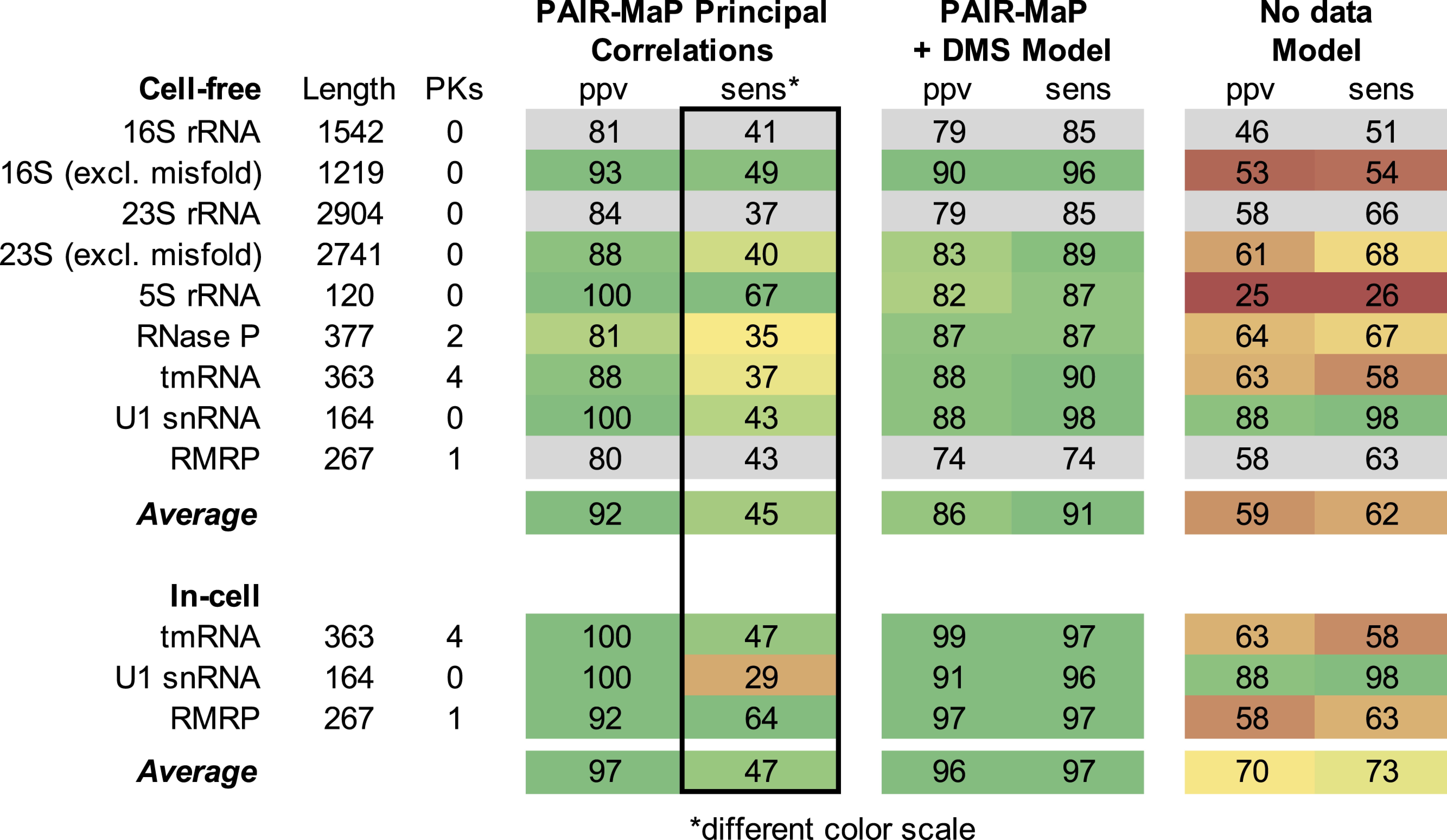
Minor PAIR-MaP correlations reveal additional complexities of ncRNA folding landscapes under cell-free conditions (Fig. 3 and SI Appendix, Figs. S2–S4); 30 to 50% of minor correlations correspond to native duplexes that are only partially folded under cell-free conditions. The other minor correlations are more challenging to evaluate. As noted above in our analysis of the adenine riboswitch, we expect some fraction of minor PAIR-MaP correlations to report indirect cooperative folding interactions. This feature is particularly evident for RNase P, where nonnative PAIR-MaP signals are best explained as indirect interactions from global unfolding/folding transitions (SI Appendix, Fig. S2). In contrast, for RNAs such as the 16S and 23S rRNA, a large fraction of the minor PAIR-MaP correlation network likely reflects alternative misfolded states (Fig. 3_A_ and SI Appendix, Fig. S4).
