The TSC1–TSC2 complex: a molecular switchboard controlling cell growth (original) (raw)
. Author manuscript; available in PMC: 2009 Aug 31.
Published in final edited form as: Biochem J. 2008 Jun 1;412(2):179–190. doi: 10.1042/BJ20080281
Abstract
TSC1 and TSC2 are the tumour-suppressor genes mutated in the tumour syndrome TSC (tuberous sclerosis complex). Their gene products form a complex that has become the focus of many signal transduction researchers. The TSC1–TSC2 (hamartin–tuberin) complex, through its GAP (GTPase-activating protein) activity towards the small G-protein Rheb (Ras homologue enriched in brain), is a critical negative regulator of mTORC1 (mammalian target of rapamycin complex 1). As mTORC1 activity controls anabolic processes to promote cell growth, it is exquisitely sensitive to alterations in cell growth conditions. Through numerous phosphorylation events, the TSC1–TSC2 complex has emerged as the sensor and integrator of these growth conditions, relaying signals from diverse cellular pathways to properly modulate mTORC1 activity. In the present review we focus on the molecular details of TSC1–TSC2 complex regulation and function as it relates to the control of Rheb and mTORCl.
Keywords: cell growth control, hamartin (TSC1), mammalian target of rapamycin complex 1 (mTORC1), Ras homologue enriched in brain (Rheb), tuberin (TSC2), tuberous sclerosis complex (TSC)
INTRODUCTION
The ability to sense and integrate diverse environmental signals is essential for both prokaryotic and eukaryotic cells to mount appropriate physiological responses. Cells have the capacity to sense two fundamental signals from their growth environment: the availability of nutrients and conditions of stress. Although many bacteria can also detect the presence of other bacteria through quorum sensing, eukaryotic cells, especially those of multicellular organisms, can perceive an enormous variety of additional signals from surrounding cells in the form of growth factors, cytokines and cell-adhesion molecules. Significant progress has been made in delineating the biochemical pathways that sense and relay individual signals within cells. However, the molecular mechanisms by which eukaryotic cells integrate multiple concurrent signals to appropriately alter cell physiology are less well understood. Underlying this problem is that, to date, few points of integration have been identified that can sense and respond to a variety of distinct signals.
Over the past decade, a protein complex consisting of the TSC1 (also known as hamartin) and TSC2 (also known as tuberin) proteins has been the focus of geneticists, cell biologists and biochemists alike. Through their collective work, a central role for this complex in linking a network of seemingly unconnected signalling pathways has been revealed. As detailed below, the TSC1–TSC2 complex has emerged as a critical integrator of growth-factor, nutrient and stress signals to control protein synthesis, cell growth and other cellular processes.
TSC (TUBEROUS SCLEROSIS COMPLEX) AND THE TSC1–TSC2 COMPLEX
The TSC1 and TSC2 genes were identified in 1997 and 1993 respectively as the genetic loci mutated in the autosomal dominant tumour syndrome TSC [1–3]. TSC is a multisystemic disorder characterized by the development of numerous benign tumours (e.g. hamartomas) most commonly affecting the brain, kidneys, skin, heart and lungs. The clinical manifestations of TSC with the highest morbidity include severe neurological disorders (i.e. epilepsy, autism and mental retardation), renal angiomyolipomas and pulmonary LAM (lymphangioleiomyomatosis). TSC2 mutations have also been found in sporadic (non-TSC-associated) cases of LAM [4], which primarily affects women and is characterized by smooth-muscle cell proliferation and cystic destruction of the lung. For more details on TSC genetics and pathology, readers should refer to the excellent review by Crino and colleagues [5].
