The roles of structural dynamics in the cellular functions of RNAs (original) (raw)
. Author manuscript; available in PMC: 2020 Nov 11.
Published in final edited form as: Nat Rev Mol Cell Biol. 2019 Aug;20(8):474–489. doi: 10.1038/s41580-019-0136-0
Abstract
RNAs fold into 3D structures that range from simple helical elements to complex tertiary structures and quaternary ribonucleoprotein assemblies. The functions of many regulatory RNAs depend on how their 3D structure changes in response to a diverse array of cellular conditions. In this Review, we examine how the structural characterization of RNA as dynamic ensembles of conformations, which form with different probabilities and at different timescales, is improving our understanding of RNA function in cells. We discuss the mechanisms of gene regulation by microRNAs, riboswitches, ribozymes, post-transcriptional RNA modifications and RNA-binding proteins, and how the cellular environment and processes such as liquid-liquid phase separation may affect RNA folding and activity. The emerging RNA-ensemble–function paradigm is changing our perspective and understanding of RNA regulation, from in vitro to in vivo and from descriptive to predictive.
A central and bold goal of biomedical sciences is to achieve a predictive understanding of living cells, tissues and, ultimately, whole organisms based on the properties of their constituent biomolecules. To date, much effort has been directed towards understanding the functions of biomolecules by determining their 3D structures at atomic resolution, but we still lack the ability to quantitatively predict from these structures key biochemical properties such as folding stability, catalytic efficiency and binding affinity and specificity. In reality, all macromolecules dynamically alternate between conformational states to carry out their biological functions. Decades ago, it was realized that the structures of biomolecules are better described as ‘screaming and kicking’1, constantly undergoing motions on timescales spanning 12 orders of magnitude, from picosecond to seconds, and that in addition to sequence and structure, these motions are important for their activities. The goal of this Review is to show that although the dynamics of biomolecules can be dizzyingly complex, they can be described, classified and, ultimately, will be used to quantitatively predict molecular behaviour.
RNA molecules have crucial functions both in normal, physiological conditions2 and in pathological conditions3 (FIG. 1). The number of RNAs targeted by drugs to treat infectious diseases, cancer and genetic disorders is rapidly growing4 and so is the number of applications in which RNA is engineered to drugs, cellular devices and tools for molecular and synthetic biology5. RNAs such as microRNAs readily fold into stem-loop secondary structures, which can be recognized by protein6,7, and other RNAs such as ribozymes and riboswitches can also fold into complex tertiary structures and exhibit activities that rival those of proteins8–10. Another subset of RNAs form large quaternary assemblies of ribonucleoprotein (RNP) machines such as the ribosome11,12 and spliceosome13,14 (FIG. 1). Moreover, RNA fine-tunes and expands its functionality through its ability to change structure in response to specific cellular cues (reviewed in REF15). Indeed, mutations that disrupt RNA structural dynamics have been linked to human diseases16–20 (BOX 1) and many antibiotics function by disrupting RNA structural dynamics21.
Fig. 1 |. RNA structural changes enable biological functions.

a | Riboswitches undergo metabolite- induced conformational changes to turn gene expression on or off. b | Conformational changes in the 5′ leader of the HIV-1 RNA genome drive genome dimerization and are proposed to regulate the switch between translation and packaging of the viral RNA genome. c | Alternative splicing (AS) factors bind cognate RNA-binding motifs as single-stranded RNA. Therefore, AS depends on the equilibrium between structured and unstructured conformations of the RNA at the binding site. d | Ribozymes undergo changes in tertiary structure during their catalytic cycles. e | Binding of ribosomal protein S15 induces a conformational change in ribosomal RNA to direct the ordered assembly of the ribosome. f | MicroRNAs interact with protein partners in specific conformations, which can be important for recognition, binding affinity and downstream activity. LIN28A binds to the primary let-7 microRNA or the precursor let-7 (pre-let-7) microRNA and induces a conformation that prevents binding of the microRNA processing factors Drosha and Dicer, respectively. g | Long non-coding RNAs have many cellular functions, including as scaffolds that direct RNA–protein, RNA–RNA and RNA–DNA interactions in epigenetic regulation. h | RNA can be found in phase-separated granules, in which it forms dynamic RNA–protein and RNA–RNA interactions. RNP, ribonucleoprotein.
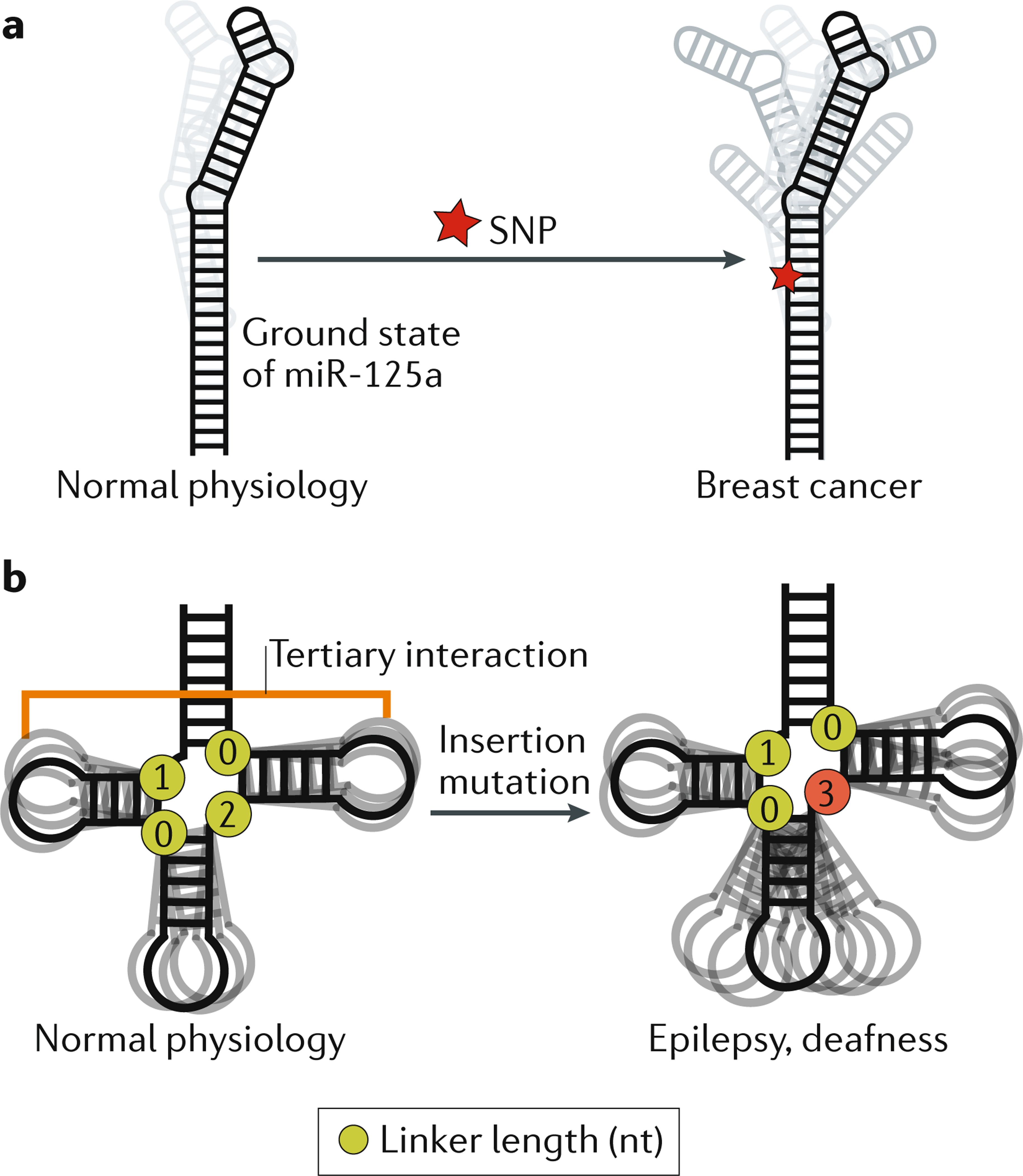
Box 1 |. The relevance of RNA ensembles to human diseases.
In addition to clarifying RNA folding and its roles in cellular processes, an ensemble perspective of RNA structure is required to understand how some mutations, including potentially disease-causing mutations, affect RNA activity. Single-nucleotide polymorphisms (SNPs) are the most common type of genetic variation in the human population and many are associated with diseases. There is mounting evidence that many disease-associated SNPs induce changes in RNA ensembles, and ensemble-based mechanisms have been proposed to explain disease phenotypes16,18,20. In retinoblastoma, SNPs in the 5′ UTR of the RBL1 mRNA, which encodes the tumour suppressor retinoblastoma-like protein 1, can collapse the ensemble of secondary structures formed by the UTR into a single secondary structure, resulting in reduced expression of the tumour suppressor18. Structural dynamics at microRNA sites that are recognized by microRNA-processing factors are proposed to be important for their maturation16,150. A SNP in the human microRNA miR-125a that interferes with its maturation is highly associated with breast cancer151. This SNP is proposed to inhibit maturation efficiency and lead to breast cancer by increasing the number of non-native secondary-structure conformations and simultaneously decreasing the population of the native, or ground state, conformation, which is thought to be the conformation most amenable to processing (see the figure part a)16. Other mutations in miR-125a affect its maturation in a manner correlated with their predicted effect on the RNA ensemble16. Thus, ensemble descriptions of microRNAs may be more predictive of their maturation efficiency than single structures, and may be required to fully model the effects of disease-causing mutations16.
Pathogenic mutations can also affect RNA junction topology and the resulting inter-helical dynamics. For example, the highly conserved topology of the tRNA four-way junction skews the orientation of helices towards tertiary conformations73 (see the figure, part b). Mammalian mitochondrial tRNAs such as tRNASer(UCN) fold into near-canonical tRNA 3D structures despite having shortened inter-helical loops, which disrupt tertiary interactions that are conserved in many tRNAs. Using a coarse grain computational model that predicts RNA global dynamics, the topological constraints encoded by the shorter inter-helical loops in tRNASer(UCN) were shown to decrease the free energy of folding by decreasing the energy cost of redistribution, thus compensating for the loss of tertiary interactions152. A pathogenic insertion mutation that changes the junction topology of mammalian mitochondrial tRNASer(UCN) decreases the concentration of the tRNA in vivo and is linked to hearing loss and epilepsy. The mutation is predicted to broaden the unfolded ensemble and thereby destabilize tertiary folding152 (see the figure, part b). These computational predictions were verified experimentally152. Because tRNA processing requires a properly folded conformation, the mutation was proposed to increase the susceptibility of tRNASer(UCN) to a pathway that degrades unfolded and misprocessed tRNAs, thereby decreasing its cellular concentration152.