Strikingly, PAIR-MaP is also highly predictive of base-paired structure in cells for tmRNA, U1, and RMRP (ppv = 92 to 100% for principal correlations; Fig. 3 D and F, SI Appendix, Fig. S3, and Table 1). Indeed, principal and minor PAIR-MaP correlations consolidate around the known structure of each RNA compared to cell-free PAIR-MaP networks, consistent with proteins stabilizing a single predominant structure in cells (SI Appendix, Figs. S2 and S3). However, we were not able to perform PAIR-MaP analysis for the in-cell rRNAs and RNase P. These RNAs are exceptionally stable such that paired nucleotides are almost never modified, precluding measurement of PAIR-MaP correlations. Such datasets are readily automatically identified and rejected by the PAIR-MaP algorithm (Materials and Methods).
Overall, ∼45% of helices are detected as principal PAIR-MaP correlations (Table 1), but helix detection sensitivity (sens) does vary with molecular context. Analysis of our cell-free datasets reveals that PAIR-MaP has greatest sens (>50%) when each duplex strand contains an A or C (for example, AAG paired to CUU). Conversely, sens is lowest (<10%) when one strand consists entirely of G residues (GGG paired to CCC). This sequence dependence is consistent with the reactivity and specificity biases of DMS defined above. Sensitivity is additionally impacted by thermodynamic stability, with duplexes containing 2 or more G–C pairs detected with lower sens (for example, CCG paired to CGG is detected with ∼30% sens). As a single-molecule method, PAIR-MaP also requires that duplexes occur in the same sequencing read, corresponding to an interduplex length limitation of ∼500 nt with current technology. Finally, sensitivity depends strongly on sequencing depth: A depth of at least ∼400,000 is required to detect duplex correlations at ppv >0.8 and sens >0.3 (SI Appendix, Fig. S5).
In sum, PAIR-MaP is a specific and sensitive technique for directly detecting duplexes in endogenous RNAs in cells and reveals significant complexity in the folding landscapes of ncRNAs that is counteracted by protein stabilization in cells.
Accurate in-Cell Structure Modeling.
While PAIR-MaP provides an important model-free strategy for detecting RNA duplexes and characterizing RNA structural complexity, one critical end goal of chemical probing analysis is to determine complete RNA structure models. Building on prior studies, we developed a strategy to use PAIR-MaP data to enable highly accurate RNA structure modeling, including in cells.
We first capitalized on our discovery that DMS reacts with all 4 nucleotides by developing nucleotide-specific pseudo free-energy functions for DMS-directed structure modeling in RNAstructure (SI Appendix, Supporting Methods) (26, 27). On its own, this DMS-directed structure modeling strategy enables highly accurate de novo structure determination with average ppv ≈ 90% and sens ≈ 90% when applied to our panel of endogenous E. coli and human RNAs (SI Appendix, Table S2). This level of accuracy is more than sufficient for mechanistic hypothesis generation. Nevertheless, some important structural features are missed, including 1 of the 4 PKs in tmRNA (Fig. 4). We therefore developed an integrated modeling strategy in which we both apply per-nucleotide DMS reactivity restraints and also provide modest energetic bonuses to base pairs directly detected by PAIR-MaP. This integrated strategy is less accurate when modeling tmRNA under cell-free conditions due to the effects of several nonnative PAIR-MaP correlations that likely reflect misfolding in noncellular contexts (Table 1 and SI Appendix, Fig. S2). However, for all other RNAs and conditions, this integrated strategy yields equivalent or higher-accuracy structure models (Table 1 and SI Appendix, Fig. S2). Notably, when using in-cell data, this integrated strategy recovers tmRNA structure with near-perfect accuracy, including all 4 PKs (ppv = 99% and sens = 97%; Fig. 4). It is worth emphasizing that tmRNA, with its mixture of long-range interactions and multiple PKs, is one of the most difficult structure modeling challenges of which we are aware. Thus, while DMS-directed structure modeling provides excellent accuracy, integrated modeling with PAIR-MaP data can provide notable improvement for RNAs with particularly challenging structures.