Upon identification of TSC1 and TSC2 as novel tumour suppressor genes, attention immediately turned to understanding the function of their distinct gene products. The 140 kDa TSC1 and 200 kDa TSC2 proteins share no homology with each other and very little with other proteins (Figure 1A). However, orthologues can be found in most eukaryotic cells, including the fission yeast Schizosaccharomyces pombe. The only putative functional domain that is apparent in these two proteins is a region of homology at the C-terminus of TSC2 to the GAP (GTPase-activating protein) domain of Rap1GAP. Early searches for a GTPase target revealed that this domain of TSC2 has weak in vitro GAP activity towards both Rap1 and Rab5 [6,7], which are two rather distinct small G-proteins. Although these targets may not be relevant in vivo, these studies were important in demonstrating that this region of TSC2 was indeed a GAP domain. Importantly, missense mutations in this domain are found at some frequency in TSC patients [8], and cell-culture and xenograft experiments have suggested that this domain contains the tumoursuppressor activity of TSC2 [9].
Figure 1. Schematic of the TSC1 and TSC2 proteins and the components and downstream targets of the two mTOR complexes.
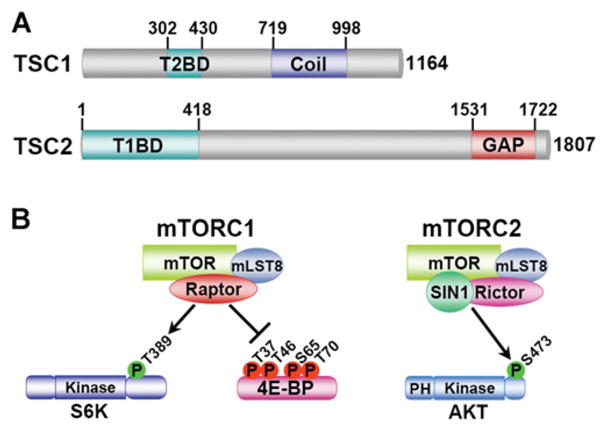
(A) The functional domains on TSC1 and TSC2 are depicted schematically with numbers representing amino acid residues on the full-length human proteins. Abbreviations: T2BD, TSC2-binding domain; T1BD, TSC1 -binding domain; Coil, predicted coiled-coil domain; GAP, GAP domain homologous with that in Rap1GAP. (B) mTOR is found in two functionally distinct complexes, mTORC1 and mTORC2, with distinct downstream substrates. mLST8, mammalian LST8; PH, pleckstrin homology domain.
An important finding in understanding the functional link between the TSC1 and TSC2 proteins in tumour suppression came when they were shown to physically associate [10,11]. With respect to nomenclature, it is worth distinguishing between this complex, the TSC1–TSC2 complex, and the multifaceted disease TSC for which its components are named. This has been a point of confusion in the literature. TSC1 and TSC2 associate through roughly mapped regions [12] (Figure 1A) to form what is believed to be a heterodimeric complex, and this is also true of orthologues in the fruitfly Drosophila [13,14] and the yeast S. pombe [15]. Genetic studies of TSC1 and TSC2 in humans, mice, Drosophila (see below) and yeast strongly suggest that their gene products are interdependent and that these proteins function primarily as a complex. Although the GAP activity of TSC2 is of obvious functional importance to the complex, TSC1 is required to stabilize TSC2 and prevent its ubiquitin-mediated degradation [16,17]. Aside from those affecting the TSC2 GAP domain, missense mutations in TSC1 or TSC2 commonly destabilize the complex, thereby leading to TSC2 degradation [12,18]. Although it remains possible that TSC1 and TSC2 have distinct functions outside of the TSC1-TSC2 complex, such putative functions are, as yet, poorly defined and will not be included in our discussion below.
GENETIC LINK TO THE INSULIN/IGF-1 (INSULIN-LIKE GROWTH FACTOR 1) PATHWAY
The major breakthrough in understanding the physiological function of the TSC1–TSC2 complex and its involvement in a well-studied signal-transduction pathway came from studies on organ growth in Drosophila. Three very similar, but independent, genetic screens for regulators of organ growth identified loss-of-function mutations in the Drosophila Tsc1 gene as causative of an eye-overgrowth phenotype [13,14,19]. The organ overgrowth resulting from Tsc1 mutations was found to result from both increased cell proliferation and, most strikingly, a cell-autonomous increase in size. Importantly, Tsc2 mutations also result in increased cell size [19], and Tsc1 Tsc2 double mutants have phenotypes identical with that of either single mutant [13]. Furthermore, overexpression of both Tsc1 and Tsc2 dramatically decreases organ and cell size, whereas overexpression of either alone does not [13,14,19]. These genetic data strongly implicate the complex between TSC1 and TSC2 as the functional unit for these tumour suppressors, at least for the control of cell proliferation and growth.