In solution, an RNA molecule dynamically samples a vast number of conformations. Some conformations may form with high probability and account for a large (~10%) proportion of the population of the RNA at any given time, whereas others, such as the unfolded state, may be extremely rare (0.00001% of the population). Some conformations may form rapidly — within pico-seconds — whereas others may form slowly (within seconds). The population-weighted distribution of all conformations of an RNA is referred to as an ‘ensemble’. (The term ensemble is often used loosely to describe any collection of structures of a given biomolecule, and these descriptions should be distinguished from the rigorous statistical and mechanical description of ensembles as the Boltzmann distribution of conformations discussed in this review15,22–24.)
In this Review, we discuss how the ensemble description of RNA structural dynamics15,25–27 is providing a more predictive understanding (compared with that provided by static structures) of how regulatory RNAs fold and function in cells, thereby illuminating their relevance to human diseases (BOX 1 ) and their therapy. We discuss the mechanisms of gene regulation by ensembles of microRNAs, riboswitches, ribozymes, post-transcriptionally modified RNAs and RNA-binding proteins (RBPs), and how the cellular environment affects RNA folding and activity. Although dynamics studies of the assembly and function of RNP machines such as the ribosome11,12 and spliceosome13,14 are important and well-established examples of the concepts discussed here, they are beyond the scope of this Review.
Activation of RNA cellular functions
The activities of many regulatory RNAs arise from changes to their structure (FIG. 1). The structural changes can be induced by the binding of proteins, metabolites, ions, DNA and other RNAs and by post-transcriptional modifications, changes in environmental conditions such as temperature and solute concentration, mutations to the RNA sequence or even by the act of RNA synthesis itself through co-transcriptional folding of the RNA. The outcomes of RNA structural changes and their dependent interactions include turning gene expression on or off, alternative splicing, regulating microRNA maturation and RNP assembly (FIG. 1). A wide variety of experimental methods have been developed to probe these functionally relevant RNA conformational dynamics (Supplementary Box 1).
Changes in RNA conformation can occur at the secondary, tertiary or quaternary structural levels. For example, riboswitches control gene expression by folding into alternative secondary structures upon ligand binding8,28–30 (FIG. 1a). The 5′ leader of the HIV-1 RNA genome undergoes changes in secondary structure that direct genome dimerization and are proposed to regulate the switch between translation and packaging of the viral genome31 (FIG. 1b). Many RBPs, including alternative splicing factors, bind single-stranded RNA motifs and thus may require melting of the RNA secondary structure before binding32 (FIG. 1c). On the other hand, ribozymes cycle through different tertiary structures to enable substrate binding, catalysis (often through multiple reaction steps) and product release33,34 (FIG. 1d). Proteins also induce changes in RNA tertiary structure that can trigger various outcomes, such as the quaternary assembly of the ribosome and other RNP machines35 (FIG. 1e). Proteins can also inhibit RNA activity by stabilizing inactive RNA conformations. For example, LIN28A binds the let-7 pre-microRNA and inhibits its maturation in part by changing its structure such that it can no longer be recognized by the microRNA maturation factors Drosha and Dicer36 (FIG. 1f).
RNA structural dynamics are likely to be important for other, less characterized processes. For example, long non-coding RNAs (lncRNAs)37–39 likely undergo conformational changes when acting as scaffolds for assembling proteins, DNA and RNA molecules37–39 (FIG. 1g). In cells, RNA is often found in phase-separated granules, where it dynamically interacts with other RNAs and proteins40–42. RNA conformational changes are likely important for nucleating such phase transitions and in defining the properties of the resulting granules (FIG. 1h).
Dynamic ensembles of RNA structures
For many RNAs, understanding their function requires understanding how cellular cues and modifiers change their conformation. What has become clear in the past two decades, and follows from first principles of statistical mechanics15,24, is that cellular modifiers change the abundance of two or more pre-existing conformations in the ensemble43–46. Furthermore, although it is commonly and reasonably assumed that the formation of RNA–ligand and RNA–protein complexes generally results in more rigid RNA structures, RNAs in both types of complexes are also more accurately described as dynamic ensembles46–48. Thus, cellular RNA modifiers do not change RNA from one structure to another; rather, they change the RNA ensemble from one distribution to another by changing the relative populations of different conformations15,43,46,49 (FIG. 2a). Some cellular modifiers, such as RNA chaperones50,51, accelerate the rates of interconversion by lowering energetic barriers. To understand how cellular modifiers activate RNA, we need to describe the RNA ensemble and understand how it is redistributed by cellular modifiers.
Fig. 2 |. Dynamic ensembles describe the roles of RNA structural changes.

a | A representative RNA free energy landscape of the transactivation response element (TAR) of HIV-1 (REF.53). The different conformations of TAR include a co-axially stacked conformation, a bent flexible conformation, three non-native secondary structures, including one with a base triple that represents a protein-bound conformation, and finally the unfolded conformation. The relative energetic stabilities (G_0_i) of each conformation i are represented by the depth of the free energy minima and the corresponding abundance of each conformer is shown as the fractional population over the entire ensemble. TAR activates transcription elongation of the HIV-1 genome by forming a complex with the viral protein Tat and host factors including positive transcription elongation factor b (P-TEFb), which is part of the super elongation complex (SEC). Binding to partner molecules, changes in the cellular environment such as in the concentration of magnesium ions (Mg2+) and/or of crowding agents or mutations can remodel the free energy landscape and redistribute the ensemble of conformations, thereby altering RNA activity. The structural features of the TAR native structure include the lower helix (green), bulge (yellow) and upper helix and apical loop (blue). The dashed orange line represents tertiary interactions. b | Illustration of key aspects of RNA activity that can be modelled using RNA ensembles but not by static RNA structures. (Left) The strength of interactions between RNAs and ligands depends on the population of the bound conformation in the free ensemble, with a lower population corresponding to higher energy cost of redistribution (Δ_G_0redist) and therefore lower binding affinity, and vice versa. The free RNA ensemble allowed by the long linker is broader and thus less likely to sample the bound conformation, resulting in weaker binding affinity (top). By contrast, a short linker results in a narrow ensemble centred around the bound conformation, which will result in tighter binding affinity (bottom). The free energy landscapes illustrate the extent of overlap between the free and bound ensembles for both examples. (Middle) Similarly, the degree of RNA activity correlates with the population of the active conformer in the ensemble. A small population of the active RNA conformation in the ligand-bound RNA ensemble will elicit low-level biological activity (top), whereas a large population of the active RNA conformation in the ligand-bound RNA ensemble will elicit high-level biological activity (bottom). The free energy landscapes illustrate the stability of the RNA in the active conformation for both examples. (Right) Selective pressure that favours dynamic ensembles (rather than a single conformation) can give rise to unique conservation patterns, which depend on the nature of the dynamics. In the example of sequence conservation, bases that form Watson–Crick pairs (paired red circles) in the native secondary structure form mismatches (paired red and yellow circles) in an alternative non-native secondary structure. The relative stabilities of these pairings determines the population of each secondary structure, and, thus, a mutation can affect this equilibrium by differently affecting the two structures. If the relative population of structures in the ensemble is important for function, there will be evolutionary pressure to maintain the relative stability of structures, rather than solely the stability of the native secondary structure. In the example of topological conservation, secondary-structure elements such as the length of junction linkers are important determinants of inter-helical dynamics. Thus, evolutionary pressure to maintain inter-helical dynamics can result in sequence-independent conservation of secondary structure.
Describing RNA ensembles
Relative to a single structure, ensembles are more difficult to describe and to experimentally characterize. The free energy landscape that was first developed to describe complex systems such as glasses and, later, proteins15,24 provides a powerful framework for describing ensembles of macromolecules by specifying the energetic stability (free energy, G_0_i) of every allowed conformation15,24 (FIG. 2a). The population size of any given conformation will depend on its energetic stability relative to other conformations, whereas the rates at which any two conformations interconvert will depend on the energetic barriers that separate them.
The experimentally determined44,52 ensemble for the transactivation response element (TAR) RNA of HIV-1 (REF53) highlights general features of the free energy landscape that appear to be common to many RNAs (FIG. 2a). TAR is a model system for studying RNA structural dynamics, and one of the few RNAs for which a detailed and comprehensive ensemble and free energy landscape is available following the application of various experimental techniques (Supplementary Box 1).
The free energy landscape is rugged, punctuated by local energetic minima that correspond to a subset of highly populated conformations (FIG. 2a). These energetic minima are separated by variable energetic barriers that reflect different rates of interconversion. In practice, it is only possible to observe a subset of dominant conformations in an ensemble with populations that fall within the detection limits of experimental techniques (Supplementary Box 1). Despite this limited view, the landscape of even a simple RNA such as TAR can be highly complex. A single native secondary structure dominates (~80%) the ensemble of free TAR in solution, but the 3D structure jitters on the picosecond-to-microsecond timescale, thereby forming various conformations, in which the two helices of TAR are either stacked and rigid (population of ~40%) or bent and flexible (population of ~40%)54 (FIG. 2a). Also present are low-abundance non-native secondary structures (populations of ~10%, ~0.1% and ~0.001%) that form on the microsecond-to-millisecond timescale and that differ with respect to base pairing in and around the non-canonical bulge and apical loop52. At even lower abundance (population of ~0.00001%) are partially or fully unstructured conformations. As we discuss below, cellular modifiers and other perturbations change the relative population of these RNA conformations rather than ‘create’ new conformations — all conformations are present on the landscape, but their abundance can vary greatly.
Cellular RNA conformational changes
The TAR example also provides a perspective on how RNA ensembles are redistributed and the biological consequences of this redistribution. Like many RNAs, TAR binds a protein, the viral _trans_activator Tat, which skews the TAR ensemble towards coaxial conformations that are stabilized by a base triple55 (FIG. 2a). This conformational change facilitates proper assembly of an RNP complex that activates transcription elongation of viral genes. In addition, divalent metal ions such as magnesium (Mg2+) skew the TAR ensemble towards coaxial conformations54 that lack the base triple by neutralizing charge repulsion in and around the bulge (FIG. 2a). As we discuss below, other cellular factors can also redistribute the RNA ensembles; when not properly accounted for, these factors can cause differences in RNA ensembles measured in vitro versus in vivo.
Mutations, which are widely used to study RNA and also occur naturally, are another important mechanism of redistributing RNA ensembles. A single point mutation can be sufficient to stabilize a non-native TAR secondary structure that differs significantly from the major conformation sampled in the wild-type sequence, thereby changing its population from ~0.1% to >99%52,56 (FIG. 2a). Likewise, increasing or decreasing the length of the TAR bulge can broaden or narrow the range of interhelical dynamics sampled by the dominant native state57. Mutation-induced changes in the RNA ensemble provide an avenue for studying the importance of structural dynamics in vivo, and are increasingly being linked to disease (BOX 1).