Fig. 4.

PAIR-MaP enables accurate modeling of tmRNA structure in cells. Minimum free-energy structure models obtained without experimental data (Top), guided by nucleotide-specific DMS reactivity restraints (Middle), and guided by DMS reactivity restraints and PAIR-MaP restraints (Bottom). True-positive (gray), false-positive (purple), and false-negative (green) predictions and overall ppv and sens are shown for each model. The 4 correctly modeled PKs are labeled.
Overall, the ∼90% accuracy of DMS+PAIR-MaP-directed structure modeling is comparable to best-in-class SHAPE-based strategies (SI Appendix, Table S2) (7). It is particularly striking that, using PAIR-MaP, accuracy remains similar or increases in cells for all RNAs. DMS reactivity has been widely used to model RNA structure de novo. To date, the accuracy of DMS-directed structure modeling has been examined only for short RNAs (27). This work provides quantitative validation that DMS can be used to guide accurate structure modeling of long, complex RNAs. Furthermore, this analysis provides important systematic validation that de novo structure modeling can be performed accurately in cells, which has not been previously validated for any reagent.
Identification of an Unnoticed Conserved Helix in RMRP.
During benchmarking of PAIR-MaP on human RMRP it became clear that the accepted RMRP structure was incomplete. RMRP is an essential ncRNA, conserved across eukaryotes, that is involved in rRNA processing and other potential functions (28). RMRP is ancestrally related to the eukaryotic RNase P RNA, and phylogenetic analyses have shown that RMRP and RNase P share similar conserved structures (29, 30). As noted above, RMRP clearly misfolds under cell-free conditions (SI Appendix, Fig. S3). This misfolding is resolved in cells, with PAIR-MaP correlations and structure modeling showing that RMRP forms the accepted base-paired structure with one notable exception. Strikingly, PAIR-MaP revealed the presence of an additional “P7” helix that closes the catalytic core domain of RMRP (Fig. 5_A_). The P7 helix is a conserved architectural feature of RNase P but, to date, has not been observed in RMRP (30). We used this updated structure model to realign the published RMRP multiple-sequence alignment (31), which newly reveals that the P7 extension is conserved from yeast to humans (Fig. 5_B_). Furthermore, single-nucleotide mutations or insertions within P7 are pathogenic in humans (Fig. 5_B_) (32). Thus, in-cell PAIR-MaP enabled discovery of a functionally important helix missed by previous analyses and provides insights into the structural basis of human disease.
Fig. 5.

PAIR-MaP identifies a previously undetected conserved helix in RNase MRP. (A) Comparison between the in-cell DMS+PAIR-MaP structure model and prior covariation-based structure model (30). Purple, DMS+PAIR-MaP-predicted pairs not present in the prior covariation model. Green, accepted pairs missed by the DMS+PAIR-MaP model. Gray, pairs present in both models. DMS reactivities and PAIR-MaP correlations are shown at bottom following the same scheme as in Fig. 2. The PAIR-MaP correlation supporting the P7 interaction is indicated by the arrow. (B) Realigned consensus structure of RMRP reveals significant covariation for the P7 helix (933 sequences, from yeast to human). Base pairs supported by significant covariation [green, assessed using R-scape (40)] or sequence conservation [red, assessed using R2R (44)] are indicated. Human disease-associated mutations in the P7 helix are indicated by orange arrows (32).