As signalling downstream of the InR (insulin/IGF-1) orthologue was known to be a critical regulator of cell size in Drosophila (for a review, see [20]), all three groups [13,14,19] performed genetic epistasis experiments to determine whether the TSC1–TSC2 complex was involved in this pathway. Indeed Tsc1 and Tsc2 mutations and co-overexpression exhibited strong genetic interactions with components of InR signalling, including InR, PTEN (phosphatase and tensin homologue deleted on chromosome 10), Akt (also known as protein kinase B) and S6K (70 kDa ribosomal protein S6 kinase; also known as p70S6K), for the control of cell growth. Most striking was the finding that the lethality of InR loss-of-function mutants could be partially rescued by loss of one functional copy of either Tsc1 or Tsc2 [13]. Collectively, these studies demonstrated that the TSC1–TSC2 complex was a critical negative regulator of signalling downstream of InR and Akt. However, these genetic analyses could not distinguish between this complex acting within the InR pathway or in a parallel pathway merging on common downstream targets.
Subsequent to these genetic studies, biochemical studies found that the TSC1–TSC2 complex acts directly downstream of the serine/threonine kinase Akt within this pathway. Employing a combination of biochemistry and bioinformatics, an unbiased screen for new Akt substrates in mammalian cells found that Akt directly phosphorylates two residues on TSC2 (Ser939 and Thr1462 in full-length human TSC2) that are conserved in the Drosophila orthologue [21]. Two additional studies, examining this possible link directly, also found that Akt phosphorylates these two sites within cells [22], and this is also true of the conserved Drosophila sites (Ser924 and Thr1518) [23]. Akt-mediated phosphorylation of these sites on TSC2 appears to be important for insulin and other growth factors to activate key downstream regulators of cell growth (see below). The molecular mechanisms of TSC1–TSC2 complex regulation by multisite phosphorylation are discussed in detail below.
CELL GROWTH CONTROL THROUGH TOR (TARGET OF RAPAMYCIN) AND S6K
The seminal studies in Drosophila, described above, and subsequent studies, strongly suggested that the TSC1–TSC2 complex inhibits cell growth by inhibiting the activation of S6K. In previous genetic studies, S6K was found to be a critical driver of cell growth, as S6k mutant fruitflies were found to be smaller, a phenomenon due exclusively to a decrease in cell size [24]. S6k loss-of-function mutations suppressed the eye-overgrowth phenotype of Tsc2 mutants [14,25,26], and S6k overexpression suppressed the decreased-cell-size phenotype caused by _Tscl a_nd Tsc2 overexpression [14,19]. Importantly, loss-of-function mutations in the gene encoding Drosophila TOR, an essential upstream activator of S6K, also suppressed Tsc2 mutant phenotypes [25], and mutations reducing TOR and S6K activity were found to rescue the lethality caused by Tsc1 mutations [26]. These studies demonstrated that the TSC1–TSC2 complex acts as an upstream inhibitor of S6K, perhaps through inhibition of TOR signalling.