Predictive value of ensembles
The dynamic ensemble description of RNA is important not just because it reflects the true structural nature of the molecule, but also because it enables describing and predicting key steps in RNA-mediated processes that involve conformational change that cannot be accurately modelled based on static RNA structures (FIG. 2b).
Energy is required to redistribute an RNA ensemble (Supplementary Box 2). This energy cost of redistribution (Δ_G_0redist)58 has to be paid for by the formation of favourable intramolecular contacts, such as tertiary interactions during folding or intermolecular interactions when an RNA binds a partner molecule. The energy cost will be large for large changes in the ensemble and will be zero when the ensemble does not change. For example, if binding a molecule leads to stabilization of a single RNA conformation, the population of that conformation in the free (unbound) ensemble will dictate the free energy cost (FIG. 2b and Supplementary Box 2). As a result, knowledge of Δ_G_0redist in addition to the energetics of binding (Δ_G_0bind) is ultimately required to determine the overall strength of any interaction, be it the binding affinity of a protein or ligand to its target RNA or the stability of a folded conformation (FIG. 2b and Supplementary Box 2). Importantly, whereas the energy cost cannot be inferred from static RNA structures, it can be determined from the original and redistributed ensembles (Supplementary Box 2).
The strength of the response to a given cellular modifier will often depend on the relative abundance of ‘active’ versus ‘inactive’ RNA conformations and/or on their kinetic rates of interconversion in the redistributed ensemble (FIG. 2b). Again, static structures do not provide this information. Finally, as we discuss below, static structures may not fully capture evolutionary conservation patterns that maintain the stabilities of multiple RNA conformations in the ensemble (FIG. 2b). For example, co-variation could exist between nucleotides that do not form base pairs in the energetically most favourable, native secondary structure because they form base pairs in alternative, higher-energy, non-native conformations that are functionally important19. This can be important when functionally annotating the transcriptome or when trying to understand the deleterious consequences of RNA mutations.
Organizing principles of RNA ensembles
Dynamic ensembles of biomolecules contain hundreds of thousands of conformations that interconvert on timescales spanning 12 orders of magnitude. Organizing principles are emerging that simplify the description and determination of RNA dynamic ensembles, while also providing a unified framework for understanding their regulatory functions and their dependence on sequence versus secondary structure. These principles also have important implications for the evolvability of RNA (FIG. 3).
Fig. 3 |. Organizing principles of RNA ensembles.

RNAs are composed of modular structural motifs with context-independent conformational preferences (ensemble modularity). Examples of RNA motifs are shown (ensemble modularity). The structural dynamics of each motif can be decomposed into a set of independent and reoccurring motional modes, which occur at different timescales and have different dependencies on sequence versus secondary structure and topology. The different modes represent transitions between conformations on different tiers in a hierarchically organized free energy landscape. Shown is an example RNA hairpin under ‘hierarchical landscapes’. In tier 0, formation and loss of tertiary interactions involving the tetraloop occurs on the slow millisecond (ms)–hour (h) timescales. In tier 1, the hairpin transitions between structures with alternative base pairing on the microsecond (μs)-ms timescale. Finally, in tier 2, the hairpin undergoes faster, inter-helical dynamics and local motions of the bases and sugars at picosecond (ps)-nanosecond (ns) timescales. Mutations (indicated with a red star) can affect different motional modes within individual motifs while minimally disrupting other motional modes or other motifs and therefore the core functionality of the RNA, thereby making RNA a highly evolvable molecule. The A-form helix represents the canonical RNA state, in which two strands of RNA are connected by Watson-Crick base pairing. A kink-turn (k-turn) is a special type of bulge that introduces a very tight kink into the backbone of the RNA. This comprises a 3-nucleotide bulge flanked by A–G and G–A base pairs.
Structural motifs and their modularity
One of the major challenges in advancing our understanding of the connections between the RNA ensemble and RNA function is that determining ensembles by experimental or computational methods is very challenging and time-consuming even for simple RNAs such as TAR (Supplementary Box 1). Recently, an RNA reconstitution model was proposed that may facilitate the reconstitution of ensembles by interrogating their component motifs59. RNA structures consist of a limited number of secondary and tertiary motif ‘building blocks’, which generally autonomously fold into similar structures in different RNAs60. For example, building blocks for tertiary structure such as tetraloops and their receptors fold into similar structures independently of the sequence or structural context outside the motifs. This principle of RNA structure modularity has been extended to thermodynamics through the concept of ensemble modularity33,59. Ensemble modularity posits that the free energy landscape of a given RNA motif is independent of the context outside the motif and that its conformational ensemble is dictated by its internal properties, which are influenced by additional geometric constraints and preferences from the remainder of the molecule (FIG. 3). In other words, the intrinsic energetic preference for or probability of forming a given conformation does not change with context even though additional intermolecular interactions could exist that redistribute the ensemble. In the RNA reconstitution model33, the free energy landscapes of constituent motifs are added to one another61 to reconstitute the thermodynamic ensemble of an RNA assembly59. This suggests that insights into the dynamic behaviour of RNA motifs in complex structural contexts such as RNPs or lncRNAs can be obtained from studies of isolated motifs. The reconstitution model also reduces the number of motifs whose dynamics need to be characterized because one ensemble can be used to model a motif in different contexts33 (FIG. 3). Thus, in the future, it may be feasible to reconstitute ensembles for a large variety of RNAs from an ‘atlas’ of motif ensembles59. As discussed below, recent advances in high-throughput technologies may provide an avenue for compiling such an atlas.
The timescales of motions
The decomposition of 3D structures into structural motifs has greatly aided structure-function studies. Analogously, decomposition of ensembles into motions that occur on different timescales may help elucidate ensemble-organizing principles that would otherwise be buried within the complexity of structural dynamics. A classification according to timescale is appealing not only because the kinetic rates of conformational change can be important determinants of resulting activity, but also because motions on different timescales often have distinct dependencies on sequence versus secondary structure. Different spectroscopic methods used to characterize RNA dynamics also tend to be sensitive to different timescale ranges15 (Supplementary Box 1).
Free energy landscapes of proteins are hierarchically organized into different ‘tiers’, which feature an increasing number of conformations that interconvert on faster timescales24. The free energy landscape of RNA also appears to be hierarchically organized15. The motions that have been characterized to date using various methods (Supplementary Box 1) can be classified into three tiers, which represent three different timescale ranges.
At the top tier, tier 0, a small number of conformations differ substantially in terms of the presence or absence of secondary or tertiary structures. Interconversion between these conformations requires the breaking of several base pairs and, consequently, they occur on slow timescales ranging from milliseconds to several hours (FIG. 3). Transitions between tier 0 conformations can sequester secondary structures from or expose them to the cellular machinery, make or break tertiary contacts that are required for tertiary folding into functional structures, or anneal or melt intramolecular or intermolecular duplexes43. Protein chaperones50 are often required to accelerate tier 0 dynamics to biological timescales (that is, faster than milliseconds)62.
Each tier 0 conformation can be subdivided into tier 1 conformations that feature more subtle differences in base pairing and secondary structure in and around non-canonical motifs52,63–65. Interconversion between these conformations typically requires the breaking of a single base pair and usually occurs on the faster timescale of microseconds to milliseconds, without assistance from protein chaperones52,63–65 (FIG. 3). Relative to tier 0, many more iso-energetic conformations are likely to be found in tier 1 because they feature finer variations around the parent tier 0 conformation. Tier 1 dynamics can function as fast switches63,66 or help break down slow tier 0 conformational transitions into multiple kinetically labile steps67,68.
Each tier 1 conformation can be subdivided into an even larger number of tier 2 conformational states, which interconvert on the picosecond to nanosecond timescale and include more continuous variations in sugar pucker, base orientation, backbone angles and the global orientation and translation of helices53 (FIG. 3). Tier 2 dynamics enable RNA structures to be readily moulded into specific conformations, for example those that optimize interactions with proteins, ligands, DNA and other RNAs69–71.
Evolutionary conservation
Dynamics that involve changes in base pairing (tiers 0 and 1) are strongly dependent on sequence. Functionally important base-pair dynamics have been inferred in riboswitches based on evolutionary co-variations of sequence that support multiple RNA secondary structures18,19. Conservation at the level of the dynamic ensemble could also explain the lack of co-variation in the UTRs of various mRNAs despite a high degree of evolutionary sequence conservation18 (FIG. 2b).
In contrast to tiers 0 and 1, the faster tier 2 motions depend more on the secondary structure of RNA than on its sequence59,72. For example, global motions of helical domains depend more on the length and asymmetry (or topology) of inter-helical junctions than on the sequence of the junctions59,72. The existence of functionally important tier 2 dynamics therefore implies that there are selective pressures to conserve RNA secondary structures independently of sequence, as indeed is the case for some tRNAs73. The topology of different junctions can also co-vary to ensure proper RNA tertiary assembly, and this can in principle result in co-variation of junction topology59. The topological requirements for certain RNA activities, such as the high cleavage efficiencies of certain microRNAs74, may in part reflect the fine-tuning of such dynamics, which optimize interactions with processing proteins.
In the future, it may be possible to infer functionally important dynamics at different tiers based on the conservations of sequence versus secondary structure and topology. For example, the conservation of interhelical junctions in lncRNAs such as COOLAIR may be owing to inter-helical motions that help lncRNAs with scaffolding functions to spatially organize protein, DNA and other RNA molecules that bind them37–39 (FIG. 1g).
Together, ensemble modularity and the hierarchical nature of RNA free energy landscapes make RNA an evolutionary highly malleable substrate. Mutations within individual motifs that affect distinct dynamics tiers can be tested and selected for while minimally disrupting other motional modes or motifs and, thus, the core functionality of the RNA (FIG. 3).
Ensemble predictions in vitro
We now examine how the description of RNA as ensembles is being put into practice and improves the modelling and prediction of biochemical properties such as the binding affinity of small molecules, the stability of tertiary assembly, the robustness of RNA regulatory functions and the fidelity (accuracy) of RNA-dependent processes. In this section we focus on studies performed in vitro, and in subsequent sections we discuss extensions of the ensemble description to predict aspects of RNA folding and activity in vivo. Because these studies are in their infancy, we focus on examples that employ experimentally verified RNA ensembles, which are still generally more reliable than computationally predicted ensembles25,75,76.
