FUS and TDP-43 Phases in Health and Disease (original) (raw)
. Author manuscript; available in PMC: 2022 Jan 1.
Published in final edited form as: Trends Biochem Sci. 2021 Jan 11;46(7):550–563. doi: 10.1016/j.tibs.2020.12.005
Abstract
The distinct prion-like domains (PrLDs) of FUS and TDP-43, modulate phase transitions that result in condensates with a range of material states. These assemblies are implicated in both health and disease. In this review, we examine how sequence, structure, post-translational modifications, and RNA can affect the self-assembly of these RNA-binding proteins (RBPs). We discuss how our emerging understanding of FUS and TDP-43 liquid–liquid phase separation (LLPS) and aggregation, could be leveraged to design new therapies for neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and limbic-predominant age-related TDP-43 encephalopathy (LATE).
Biology on the Mesoscale
A revolution in understanding cellular organization is occurring on the mesoscale, between the largest protein complexes and membrane-encompassed organelles. In this vast space, an increasingly large number of cellular assemblies reside without precise stereospecific interfaces, defined stoichiometric ratios of components, or a delimiting membrane [1]. Such assemblies, dubbed condensates, include the nucleolus and various ribonucleoprotein (RNP) granules such as stress granules (SGs) (see Glossary), paraspeckles, DNA-damage foci, signalasomes, and the pyrenoid, among others. Not only are condensates widespread, some posit they are as old as life itself, possibly facilitating biochemistry in the RNA world [2].
Many of these condensates are thought to arise through the process of liquid–liquid phase separation (LLPS) or liquid–gel phase transitions, and can be described by a phase diagram [2–4] (Figure 1A). LLPS results in the switch-like demixing of biomolecules into concentrated liquid droplets. The resulting condensates can retain the properties of a liquid, readily exchanging components with the bulk solution, fusing with other droplets, and wetting surfaces upon collision [3,4]. The high local concentration of specific biomolecules within a condensate can facilitate further transitions in material state, resulting in gelatinous or solid assemblies, or nucleating the growth of fibrillar structures [5]. A central principle underlying LLPS is multivalency, the ability of one biomolecule to simultaneously interact with multiple other copies of itself (homotypic phase separation) or with multiple other biomolecules (heterotypic phase separation) [6]. In the case of proteins, multivalency is frequently, but not exclusively, facilitated by intrinsically-disordered regions (IDRs), which lack a stable 3D structure and whose structural heterogeneity allows interacting motifs to be displayed in multiple orientations [7]. The contribution of IDRs to LLPS, including those of FUS and TDP-43, have been the subject of multiple studies and reviews [8–10]. Often encoded in these domains are ‘stickers’, residues or motifs that enable protein-protein or protein–nucleic acid interactions, interspersed by flexible ‘spacers’ that modulate sticker interactions [11]. Here, we describe the role of a specific subclass of IDR, prion-like domains (PrLDs) (Box 1) in LLPS function and dysfunction, with specific focus on archetypal yet contrasting examples of PrLD proteins, FUS and TDP-43 [12–15].
Figure 1. Model Phase Transitions.
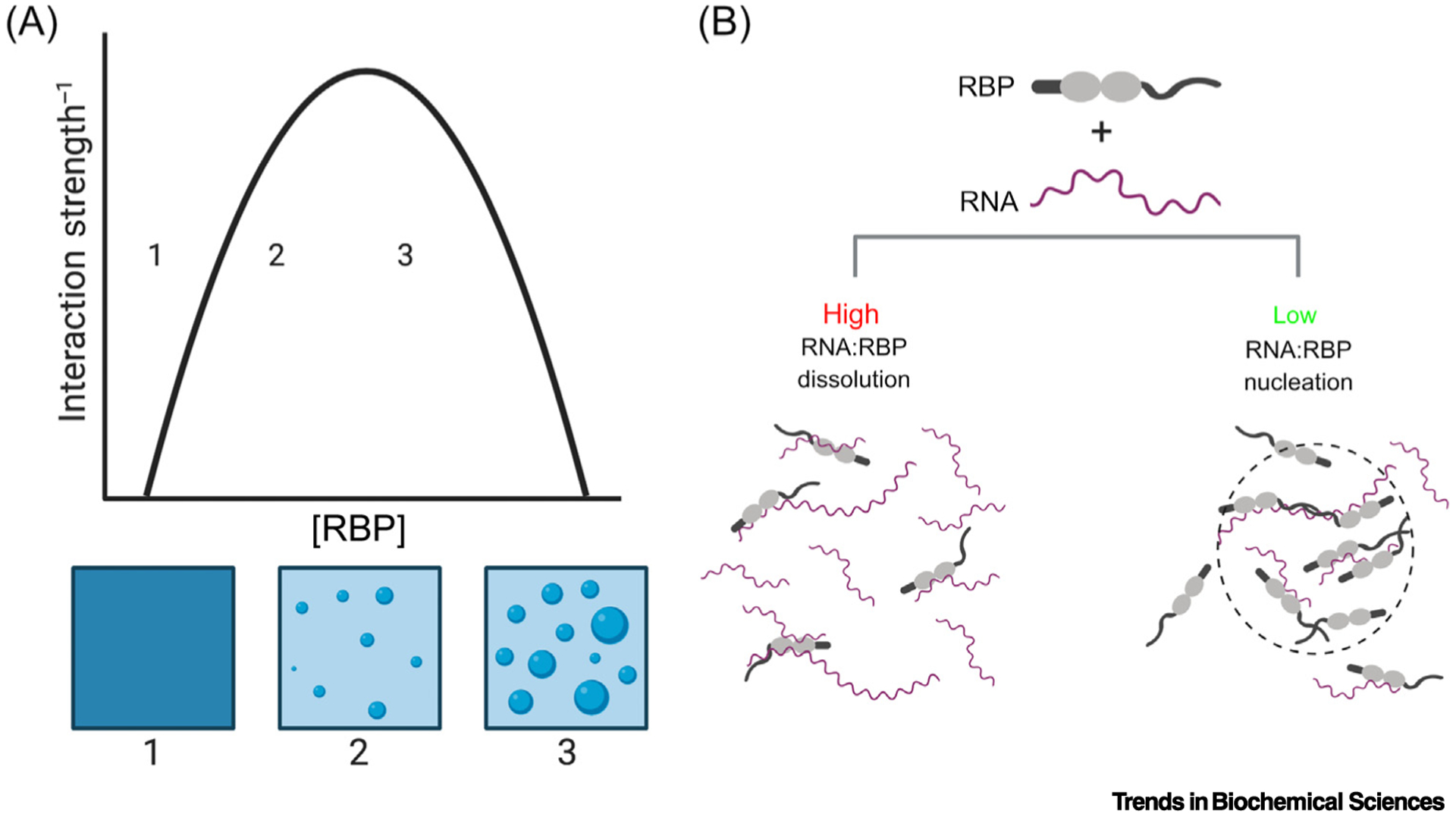
(A) A phase diagram describing homotypic phase separation. Crossing the phase boundary leads to a transition from a well-mixed solution (position 1) to a biphasic solution, with light and dense phases (position 2). Further increasing the concentration (position 3) increases the volume fraction of the dense phase, without altering the light or dense phase concentrations. (B) High ratios of RNA:RNA binding proteins (RBPs), including FUS and TDP-43, can prevent phase separation and aggregation [21,32]. Conversely, lower ratios of RNA to RBPs can lead multiple copies of a protein bound to a single RNA, creating a high local protein concentration and facilitating intermolecular interactions between RBPs, thus nucleating a phase transition [20,139].
Box 1. Prions versus Prion-like Domains.