TOR proteins are serine/threonine kinases of the PIKK (phosphoinositide 3-kinase-related kinase) family, with orthologues found in all eukaryotes. TOR proteins play an evolutionarily conserved role in the control of cell growth (i.e. an increase in cell size), but they have also been found to regulate aspects of cell proliferation, survival and metabolism in specific settings (for focused reviews on the function of TOR proteins, see [27–30]). TOR proteins are found in two functionally distinct complexes that coexist in cells from yeast to humans [31] (Figure 1B). The best characterized of the two complexes is TORC1 (TOR complex 1), which comprises TOR, Raptor [regulatory associated protein of mTOR (mammalian target of rapamycin)] and LST8 (lethal with SEC13 protein 8). TORC2 is comprised of TOR, Rictor (rapamycin-insensitive companion of mTOR), SIN1 (stress-activated-protein-kinase-interacting protein 1) and LST8 (reviewed in [29,30]). Little is currently known regarding the regulation and function of TORC2. Although it has been shown to regulate aspects of the actin cytoskeleton [32,33], the only direct target of mTORC2 (mammalian TORC2) identified to date is Akt. mTORC2 phosphorylates Akt on a residue (Ser473) within a hydrophobic motif C-terminal to its kinase domain, and this contributes to the full activation of Akt downstream of growth factors [34]. TORC1 is strongly sensitive to inhibition by the naturally occurring compound rapamycin and by a large number of synthetic analogues (e.g. RAD001 and CCI779), whereas TORC2 is insensitive to acute rapamycin treatment. Rapamycin binds to FKBP12 (FK506-binding protein 12), a highly conserved peptidyl-prolyl isomerase of the immunophilin family [35], and the FKBP12–rapamycin complex binds to and inhibits TOR within TORC1, but not TORC2 (reviewed in [29]). Conclusions regarding TOR protein function have relied heavily on the use of rapamycin and are therefore probably specific to TORC1. However, prolonged rapamycin treatment affects mammalian TORC2 (mTORC2) assembly and can block mTORC2 function in some cells [36].
mTORC1 (mammalian TORC1) has two well-characterized classes of downstream targets, the ribosomal S6 kinases S6K1 and S6K2, and 4E-BP1 and 4E-BP2 [eIF4E (eukaryotic translation initiation factor 4E)-binding proteins 1 and 2], which possess residues found to be directly phosphorylated by the mTOR kinase within this complex (Figure 1B; reviewed in [27–30]). S6K1 and S6K2 (referred to collectively here as S6K) are phosphorylated by mTORC1 on a hydrophobic motif that shows high similarity to that of Akt. Therefore S6K and Akt, which are members of the same kinase family, are phosphorylated on a similar motif by mTOR, but mTOR regulates these two kinases as part of two distinct complexes, namely mTORC1 and mTORC2 respectively. Phosphorylation of S6K on Thr389 (numbering according to the 70 kDa isoform of S6K1) within this motif is required for its activation, and rapamycin treatment of cells leads to rapid loss of S6K phosphorylation and activity [37,38]. Once active, S6K phosphorylates downstream substrates, such as ribosomal S6 and eIF4B, to promote mRNA translation. 4E-BP1 is phosphorylated by mTORC1 on residues within a distinct motif from that found in S6K. Four separate proline-directed sites (Ser/Thr-Pro) on 4E-BP1 (Thr37, Thr47, Ser65 and Thr70) can be phosphorylated by mTORC1 and are sensitive to rapamycin treatment [39,40]. Phosphorylation of the 4E-BPs triggers their release from eIF4E at the 5′ 7-methyl-GTP cap of mRNAs, thereby precipitating events initiating cap-dependent translation. Therefore mTORC1 activation promotes cell growth, at least in part, by increasing the anabolic process of protein synthesis through activation of S6K and inhibition of 4E-BP [41].
It is now recognized that the primary function of the TSC1–TSC2 complex is as a critical negative regulator of mTORC1 activation. The Drosophila genetic experiments described above demonstrated that the TSC1–TSC2 complex inhibits S6K activation, suggesting that the complex might specifically inhibit TORC1 signalling. Indeed, subsequent experiments in mammalian cells demonstrated that mTORC1-mediated (i.e. rapamycin-sensitive) phosphorylation of both S6K1 and 4E-BP1 is inhibited by TSC1–TSC2 complex overexpression and is activated in cells lacking the TSC1–TSC2 complex [21,22,25,42–45]. Most striking is the effect of TSC gene disruption on mTORC1 signalling, which becomes robustly activated in a growth-factor-independent manner. Elevated mTORC1 activity has also been detected in all rodent and human tumours and tumour-derived cell lines lacking TSC1 or TSC2 (see, for example, [42,45–50]).