Prediction of ligand binding
A growing number of RNAs have been linked to diseases, which has spurred great interest in developing small-molecule inhibitors that target the 3D structure of RNA rather than conventionally targeting the RNA sequence3,4. Changes in RNA structure upon small-molecule binding present significant challenges to structure-based drug discovery using, for example, computational docking77, but they also present opportunities, as stabilizing inactive RNA conformations with small molecules is a proven concept and the functional mechanism of many antibiotics that target the ribosome21. For example, the binding affinity of 38 small-molecule TAR binders was calculated with reasonable accuracy by computationally docking small molecules to an NMR-derived dynamic ensemble of TAR containing 20 conformations (FIG. 4a). The predictions deteriorated considerably when docking to static NMR or X-ray structures. This ensemble-based virtual screen identified a compound that binds TAR with high selectivity and that inhibits HIV replication in a cell line with half-maximal inhibitory concentration of ≈23 μM (REF78). In a retrospective study, ensemble-based docking was able to distinguish 26 hits from 102,307 non-hits79. By contrast, when docking to static structures or lower-quality ensembles, the predictions were of lower quality and exhibited large variability (FIG. 4a). The computational-docking prediction of TAR ensemble redistribution following small-molecule binding was in reasonable agreement with experimentally determined structures of TAR-small-molecule complexes79. Further improvements in virtual screening may enable the rational identification of compounds that specifically bind and stabilize inactive conformations in the RNA ensemble.
Fig. 4 |. Ensemble-based modelling of RNA activity in vitro.

a | The RNA-binding affinity of small molecules can be calculated computationally by virtually docking small molecules to dynamic RNA ensembles. The affinity predictions deteriorate considerably when static structures are used for the screening78,79. b | The tectoRNA host-guest system is a heterodimer of two structured RNAs (the host and the guest) connected by two tetraloop–tetraloop receptor tertiary contacts. The energetics of tertiary assembly of the tectoRNA host–guest system can vary considerably depending on the relative alignment of the tertiary receptors, which is dictated by helix-junction-helix (HJH) motifs such as mismatches, bulges and internal loops. The energetics of tertiary assembly is much better modelled by considering the guest HJH motifs as dynamic ensembles compared with static structures. c | G·T and G·U mismatches form wobble conformations that differ from the Watson-Crick geometry. On this basis, polymerases and ribosomes can discriminate against mispairing and reduce the error frequency during DNA replication, transcription and translation. However, the ensembles of G·T and G·U mismatches also include low-abundance, short-lived Watson-Crick-like conformations that are stabilized by rare tautomeric (blue) and anionic (green) bases. The population and lifetime of these rare species were integrated into a kinetic model, which could predict the probability of dGdT misincorporation over a wide range of conditions and for modified mutagenic bases63,64. Part c is adapted from REF63, Springer Nature Limited.
Predicting tertiary assembly
Modelling tertiary interactions requires consideration of the nucleotides involved in the tertiary contact and the relative alignment of the tertiary receptors, which is often dictated by helix-junction-helix (HJH) motifs such as bulges, internal loops and higher-order junctions that adjoin RNA helical stems72,80. The tectoRNA host-guest system is a tertiary-interactions modelling system, which includes a heterodimer composed of two structured RNAs connected by two intermolecular tetraloop–tetraloop receptor tertiary contacts (FIG. 4b). The ‘guest’ RNA contains variable HJH motifs that alter their alignment to enable the formation of tertiary contacts with different energetic penalties for tertiary assembly. Ensemble models of HJH motifs were successfully used to predict the energetics of tertiary assembly for thousands of tectoRNA variants encompassing different HJH motifs, measured with the quantitative high-throughput method RNA-MaP59 (Supplementary Box 1). A dynamic ensemble description of RNA helices and HJH motifs was more predictive of tertiary assembly energetics compared with single structures59,81 (FIG. 4b).
By providing ensemble information in a high-throughput manner (Supplementary Box 1), RNA-MaP brings within reach the ability to determine experimentally verified ensembles of a vast number of RNA motifs. As stated above, such an atlas can be used with a reconstitution model to streamline and accelerate the determination of ensembles for more complex RNAs and to provide rich data that can guide the development of next-generation nucleic acid force fields for computer modelling75,76.
Robustness of RNA regulatory functions
Ensembles are helping to model the robustness of RNA-based regulation. For example, a three-state ensemble description of the adenine-sensing translation riboswitch from the pathogenic bacterium Vibrio vulnificus helped explain how it maintains robust switching efficiency over a wide range of biologically relevant temperatures82. The riboswitch is present in the 5′ UTR of the add gene and activates translation through a conformational change that is stabilized by binding to adenine. NMR studies found that, before ligand binding, the apo (ligand-free) ensemble of this riboswitch consists of two major conformations, only one of which is binding competent. The relative populations of these conformations are sensitive to temperature such that it counteracts the temperature-dependent ligand-binding affinity, thereby leading to robust switching of the riboswitch and translation regulation82. Specifically, at low temperature, the population of the binding-competent conformation is small, which counteracts high-affinity binding, and vice versa at high temperatures. Computational simulations showed how the observed temperature-dependent pre-equilibrium could maintain more robust switching efficiency than a simple single-state binding model.
Predicting fidelity
The mechanisms that ensure high fidelity of DNA replication, transcription and translation highlight the biological importance of very low-abundance conformations. NMR30,31 and X-ray41,42 studies show that G-T and G-U wobble mismatches in nucleic acid duplexes exist in vitro as ensembles, which include Watson-Crick-like conformations that form through tautomerization or ionization of the bases (FIG. 4c). By mimicking the Watson-Crick geometry, these low abundance and short-lived Watson-Crick-like conformations (populations as small as 0.001% and lifetimes as short as microseconds), which feature sub-angstrom differences in structure relative to the dominant wobble conformation, are proposed to evade fidelity checkpoints and contribute to the introduction of errors during replication, transcription and translation. An ensemble-based kinetic model (FIG. 4c) could predict the frequency of G-T misincorporation during replication across a wide range of pH conditions and types of polymerases, as well as the misincorporation frequencies of modified mutagenic bases63. Mutagenic modifications can bias the ensemble of DNA duplexes towards the tautomeric or anionic Watson–Crick-like conformations63. Likewise, the tRNA modification uridine 5-oxyacetic acid (cmo5U), which is found at the tRNA wobble position, redistributes the G–cmo5U ensemble in the minihelix formed by pairing the mRNA codon and tRNA anticodon towards anionic Watson–Crick-like conformations, thereby expanding the translational decoding capacity to include this G–cmo5U mismatch83.
Effects of the cellular environment
It is currently not feasible to determine high-resolution ensembles for RNA within the complex cellular environment. Therefore, to extend the ensemble description to cells, we need to determine what aspects unique to the cellular environment affect the RNA ensemble and/or its redistribution, and to devise ways to describe and reconstitute these contributions in vitro (FIG. 5).
Fig. 5 |. The effect of cellular environments on RNA behaviour.

a | In the unbound, ground state of the fluoride riboswitch, an unstable RNA linchpin connects the upper and middle riboswitch helices. (Bottom) If fluoride is present, it binds and stabilizes the riboswitch, resulting in transcription. (Top) If fluoride is not present, the riboswitch can enter an unbound excited state, in which the unstable linchpin breaks. Linchpin breakage allows the invasion of the mRNA part of the RNA molecule and causes the riboswitch to refold and form a terminator helix, which terminates transcription. b | As mRNA is being transcribed by an RNA polymerase (RNAP), its free energy landscape changes. The elongating transcript may fold into an initial ensemble, which redistributes following the synthesis of additional nucleotides. c | The RNA post-transcriptional modification _N_6-methyladenosine (m6A) reduces the energetic cost (Δ_G_0redist) of conformational changes, thereby enabling the opening of the RNA duplex structure and promoting the binding of heterogeneous nuclear ribonucleoprotein C (HNRNPC) to mRNAs at single-stranded RNA regions124. Apo, ligand free; F−, fluoride ion. Part a is adapted from REF 66, Springer Nature Limited.
Cellular constituents
The cellular environment is heterogeneous and consists of many microenvironments that differ in pH, molecule crowding and composition of metals and co-solutes such as osmolytes; these constituents can also vary between cell types and growth conditions. In vitro studies have examined how these different constituents affect the RNA ensemble and its redistribution, and have provided guidelines for optimizing in vitro conditions to mimic the in vivo environment.
Metal ions.
Every RNA is surrounded by an ion atmosphere, which profoundly affects its folding and interactions84–86. In particular, divalent metals such as Mg2+ are important for the proper folding and function of RNAs87–89. The nature of the ion atmosphere and how it affects RNA behaviour may differ in cells compared with typical in vitro conditions. For example, in cells, the free Mg2+ concentration is ~1 mM, but additional chelated Mg2+ ions exist that raise the total concentration to ~50 mM. Using amino-acid chelated Mg2+ to model cellular chelated Mg2+, a recent study found that weakly chelated Mg2+ biases the ensemble towards compactly folded conformations to a similar degree as free Mg2+ does, thus decreasing RNA degradation and increasing ribozyme activity90.
Molecular crowding.
Cells contain 20–40% (w/v) macromolecules that can exclude volume, interact with RNA and alter the solution properties. The effect of molecular crowding on RNA ensembles has been empirically examined using reagents that mimic different aspects of crowding in vitro (reviewed in REF91). Crowding agents with high molecular weight have been suggested to bias RNA ensembles towards tertiary folding through simple excluded-volume effects92,93, and this simple excluded-volume effect has been proposed to explain the enhanced catalytic activities of ribozymes94–96 and enhanced binding affinities of ligands to riboswitches in crowded conditions97. Molecular crowding can stabilize tertiary folding of the Azoarcus group I ribozyme and compensate for loss of activity owing to destabilizing mutations, which possibly explains differences in the activity of these mutants when measured in vitro versus in vivo98. The cellular environment can therefore alter the energy cost of redistribution relative to in vitro conditions, where crowding is not taken into consideration.
Cellular solutes.
Small cellular co-solutes such as osmolytes, including methylamines, amino acids, sugars and alcohols, appear to have different effects on the RNA ensemble compared to crowding agents with high molecular weight, and the effects can vary significantly in different Mg2+ concentrations. Nine osmolytes were found to destabilize the RNA secondary structure to varying extents and to either stabilize or destabilize the RNA tertiary structure99,100. The stabilization of the tertiary structure was attenuated or even reversed in the presence of high Mg2+ concentration99,100. Osmolytes appear to have such complex effects because they form favourable interactions with exposed nucleobases and also form unfavourable interactions with the exposed ribose-phosphate backbone99. Other small cellular solutes such as metabolites have been found within RNA ligand-binding sites and shown to decrease the apparent binding affinity to cognate ligands101. This is an important example of how the cellular environment can affect RNA activity through mechanisms that do not necessarily involve ensemble redistribution.
Liquid-liquid phase separation
An important cellular phenomenon is the organization of specific RNAs, proteins and other molecules into phase-separated granules102,103. These biological condensates are thought to regulate gene expression by compartmentalizing and concentrating specific molecules and machineries to change reaction specificities or kinetics, or by direct subcellular relocalization102,104,105. RNA-containing biological condensates, or RNA granules, which include, among others, processing bodies, stress granules, nucleoli, paraspeckles and germ granules, form through complex multivalent RNA-RNA interactions and RNA interactions with proteins, often through intrinsically disordered regions of proteins105,106.