Mechanistic Insights into Bacterial mRNA Autoregulatory Elements.
We next applied PAIR-MaP to examine the structures of RNAs that have proven challenging to characterize via traditional approaches. We focused on two E. coli 5′ untranslated regions (5′-UTRs) that contain regulatory elements that bind ribosomal proteins (r-proteins) to inhibit translation of downstream genes, forming a feedback loop that ensures balanced synthesis of r-proteins and rRNA (33). These 5′-UTRs are good exemplars of the functional relationships between RNA structure and protein binding that govern regulation of many RNAs in vivo.
We first applied PAIR-MaP to characterize the S4-binding element (S4E) located upstream of the E. coli rpsM gene (also termed the α-operon). Prior studies have suggested that S4 induces a conformational change in the S4E, stabilizing a double PK structure that inhibits translation of rpsM and downstream genes (34, 35). However, the proposed double PK is not fully consistent with biochemical and genetic data, and the structure of the S4E in the absence of the S4 protein is unknown (SI Appendix, Supporting Discussion and Fig. S6). Cell-free PAIR-MaP data show that the rpsM 5′-UTR and coding sequence fold into 4 stem–loop helices (Fig. 6_A_). Of particular note, we identify an unstable helix (H3) formed between the Shine–Dalgarno sequence and the rpsM coding sequence. Strikingly, in-cell experiments reveal stabilization of H3 as well as appearance of signals indicating loop–loop pairing between H2 and H3, consistent with S4 protein binding and stabilizing a kissing loop structure in cells (Fig. 6_B_). This kissing loop structure is more consistent with S4E sequence conservation compared to the previously proposed double PK and uniquely explains the impact of Shine–Dalgarno sequence mutations on S4 binding (Fig. 6 B and C and SI Appendix, Supporting Discussion and Fig. S6) (35). There is also potential structural homology between the kissing loop structure and the S4 binding site on the 16S rRNA (SI Appendix, Fig. S6). Thus, our data support a model in which S4 binds a kissing loop structure, which stabilizes the H3 stem and thereby prevents translation initiation on rpsM (SI Appendix, Supporting Discussion).
Fig. 6.

Structural features of r-protein regulatory elements. (A) Cell-free structure of the E. coli rpsM 5′-UTR containing the S4E. The sequence is numbered relative to the _rpsM_-specific transcription start site consistent with prior studies; however, our data are specific to the intergenic form of the rpsM 5′-UTR transcribed from upstream promoters. CDS, coding sequence. (B) In-cell structure of the S4E. The kissing loop interaction (KL; purple) is not predicted by minimum free-energy structure modeling but is clearly supported by PAIR-MaP correlations. The SD mutation, which disrupts H3 and abrogates S4 binding (35), is labeled. (C) Revised consensus structure of the S4E across Gammaproteobacteria. Covariation and base-pairing conservation were assessed using R-scape and R2R, respectively (40, 44). (D) Cell-free structure of the E. coli rpsB 5′-UTR containing the S2E. The previously undetected P1 interaction (purple) is predicted by DMS+PAIR-MaP minimum free-energy modeling. (E) In-cell structure of the S2E. The P1 and PK interactions (in purple and green, respectively) are not predicted by minimum free-energy modeling but are clearly supported by PAIR-MaP correlations. A deletion that disrupts P1 and abrogates S2 regulation is labeled (36). (F) Homology between the S2E (Left) and the S2 binding site in the 16S rRNA (Middle), and the crystal structure of the S2 ribosome binding site (Right; Protein Data Bank ID code 4YBB). P1 and PK nucleotides are highlighted in purple and green, respectively, and homologous nucleotides are highlighted in orange. The key for PAIR-MaP plots is provided in Fig. 2.