Aberrant mTORC1 signalling is believed to be the driving force behind tumour formation triggered by loss of these tumour suppressors. In rodent models of TSC, short-term inhibition of mTORC1 with rapamycin (4 days or less) initiates apoptosis within tumours [46,51], and prolonged treatment with rapamycin, or its analogue CCI779, blocks tumour formation and causes tumour regression [52,53]. On the basis of our molecular understanding of this critical connection between the TSC1–TSC2 complex and mTORC1 inhibition, several clinical trials have been initiated over the past few years to test the efficacy of rapamycin and its analogues for the treatment of specific clinical manifestions of the TSC and LAM diseases. Although it is still early days with these trials, the results thus far demonstrate that mTORC1 inhibitors reproducibly decrease the size of tumours with TSC gene mutations [54–56].
Rheb (Ras HOMOLOGUE ENRICHED IN BRAIN): THE SMALL G-PROTEIN TARGET OF THE TSC2 GAP DOMAIN
As with the TSC–TOR connection, the small G-protein regulated by the TSC1–TSC2 complex to control mTORC1 activation was identified and characterized through a combination of genetics and biochemistry. Rheb is a member of the Ras superfamily that appears to be conserved in all eukaryotes and, despite the term ‘brain’ in its name, is in fact ubiquitously expressed in mammals. In Drosophila genetic studies, Rheb was found to promote cell growth in a TOR- and S6K-dependent manner [57–59] and to act downstream of TSC 1 and TSC2 [58–60]. Biochemical studies demonstrated that the TSC1–TSC2 complex directly regulates Rheb in both Drosophila and mammalian cells [60–64]. The GAP domain of TSC2 within this complex stimulates the intrinsic GTPase activity of Rheb [60–62], which is low relative to that of other small G-proteins [65], thereby expediting the conversion of Rheb-GTP into Rheb-GDP. In mammalian cells, a Rheb homologue exists, named Rhebl1 (Rheb like-1) or Rheb2, that appears to have overlapping functions with Rheb in controlling mTORC1 signalling downstream of TSC1 and TSC2 [66,67]. Collectively, these studies demonstrate that the TSC1–TSC2 complex inhibits mTORC1 signalling through its GAP activity towards Rheb (Figure 2A). Whether a GEF (guanine-nucleotide exchange factor) exists that counters the activity of the TSC1-TSC2 complex in the control of Rheb remains unknown. A study of the role of TCTP (translationally controlled tumour protein) in Drosophila development suggested that it might function as a Rheb GEF [68]. However, whether this very abundant, often secreted, chaperone-like protein plays any in vivo role in Rheb regulation is unclear.
Figure 2. Two models of the regulation of mTORC1 by TSC1–TSC2 and Rheb.

(A) Model 1. Under poor growth conditions, the TSC1–TSC2 complex acts as a GAP for Rheb, thereby stimulating the conversion of Rheb–GTP into Rheb–GDP. Under optimal growth conditions, Rheb–GTP accumulates and binds directly to mTOR within mTORC1 to somehow activate it. Whether a GEF exists that counters the activity of the TSC1–TSC2 complex for Rheb regulation is not known. mLST8, mammalian LST8. (B) Model 2. The TSC1–TSC2 complex acts as described in (A) above, but Rheb–GTP activates mTORC1 through binding to FKBP38, thereby triggering its release from mTOR. In this model, FKBP38 inhibits mTORC1 under poor growth conditions in a manner analogous to the rapamycin–FKBP12 complex.