We expect RNA ensembles to help determine the strength and dynamics of RNA-RNA and RNA-protein multivalent interactions, and, therefore, whether a given RNA undergoes liquid-liquid phase separation. Dynamic RNA-protein interactions are also proposed to contribute to the properties of the droplets, such as their fluidity41,42. Indeed, studies show that the identity of droplet populations is at least in part defined by the RNA secondary structure, as two functionally disparate mRNAs that bind the same protein but form different secondary structures were shown to form distinct droplets that do not mix in vitro and in vivo107. By contrast, RNAs that form non-specific interactions can prevent phase separation in vitro and in vivo108.
In turn, the biological condensates create unique environments that may affect the RNA ensemble and its redistribution energies. For example, the activity of the hammerhead ribozyme increased 70-fold in phase-separated droplets formed by crowding agents of high molecular weight in vitro, likely due to stabilization of the folded RNA conformation40. Furthermore, conditions that favour droplet formation can increase the dynamics of protein-bound RNA, measured using single-molecule fluorescence microscopy41,42.
Examining how RNA ensembles influence the formation of granules and in turn are affected by them will benefit from reconstituting in vitro at least certain aspects of RNA granules107–109. The formation in vitro of RNA foci that are common in many neurological diseases was shown to be inhibited by the same RNA intercalating molecule that inhibits the formation of these foci in cells and patient primary tissues109. Although having the ability to reconstitute droplets in vitro is an important step towards their in-depth characterization, new techniques will likely have to be developed to shed light on the behaviour of RNA and its ensemble within these environments.
Co-transcriptional folding
Within cells, many RNAs fold as they are being synthesized in a process termed ‘co-transcriptional folding’. Many regulatory processes that are coupled to co-transcriptional folding require descriptions of ensembles and free energy landscapes, which can change over time as the transcript is elongated. The time available to redistribute the ensemble during co-transcriptional folding is crucial for predicting the structural landscape and therefore the functional output of such processes.
Riboswitches are highly structured regulatory RNA elements typically found in 5′ UTRs, which regulate gene expression in response to cellular modifiers such as metabolites8,10. Riboswitch folding illustrates the importance of co-transcriptional folding to RNA function. Many riboswitches are activated in vivo at metabolite concentrations that exceed the ligand-binding affinities measured in vitro for pre-folded RNAs. For example, the thiamine pyrophosphate binding riboswitch binds thiamine pyrophosphate in vitro with an apparent dissociation constant (_K_d) of 50 nM (REF8), whereas the thiamine pyrophosphate concentration that causes the riboswitch to terminate transcription is in the micromolar range28. For riboswitches that regulate gene expression at the transcriptional level, co-transcriptional mRNA folding defines a time window during which ligand binding can redistribute the riboswitch ensemble, leading to the formation of conformations that either promote or terminate transcription of the rest of the mRNA. Because ligand-binding kinetics can be slow relative to this time window110, binding does not always reach thermodynamic equilibrium and riboswitch activation requires ligand concentrations that exceed the dissociation constant111.
Another striking difference between riboswitch activity in vitro and in vivo is that although ligands activate riboswitches during co-transcriptional mRNA folding, at equilibrium in physiological concentrations of Mg2+, the riboswitches often fold into identical conformations in the presence or absence of ligand66,112–114. How, then, do these riboswitches switch between the active and inactive conformations? Studies increasingly indicate that metastable conformations in the ensemble that form during co-transcriptional folding are stabilized or destabilized in a ligand-dependent manner and thus bias riboswitch folding towards active or inactive conformations66,112–115. For example, the sensing aptamer domain of the Bacillus cereus fluoride riboswitch folds into nearly identical structures in the presence or absence of its cognate ligand fluoride66. However, NMR studies show that the dynamics of the ligand-free (apo) and ligand-bound ensembles differ: the apo ensemble uniquely includes a low-abundance (population of ~1%) and short-lived (lifetime of ~3 ms) conformational state, which is proposed to increase the vulnerability of the riboswitch to mRNA-strand invasion, thereby biasing riboswitch folding towards supporting transcription termination66 (FIG. 5a). Ligand binding decreases the population of this conformational state, thereby protecting the riboswitch from strand invasion and redirecting folding towards supporting transcription. A kinetic mechanism that integrates such a two-state ensemble predicted the dependence of riboswitch activity on transcription rates66.
Unique to co-transcriptional folding are the changes to the free energy landscape and RNA ensembles that can occur during transcription elongation, as new sequence elements are transcribed (FIG. 5b). The time available to redistribute the ensemble will vary depending on the rate of transcription and the absence or presence of transcription pausing sites111,116. Insights into the changing RNA ensemble can be obtained from studying the ensemble behaviour of variable-length transcripts. For example, for both a 2′-deoxyguanosine-sensing transcription riboswitch from Mesoplasma florum115 and a guanine-sensing riboswitch from Bacillus subtilis112, the relevant active conformations are favoured for a particular range of nascent-mRNA length in the absence of ligand; when the length of the nascent transcript exceeds the threshold, the riboswitch folds into the alternative, inactive conformation. During co-transcriptional mRNA folding, these length-dependent metastable active conformations are sufficiently long-lived to avoid transcription termination. Ligand binding prevents the formation of these metastable states and stabilizes the inactive conformations (FIG. 5b). Because the conformational changes and ligand binding are all under kinetic control, transcription rates and pause sites are crucial for riboswitch regulation112,113. Other methods for reconstituting co-transcriptional folding now provide a basis for further exploring this fascinating and highly complex process114,117–119.
Post-transcriptional modifications
Many RNAs in cells, including mRNAs, microRNAs and lncRNAs, are post-transcriptionally modified through different chemical modifications120. Post-transcriptional modifications such as _N_6-methyladenosine (m6A) can affect the cellular function of RNA by recruiting specific m6A-reader proteins or by blocking key mRNA interactions during translation, and there is also evidence that they can modulate RNA activity by redistributing RNA ensembles121–126. For example, by destabilizing RNA helices, m6A can increase the binding affinity of an RBP to its RNA target127,128 (FIG. 5c). Conversely, m6A can strongly inhibit RNA-protein interactions by biasing the ensemble of a non-canonical A-G mismatch of some box C/D small-nucleolar RNAs away from the conformation required for their proper folding129. By contrast, _N_1-methyladenosine facilitates proper folding of certain tRNAs130 by potently destabilizing base pairs in non-native conformations130; similar mechanisms could explain how _N_1-methyladenosine promotes mRNA translation131.
Understanding RNA activity in vivo
Although we are still far from determining RNA ensembles in vivo and using them to better understand and predict cellular RNA activity, transcriptome-wide studies of RNA structure and of RNA-protein binding are helping us to understand RNA behaviour in vivo. Different perspectives are emerging from these studies that appear to depend on the method used or the system studied, thereby highlighting important challenges that need to be addressed before we can describe RNA ensembles in vivo.
Transcriptome ensembles
Transcriptome-wide structure probing experiments, which measure the reactivity of nucleotides to various chemical reagents, are beginning to illuminate how the cellular environment affects RNA folding. The reactivity of nucleotides provides low-resolution information on RNA structure, for example on whether a nucleotide is paired or not. Because the reactivity of nucleotides is averaged over all RNA conformations present during the reaction time, they carry information regarding the RNA ensemble within cells, although many studies interpret the data assuming a single RNA conformation.
Early structure-probing studies indicated that the reactivity of cellular RNAs, particularly mRNAs, to chemical probes is higher in cells compared with in vitro experiments, which typically remove any bound proteins and also include denaturing and refolding, indicating that mRNAs are less structured in vivo than in vitro125,132,133. Recent improvements in structure probing, which focus on modelling RNA structures of individual transcripts, suggest that the structures of mRNAs in vivo are transiently destabilized by active translation134,135. Other than translation-dependent unfolding, mRNA reactivity was found to be generally similar in vitro and in vivo, with lowly translated mRNAs being minimally perturbed, suggesting that RNA is similarly structured in vivo as it is in vitro134. Another recent study identified some differences in RNA structure in vitro and in vivo at different cellular compartments, which were linked to cellular processes including RBP binding, transcription, translation and RNA decay136.
Similarly to studying the occurrence of structural dynamics on different timescales, structure-probing data can also provide different views of the RNA ensemble, which depend on the experimental technique used to produce them. For example, dimethyl sulfate profiling of mammalian RNA found that although thousands of RNA G-quadruplex structures (RG4) in G-rich sequences form stably in vitro, they are almost entirely unfolded in vivo137. Cellular factors such as helicases and RBPs were proposed to either destabilize or to tightly regulate the formation of RG4 (REF137). Although these data strongly suggest that in vivo the RG4 ensemble is very biased towards unfolded conformations, they do not preclude the possibility that RG4 form transiently and to a degree that is undetectable by dimethyl sulfate profiling. Indeed, a recent study using a crosslinking technique to obtain global snapshots of RNA structure suggests that RG4 form transiently138. Considering that there are many examples of rare but important RNA conformational states, transient RG4 could indeed have important biological roles.
Interpreting chemical probing data in terms of dynamic ensembles is challenging. In addition to low sensitivity to rare transient structures, the quantity and complexity of the data are low relative to the number of parameters needed to define an ensemble, and the nature of the dependence of chemical reactivity on structure is not entirely understood139. Nevertheless, methodologies are being developed to interpret chemical probing data in terms of secondary structure ensembles139–142, using strategies similar to those developed to generate NMR 3D ensembles143 (Supplementary Box 1). This in turn provides insights into how the cellular environment redistributes ensembles compared with the ensembles determined in vitro. Chemical probing data were used to compare the ensemble of the human ACTB mRNA (encoding β-actin) in vitro and in cells144. Based on differences in SHAPE (selective 1′-hydroxyl acylation analysed by primer extension) reactivities144,145, a region that harbours a protein-binding site was proposed to form distinct ensembles in vitro and in vivo. In the in vivo ensemble, the dominant structure exposes the protein-binding site, whereas it is more occluded in the structure dominating the in vitro ensemble. Interestingly, the dominant in vitro structure was a minor population in the in vivo ensemble, suggesting that the same underlying ensemble is redistributed under cellular conditions (FIG. 6a).
Fig. 6 |. RNA ensembles in cells.