We next examined the S2-binding element (S2E) located in the 5′-UTR of the E. coli rpsB-tsf transcript (36). Phylogenetic analyses have predicted that the S2E folds into a PK (33), but the PK interaction has not been directly confirmed and prior SHAPE experiments were ambiguous (37). Significantly, while no PK is observed under cell-free conditions, in-cell PAIR-MaP experiments reveal multiple minor signals consistent with S2-induced stabilization of the PK in cells (Fig. 6 D and E). Our data also reveal a previously undetected “P1” helix (Fig. 6 D and E). The functional importance of P1 is supported by prior genetic studies, which observed that deletion of P1-involved sequences abrogate S2 regulation (Fig. 6_E_) (36). The discovery of the P1 helix also illuminates how the S2 protein recognizes the S2E RNA. Whereas it was previously thought that the S2E lacked homology to the 16S rRNA binding site of S2 (33), identification of P1 makes it clear that S2 recognizes a common architecture in both the S2E and 16S rRNA (Fig. 6_F_). Interestingly, this architecture only appears to be conserved among enterobacterial S2Es, with S2Es from more distant bacterial species lacking the capacity to form P1 and thus potentially using a different mode of S2 recognition (SI Appendix, Supporting Methods). Because P1 sequences show little variability within the enterobacterial clade, this structural element has gone undiscovered by de novo covariation analysis. Moreover, the moderate DMS reactivities for both P1 and the PK indicate that these elements are only partially formed in cells (Fig. 6_E_), making them challenging to discover via traditional chemical probing strategies. Thus, in-cell PAIR-MaP analysis reveals divergent, yet functionally important, structural features of the E. coli S2E RNA missed by conventional analyses.
Discussion
Chemical probing is a central tool in RNA structural biology, providing nucleotide-resolution insight into local RNA structure in an adaptable, experimentally concise manner. However, the inability to measure base-pairing interactions directly had remained a fundamental limitation of these approaches. We show that single-molecule correlated chemical probing coupled with PAIR-MaP analysis resolves this limitation, allowing direct visualization of RNA duplexes in living bacterial and human cells. The correlation data obtained from PAIR-MaP experiments are typically sufficiently dense to define global RNA architecture, providing direct evidence of complex structural features such as PKs and long-range pairing. Equally valuable, PAIR-MaP data provide insight into the complexity of the RNA structural landscape, revealing alternative and unstable pairing interactions that are difficult to measure via conventional means. Finally, PAIR-MaP data can be used in combination with automated computational modeling strategies to derive complete, accurate models of RNA structure as it exists in cells.
PAIR-MaP offers significant advantages compared to alternative in vivo duplex-detection strategies. Most importantly, PAIR-MaP resolves base pairs at nucleotide resolution with superior ppv (>90%) and sens (∼45%) (Table 1). PAIR-MaP is straightforward to implement; the innovation of the strategy lies in improved conditions enabling pan-RNA modification by DMS, the MaP readout, and algorithmic interpretation of the single-molecule correlated chemical probing signal. DMS-MaP experiments are already broadly used throughout the RNA community, and focused sequencing libraries of even rare RNAs are easily prepared using PCR amplification without the need to enrich for or pull down target RNAs (38). Finally, a single PAIR-MaP experiment reports both local reactivity and pairwise interaction information, obviating the need for multiple experiments.
PAIR-MaP does have several limitations. Due to high sequencing read-depth requirements, PAIR-MaP is poorly suited for transcriptome-wide profiling. In contrast to cross-linking and ligation strategies, PAIR-MaP requires duplexes to be self-contained within a contiguous sequencing read, currently ∼500 nt, and cannot detect intermolecular duplexes. PAIR-MaP also cannot detect duplexes in a few exceptionally stable, protein-coated RNP complexes such as the ribosome and RNase P. More generally, PAIR-MaP correlations are innately “nonnative” measurements—in a sense, PAIR-MaP measures DMS-induced sequential unfolding of RNA molecules. The progressive accumulation of DMS adducts could promote formation of misfolded states, or shift the native equilibrium of dynamic RNAs. However, because of the stochasticity of the DMS modification process, every molecule is perturbed in a unique and noncoherent manner, and hence perturbations should average out over a population of molecules. Our extensive benchmarking supports that DMS-induced perturbations do not significantly impact PAIR-MaP accuracy.