Although genetic and biochemical studies strongly suggest that GTP-bound Rheb potently activates mTORC1, the molecular mechanism has been somewhat elusive. Purified Rheb loaded with GTP, but not GDP, stimulates mTORC1’s in vitro kinase activity in a dose-dependent manner [69], suggesting that the regulation is rather direct (Figure 2A). Rheb has been found to bind to mTOR in overexpression studies [67,70]. However, associations between endogenous Rheb and components of mTORC1 have not been detected. In general, Ras-related small G-proteins bind to their downstream effectors only in the GTP-bound state. Paradoxically, Rheb has been found to bind more tightly to mTOR in its GDP-bound or nucleotide-free states [70]. However, the interaction between Rheb and TOR from fission yeast appears to be dependent on Rheb binding to GTP [71]. Recently, Rheb was found to directly associate with the FKBP12 homologue FKBP38 (also known as FKBP8), and this interaction appears to be stronger with GTP-loaded Rheb [72]. In addition, FKBP38 binds to mTORC1 preferentially under poor growth conditions (i.e. serum or amino acid withdrawal), when mTORC1 is inactive, and FKBP38 appears to associate through the FRB (FKBP12rapamycin-binding) domain of mTOR. That study suggests an intriguing model in which Rheb-GTP binds to FKBP38 and triggers its release from mTORC1, thereby stimulating mTORC1 activation (Figure 2B). In support of this model, an independent study carried out before that one found that decreasing FKBP38 expression with antisense oligonucleotides blocked the growth-inhibitory effects of TSC1–TSC2 overexpression [73]. Although more studies are needed, these findings suggest that FKBP38 might be a Rheb effector that regulates mTORC1 and, perhaps, unknown targets downstream of the TSC1–TSC2 complex and Rheb.
SIGNAL INTEGRATION BY THE TSC1–TSC2 COMPLEX
Through its downstream targets, TORC1 activity leads to acute activation of cap-dependent translation, but also leads to a more prolonged increase in the protein-synthetic capacity of the cell by up-regulating ribosome biogenesis (reviewed in [29]). As protein synthesis is the most energy-consuming anabolic process within eukaryotic cells, it is not surprising that TORC1 has evolved the ability to sense perturbations in optimal growth conditions. Through upstream signalling events, TORC1 senses the availability of intracellular nutrients, the energy status of the cell, conditions of cellular stress, and, in higher eukaryotes, the state of the whole organism through growth factors and cytokines. With the discovery of a small-G-protein switch upstream of TORC1 in most eukaryotes, many researchers have now addressed the possibility that TORC1 might sense some of these signals through pathways affecting the TSC1–TSC2 complex and Rheb. What has emerged from these studies is that the TSC1–TSC2 complex, especially in higher eukaryotes, is a central hub of signal transduction within the cell. Through numerous phosphorylation events on TSC1 and TSC2 (Figure 3 and Table 1), this complex senses signals from a large number of distinct signalling pathways to modulate mTORC1 activity. Although the TSC1–TSC2 complex and Rheb are likely to have additional cellular functions, our discussion below is focused on mechanisms of mTORC1 regulation through these proteins.
Figure 3. Direct phosphorylation sites on TSC1 and TSC2.

(A) Phosphorylation of TSC1 at specific residues by different protein kinases. Among these, only the Thr417 site is conserved between human and Drosophila TSC1. For the alignment, the phosphorylation site is indicated in yellow, identical residues in green, strongly similar residues in blue and semi-conserved residues in grey. (B) Same as above, but for TSC2. Among these sites, only the aligned sites are conserved between human and Drosophila TSC2. It should be noted that the AMPK/GSK3 sites in Drosophila (Ser1107/Ser1103) are not found in the same region of the primary sequence as in human TSC2.
Table 1.