a | Comparison of in vitro and in vivo ensembles described using selective 1′-hydroxyl acylation analysed by primer extension (SHAPE) data for a region of the human ACTB mRNA that contains two binding sites for zipcode binding protein 1 (ZBP1; also known as Insulin-like growth factor 2 mRNA-binding protein 1). Circle areas are proportional to the population of the conformation they represent. The data demonstrate a redistribution of the ensemble away from the dominant structure in vitro (green), in which a ZBP1 binding site is occluded towards another structure in vivo (purple), in which the ZBP1 binding site is exposed. b | RNA secondary structures can determine whether or not a recognition motif for an RNA-binding protein will be bound and active in vivo. For example, the splicing factor RNA binding fox-1 homologue 2 (RBFOX2) enhances splicing by preferentially binding its target RNAs in less structured regions. c | A new method identifies compounds that bind their target RNAs specifically within the cellular context by self-assembling into multivalent compounds using click chemistry148. This has been applied to inhibit muscleblind-like 1 protein (MBNL1) binding to expanded CCUG repeats in the myotonic dystrophy type 2 mRNA. When the compound self-assembles along the repeats, the molecules link to form a single polymer (yellow lines zig-zag to indicate covalent binding). When this occurs, MBNL1 is not sequestered by the repeats and functions normally, thereby eliminating disease symptoms. Part a is adapted with permission from REF144, Elsevier.
Methods such as ‘mutate and map’65,146 (Supplementary Box 1), which increase the information content of chemical probing data by examining how point mutations affect reactivity, hold great promise in determining transcriptome-wide RNA ensembles in vivo, although technological advances will be required to maximize their potential.
RNA-binding molecules
The complex cellular environment could in principle affect the RNA-binding preferences of RBPs compared with the in vitro environment; inversely, the binding of RBPs in vivo could alter RNA behaviour compared with the behaviour in vitro. Many RBPs bind specific RNA motifs with high affinity in vitro. By contrast, some crosslinking experiments in vivo indicate that most RNA motifs are not bound by their respective RBPs, raising the question of why the sites are differently bound in cells147. One possible reason for this different binding of sites is differences in the formation of RNA secondary structures around the motif, which can occlude binding sites32 (FIG. 6b). Better agreement between in vitro and in vivo binding was obtained when longer RNA transcripts of ~100 nucleotides were used in vitro, which allow for the formation of local secondary structures. These results suggest that similar ensemble-redistribution energetic penalties are required to access the binding motif in vitro and in vivo when the relevant structural context is included and that RNA structure is the major determinant of protein binding as opposed to other possible factors such as cellular localization or competitive binding32.
By contrast, a study comparing the RNA-binding preferences of the human RBPs Pumilio homologue 1 and Pumilio homologue 2 in vitro with their transcriptome binding preferences in vivo found that RNA secondary structures can limit the accessibility of binding sites in vitro but have negligible effects in vivo148. Thus, the cellular environment — possibly the binding of RBPs or helicases — seems to bias the RNA ensemble towards the unfolded conformation. Using data for binding of these proteins to thousands of RNAs, a thermodynamic model was developed that could accurately predict transcriptome-wide in vivo crosslinking data148.
The cellular environment can also affect the binding of in vitro-identified therapeutic small molecules to their intended RNA targets4. A new method selects for compounds that bind their target RNAs specifically within the cellular context using self-assembling small-molecule inhibitors that bind to sequence repeats in an RNA target and are assembled into multivalent compounds using click chemistry149 (FIG. 6c). Performing this reaction in vitro and in vivo on RNA CCUG repeats, which cause myotonic dystrophy type 2, resulted in the same strong binding of a multivalent compound to the targeted repeat. Thus, in this case, the RNA structure is likely unchanged between the cellular environment and in vitro conditions, and retains the ability to bind and assemble an inhibitor even though the repeats are much longer in vivo than those tested in vitro. This further demonstrates ensemble modularity, because increasing the number of repeat motifs in a single RNA does not change the binding properties of the individual motifs.
Conclusion and future perspectives
The description of RNA in terms of dynamic ensembles is changing our understanding of RNA-regulated processes in vivo. Determining ensembles remains far more difficult than solving structures, and doing so in vivo is a daunting challenge. Meeting this challenge will require an integrated approach through close collaborations between experimentalists and computational scientists to facilitate the continued development of force fields75,76 for RNA ensemble modelling in the cellular environment to a level that allows predictions to be made and tested experimentally. We propose four principles of ensemble redistribution to help guide these future efforts.
- Principle #1. Cellular factors redistribute the ensemble of full-length RNAs by changing the relative populations of pre-existing conformations, which correspond to local energetic minima along the free energy landscape. Cellular factors do not reshape the free energy landscape by creating new energetic minima. This limits the ways with which cellular factors can act on RNA to alter its behaviour within cells.
- Principle #2. Although the unique cellular environment can redistribute RNA ensembles compared to in vitro conditions, the physiological ensemble can be reconstituted in vitro using proper RNA samples (length, absence or presence of modifications, and so forth) and including relevant cellular components within the buffer. Reconstituting the cellular environment in vitro provides a means to test for and determine physiologically relevant ensembles at high resolution, which complements approaches that determine RNA ensembles using lower-resolution measurements performed in vivo.
- Principle #3. RNAs that function during co-transcriptional mRNA folding require a time-dependent ensemble description, in which new transient conformations can be created that carry out unique functions. Such conformations may not be observed in studies of the full-length RNA ensemble and can only be characterized by analysing the evolution of the ensemble during co-transcriptional folding or through studies of RNA transcripts of variable lengths.
- Principle #4. Conservation of RNA functional dynamics can result in unique evolutionary conservation patterns. Thus, searching for evolutionary conservation of RNA ensembles, in addition to structure, may aid the functional annotation of non-coding RNAs and help to discriminate functional RNAs from transcriptional noise.
As applications of RNA ensembles in drug discovery and synthetic biology already appear on the horizon, there is little doubt that higher-level understanding of RNA ensembles will lead to other, unexpected discoveries.
Supplementary Material
Supplementary Box 1
Supplementary Box 2
Acknowledgements
The authors thank A. Mustoe, B. Liu, A. K. Rangadurai, N. Orlovsky and other members of the Al-Hashimi laboratory for critical input and help in making figures, and R. Das (Stanford University, CA, USA), P. Z. Qin (USC), Q. Zhang (UNC Chapel Hill, NC, USA), A. Bartesaghi (Duke University), S. Bonilla (Stanford University) and T. Oas (Duke University) for critical input. This work was supported by the US National Institutes of Health (P50 GM103297 and P01 GM0066275 to H.M.A. and D.H..; F31 GM119306 to L.R.G.).
Glossary
Secondary structures
RNA structures described in terms of nucleotide pairing
Ribozymes
RNA structures capable of catalysing specific biochemical reactions such as cleaving the RNA phosphodiester backbone
Riboswitches
RNA structures typically found in 5′-untranslated regions of bacterial mRNAs, which regulate transcription or translation through a ligand-induced conformational change
Tertiary structures
Typically long-range interactions between distal RNA structural elements or nucleotides involved in base pairing
Quaternary assemblies
Higher-order organizations of RNA molecules in complex with other molecules, including with other RNAs, proteins and DNA
Boltzmann distribution
A probability distribution that describes the likelihood that a system will be in a specific state based on the relative energy of that state and the temperature of the system
Ground state
The lowest-energy and therefore most populated structural conformation of an RNA molecule
RNA junction topology
In a structured RNA molecule, the lengths of the different single strands that adjoin helices
Four-way junction
A structural element in which four helices come together
Native secondary structure
The lowest-energy and therefore most populated secondary structure adopted by a particular RNA molecule
Dynamic ensembles
The many conformations adopted by an RNA molecule over time and their abundance or probabilities of formation as described by the Boltzmann distribution
Free energy landscape
A depiction of the free energy of every conformational state in a macromolecule
Non-native secondary structures
Alternative secondary structures of RNA that are of higher energy than the native structure, but still form in solution with non-negligible probability
Apical loop
A single-stranded RNA loop of variable length at the end of a helical region
Coaxial conformations
Conformations in which two helices stack on each other across inter-helical junctions
Base triple
A structural element in which three nucleotides are hydrogen bonded to one another
Inter-helical dynamics
Conformational changes at junctions between two helices that lead to changes in the bend and twist angles between the two helices, thus greatly affecting the global conformation of the RNA
Tetraloops
Apical loops composed of four nucleotides
Sugar pucker
A conformational description of the ribose sugar ring in nucleic acids. The sugar pucker tends to be predominately C3′-endo for the helical A-form RNA conformation
Computational docking
The use of computational algorithms to predict the lowest-energy conformation for the binding of a small molecule to a receptor molecule (RNA or protein)
Ensemble redistribution
Changes in the abundance of two or more conformations in the RNA ensemble, which are induced by a cellular cue such as the binding of a protein or ligand or by post-transcriptional modifications
Tertiary receptors
Regions of RNA molecules that are involved in tertiary, long-range interactions
Nucleic acid force fields
The physical models used to computationally predict nucleic acid dynamics in molecular dynamics simulations
Tautomerization
The interconversion between two molecules with the same molecular formula but different connectivity
Metastable conformations
RNA conformations that are only stable within a short range of nucleotide lengths during co-transcriptional folding
SHAPE
Selective 1′-hydroxyl acylation analysed by primer extension (SHAPE) is a common chemical-probing technique used to elucidate RNA secondary structures
Click chemistry
Simple and robust chemical reactions commonly used to covalently join specific substrates
Footnotes
Competing interests
H.M.A. is an adviser to and holds an ownership interest in Nymirum Inc., an RNA-based drug discovery company. Some of the technology used to generate ensembles described in this Review has been licensed to Nymirum, Inc.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Molecular Cell Biology thanks A. Laederach, J. Lucks and K. Weeks for their contribution to the peer review of this work.