Prions are infectious proteins that underlie transmissible spongiform encephalopathies in mammals [119–121]. However, prions are also found in yeast, where they can confer beneficial, heritable phenotypes [119,122–126]. Proteins that form prions in yeast often harbor a prion domain, a distinctive low-complexity domain enriched in uncharged, polar residues (especially glutamine, asparagine, tyrosine, and serine) and glycine, which encodes the ability to form a prion [14,119]. PrLDs represent a specific class of disordered, low-complexity protein domains, with sequence composition reminiscent of canonical yeast prion domains [12,13,15,91,127]. In yeast, prions enable highly penetrant, protein-based epigenetic inheritance, which can be advantageous [12,119,123]. Prion proteins stochastically sample a self-templating ‘[PRION+]’ conformation, that is typically a stable amyloid state, which can alter gene expression and metabolic programs transgenerationally [12,119,123]. However, not all yeast prions are amyloids. Recently, [SMAUG+] has been defined as a yeast prion that is embodied by a non-amyloid condensate [128]. Notably, condensates in human cells are rich in proteins with PrLDs, many of which undergo LLPS both in vivo and in vitro, adopting a range of material states [22,30,66,129]. Similar to the gene ontology of yeast prions, human PrLD proteins are enriched among RNA-binding proteins (RBPs), chromatin-modifying enzymes, and transcription factors; classes of proteins that can exert highly pleiotropic control of gene expression [12,14]. In another similarity to prions, there is mounting evidence to suggest that prion-like TDP-43 aggregation can spread from cell-to-cell in a seed-dependent manner in cultured cells and mouse brains, that mimics features of human disease [130–132]. Together, similarities in the sequence composition, ability to self-assemble into a range of material states, gene ontology, and aggregation propensity between yeast prion proteins and human PrLD-containing proteins, raises tantalizing possibilities that metastable control of gene expression may be driven by PrLD-mediated phase transitions to various material states in human cells [12,14,66,133,134].
Functional Phase Separation
LLPS enables the switch-like condensation of biomolecules resulting in a dilute (light) phase and concentrated (dense) phase that can differ in concentration by >50-fold (Figure 1A) [16]. This switch-like behavior enables highly nonlinear changes in protein localization to arise from small changes in concentration or environmental conditions [17]. This nonlinearity could potentially poise biological systems to: (i) effect rapid and dramatic changes in the subcellular availability of particular molecules; or (ii) alter effective stoichiometry of interacting proteins via selective partitioning into condensates [17]. LLPS can be tuned by altering the interaction strength between molecules (e.g., via post-translational modification) [18,19] or by nucleating high local concentrations (e.g., by docking multiple molecules on a scaffold, such as RNA, or poly(ADP-ribose) (PAR)) [17,20–23] (Figure 1B). In this way, cells can quickly alter effective protein concentrations without the time and energy expenses inherent to protein synthesis or degradation. For regulatory proteins like FUS and TDP-43, this phase separation could facilitate stimulus responsive gene-expression programs. Conversely, aberrant phase separation could potentially result from alterations to the sensitivity, duration, reversibility, localization, or composition of dense phases.
FUS and TDP-43 as Case Studies
The PrLD-containing RNA-binding proteins (RBPs), FUS, and TDP-43, provide some of the most well-studied examples of RBPs that undergo LLPS. FUS and TDP-43 can form condensates with a range of material states implicated in both normal function and disease. Both RBPs are involved in RNA metabolism at multiple stages, including transcriptional regulation, pre-mRNA splicing, RNA processing, and RNA localization. Further, both are components of stress-induced RNP granules known as SGs and are found in cytoplasmic inclusions in degenerating neurons that are key pathological hallmarks of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [13]. Cytoplasmic TDP-43 inclusions are also a key feature of limbic-predominant age-related TDP-43 encephalopathy (LATE), a newly recognized neurodegenerative disorder that mimics Alzheimer’s disease [24]. FUS and TDP-43 have a modular architecture shared among a family of proteins known as heterogeneous nuclear ribonucleoproteins (hnRNPs) [13], including disordered PrLDs as well as folded RNA-recognition motifs (RRMs) and other RNA-binding domains (RBDs) such as the disordered arginine–glycine rich regions (RGG boxes) of FUS and the ordered N-terminal region of TDP-43 (Figures 2 and 3). This architecture is important; PrLDs can tune both the critical concentration required for phase separation as well as the material state of the resulting condensate, and the RBDs can interact with nucleic acids and other polymers that may serve as nucleators and regulators of condensation [20–22,25] (Figure 1B). Importantly, PrLDs enable both homotypic interactions and heterotypic interactions, giving rise to RNP granules with complex proteomes [26,27]. The relative ratios of codemixing proteins govern the collective saturation concentration (C sat), 3D organization, and material state of the resulting condensate [28,29].
Figure 2. Interaction Interfaces Regulating FUS LLPS.
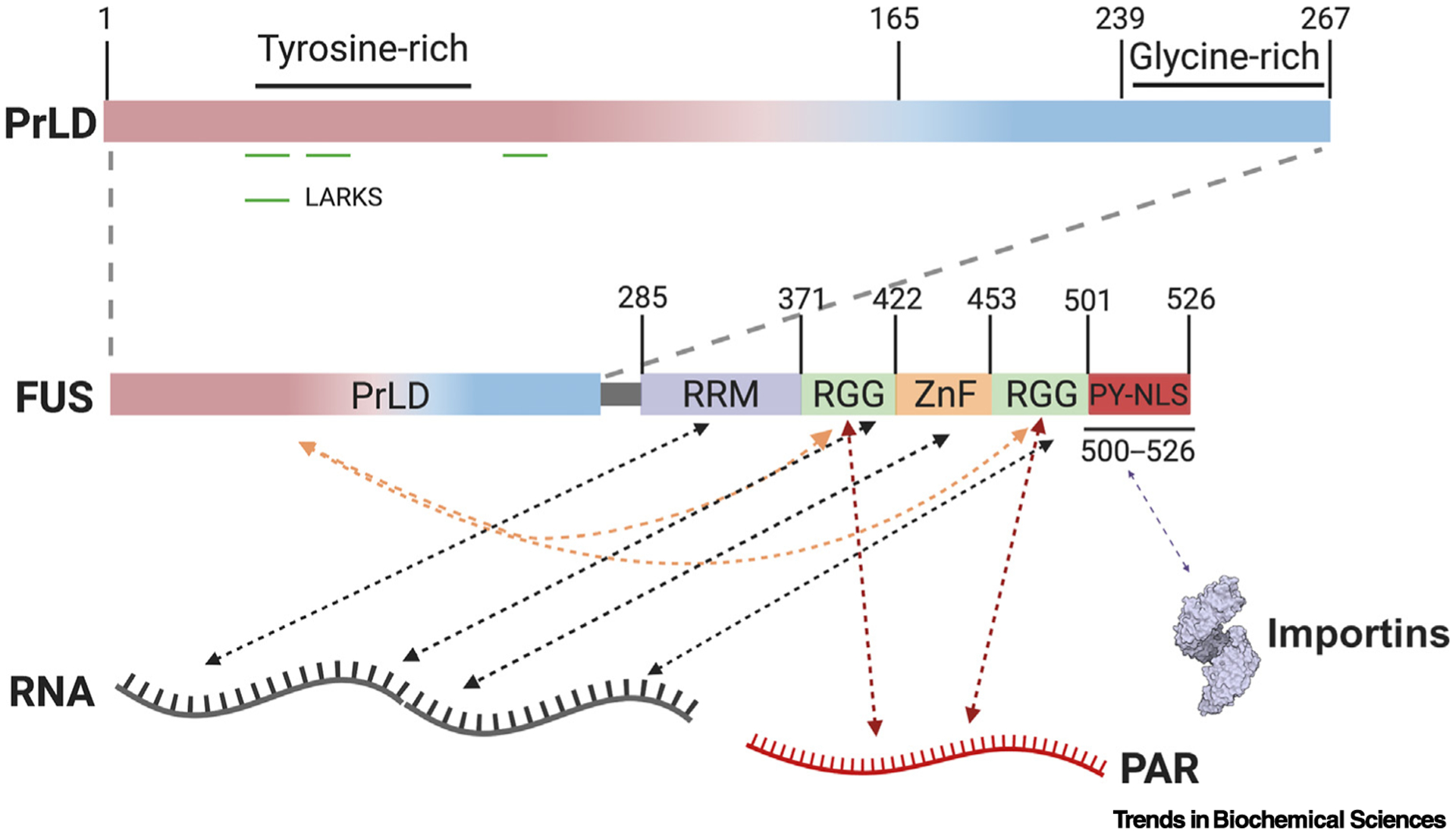
Residues 1–239 of FUS harbor the prion-like domain (PrLD), which is followed by a glycine-rich region extending to residue 267, a RNA-recognition motif (RRM), two arginine-glycine rich regions (RGGs), a zinc finger (ZnF) domain, and a PY-nuclear localization signal (PY-NLS) [13]. Low complexity aromatic-rich kinked segments (LARKS) in the PrLD are capable of forming homotypic cross-β interactions [52–54]. Arginine residues in RGG domains and tyrosine residues in the PrLD can form intramolecular (orange arrows) and intermolecular interactions [13,46]. RRM, RGG, and ZnF domains, further mediate interactions between FUS and RNA molecules (black arrows) [13]. The RGG domains also interact with poly(ADP Ribose) (PAR) (red arrows) [55]. The PY-NLS interacts with importins which regulate FUS condensation (purple arrow) [39,48,57,58]. Collectively, these interactions govern FUS assembly via competition between inter- and intramolecular interactions tuned by post-translational modifications and scaffolds like RNA and PAR.