Sites on TSC1 and TSC2 with evidence of in vivo phosphorylation by known kinases
| Site* | Kinase | Conservation | Proposed function† | Reference(s) |
|---|---|---|---|---|
| TSC1 | ||||
| Thr417 | CDK1 | Metazoant‡ | Inhibit | [108] |
| Ser487 | IKKβ | Mammal | Inhibit (dissociate complex?) | [96] |
| Ser511 | IKKβ | Vertebrate | Inhibit (dissociate complex?) | [96] |
| Ser584 | CDK1 | Vertebrate | Inhibit | [108] |
| Ser1047 | CDK1 | Mammal | Inhibit | [108] |
| TSC2 | ||||
| Ser540 | ERK | Vertebrate | Inhibit (dissociate complex) | [95] |
| Ser664 | ERK | Vertebrate | Inhibit (dissociate complex) | [95] |
| Ser939 | Akt/RSK1 | Metazoan | Inhibit (14-3-3 binding) | [21,22,93] |
| Ser981 | Akt? | Vertebrate | Inhibit (14-3-3 binding) | [82] (in vitro) |
| Ser1130 | Akt | Vertebrate | Inhibit | [22,82] |
| Ser1132 | Akt | Vertebrate | Inhibit | [22,82] |
| Ser1254 | MAPKAP kinase-2 | Vertebrate | Unknown (14-3-3 binding) | [110] |
| Thr1271 | AMPK | Vertebrate | Activate | [98] |
| Ser1371 | GSK3 | Mammal‡ | Activate | [101] |
| Ser1375 | GSK3 | Mammal | Activate | [101] |
| Ser1379 | GSK3 | Mammal | Activate | [101] |
| Ser1383 | GSK3 | Metazoan | Activate | [101] |
| Ser1387 | AMPK | Metazoan | Activate | [98] |
| Thr1462 | Akt/RSK1 | Metazoan | Inhibit (14-3-3 binding) | [21,22,93] |
| Ser1798 | RSK1 | Vertebrate | Inhibit | [93] |
Amino acids
As the essential building blocks of proteins, amino acids profoundly affect every step of protein translation, including the events required for translation initiation that are regulated by TORC1. Amino acid sensing is one of the more ancient regulatory mechanisms feeding into TORC1, and in higher eukaryotes, the requirement for amino acids is dominant over inputs from growth factors for TORC1 activation [74]. Unlike the fission yeast S. pombe, the budding yeast Saccharomyces cerevisiae does not encode homologues of TSC1 or TSC2, suggesting that at least some signals emanating from amino acids affect TORC1 in a manner that is independent of these proteins. However, several studies have suggested roles for this complex in the acute attenuation of TORC1 signalling upon amino acid withdrawal from Drosophila or mammalian cells [22,25,44,58]. It is clear from the results obtained from these studies and from additional studies concluding that amino acid signalling occurs independently of TSC2 [75], that TORC1 signalling is significantly more resistant to amino acid withdrawal in cells lacking the TSC1–TSC2 complex compared with their wild-type counterparts. However, some decrease in TORC1 signalling is reproducibly detected in TSC1- and TSC2-deficient cells, especially if the cells are starved of both serum and all amino acids. Therefore it is fair to state that at least some amino acid sensing by TORC1 occurs independently of the TSC1–TSC2 complex.
It is possible that the distinct conclusions made in this regard are due to aberrantly high levels of Rheb-GTP providing a strong stimulatory signal to TORC1 that supercedes parallel signals from amino acids in cells deficient in TSC1 or TSC2.
The role of Rheb in amino acid signalling to TORC1 has been difficult to ascertain. A large number of studies have demonstrated that Rheb overexpression can override the effects of amino acid starvation on TORC1 inhibition [58,59,61,62,64,70,75,76]. Furthermore, knockdown experiments have revealed that Rheb is essential for activation of TORC1 by amino acid refeeding [58,77], and amino acids enhance the ability of overexpressed Rheb to associate with mTOR [76]. However, conflicting data exists as to whether or not Rheb-GTP levels change in response to amino acid starvation and refeeding [60,75,77,78]. In general, the inability of the TSC and TOR research communities to reliably assay Rheb-GDP/GTP ratios in cells has been a limiting factor in determining whether a given signal goes through the TSC–Rheb circuit or through a parallel pathway to regulate TORC1. This is due to both artificially high levels of GTP-bound Rheb observed upon exogenous overexpression of the protein [65] and a lack of antibodies that can immunoprecipitate a significant fraction of endogenous Rheb from cell lysates. Therefore, although Rheb is required for amino acid signalling to TORC1, whether it is involved in amino acid sensing pathways remains unknown.
It seems likely that multiple pathways exist that sense and signal the availability of specific amino acids to TORC1. Recent studies have linked the class III PI3K (phosphoinositide 3-kinase) VPS34 (vacuolar protein sorting 34) [77,79] and the Ste20 (sterile20)-related kinase MAP4K3 (mitogen-activated protein kinase kinase kinase kinase 3) [80] to TORC1 activation by amino acids. Although these are significant breakthroughs, there is much we still do not know regarding the regulation of TORC1 by amino acids in any system. It is striking that genetic systems such as those of yeasts, nematode worms and fruitflies have yet to reveal significant insights into this ancient nutrient-sensing pathway.