References
- 1.Weber G. Energetics of ligand binding to proteins. Adv. Protein Chem 29, 1–83 (1975). [DOI] [PubMed] [Google Scholar]
- 2.Cech TR & Steitz JA The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Esteller M. Non-coding RNAs in human disease. Nat. Rev. Genet 12, 861–874 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Childs-Disney JL & Disney MD Approaches to validate and manipulate RNA targets with small molecules in cells. Annu. Rev. Pharmacol. Toxicol 56, 123–140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKeague M, Wong RS & Smolke CD Opportunities in the design and application of RNA for gene expression control. Nucleic Acids Res. 44, 2987–2999 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartel DP Metazoan microRNAs. Cell 173, 20–51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee RC, Feinbaum RL & Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Winkler W, Nahvi A & Breaker RR Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 419, 952–956 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Cate JH et al. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science 273, 1678–1685 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Serganov A & Patel DJ Ribozymes, riboswitches and beyond: regulation of gene expression without proteins. Nat. Rev. Genet 8, 776–790 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loveland AB, Demo G, Grigorieff N & Korostelev AA Ensemble cryo-EM elucidates the mechanism of translation fidelity. Nature 546, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prabhakar A, Choi J, Wang J, Petrov A & Puglisi JD Dynamic basis of fidelity and speed in translation: coordinated multistep mechanisms of elongation and termination. Protein Sci. 26, 1352–1362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fica SM & Nagai K. Cryo-electron microscopy snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat. Struct. Mol. Biol 24, 791–799 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanco MR et al. Single molecule cluster analysis dissects splicing pathway conformational dynamics. Nat. Methods 12, 1077–1084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mustoe AM, Brooks CL & Al-Hashimi HM Hierarchy of RNA functional dynamics. Annu. Rev. Biochem 83, 441–466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dallaire P. et al. Structural dynamics control the microRNA maturation pathway. Nucleic Acids Res. 9956–9964 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salari R, Kimchi-Sarfaty C, Gottesman MM & Przytycka TM Sensitive measurement of single-nucleotide polymorphism-induced changes of RNA conformation: application to disease studies. Nucleic Acids Res. 41, 44–53 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kutchko KM et al. Multiple conformations are a conserved and regulatory feature of the RB1 5′ UTR. RNA 21, 1274–1285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ritz J, Martin JS & Laederach A. Evolutionary evidence for alternative structure in RNA sequence co-variation. PLOS Comput. Biol 9, e1003152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halvorsen M, Martin JS, Broadaway S & Laederach A. Disease-associated mutations that alter the RNA structural ensemble. PLOS Genet. 6, e1001074 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hermann T. Strategies for the design of drugs targeting RNA and RNA-protein complexes. Angew. Chem. Int. Ed. Engl 39, 1890–1904 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Boehr DD, Nussinov R & Wright PE The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol 5, 789–796 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei G, Xi W, Nussinov R & Ma B. Protein ensembles: how does nature harness thermodynamic fluctuations for life? The diverse functional roles of conformational ensembles in the cell. Chem. Rev 116, 6516–6551 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frauenfelder H, Sligar SG & Wolynes PG The energy landscapes and motions of proteins. Science 254, 1598–1603 (1991). [DOI] [PubMed] [Google Scholar]
- 25.Schroeder SJ Challenges and approaches to predicting RNA with multiple functional structures. RNA 24, 1615–1624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cruz JA & Westhof E. The dynamic landscapes of RNA architecture. Cell 136, 604–609 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Leulliot N & Varani G. Current topics in RNA-protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry 40, 7947–7956 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Mironov AS et al. Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell 111, 747–756 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Breaker RR Prospects for riboswitch discovery and analysis. Mol. Cell 43, 867–879 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nudler E. Flipping riboswitches. Cell 126, 19–22 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Keane SC et al. NMR detection of intermolecular interaction sites in the dimeric 5′-leader of the HIV-1 genome. Proc. Natl Acad. Sci. USA 113, 13033–13038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taliaferro JM et al. RNA sequence context effects measured in vitro predict in vivo protein binding and regulation. Mol. Cell 64, 294–306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bisaria N. et al. Kinetic and thermodynamic framework for P4-P6 RNA reveals tertiary motif modularity and modulation of the folding preferred pathway. Proc. Natl Acad. Sci. USA 113, E4956–E4965 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ke A, Zhou K, Ding F, Cate JH & Doudna JA A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature 429, 201–205 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Shajani Z, Sykes MT & Williamson JR Assembly of bacterial ribosomes. Annu. Rev. Biochem 80, 501–526 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Nam Y, Chen C, Gregory RI, Chou JJ & Sliz P. Molecular basis for interaction of let-7 microRNAs with Lin28. Cell 147, 1080–1091 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X. et al. Maternally expressed gene 3 (MEG3) noncoding ribonucleic acid: isoform structure, expression, and functions. Endocrinology 151, 939–947 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Somarowthu S. et al. HOTAIR forms an intricate and modular secondary structure. Mol. Cell 58, 353–361 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hawkes EJ et al. COOLAIR antisense RNAs form evolutionarily conserved elaborate secondary structures. Cell Rep. 16, 3087–3096 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strulson CA, Molden RC, Keating CD & Bevilacqua PC RNA catalysis through compartmentalization. Nat. Chem 4, 941–946 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Kim Y & Myong S. RNA remodeling activity of DEAD box proteins tuned by protein concentration, RNA length, and ATP. Mol. Cell 63, 865–876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elbaum-Garfinkle S. et al. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl Acad. Sci. USA 112, 7189–7194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dethoff EA, Chugh J, Mustoe AM & Al-Hashimi HM Functional complexity and regulation through RNA dynamics. Nature 482, 322–330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Q, Stelzer AC, Fisher CK & Al-Hashimi HM Visualizing spatially correlated dynamics that directs RNA conformational transitions. Nature 450, 1263–1267 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Salmon L, Yang S & Al-Hashimi HM Advances in the determination of nucleic acid conformational ensembles. Annu. Rev. Phys. Chem 65, 293–316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi X, Huang L, Lilley DM, Harbury PB & Herschlag D. The solution structural ensembles of RNA kink-turn motifs and their protein complexes. Nat. Chem. Biol 12, 146–152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansen AL & Al-Hashimi HM Dynamics of large elongated RNA by NMR carbon relaxation. J. Am. Chem. Soc 129, 16072–16082 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Shajani Z, Drobny G & Varani G. Binding of U1A protein changes RNA dynamics as observed by 13C NMR relaxation studies. Biochemistry 46, 5875–5883 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Kumar S, Ma B, Tsai CJ, Sinha N & Nussinov R. Folding and binding cascades: dynamic landscapes and population shifts. Protein Sci. 9, 10–19 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herschlag D. RNA chaperones and the RNA folding problem. J. Biol. Chem 270, 20871–20874 (1995). [DOI] [PubMed] [Google Scholar]
- 51.Russell R. RNA misfolding and the action of chaperones. Front. Biosci 13, 1–20 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dethoff EA, Petzold K, Chugh J, Casiano-Negroni A & Al-Hashimi HM Visualizing transient low-populated structures of RNA. Nature 491, 724–728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salmon L, Bascom G, Andricioaei I & Al-Hashimi HM A general method for constructing atomic-resolution RNA ensembles using NMR residual dipolar couplings: the basis for interhelical motions revealed. J. Am. Chem. Soc 135, 5457–5466 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merriman DK et al. Shortening the HIV-1 TAR RNA bulge by a single nucleotide preserves motional modes over a broad range of time scales. Biochemistry 55, 4445–4456 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Puglisi JD, Tan R, Calnan BJ, Frankel AD & Williamson JR Conformation of the TAR RNA-arginine complex by NMR spectroscopy. Science 257, 76–80 (1992). [DOI] [PubMed] [Google Scholar]
- 56.Lee J, Dethoff EA & Al-Hashimi HM Invisible RNA state dynamically couples distant motifs. Proc. Natl Acad. Sci. USA 111, 9485–9490 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Merriman DK et al. Increasing the length of poly-pyrimidine bulges broadens RNA conformational ensembles with minimal impact on stacking energetics. RNA 24, 1363–1376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qi Y. et al. Continuous interdomain orientation distributions reveal components of binding thermodynamics. J. Mol. Biol 430, 3412–3426 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Denny SK et al. High-throughput investigation of diverse junction elements in RNA tertiary folding. Cell 174, 377–390 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tinoco I Jr & Bustamante C. How RNA folds. J. Mol. Biol 293, 271–281 (1999). [DOI] [PubMed] [Google Scholar]
- 61.Dill KA Additivity principles in biochemistry. J. Biol. Chem 272, 701–704 (1997). [DOI] [PubMed] [Google Scholar]
- 62.Rist MJ & Marino JP Mechanism of nucleocapsid protein catalyzed structural isomerization of the dimerization initiation site of HIV-1. Biochemistry 41, 14762–14770 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Kimsey IJ et al. Dynamic basis for dG*dT misincorporation via tautomerization and ionization. Nature 554, 195–201 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kimsey IJ, Petzold K, Sathyamoorthy B, Stein ZW & Al-Hashimi HM Visualizing transient Watson-Crick-like mispairs in DNA and RNA duplexes. Nature 519, 315–320 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tian S, Cordero P, Kladwang W & Das R. High-throughput mutate-map-rescue evaluates SHAPE- directed RNA structure and uncovers excited states. RNA 20, 1815–1826 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao B, Guffy SL, Williams B & Zhang Q. An excited state underlies gene regulation of a transcriptional riboswitch. Nat. Chem. Biol 13, 968–974 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xue Y, Gracia B, Herschlag D, Russell R & Al-Hashimi HM Visualizing the formation of an RNA folding intermediate through a fast highly modular secondary structure switch. Nat. Commun 7, ncomms11768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gracia B. et al. Hidden structural modules in a cooperative RNA folding transition. Cell Rep. 22, 3240–3250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang J, Jones CP & Ferre-D’Amare AR Global analysis of riboswitches by small-angle X-ray scattering and calorimetry. Biochim. Biophys. Acta 1839, 1020–1029 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Williamson JR Induced fit in RNA-protein recognition. Nature Struct. Biol 7, 834–837 (2000). [DOI] [PubMed] [Google Scholar]
- 71.Borkar AN et al. Structure of a low-population binding intermediate in protein-RNA recognition. Proc. Natl Acad. Sci. USA 113, 7171–7176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bailor MH, Sun X & Al-Hashimi HM Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science 327, 202–206 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Mustoe AM, Brooks CL 3rd & Al-Hashimi HM Topological constraints are major determinants of tRNA tertiary structure and dynamics and provide basis for tertiary folding cooperativity. Nucleic Acids Res. 42, 11792–11804 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fang W & Bartel DP The menu of features that define primary microRNAs and enable de novo design of microRNA genes. Mol. Cell 60, 131–145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sponer J. et al. RNA structural dynamics as captured by molecular simulations: a comprehensive overview. Chem. Rev 118, 4177–4338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tan D, Piana S, Dirks RM & Shaw DE RNA force field with accuracy comparable to state-of-the-art protein force fields. Proc. Natl Acad. Sci. USA 115, E1346–E1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin JH, Perryman AL, Schames JR & McCammon JA Computational drug design accommodating receptor flexibility: the relaxed complex scheme. J. Am. Chem. Soc 124, 5632–5633 (2002). [DOI] [PubMed] [Google Scholar]