Figure 3. Interaction Interfaces Regulating TDP-43 Liquid–Liquid Phase Separation (LLPS).
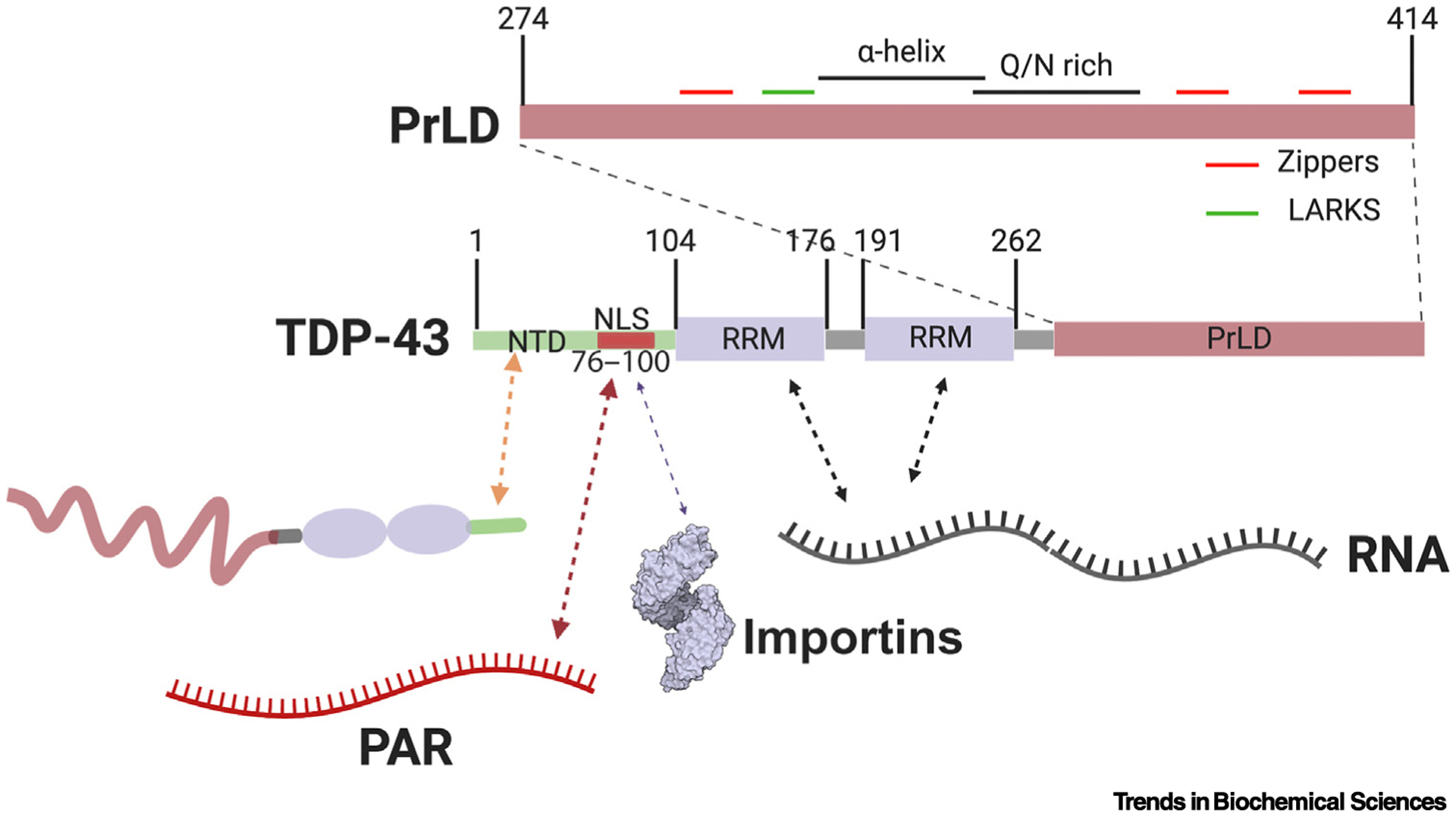
TDP-43 consists of an N-terminal domain (NTD) that can form homotypic interactions (orange arrow) [18,76], and which contains a nuclear localization signal (NLS) harboring two poly(ADP Ribose) (PAR)-binding motifs (red arrow) [13,22]. The NLS also engages importins, which can regulate TDP-43 condensation (purple arrow) [39]. The NTD is followed by two RNA-recognition motif (RRM) domains and a prion-like domain (PrLD). The two RRM domains facilitate the interactions between TDP-43 and RNA molecules (black arrows), which are important for regulation of TDP-43 LLPS [32]. The PrLD itself contains various subdomains capable of forming intermolecular interactions with other copies of TDP-43 (top). These subdomains include a region with α-helical propensity, a Q/N-rich region, numerous Zippers, and low complexity aromatic-rich kinked segments (LARKS) [78,80,83].
Evolution has likely tuned the Csat of various RBPs to achieve regulated and functional condensation. For example, the respective Csat for in vitro LLPS and estimated cellular concentrations of FUS and TDP-43, are very similar [22,30]. Evidence of more nuanced tuning of saturation conditions resulting from interactions with polymeric scaffolds, including PAR [22,31] and RNA [20,21,32,33], post-translational modifications [18,19], oligomerization state [34], and the speed at which scaffold complexes diffuse through the cell [34], all suggest ways in which FUS and TDP-43 could be poised to condense as conditions warrant. These additional layers of regulation can divorce Csat from total cellular concentration of the protein, enabling local condensation or dissolution [34]. This divorce might have distinct consequences for function and dysfunction such as the trafficking of RNAs in RNP granules to regulate local translation [35]. Moreover, FUS and TDP-43 appear to be supersaturated in motor neurons (the selectively vulnerable neuron in ALS), which may make them even more prone to LLPS and pathological aggregation [36].
FUS and TDP-43 Partition into Heterotypic Condensates
Csat is a straightforward concept when applied to homotypic condensates, including many of the test-tube condensates that facilitated a detailed understanding of phase behavior for PrLD proteins [30,37]. Cellular condensates, by contrast, contain a host of factors. For example, SGs contain both FUS and TDP-43 along with hundreds of proteins and diverse translationally-arrested mRNAs [22,26,27,33,38,39]. As such, particular proteins may partition into a condensate exhibiting liquid-like properties (e.g., a spherical shape, the ability to fuse, and rapid recovery from photobleaching), while lacking a clearly defined Csat. Such observations may be reconciled by the fact that the specific protein under study may not itself be the scaffold responsible for nucleating the condensate, but rather may be a client that preferentially partitions into a heterotypic phase [40]. Further, if two or more proteins ‘co-scaffold’ the same dense phase with one another, the Csat for such condensates would be a function of the concentration of each protein and the stoichiometric ratios between the two. Recent work explores this concept via both theory and experiment [28,29]. The nucleolus forms via heterotypic LLPS in a manner that depends on the concentrations of and interactions between multiple protein and RNA components [29], and this behavior can be modeled for other condensates [28,29]. Together, these studies elegantly demonstrate the intuitive concept that heterotypic condensates arise from heterogeneous interactions [41,42]. Importantly, these studies dispel the notion that individual proteins that undergo LLPS in cells must display a fixed Csat [43]. This concept is likely to be of central importance to understanding: (i) how different cell types with varied gene-expression programs form and regulate condensates; and (ii) how changes in the relative ratios of co-condensing proteins could sensitize certain cell types to aberrant phase transitions (Box 2) and disease.
Box 2. Defining Aberrant Phase Transitions Beyond Aggregation.
Proposed roles for phase transitions as organizers and drivers of biochemical reactions, concentration buffers, and a means by which the cell can compute responses to changing environmental conditions, motivate us to consider how aberrant phase separation may manifest itself as a function of changes in Csat that go beyond simple hastening of solidification [17]. Mutations that constitutively lower the Csat of a phase-separating protein would reduce the light phase concentration of the protein, and correspondingly increase the proportion of total protein found in the dense phase. If the functional phase of the protein in a given cellular context were the light phase, the result could be loss-of-function. Conversely, if a given function took place in or was otherwise facilitated by the dense phase, this shift could result in a potentially adverse gain-of-function. A phase-separating protein with distinct functions in both the light and dense phase further expands the potential definition of aberrant LLPS, as distinct light and dense phase functionalities could become unbalanced as a result of mutations altering Csat. Imbalanced function of proteins exerting highly pleiotropic control of gene expression, such as TDP-43 and FUS, could in turn elicit systemic dysfunction resulting from global changes in gene expression, a maladaptive version of prion behavior observed in yeast [128,135].