Overall, our study highlights the extensive potential of PAIR-MaP for characterizing RNA structure and dynamics and ultimately for understanding biology. PAIR-MaP allowed us to determine the in-cell structure of human RMRP, revealing a universally conserved and disease-linked RNA helix that has been missed by prior phylogenetic and chemical probing analyses. Our analysis of bacterial mRNA regulatory motifs further uncovered dynamic helices that are essential for understanding protein-binding and regulatory function, but which are only appreciably formed in cells and are invisible to lower-resolution methods. In recent years, high-throughput chemical probing methodologies have helped open up a new frontier of in-cell structure–function studies in complex RNAs such as mRNAs and long ncRNAs (2, 39). However, the structural data obtained from many of these studies remain difficult to evaluate, particularly for evolutionarily divergent long ncRNAs (40). We anticipate that PAIR-MaP will broadly facilitate the next generation of high-resolution, in-cell structural insights into these and many other RNAs that continue to challenge conventional characterization.
Materials and Methods
Experimental Methods.
DMS probing experiments were performed on total RNA gently extracted (8, 41) from E. coli K-12 MG1655 and human Jurkat cells (cell-free) and on intact cells (in-cell) buffered with 200 or 300 mM bicine (pH 8.0), 200 mM potassium acetate (pH 8.0), and 5 mM MgCl2 at 37 °C. In vitro transcribed adenine riboswitch RNA was probed at 30 °C in the absence or presence of 100 μM adenine ligand in 300 mM bicine (pH 8.0), 100 mM NaCl, and 5 mM MgCl2. Unmodified control samples were also obtained for all RNAs. MaP reverse transcription was used to convert DMS adducts into mutations in complementary DNA and is compatible with nearly all protocols for creating libraries for massively parallel sequencing (14, 38). Here, sequencing libraries were prepared by both randomly primed Nextera (E. coli 16S and 23S rRNA) and gene-specific PCR (other RNAs) (38) and sequenced on an Illumina MiSeq instrument (SI Appendix, Tables S4 and S5). ShapeMapper was used to align and parse mutations from DMS-MaP sequencing data (17).
The PAIR-MaP Algorithm.
PAIR-MaP analysis begins by computing the correlation between joint mutations of all pairs of 3-nt windows in the DMS-modified sample (SI Appendix, Fig. S1). Nucleotide windows separated by 8 or fewer nucleotides, having insufficient read depth or low modification rates in the modified sample, or exhibiting high mutation rates or significant correlations in the unmodified background control dataset are excluded. Correlation is quantified using the average product corrected G test (GijAPC) (40, 42), and a pair of nucleotide windows is considered significantly correlated if GijAPC>20, corresponding to P < 10−5. High-confidence base-pairing signals are then identified from the set of significantly correlated windows by filtering by sequence complementarity, correlation strength, and reactivity. Specifically, correlated windows must be able to form 3 Watson–Crick or G–U pairs, the windows must be positively correlated, GijAPC must be greater than 2 SDs above the mean, and both windows must have mean normalized DMS reactivities <0.2 (for principal correlations) or <0.5 (for minor correlations). Complementary correlations that are unambiguously the strongest correlation for each interacting nucleotide window, with each window passing the stricter 0.2 reactivity cutoff, are classified as principal. The remaining set of complementary correlations are classified as minor. PAIR-MaP analysis can only be performed if RNAs are modified by DMS at a sufficiently high level; datasets exhibiting median comodification rates <0.0005 are automatically disqualified (for example, the in-cell rRNA datasets). In PAIR-MaP plots, principal and minor correlations are colored with varying intensity based on Z-score significance on a scale from Z = 2 to Z ≥ 6. PAIR-MaP analysis code is available as part of the RingMapper/PairMapper software suite (v1.0), available for download at https://github.com/Weeks-UNC/RingMapper.