- 78.Stelzer AC et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol 7, 553–559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ganser LR et al. High-performance virtual screening by targeting a high-resolution RNA dynamic ensemble. Nat. Struct. Mol. Biol 25, 425–434 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chu VB et al. Do conformational biases of simple helical junctions influence RNA folding stability and specificity? RNA 15, 2195–2205 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yesselman JD et al. RNA tertiary structure energetics predicted by an ensemble model of the RNA double helix. Preprint at bioRxiv 10.1101/341107 (2018). [DOI] [Google Scholar]
- 82.Reining A. et al. Three-state mechanism couples ligand and temperature sensing in riboswitches. Nature 499, 355–359 (2013). [DOI] [PubMed] [Google Scholar]
- 83.Strebitzer E. et al. 5-Oxyacetic acid modification destabilizes double helical stem structures and favors anionic Watson-Crick like cmo5 U-G base pairs. Chemistry 24, 18903–18906 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Allred BE, Gebala M & Herschlag D. Determination of ion atmosphere effects on the nucleic acid electrostatic potential and ligand association using AH+YC wobble formation in double-stranded DNA. J. Am. Chem. Soc 139, 7540–7548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gebala M. et al. Cation-anion interactions within the nucleic acid ion atmosphere revealed by ion counting. J. Am. Chem. Soc 137, 14705–14715 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lipfert J, Doniach S, Das R & Herschlag D. Understanding nucleic acid-ion interactions. Annu. Rev. Biochem 83, 813–841 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Woodson SA Compact intermediates in RNA folding. Annu. Rev Biophys 39, 61–77 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Draper DE A guide to ions and RNA structure. RNA 335–343 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chu VB, Bai Y, Lipfert J, Herschlag D & Doniach S. A repulsive field: advances in the electrostatics of the ion atmosphere. Curr. Opin. Chem. Biol 12, 619–625 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamagami R, Bingaman JL, Frankel EA & Bevilacqua PC Cellular conditions of weakly chelated magnesium ions strongly promote RNA stability and catalysis. Nat. Commun 9, 2149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leamy KA, Assmann SM, Mathews DH & Bevilacqua PC Bridging the gap between in vitro and in vivo RNA folding. Q. Rev. Biophys 49, e10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dupuis NF, Holmstrom ED & Nesbitt DJ Molecular-crowding effects on single-molecule RNA folding/unfolding thermodynamics and kinetics. Proc. Natl Acad. Sci. USA 111, 8464–8469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kilburn D, Roh JH, Guo L, Briber RM & Woodson SA Molecular crowding stabilizes folded RNA structure by the excluded volume effect. J. Am. Chem. Soc 132, 8690–8696 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Strulson CA, Yennawar NH, Rambo RP & Bevilacqua PC Molecular crowding favors reactivity of a human ribozyme under physiological ionic conditions. Biochemistry 52, 8187–8197 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nakano S, Karimata HT, Kitagawa Y & Sugimoto N. Facilitation of RNA enzyme activity in the molecular crowding media of cosolutes. J. Am. Chem. Soc 131, 16881–16888 (2009). [DOI] [PubMed] [Google Scholar]
- 96.Desai R, Kilburn D, Lee HT & Woodson SA Increased ribozyme activity in crowded solutions. J. Biol. Chem 289, 2972–2977 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rode AB, Endoh T & Sugimoto N. Crowding shifts the FMN recognition mechanism of riboswitch aptamer from conformational selection to induced fit. Angew. Chem. Int. Ed. Engl 57, 6868–6872 (2018). [DOI] [PubMed] [Google Scholar]
- 98.Lee HT, Kilburn D, Behrouzi R, Briber RM & Woodson SA Molecular crowding overcomes the destabilizing effects of mutations in a bacterial ribozyme. Nucleic Acids Res. 43, 1170–1176 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lambert D & Draper DE Effects of osmolytes on RNA secondary and tertiary structure stabilities and RNA-Mg2+ interactions. J. Mol. Biol 370, 993–1005 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nakano S, Miyoshi D & Sugimoto N. Effects of molecular crowding on the structures, interactions, and functions of nucleic acids. Chem. Rev 114, 2733–2758 (2014). [DOI] [PubMed] [Google Scholar]
- 101.Kim YB et al. Ligand binding to 2-deoxyguanosine sensing riboswitch in metabolic context. Nucleic Acids Res. 45, 5375–5386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Banani SF, Lee HO, Hyman AA & Rosen MK Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hyman AA, Weber CA & Jülicher F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol 39–58 (2014). [DOI] [PubMed] [Google Scholar]
- 104.Nielsen FC, Hansen HT & Christiansen J. RNA assemblages orchestrate complex cellular processes. Bioessays 38, 674–681 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anderson P & Kedersha N. RNA granules: post- transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol 10, 430–436 (2009). [DOI] [PubMed] [Google Scholar]
- 106.Spector DL SnapShot: cellular bodies. Cell 127, 1071 (2006). [DOI] [PubMed] [Google Scholar]
- 107.Langdon EM et al. mRNA structure determines specificity of a polyQ-driven phase separation. Science 360, 922–927 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maharana S. et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jain A & Vale RD RNA phase transitions in repeat expansion disorders. Nature 546, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gleitsman KR, Sengupta RN & Herschlag D. Slow molecular recognition by RNA. RNA 23, 1745–1753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wickiser JK, Winkler WC, Breaker RR & Crothers DM The speed of RNA transcription and metabolite binding kinetics operate an FMN riboswitch. Mol. Cell 18, 49–60 (2005). [DOI] [PubMed] [Google Scholar]
- 112.Steinert H. et al. Pausing guides RNA folding to populate transiently stable RNA structures for riboswitch-based transcription regulation. eLife 6, e21297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Helmling C. et al. Life times of metastable states guide regulatory signaling in transcriptional riboswitches. Nat. Commun 9, 944 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Uhm H, Kang W, Ha KS, Kang C & Hohng S. Single-molecule FRET studies on the cotranscriptional folding of a thiamine pyrophosphate riboswitch. Proc. Natl Acad. Sci. USA 115, 331–336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Helmling C. et al. NMR structural profiling of transcriptional intermediates reveals riboswitch regulation by metastable RNA conformations. J. Am. Chem. Soc 139, 2647–2656 (2017). [DOI] [PubMed] [Google Scholar]
- 116.Landick R. The regulatory roles and mechanism of transcriptional pausing. Biochem. Soc. Trans 34, 1062–1066 (2006). [DOI] [PubMed] [Google Scholar]
- 117.Hua B, Panja S, Wang Y, Woodson SA & Ha T. Mimicking co-transcriptional RNA folding using a superhelicase. J. Am. Chem. Soc 140, 10067–10070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Watters KE, Strobel EJ, Yu AM, Lis JT & Lucks JB Cotranscriptional folding of a riboswitch at nucleotide resolution. Nat. Struct. Mol. Biol 23, 1124–1131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Strobel EJ, Watters KE, Nedialkov Y, Artsimovitch I & Lucks JB Distributed biotin- streptavidin transcription roadblocks for mapping cotranscriptional RNA folding. Nucleic Acids Res. 45, e109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Harcourt EM, Kietrys AM & Kool ET Chemical and structural effects of base modifications in messenger RNA. Nature 541, 339–346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Meyer KD & Jaffrey SR Rethinking m6A readers, writers, and erasers. Annu. Rev. Cell Dev. Biol 33, 319–342 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Choi J. et al. 2′-O-methylation in mRNA disrupts tRNA decoding during translation elongation. Nat. Struct. Mol. Biol 25, 208–216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Choi J. et al. N6-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat. Struct. Mol. Biol 23, 110–115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Roost C. et al. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J. Am. Chem. Soc 137, 2107–2115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Spitale RC et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu B. et al. A potentially abundant junctional RNA motif stabilized by m6A and Mg2. Nat. Commun 9, 2761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu N. et al. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vaidyanathan PP, AlSadhan I, Merriman DK, Al-Hashimi HM & Herschlag D. Pseudouridine and N6-methyladenosine modifications weaken PUF protein/RNA interactions. RNA 23, 611–618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Huang L, Ashraf S, Wang J & Lilley DM Control of box C/D snoRNP assembly by N6-methylation of adenine. EMBO Rep. 18, 1631–1645 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou H. et al. m1A and m1G disrupt A-RNA structure through the intrinsic instability of Hoogsteen base pairs. Nat. Struct. Mol. Biol 23, 803–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li X. et al. Base-resolution mapping reveals distinct m1a methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell 68, 993–1005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ding Y. et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505, 696–700 (2014). [DOI] [PubMed] [Google Scholar]
- 133.Rouskin S, Zubradt M, Washietl S, Kellis M & Weissman JS Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505, 701–705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mustoe AM et al. Pervasive regulatory functions of mRNA structure revealed by high-resolution SHAPE probing. Cell 173, 181–195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Beaudoin JD et al. Analyses of mRNA structure dynamics identify embryonic gene regulatory programs. Nat. Struct. Mol. Biol 25, 677–686 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sun L. et al. RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol 26, 322–330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Guo JU & Bartel DP RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 353, aaf5371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang SY et al. Transcriptome-wide identification of transient RNA G-quadruplexes in human cells. Nat. Commun 9, 4730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Spasic A, Assmann SM, Bevilacqua PC & Mathews DH Modeling RNA secondary structure folding ensembles using SHAPE mapping data. Nucleic Acids Res. 46, 314–323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li H & Aviran S. Statistical modeling of RNA structure profiling experiments enables parsimonious reconstruction of structure landscapes. Nat. Commun 606 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ding Y, Chan CY & Lawrence CE Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 32, W135–W141 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rogers E & Heitsch CE Profiling small RNA reveals multimodal substructural signals in a Boltzmann ensemble. Nucleic Acids Res. 42, e171 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Frank AT, Stelzer AC, Al-Hashimi HM & Andricioaei I. Constructing RNA dynamical ensembles by combining MD and motionally decoupled NMR RDCs: new insights into RNA dynamics and adaptive ligand recognition. Nucleic Acids Res. 37, 3670–3679 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Woods CT et al. Comparative visualization of the RNA suboptimal conformational ensemble in vivo. Biophys. J 113, 290–301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Merino EJ, Wilkinson KA, Coughlan JL & Weeks KM RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc 4223–4231 (2005). [DOI] [PubMed] [Google Scholar]
- 146.Cordero P & Das R. Rich RNA structure landscapes revealed by mutate-and-map analysis. PLOS Comput. Biol 11, e1004473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Van Nostrand EL et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jarmoskaite I. et al. A quantitative and predictive model for RNA binding by human Pumilio proteins. Preprint at bioRxiv 10.1101/403006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rzuczek SG, Park H & Disney MD A toxic RNA catalyzes the in cellulo synthesis of its own inhibitor. Angew. Chem. Int. Ed. Engl 53, 10956–10959 (2014). [DOI] [PubMed] [Google Scholar]
- 150.Quarles KA et al. Ensemble analysis of primary microRNA structure reveals an extensive capacity to deform near the Drosha cleavage site. Biochemistry 52, 795–807 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li W. et al. Germline mutation of microRNA-125a is associated with breast cancer. J. Med. Genet 46, 358–360 (2009). [DOI] [PubMed] [Google Scholar]
- 152.Mustoe AM et al. Noncanonical secondary structure stabilizes mitochondrial tRNA(Ser(UCN)) by reducing the entropic cost of tertiary folding. J. Am. Chem. Soc 3592–3599 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Box 1
Supplementary Box 2