If a protein is only functioned in a given phase, biological decision-making could be altered through alterations in Csat. If only the light phase were functional, environmentally-triggered LLPS would result in marked reductions in the available pool of functional protein via titration into the dense phase. This temporarily reduced functionality could facilitate a particular reversible response. Here, mutations increasing Csat could prevent or delay the response, while mutations lowering Csat could render cells environmentally hypersensitive, invoking costly transitions to gene regulatory programs. Two recent studies demonstrating increased enzymatic activity as a function of liquid–liquid and liquid-to-solid transitions, lend credence to the notion that alterations in Csat could adversely impact cellular health [136,137]. In these examples, a reduction in Csat could lead to increased condensation, with a corresponding gain-of-function. Conversely, the loss of liquid and solid phases in these instances could lead to a loss-of-function. With the exception of an extremely cooperative process of aggregation, where nearly all available material enters the aggregated phase, the definitions of aberrant phase transitions resulting from alterations in Csat offered here are largely divorced from the material state of the dense phase. Thus, these definitions are fully compatible with the possibility not just of pathology via accelerated gelation or solidification, but the existence of toxic liquids [65,96].
Molecular Grammar of FUS LLPS
Both FUS and TDP-43 mislocalize to the cytoplasm and aggregate in certain forms of ALS and FTD, yet they do so in a mutually-exclusive manner [44]. Thus, ALS and FTD cases with FUS inclusions lack TDP-43 inclusions, and vice versa [45]. Relatedly, the ‘molecular grammar’ linking primary sequence to condensation has been dissected for both FUS and TDP-43, revealing distinct rules of engagement imparted by different sequence features (Figures 2 and 3). The FUS PrLD renders FUS intrinsically aggregation-prone and in isolation can undergo LLPS driven by tyrosine–tyrosine interactions, albeit at concentrations orders of magnitude higher than the full-length protein [21,46,47]. Notably, in vitro this synergy persists even in trans, when the PrLD and RGGs are added as separate recombinant proteins [46]. The PrLD of FUS contains multiple tyrosines, which form π–π and cation–π interactions with arginine residues found outside the PrLD in RGGs [46,48]. In addition to enabling interdomain contacts within and between FUS molecules, RGG domains also function as RNA-interaction motifs with degenerate specificity [49,50]. RNA itself regulates FUS assembly, acting as a nucleator at lower RNA:FUS ratios, while antagonizing LLPS at higher RNA:FUS ratios [20,21,51].
Together, interdomain contacts within and between FUS molecules, and between FUS and RNA, create a multitude of possible points of regulation of FUS LLPS and the material state of the resulting condensates. RNA–RGG interactions could obscure RGG arginines from PrLD tyrosines, thus causing the PrLD to interact with the RBD of other FUS molecules not bound to RNA (Figure 2). Certain FUS:RNA ratios could drive multimerization of FUS along a single RNA (Figure 1B) [20]. Multiple copies of FUS bound to a single RNA via the RRM or zinc finger domains would create a high local concentration that could nucleate PrLD–PrLD interactions, such as those observed in FUS fragments that form kinked cross-β polymers [20,52]. Among such cross-β forming stretches are members of a class of motifs dubbed ‘low complexity aromatic-rich kinked segments’ (LARKS), which may assemble the core of some FUS fibril polymorphs [52–54]. Conversely, less dense FUS nucleation could disfavor proximity-driven PrLD interactions, instead favoring tyrosine–arginine driven RGG–PrLD interactions (Figure 2). Similar regulation could be achieved via RGG domain interactions with PAR, and PAR has been shown to nucleate FUS condensates at sites of DNA damage [55,56]. Likewise, the interspersal of other RBPs along a nucleating RNA or PAR and in the surrounding liquid may all compete with FUS–FUS interactions [21,46,51]. In these ways and others, the structure of bound RNA, and the relative density and spacing of motifs recognized by FUS and other RBPs, could alter the properties of the resulting condensates.
Regulation of FUS LLPS
The ability of FUS to make interdomain contacts in cis and trans, involving RGG domains that are also capable of interacting with RNA, suggests that FUS LLPS has multiple modes of potential regulation. Recently, multiple groups reported that the nuclear-import receptor, Karyopherin-β2 (Kapβ2), also chaperones FUS phase transitions, preventing [39,48,57–59] and even reversing [39,57,60] FUS LLPS, gelation, and fibrilization. Kapβ2 binds primarily to the PY-nuclear localization signal (PY-NLS) of FUS, which then enables extensive additional contacts across FUS, which effectively disrupts FUS–FUS interactions [39,57,58]. Post-translational modifications of FUS can also regulate both interdomain contacts and binding to Kapβ2, which in turn regulate FUS LLPS [58]. One such example is phosphorylation of the critical tyrosine of the FUS PY-NLS, which can alter the association of FUS and Kapβ2, and in turn regulate nucleocytoplasmic partitioning [61].
Related to the important role of RGG domains in FUS LLPS, post-translational modifications to arginine residues are also important regulators of FUS condensation. Arginine methylation of FUS reduces condensation and SG association [58]. Conversely, FUS hypomethylation, a molecular phenotype of FUS inclusions in FTD, drives FUS gelation to more stable cross-β structures [48]. In addition to arginine methylation, a number of other FUS post-translational modifications have been cataloged and implicated in tuning LLPS [62]. The role of RNA as a modulator of FUS LLPS may itself be tuned by tyrosine phosphorylation of the FUS RRM, which has been identified as a site of phosphorylation [62,63]. Aside from the RRM, phosphorylation of the PrLD of FUS has also been observed to antagonize phase separation and aggregation [19,64].
Molecular Grammar of TDP-43 LLPS
Like FUS, TDP-43 also undergoes LLPS in vitro at near physiological concentrations, though it may require the addition of a crowding agent such as dextran [18,22,32]. It is a component of cellular condensates with a range of material states, including liquid-like SGs [26,27], various nuclear foci [65], and amyloid-like myogranules in muscle [66]. TDP-43 is also a component of nuclear RNP granules including paraspeckles [67] and nuclear stress bodies [68], which both contain specific, stress-induced, noncoding RNAs (ncRNAs). In vitro, full-length TDP-43 undergoes LLPS at physiological concentrations [22,32], whereas the C-terminal fragments harboring the PrLD tend to aggregate [22,69,70]. Similarly, cytoplasmic aggregates containing TDP-43, and TDP-43 fragments retaining the C-terminal PrLD, are a pathological hallmark of ALS and FTD [71], and the vast majority of TDP-43 mutations associated with ALS are found in the PrLD [13,72]. The PrLD is also required for TDP-43 recruitment to SGs [73], which are enriched in numerous disease-associated, aggregation-prone RBPs [26,27]. Notably, C-terminal fragments of TDP-43 lack the N-terminal PAR-binding motifs, which are important for both LLPS and SG recruitment [22].
It has been proposed that dysregulated TDP-43 LLPS may cause or promote TDP-43 aggregation, which can be toxic [32]. For this reason, the molecular determinants of TDP-43 LLPS are an area of intense study, and like FUS, multiple regions of TDP-43 beyond the PrLD are implicated in its self-assembly. However, likely owing to sequence differences in the PrLD, TDP-43 does not appear to obey the same molecular grammar rules for LLPS deduced for other PrLD-containing RBPs, including FUS [32,46] and hnRNAPA1 [74]. By contrast, TDP-43 lacks extensive RGG domains and thus has a lesser capacity for RGG–RNA interactions and intramolecular cation–π interactions between RGG domain arginines and tyrosines in the PrLD.
TDP-43 relies on multiple intermolecular contacts facilitated by distinct and nonmutually exclusive regions in order to condense. The structured N-terminal domain (NTD) of TDP-43 is capable of forming intermolecular interactions [18] and multiple reports suggest NTD dimerization is important for the role of TDP-43 in splicing regulation [75,76]. The role of TDP-43 LLPS in splicing regulation is beginning to be explored and recent work suggests TDP-43 is capable of regulating the splicing of at least some transcripts independent of phase separation [77], but in other cases splicing is enhanced by TDP-43 LLPS [78]. NTD–NTD interactions resulting in head-to-tail TDP-43 oligomers antagonize pathologic fibrillization of TDP-43 PrLDs [76] (Figure 3). Other NTD–NTD interactions, or nucleation along RNA polymers, may instead result in the close apposition of PrLDs, facilitating specific intermolecular interactions, including those that drive LLPS (Figure 3).