Detailed descriptions of experimental methods, the PAIR-MaP algorithm, DMS reactivity normalization, structure modeling, sensitivity and specificity analysis, and covariation analysis is provided in SI Appendix, Supporting Methods.
Supplementary Material
Supplementary File
Acknowledgments
We thank S. Busan for helpful discussions and ongoing development of the ShapeMapper software, C. Weidmann for helpful discussions, and the K.M.W. laboratory for testing and feedback on PAIR-MaP. We thank D. Mathews (University of Rochester) for sharing access to the RNAstructure source code. This work was supported by the Arnold and Mabel Beckman Foundation (postdoctoral fellowship to A.M.M.) and the National Institutes of Health (R35 GM122532 to K.M.W.).
Footnotes
Competing interest statement: A.M.M. is a consultant to and K.M.W. is an advisor to and holds equity in Ribometrix, to which correlated chemical probing technologies have been licensed.
This article is a PNAS Direct Submission.
Data deposition: The PAIR-MaP datasets reported in this study have been deposited in the Gene Expression Omnibus (GEO) database, https://www.ncbi.nlm.nih.gov/geo (accession no. GSE135211). The RingMapper/PairMapper software suite has been deposited at https://github.com/Weeks-UNC/RingMapper. Supplementary Dataset S1 contains all processed PAIR-MaP data, structure models, and multiple sequence alignments and is available from the corresponding author's website and at https://doi.org/10.6084/m9.figshare.9978596.
References
- 1.Cech T. R., Steitz J. A., The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Mustoe A. M., Corley M., Laederach A., Weeks K. M., Messenger RNA structure regulates translation initiation: A mechanism exploited from bacteria to humans. Biochemistry 57, 3537–3539 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dethoff E. A., Chugh J., Mustoe A. M., Al-Hashimi H. M., Functional complexity and regulation through RNA dynamics. Nature 482, 322–330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ehresmann C., et al. , Probing the structure of RNAs in solution. Nucleic Acids Res. 15, 9109–9128 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weeks K. M., Advances in RNA structure analysis by chemical probing. Curr. Opin. Struct. Biol. 20, 295–304 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leamy K. A., Assmann S. M., Mathews D. H., Bevilacqua P. C., Bridging the gap between in vitro and in vivo RNA folding. Q. Rev. Biophys. 49, e10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siegfried N. A., Busan S., Rice G. M., Nelson J. A. E., Weeks K. M., RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 11, 959–965 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deigan K. E., Li T. W., Mathews D. H., Weeks K. M., Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. U.S.A. 106, 97–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hajdin C. E., et al. , Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc. Natl. Acad. Sci. U.S.A. 110, 5498–5503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng C. Y., Kladwang W., Yesselman J. D., Das R., RNA structure inference through chemical mapping after accidental or intentional mutations. Proc. Natl. Acad. Sci. U.S.A. 114, 9876–9881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weidmann C. A., Mustoe A. M., Weeks K. M., Direct duplex detection: An emerging tool in the RNA structure analysis toolbox. Trends Biochem. Sci. 41, 734–736 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graveley B. R., RNA matchmaking: Finding cellular pairing partners. Mol. Cell 63, 186–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziv O., et al. , COMRADES determines in vivo RNA structures and interactions. Nat. Methods 15, 785–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Homan P. J., et al. , Single-molecule correlated chemical probing of RNA. Proc. Natl. Acad. Sci. U.S.A. 111, 13858–13863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krokhotin A., Mustoe A. M., Weeks K. M., Dokholyan N. V., Direct identification of base-paired RNA nucleotides by correlated chemical probing. RNA 23, 6–13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thaplyal P., Bevilacqua P. C., Experimental approaches for measuring pKa’s in RNA and DNA. Methods Enzymol. 