Within the TDP-43 PrLD is a conserved stretch of amino acids that has been shown to form a transient α-helix amidst an otherwise disordered region [79–81]. This region is important for LLPS in the context of the isolated PrLD [80]. Notably, the helical propensity of this region increases in the context of TDP-43–TDP-43 interactions. Mutations in the helix are associated with ALS and contribute to a liquid-to-solid transition in vitro [80]. Together, these observations implicate transient structure in TDP-43 LLPS, and suggest that cellular mechanisms capable of altering the TDP-43 structural ensemble could in turn alter its phase behavior. More recently, methionine residues in a region overlapping the conserved α-helical subdomain have been implicated in LLPS and gelation via cross-β polymerization, tuned by the oxidation state of the methionine residues [81]. Thus, the helical region may enable different interactions with a spectrum of stabilities, which may relate to the range of material states adopted by various TDP-43 condensates.
Mutagenesis of TDP-43 has revealed residues in addition to those in the α-helical region that are important for LLPS, which has enabled elucidation of general rules. For example, aromatic residues adjacent to glycines or serines contribute to TDP-43 LLPS in vitro [82], and the spacing of hydrophobic ‘sticker’ residues interspersed by flexible linkers have also proven important [77]. This feature is likely applicable to other PrLD-containing systems [11]. The PrLD also harbors short stretches of amino acids that in isolation are capable of ordered oligomerization in vitro, including LARKS. The structure of the TDP-43 LARK oligomer has been determined using X-ray diffraction and microcrystal electron diffraction. These structures reveal more labile β-sheets than those adopted by classical cross-β forming prion proteins, as well as motifs forming more stable steric zippers [83]. The range of stabilities impacted by LARKS and steric zippers could in theory relate to the range of material states adopted by TDP-43 in cells. Notably, some ALS-linked mutations disrupt the formation of these kinked structures, motivating future studies aimed at determining the extent to which these structures form in the context of full-length TDP-43 [83]. Together, emerging evidence supports a model whereby different TDP-43–TDP-43 interactions spanning the structured NTD to the disordered C-terminal PrLD, collectively modulate the self-assembly of TDP-43.
Regulation of TDP-43 LLPS
The possibility of multiple intermolecular interactions across TDP-43 likely enables regulation. Phosphorylation of multiple residues in the PrLD is associated with TDP-43 aggregation in diseased neurons [84]. Conversely, phosphomimetic mutations in the folded N-terminal domain of TDP-43 disrupt LLPS and reduce the viscosity of droplets in vitro [18]. The extent to which TDP-43 phosphorylation in diseased neurons represents a pathologic mechanism or a failed effort by the cell to dissolve TDP-43 condensates remains to be elucidated. Experiments where TDP-43 is phosphorylated in vitro, prior to and after initiation of phase separation and aggregation, could aid in determining the contributions of PrLD phosphorylation to TDP-43 condensation. It is also possible that different phosphorylation sites on TDP-43 exert differential effects on condensation. TDP-43 is also acetylated on lysines in the RRMs [85]. Acetylation is associated with cell stress and leads to reduced RNA binding, which in turn may contribute to cytoplasmic localization, hyperphosphorylation, and insoluble inclusion formation [85,86]. As such, acetylation could be an upstream step in the development of TDP-43 pathology that may be targeted by drugs.
The oligomerization state of TDP-43 can regulate the onset of LLPS and aggregation. The N-terminal domain provides an oligomerization interface, which along with recruitment to RNA via the RRMs of TDP-43, could enable nucleation of TDP-43 condensation or aggregation via regulated or dysregulated complex formation [18]. To model the effects of TDP-43 nucleation on cell survival, Donnelly and colleagues employed a light-inducible oligomerization system that has been successfully used to study LLPS of the FUS IDR [34] and SG formation [87] by fusing the light-inducible oligomerization domain, Cry2, to full-length TDP-43 [32]. This approach yielded a construct that rapidly coalesced in response to light stimulation [32]. This ‘opto-TDP-43’ system recapitulates important features of ALS and FTD pathology, including the formation of cytoplasmic TDP-43 inclusions that kill neurons [32]. Notably, aggregation was inhibited by the addition of specific RNAs recognized by TDP-43, which the authors dubbed ‘bait RNAs’ [32] (Box 3), consistent with prior studies indicating that RNA buffers the phase separation of PrLD-containing proteins, including FUS [21,51].
Box 3. ‘Bait RNAs’ as a Potential New Therapeutic Modality.
The role of RNAs in regulating PrLD-containing RBP phase separation, and recent success delivering short RNA drugs to the central nervous system (CNS) to target neurodegenerative disease, motivate efforts to design oligo therapeutics [138]. Donnelly and colleagues have shown that a short bait RNA that binds TDP-43 prevents aggregation when delivered to cultured cells [32]. Notably, both toxic gain-of-function of FUS and TDP-43 aggregates, and loss-of-function due to sequestration within aggregates, are potential mechanisms of pathology in ALS and FTD. For this reason, bait RNAs represent compelling therapeutic candidates. In contrast to antisense oligonucleotides that reduce protein expression, the bait RNA approach modulates protein solubility through reversible binding, not reductions in cellular protein concentration. Targeting one mRNA using antisense oligos can prevent the expression of multiple proteins. Thus, a potential challenge to a bait RNA development is the requirement that they be delivered in sufficient quantity, or be sufficiently potent, to abrogate aggregation. However, bait RNAs could function catalytically in collaboration with cellular factors including nuclear-import receptors, which have been shown to cause FUS to release RNA, enabling bait RNAs to be recycled for further rounds of activity [57,58].
More recent work has also implicated specific post-translational modifications in regulating the assembly of TDP-43, resulting in co-occurring liquid and liquid-crystalline TDP-43 phases [86], in contrast to the polymers proposed for myogranules or redox-sensitive RNA-transport granules [81]. In this work, RRM acetylation, known to reduce RNA binding [85], causes RNA-depleted TDP-43 condensates to form. However, these condensates are layered, containing a more ordered liquid-crystalline shell that encompasses a liquid core [86]. The shell forms from acetylated TDP-43, or when a RNA-binding defective mutant TDP-43 is expressed, which recruits wild-type TDP-43 [86]. The liquidity of the core is maintained by various members of the Hsp70 family of ATP-dependent chaperones [86]. This work reveals the complexity of TDP-43 LLPS regulation, with post-translational modifications regulating RNA binding, which in turn regulates self-assembly that is further regulated by protein chaperones. In the cytoplasm, nuclear-import receptors that engage the TDP-43 NLS, Importin-α/β, can prevent and reverse TDP-43 aggregation [39,88,89]. The range of material states adopted by TDP-43- liquid, liquid-crystalline, labile cross-β, amyloid-fibrillar, and amorphous aggregates, represents the diversity of mesoscale assemblies PrLD proteins can adopt and motivates a reconsideration of an early paradigm at the intersection of the LLPS and neurodegeneration fields, (i.e., that solids are ‘bad’ for cells while liquids are ‘good’).
Good Liquids and Bad Solids?
In vitro, condensates of PrLD-containing RBPs mature over time into less dynamic gels, fibers, and aggregates, and this liquid-to-solid transition is hastened by ALS- and FTD-associated mutations [30,32,37,70,90,91]. A compelling paradigm has emerged that posits liquid-like condensates are functional, or at least nonpathogenic, and solid aggregates are a toxic endpoint of aberrant phase transitions. However, recent work challenges the exclusivity of this premise while providing preliminary evidence for pathogenic liquid phases (Box 2).
In human cells, including induced pluripotent stem cell (iPSC) derived neurons, chronic phase separation of TDP-43 in the cytoplasm is toxic, even without a transition to amorphous aggregates or fibrils [65]. High-throughput screens in simpler models suggest similar routes to toxicity. The single cell brewer’s yeast, Saccharomyces cerevisiae, lacks direct orthologs of TDP-43 and FUS, but ectopic expression of these genes recapitulates many salient features of human pathology, including cytoplasmic mislocalization, aggregation, and cytotoxicity [47,70]. These features have been leveraged in screens identifying disease modifiers [47,92] and therapeutic protein disaggregases [93–95]. Deep mutational scanning of the PrLD of TDP-43 in yeast, revealed thousands of genetic variants that modified TDP-43 toxicity [96]. Curiously, mutations predicted to increase aggregation propensity yielded larger cytoplasmic aggregates but reduced toxicity. Conversely, some mutations predicted to disfavor aggregation, generated smaller more dynamic TDP-43 assemblies with fluorescence recovery after photobleaching (FRAP) kinetics consistent with liquidity, but actually increased toxicity [96]. A detailed assessment of the in vitro propensity of disease-linked TDP-43 variants to phase-separate into liquid versus solid assemblies, juxtaposed with follow-up studies in newly developed cell-based assays that report on toxicity [32,65], could shed light on the prevalence of toxic liquid phases of TDP-43.
SGs: Protective, Problematic, or Both?
Another important question, as it pertains to mechanisms of FUS and TDP-43 toxicity, involves the role of SGs [15,41,42,87,97,98]. The overlap between ALS- and FTD-associated proteins and their local enrichment in SGs, led to the logical hypothesis that SGs may be crucibles of cytoplasmic PrLD aggregation that is the hallmark of these diseases [15]. However, it has also emerged that toxic TDP-43 aggregation can arise independently of SGs, and the RNA and PAR environment in SGs may be protective [22,32,65,99–101]. However, misregulated SG dynamics may still contribute to toxic aggregation. One possibility is that incomplete SG dissolution after the cessation of stress, results in small aggregates capable of nucleating further aggregation outside the previously protective environment of the SG [22]. If such nuclei were below the diffraction limit, they could escape detection upon SG dissolution, only to reappear later after a critical size was reached [102]. Indeed, using a light-induced oligomerization domain fused to the SG nucleating protein, G3BP1, Taylor and colleagues found that repetitive or persistent SG formation directly leads to TDP-43 aggregates [87]. Notably, like the light-driven TDP-43 aggregates formed independently of SGs by Donnelly and colleagues, these condensates contained C-terminal TDP-43 fragments and TDP-43 phosphorylated at serines 409 and 410, both hallmarks of pathological TDP-43 inclusions found in ALS and FTD patients [32,84,87]. Collectively, these studies suggest there are likely to be multiple routes to TDP-43 proteinopathy, with or without SGs as an intermediate [22,100]. It will be important to determine if interventions targeting TDP-43, such as RNA oligonucleotide therapies [32] or protein disaggregases [39,93–95,103–105], can target multiple paths to TDP-43 dysfunction. Additionally, TDP-43 is a component of other RNP granules, including NEAT1-nucleated paraspeckles [67], nuclear stress bodies [68], and transport granules in neuronal cells [90,106], raising the possibility that misregulation of other RNP granules could be the culprit in nucleating toxic PrLD aggregation. Dissecting the relationship between RNP granules and neurodegeneration will require improved understanding of granule dissolution [107,108], or as a failsafe, degradation [109], in addition to ongoing efforts to understand nucleation and to catalog constituent RNAs and proteins.
Drugging LLPS
Csat is partly a function of interaction strength which is itself a function of the solvent environment. A variety of compounds have been shown to dissolve membraneless organelles in cells and liquid droplets or hydrogels in vitro. The aliphatic alcohol, 1,6-hexanediol, was first observed to dissolve the nuclear pore, a membraneless organelle, and this observation was later extended to a multitude of RNP granules and in vitro condensates [110–112]. More recently, cellular concentrations of ATP, the crucial cellular metabolite, were shown to prevent protein phase separation in vitro, leading to the hypothesis that ATP acts as a ‘hydrotrope’ in cells to create a solvent favoring solubility [113]. Remarkably, the nontoxic and bioavailable molecules lipoic acid and lipoamide can modulate FUS LLPS in vitro and antagonize FUS LLPS in cells, which could in turn reduce FUS aggregation [114]. Likewise, tool compounds have emerged to combat TDP-43 LLPS and aggregation by binding interfaces important for self-assembly [115–117]. Compound screens have also revealed drugs that reduce SG assembly, including in motor neurons derived from iPSCs from patients harboring ALS-associated mutations [33]. One hit from this screen, mitoxantrone, was able to increase nuclear localization, and decrease SG accumulation, for a mutant form of TDP-43 lacking an NLS [33]. Together, these studies suggest that small-molecules could target PrLD LLPS, despite longstanding challenges inherent to drugging IDRs.
Our broadened definition of aberrant phase separation (Box 2) expands our imagination with respect to therapeutic interventions beyond dissolving condensates. Cells are revealing ways in which Csat is modulated through alterations in the valency of polymeric scaffolds [20–22,56] and post-translational modifications [18,48,85,86], and the concerted ‘co-scaffolding’ of dense phases [28,29,41]. Each suggests strategies for pharmacological intervention. In cases where condensation may alter function, partial modulation of enzymes that alter Csat through post-translational modifications or polymeric scaffolds, could conceivably effect highly nonlinear changes in cell behavior. Instead of targeting the disordered PrLDs central to aberrant LLPS directly, focusing on upstream enzymatic modifiers of LLPS provides targets such as folded active sites, which are often effectively targeted by existing classes of drugs. Could targeting enzymes that tune Csat, leverage small changes in enzyme output to exert highly nonlinear changes in phase behavior, while avoiding pleiotropic and off-target effects associated with more complete enzymatic inhibition? Recent work shows that some anticancer drugs selectively partition into condensates, in which their targets reside in a manner that appears independent from binding the drug target itself [118]. The extent to which small-molecule drugs may specifically partition into discrete condensates in cells, and how such partitioning impacts efficacy, are likely to be areas of considerable interest. A crucial question remains whether deleterious phases, including aggregates, can be reversed by small-molecule drugs, or whether proteins must be targeted prior to undergoing an aberrant phase transition. An orthogonal approach to drugging condensates involves mimicking their cellular regulation, including novel RNA therapeutic approaches (Box 3).
Concluding Remarks
FUS and TDP-43 are archetypes for PrLD-mediated self-assembly into condensates with a range of material properties. Further study of these proteins is likely to continue to uncover new biology that extends to other PrLD RBPs (see Outstanding Questions). Despite their similarities, they obey a distinct molecular grammar, are subject to distinct modes of regulation, and aggregate in distinct forms of disease. Thus, they suggest the complexity of cellular condensates that could be formed from the diverse collection of PrLD-containing proteins in the human proteome [12,14,15]. Together, they point towards the exploration of altered RNP granule dynamics as a biological framework to explore new therapeutic approaches for fatal neurodegenerative diseases. Systems-scale integration of proteomic, transcriptomic, imaging, genetic, and epidemiologic data will be required to inform hypotheses and guide drug-screening strategies to combat these diseases.
Outstanding Questions.
FUS and TDP-43 can condense into assemblies with a range of material states. How does the position of a condensate, on a continuum of liquid to aggregate, relate to function in various cellular contexts?
Which phase or phases, light or dense, represent the functional phase for FUS and TDP-43 for a given process? Does the functional phase vary between cell types?
What role do altered SG dynamics play in neurodegenerative disease onset?
What proteins or pathways are involved in RNP granule dissolution or degradation?
Do sequence and structural features of specific RNAs result in the nucleation of functional and materially distinct condensates?
Does modulating phase separation represent a viable therapeutic strategy for fatal neurodegenerative diseases involving PrLD aggregation?
Highlights.
Phase separation by proteins containing intrinsically-disordered regions (IDRs) underpins the biogenesis of functional membraneless organelles, as well as the formation of aggregated structures linked to neurodegenerative disease.
One class of IDR, termed a prion-like domain (PrLD), is frequently found in RNA-binding proteins, such as FUS and TDP-43, which form condensates with a range of material states.
FUS and TDP-43 form condensates in health and disease, and their phase separation is governed by distinct molecular grammar and regulation.
Aberrant phase separation is likely more diverse than liquid-to-solid transitions and may include inappropriate liquid phases.
Drugging condensates to treat disease is a promising strategy that may involve altering saturation concentrations of key scaffolds, or the specific partitioning of disruptive agents into discrete condensates.
Acknowledgments
We thank Hana Odeh, Katie Copley, and Charlotte Fare for helpful feedback on the manuscript. B.P. is supported by the BrightFocus Foundation Postdoctoral Fellowship A2019612F. J.S. is supported by grants from Target ALS (USA), ALSA, the Office of the Assistant Secretary of Defense for Health Affairs (USA), through the Amyotrophic Lateral Sclerosis Research Program (W81XWH-20-1-0242), the G. Harold & Leila Y. Mathers Foundation (USA), and National Institutes of Health (NIH) (R01GM099836, R21AG061784, and R21AG065854). J.S. is a consultant for Dewpoint Therapeutics and Maze Therapeutics.