549, 189–219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Busan S., Weeks K. M., Accurate detection of chemical modifications in RNA by mutational profiling (MaP) with ShapeMapper 2. RNA 24, 143–148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zubradt M., et al. , DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 14, 75–82 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sexton A. N., Wang P. Y., Rutenberg-Schoenberg M., Simon M. D., Interpreting reverse transcriptase termination and mutation events for greater insight into the chemical probing of RNA. Biochemistry 56, 4713–4721 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reining A., et al. , Three-state mechanism couples ligand and temperature sensing in riboswitches. Nature 499, 355–359 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Tian S., Kladwang W., Das R., Allosteric mechanism of the V. vulnificus adenine riboswitch resolved by four-dimensional chemical mapping. eLife 7, e29602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian S., Cordero P., Kladwang W., Das R., High-throughput mutate-map-rescue evaluates SHAPE-directed RNA structure and uncovers excited states. RNA 20, 1815–1826 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cordero P., Das R., Rich RNA structure landscapes revealed by mutate-and-map analysis. PLoS Comput. Biol. 11, e1004473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogler L. E., et al. , Small RNAs derived from lncRNA RNase MRP have gene-silencing activity relevant to human cartilage-hair hypoplasia. Hum. Mol. Genet. 23, 368–382 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGinnis J. L., et al. , In-cell SHAPE reveals that free 30S ribosome subunits are in the inactive state. Proc. Natl. Acad. Sci. U.S.A. 112, 2425–2430 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reuter J. S., Mathews D. H., RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11, 129 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cordero P., Kladwang W., VanLang C. C., Das R., Quantitative dimethyl sulfate mapping for automated RNA secondary structure inference. Biochemistry 51, 7037–7039 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldfarb K. C., Cech T. R., Targeted CRISPR disruption reveals a role for RNase MRP RNA in human preribosomal RNA processing. Genes Dev. 31, 59–71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X., Frank D. N., Pace N., Zengel J. M., Lindahl L., Phylogenetic analysis of the structure of RNase MRP RNA in yeasts. RNA 8, 740–751 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dávila López M., Rosenblad M. A., Samuelsson T., Conserved and variable domains of RNase MRP RNA. RNA Biol. 6, 208–220 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Kalvari I., et al. , Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 46, D335–D342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattijssen S., Welting T. J. M., Pruijn G. J. M., RNase MRP and disease. Wiley Interdiscip. Rev. RNA 1, 102–116 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Fu Y., Deiorio-Haggar K., Anthony J., Meyer M. M., Most RNAs regulating ribosomal protein biosynthesis in Escherichia coli are narrowly distributed to Gammaproteobacteria. Nucleic Acids Res. 41, 3491–3503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang C. K., Draper D. E., Unusual mRNA pseudoknot structure is recognized by a protein translational repressor. Cell 57, 531–536 (1989). [DOI] [PubMed] [Google Scholar]
- 35.Schlax P. J., Xavier K. A., Gluick T. C., Draper D. E., Translational repression of the Escherichia coli alpha operon mRNA: Importance of an mRNA conformational switch and a ternary entrapment complex. J. Biol. Chem. 276, 38494–38501 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Aseev L. V., Levandovskaya A. A., Tchufistova L. S., Scaptsova N. V., Boni I. V., A new regulatory circuit in ribosomal protein operons: S2-mediated control of the rpsB-tsf expression in vivo. RNA 14, 1882–1894 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mustoe A. M., et al. , Pervasive regulatory functions of mRNA structure revealed by high-resolution SHAPE probing. Cell 173, 181–195.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smola M. J., Rice G. M., Busan S., Siegfried N. A., Weeks K. M., Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat. Protoc. 10, 1643–1669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strobel E. J., Yu A. M., Lucks J. B., High-throughput determination of RNA structures. Nat. Rev. Genet. 19, 615–634 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rivas E., Clements J., Eddy S. R., A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods 14, 45–48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smola M. J., Calabrese J. M., Weeks K. M., Detection of RNA–protein interactions in living cells with SHAPE. Biochemistry 54, 6867–6875 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunn S. D., Wahl L. M., Gloor G. B., Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics 24, 333–340 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Serganov A., et al. , Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem. Biol. 11, 1729–1741 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weinberg Z., Breaker R. R., R2R–software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinformatics 12, 3 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File