Glossary
Heterotypic phase separation
a phase transition by two or more components, such as FUS and RNA, resulting in a dense phase composed of each component. Cellular condensates, such as SGs, are heterotypic.
Homotypic phase separation
a phase transition by a single component, such as purified FUS in an in vitro experiment. Homotypic phases are less likely to occur in cells.
Poly(ADP Ribose) (PAR)
a polymer of two or more ADP-ribose units, which can regulate phase separation. PAR can seed FUS condensates at sites of DNA damage, and aids in TDP-43 recruitment to SGs.
Saturation concentration (Csat)
the concentration above which a solution will demix into dense and light phases. The effective Csat of an RBP can be tuned by factors such as RNA, PAR, and post-translational modifications.
Spacers
the intervening residues in a protein, or nucleobases in a RNA, between the stickers. The length and flexibility of the spacers can affect the material properties of the condensate.
Stickers
amino acid residues or motifs in a protein, or nucleobases in a RNA, that engage in noncovalent interactions with other molecules to stabilize a condensate. The number and spacing of stickers can affect the material property of the condensate.
Stress granules (SGs)
highly heterotypic condensates that form in the cytoplasm in response to stress. SGs are enriched in translationally arrested mRNAs and PrLD-containing RBPs, including FUS and TDP-43.
References
- 1.Boeynaems S et al. (2018) Protein phase separation: a new phase in cell biology. Trends Cell Biol. 28, 420–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poudyal RR et al. (2018) Physical principles and extant biology reveal roles for RNA-containing membraneless compartments in origins of life chemistry. Biochemistry 57, 2509–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brangwynne CP et al. (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732 [DOI] [PubMed] [Google Scholar]
- 4.Hyman AA et al. (2014) Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol 30, 39–58 [DOI] [PubMed] [Google Scholar]
- 5.Elbaum-Garfinkle S and Brangwynne CP (2015) Liquids, fibers, and gels: the many phases of neurodegeneration. Dev. Cell 35, 531–532 [DOI] [PubMed] [Google Scholar]
- 6.Li P et al. (2012) Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin EW and Mittag T (2018) Relationship of sequence and phase separation in protein low-complexity regions. Biochemistry 57, 2478–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pak CW et al. (2016) Sequence determinants of intracellular phase separation by complex coacervation of a disordered protein. Mol. Cell 63, 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandopulle M et al. (2019) Inherited and sporadic amyotrophic lateral sclerosis and fronto-temporal lobar degenerations arising from pathological condensates of phase separating proteins. Hum. Mol. Genet 28, R187–R196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.St George-Hyslop P et al. (2018) The physiological and pathological biophysics of phase separation and gelation of RNA binding proteins in amyotrophic lateral sclerosis and fronto-temporal lobar degeneration. Brain Res. 1693, 11–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harmon TS et al. (2017) Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. Elife 6, e30294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.March ZM et al. (2016) Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 1647, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison AF and Shorter J (2017) RNA-binding proteins with prion-like domains in health and disease. Biochem. J 474, 1417–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King OD et al. (2012) The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li YR et al. (2013) Stress granules as crucibles of ALS pathogenesis. J. Cell Biol 201, 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brady JP et al. (2017) Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc. Natl. Acad. Sci. U. S. A 114, E8194–E8203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holehouse AS and Pappu RV (2018) Functional implications of intracellular phase transitions. Biochemistry 57, 2415–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang A et al. (2018) A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 37, e97452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monahan Z et al. (2017) Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 36, 2951–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz JC et al. (2013) RNA seeds higher-order assembly of FUS protein. Cell Rep. 5, 918–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burke KA et al. (2015) Residue-by-residue view of in vitro FUS granules that bind the C-terminal domain of RNA polymerase II. Mol. Cell 60, 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGurk L et al. (2018) Poly(ADP-Ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol. Cell 71, 703–717.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H et al. (2015) RNA controls polyQ protein phase transitions. Mol. Cell 60, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson PT et al. (2019) Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142, 1503–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin Y et al. (2015) Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jain S et al. (2016) ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164, 487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Markmiller S et al. (2018) Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172, 590–604.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi J-M et al. (2019) LASSI: A lattice model for simulating phase transitions of multivalent proteins. PLoS Comput. Biol 15, e1007028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riback JA et al. (2020) Composition-dependent thermodynamics of intracellular phase separation. Nature 581, 209–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel A et al. (2015) A Liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 [DOI] [PubMed] [Google Scholar]
- 31.Leung AKL (2020) Poly(ADP-ribose): a dynamic trigger for biomolecular condensate formation. Trends Cell Biol. 30, 370–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mann JR et al. (2019) RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102, 321–338.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang MY et al. (2019) Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 103, 802–819.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shin Y et al. (2017) Spatiotemporal control of intracellular phase transitions using light-activated optoDroplets. Cell 168, 159–171.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong C-E and Tsai K-J (2019) TDP-43 proteinopathy impairs neuronal mRNP granule mediated postsynapticlocal translation and mRNA metabolism. bioRxiv 10.1101/589416 Published online April 04,2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ciryam P et al. (2017) Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci 114, E3935–E3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molliex A et al. (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khong A et al. (2017) The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol. Cell 68, 808–820.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo L et al. (2018) Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173, 677–692.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banani SF et al. (2016) Compositional control of phase-separated cellular bodies. Cell 166, 651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanders DW et al. (2020) Competing protein-RNA interaction networks control multiphase intracellular organization. Cell 181, 306–324.e28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang P et al. (2020) G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell 181, 325–345.e28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McSwiggen DT et al. (2019) Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev. 33, 1619–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ratti A and Buratti E (2016) Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem 138, 95–111 [DOI] [PubMed] [Google Scholar]
- 45.Ling S-C et al. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J et al. (2018) A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 174, 688–699.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun Z et al. (2011) Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 9, e1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qamar S et al. (2018) FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell 173, 720–734.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ozdilek BA et al. (2017) Intrinsically disordered RGG/RG domains mediate degenerate specificity in RNA binding. Nucleic Acids Res. 45, 7984–7996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chong PA et al. (2018) RGG/RG motif regions in RNA binding and phase separation. J. Mol. Biol 430, 4650–4665 [DOI] [PubMed] [Google Scholar]
- 51.Maharana S et al. (2018) RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murray DT et al. (2017) Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 171, 615–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo F et al. (2018) Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat. Struct. Mol. Biol 25, 341–346 [DOI] [PubMed] [Google Scholar]
- 54.Hughes MP et al. (2018) Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 359, 698–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mastrocola AS et al. (2013) The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem 288, 24731–24741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altmeyer M et al. (2015) Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun 6, 8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshizawa T et al. (2018) Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell 173, 693–705.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hofweber M et al. (2018) Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 173, 706–719.e13 [DOI] [PubMed] [Google Scholar]
- 59.Niaki AG et al. (2020) Loss of dynamic RNA interaction and aberrant phase separation induced by two distinct types of ALS/FTD-linked FUS mutations. Mol. Cell 77, 82–94.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rhine K et al. (2020) ALS/FTLD-linked mutations in FUS glycine residues cause accelerated gelation and reduced interactions with wild-type FUS. Mol. Cell 80, 666–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Darovic S et al. (2015) Phosphorylation of C-terminal tyrosine residue 526 in FUS impairs its nuclear import. J. Cell Sci 128, 4151–4159 [DOI] [PubMed] [Google Scholar]
- 62.Rhoads SN et al. (2018) The role of post-translational modifications on prion-like aggregation and liquid-phase separation of FUS. Int. J. Mol. Sci 19, 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sloutsky R and Naegle KM (2018) Proteome-level analysis indicates global mechanisms for post-translational regulation of RRM domains. J. Mol. Biol 430, 41–44 [DOI] [PubMed] [Google Scholar]
- 64.Owen I et al. (2020) The prion-like domain of fused in sarcoma is phosphorylated by multiple kinases affecting liquid-and solid-phase transitions. Mol. Biol. Cell 31, 2522–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gasset-Rosa F et al. (2019) Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102, 339–357.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vogler TO et al. (2018) TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 563, 508–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nishimoto Y et al. (2013) The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Udan-Johns M et al. (2014) Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum. Mol. Genet 23, 157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McGurk L et al. (2018) Poly(ADP-ribose) engages the TDP-43 nuclear-localization sequence to regulate granulo-filamentous aggregation. Biochemistry 57, 6923–6926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnson BS et al. (2009) TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem 284, 20329–20339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neumann M et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 72.Buratti E (2015) Functional significance of TDP-43 mutations in disease. Adv. Genet 91, 1–53 [DOI] [PubMed] [Google Scholar]
- 73.Bentmann E et al. (2012) Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem 287, 23079–23094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Martin EW et al. (2020) Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 367, 694–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jiang L-L et al. (2017) The N-terminal dimerization is required for TDP-43 splicing activity. Sci. Rep 7, 6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Afroz T et al. (2017) Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun 8, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmidt HB et al. (2019) Phase separation-deficient TDP43 remains functional in splicing. Nat. Commun 10, 4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Conicella AE et al. (2020) TDP-43 α-helical structure tunes liquid–liquid phase separation and function. Proc. Natl. Acad Sci. U.S.A 117, 5883–5894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lim L et al. (2016) ALS-causing mutations significantly perturb the self-assembly and interaction with nucleic acid of the intrinsically disordered prion-like domain of TDP-43. PLoS Biol. 14, e1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Conicella AE et al. (2016) ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24, 1537–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin Y et al. (2020) Redox-mediated regulation of an evolutionarily conserved cross-β structure formed by the TDP43 low complexity domain. Proc. Natl. Acad. Sci. U. S. A 117, 28727–28734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li H-R et al. (2018) TAR DNA-binding protein 43 (TDP-43) liquid–liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem 293, 6090–6098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guenther EL et al. (2018) Atomic structures of TDP-43 LCD segments and insights into reversible or pathogenic aggregation. Nat. Struct. Mol. Biol 25, 463–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neumann M et al. (2009) Phosphorylation of S409/410 of TDP-43 is a consistent feature in allsporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 117, 137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cohen TJ et al. (2015) An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun 6, 5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu H et al. (2020) HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science, eabb4309 10.1126/science.abb4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang P et al. (2019) Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. Elife 8, e39578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chou C-C et al. (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/ FTD. Nat. Neurosci 21, 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hutten S et al. (2020) Nuclear import receptors directly bind to arginine-rich dipeptide repeat prtoeins and suppress their pathologicalinteractions. Cell Rep. 33, 108538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gopal PP et al. (2017) Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc. Natl. Acad. Sci 114, E2466–E2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim HJ et al. (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Elden AC et al. (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jackrel ME et al. (2014) Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 156, 170–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tariq A et al. (2018) Potentiating Hsp104 activity via phosphomimetic mutations in the middle domain. FEMS Yeast Res. 18, foy042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tariq A et al. (2019) Mining disaggregase sequence space to safely counter TDP-43, FUS, and α-synuclein proteotoxicity. Cell Rep. 28 e6, 2080–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bolognesi B et al. (2019) The mutational landscape of a prion-like domain. Nat. Commun 10, 4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Riback JA et al. (2017) Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell 168, 1028–1040.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guillén-Boixet J et al. (2020) RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell 181, 346–361.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hans F et al. (2020) Multiple distinct pathways lead to hyperubiquitylated insoluble TDP-43 protein independent of its translocation into stress granules. J. Biol. Chem 295, 673–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Boeynaems S and Gitler AD (2018) Pour some sugar on TDP(−43). Mol. Cell 71, 649–651 [DOI] [PubMed] [Google Scholar]
- 101.Zhang J et al. (2020) Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature Published online August 31, 2020. 10.1038/s41586-020-2709-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Narayanan A et al. (2019) A first order phase transition mechanism underlies protein aggregation in mammalian cells. Elife 8, e39695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jackrel ME and Shorter J (2015) Engineering enhanced protein disaggregases for neurodegenerative disease. Prion 9, 90–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shorter J (2016) Engineering therapeutic protein disaggregases. Mol. Biol. Cell 27, 1556–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cupo RR and Shorter J (2020) Skd3 (human ClpB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations. Elife 9, e55279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alami NH et al. (2014) Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang B et al. (2019) ULK1 and ULK2 regulate stress granule disassembly through phosphorylation and activation of VCP/ p97. Mol. Cell 74, 742–757.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kroschwald S et al. (2015) Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. Elife 4, e06807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Buchan JR et al. (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153, 1461–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lin Y et al. (2016) Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 167, 789–802.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ribbeck K and Görlich D (2002) The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J. 21, 2664–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kroschwald S et al. (2017) Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters Published online May 22, 2017. 10.19185/matters.201702000010 [DOI] [Google Scholar]
- 113.Patel A et al. (2017) ATP as a biological hydrotrope. Science 356, 753–756 [DOI] [PubMed] [Google Scholar]
- 114.Wheeler RJ et al. (2019) Small molecules for modulating protein driven liquid-liquid phase separation in treating neurodegenerative disease. BioRxiv Published online August 21, 2019. 10.1101/721001 [DOI] [Google Scholar]
- 115.Babinchak WM et al. (2020) Small molecules as potent biphasic modulators of protein liquid-liquid phase separation. Nat. Commun 11, 5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.François-Moutal L et al. (2019) Small molecule targeting TDP-43’s RNA recognition motifs reduces locomotor defects in a Drosophila model of amyotrophic lateral sclerosis (ALS). ACS Chem. Biol 14, 2006–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mollasalehi N et al. (2020) An allosteric modulator of RNA binding targeting the N-terminal domain of TDP-43 yields neuroprotective properties. ACS Chem. Biol 15, 2854–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Klein IA et al. (2020) Partitioning of cancer therapeutics in nuclear condensates. Science 368, 1386–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shorter J and Lindquist S (2005) Prions as adaptive conduits of memory and inheritance. Nat. Rev. Genet 6, 435–450 [DOI] [PubMed] [Google Scholar]
- 120.Aguzzi A and De Cecco E (2020) Shifts and drifts in prion science. Science 370, 32–34 [DOI] [PubMed] [Google Scholar]
- 121.Cushman M et al. (2010) Prion-like disorders: blurring the divide between transmissibility and infectivity. J. Cell Sci 123, 1191–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Newby GA and Lindquist S (2013) Blessings in disguise: biological benefits of prion-like mechanisms. Trends Cell Biol 23, 251–259 [DOI] [PubMed] [Google Scholar]
- 123.Chakravarty AK and Jarosz DF (2018) More than just a phase: prions at the crossroads of epigenetic inheritance and evolutionary change. J. Mol. Biol 430, 4607–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jarosz DF et al. (2014) Cross-kingdom chemical communication drives a heritable, mutually beneficial prion-based transformation of metabolism. Cell 158, 1083–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jarosz DF et al. (2014) An evolutionarily conserved prion-like element converts wild fungi from metabolic specialists to generalists. Cell 158, 1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Harvey ZH et al. (2020) A prion epigenetic switch establishes an active chromatin state. Cell 180, 928–940.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lancaster AK et al. (2014) PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 30, 2501–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chakravarty AK et al. (2020) A non-amyloid prion particle that activates a heritable gene expression program. Mol. Cell 77, 251–265.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Han TW et al. (2012) Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149, 768–779 [DOI] [PubMed] [Google Scholar]
- 130.Porta S et al. (2018) Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun 9, 4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nonaka T et al. (2013) Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134 [DOI] [PubMed] [Google Scholar]
- 132.Laferrière F et al. (2019) TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci 22, 65–77 [DOI] [PubMed] [Google Scholar]
- 133.Jakobson CM and Jarosz DF (2018) Organizing biochemistry in space and time using prion-like self-assembly. Curr. Opin. Syst. Biol 8, 16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Halfmann R (2016) A glass menagerie of low complexity sequences. Curr. Opin. Struct. Biol 38, 18–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chakrabortee S et al. (2016) Intrinsically disordered proteins drive emergence and inheritance of biological traits. Cell 167, 369–381.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Peeples W and Rosen MK (2020) Phase separation can increase enzyme activity by concentration and molecular organization. bioRxiv Published online September 17, 2020. 10.1101/2020.09.15.299115 [DOI] [Google Scholar]
- 137.Loring HS and Thompson PR (2020) A liquid-to-solid phase transition enhances the catalytic activity of SARM1. bioRxiv Published online August 29, 2020. 10.1101/2020.08.28.272377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Leavitt BR and Tabrizi SJ (2020) Antisense oligonucleotides for neurodegeneration. Science 367, 1428–1429 [DOI] [PubMed] [Google Scholar]
- 139.Wang C et al. (2020) Stress induces dynamic, cytotoxicity-antagonizing TDP-43 nuclear bodies via paraspeckle LncRNA NEAT1-mediated liquid-liquid phase separation. Mol. Cell 79, 443–458.e7 [DOI] [PubMed] [Google Scholar]