Growth-factor signalling
PI3K–Akt
The responsiveness of mTORC1 signalling to growth factors has long been linked to activation of PI3K and Akt, but the mechanism was unknown. Through PI3K signalling, Akt is activated by most growth factors to phosphorylate several downstream substates (reviewed in [81]). Akt was the first kinase demonstrated to directly phosphorylate the TSC1–TSC2 complex in cells in response to growth factors. Two independent studies found that Akt phosphorylates endogenous TSC2 on multiple residues [21,22]. Human TSC2 contains five predicted Akt sites (Ser939, Ser981, Ser1130, Ser1132 and Thr1462 on full-length human TSC2), all of which have been suggested to be subject to phosphorylation by Akt (Figure 3). Importantly, the two sites shown definitively to be targeted by Akt in mammalian cells, Ser939 and Thr1462, are conserved and phosphorylated in Drosophila TSC2, corresponding to Ser924 and Thr1518 [23]. There is also evidence that either Ser1130 or Ser1132 is phosphorylated by Akt in vivo [22]. Finally, Akt can phosphorylate a peptide corresponding to the sequence surrounding Ser981 in vitro [82], and Ser981 has been identified as an in vivo phosphorylation site on TSC2 by tandem-MS analyses [83]. However, whether Akt phosphorylates Ser981 on full-length TSC2 within cells has not been conclusively demonstrated. Interestingly, this site is very similar to Ser939 and is present on exon 25, which can be removed by alternative splicing. It is possible that this site represents a duplication of the ancient form found in Drosophila TSC2, as both Ser939 and Ser981 align well with Drosophila Ser924 (Figure 3B). Furthermore, there are results suggesting that Ser939 and Ser981 might play a similar function in TSC2 regulation [84] (see below). Consistent with Akt activating mTORC1 signalling through phosphorylation of TSC2 on some or all of these sites, alanine-substitution mutants of TSC2 lacking different combinations of these sites were found to dominantly block Akt-mediated activation of mTORC1 when overexpressed [21,22,84]. However, what the most critical sites are in vivo and whether individual sites differentially affect downstream regulation of Rheb are currently unknown.
Although the data obtained using phosphorylation-site mutants of TSC2 demonstrate that Akt-mediated phosphorylation of these sites inhibits the function of the TSC1–TSC2 complex in cells, the molecular mechanism of this inhibition has been the subject of much debate. It is important to note that no difference in GAP activity towards recombinant Rheb is detected between TSC1–TSC2 complexes immunoprecipitated from serum-starved or growth-factor-stimulated cells, despite a significant difference in the phosphorylation status of Ser939 and Thr1462 on TSC2 (B. D. Manning, unpublished work). Furthermore, there is no difference in GAP activity with the S939A/T1462A (B. D. Manning, unpublished work) or S939A/S981A [84] mutants relative to wild-type TSC2. Therefore, either these immunoprecipitation–GAP assays are not sensitive enough to detect relevant differences, or Akt-mediated phosphorylation of TSC2 inhibits its ability to regulate Rheb through an alternative mechanism without affecting its GAP activity per se. One proposed mechanism involves disruption of the TSC1–TSC2 complex. This was first proposed from work in Drosophila S2 cells that demonstrated an insulin-stimulated dissociation of TSC1 and TSC2 that was dependent on the Akt phosphorylation sites [23]. However, another group of researchers [85] has since repeated these experiments without seeing any effect of insulin or the Akt phosphorylation sites on the interaction between TSC1 and TSC2. Furthermore, two studies in mammalian cells found that Akt-mediated phosphorylation of TSC2 had no effect on the association between TSC1 and TSC2 [21,22]. Two additional studies have found that TSC2 phosphorylation downstream of Akt targets both TSC1 and TSC2 for degradation [82,86]. However, this does not occur rapidly and, although it might contribute to the long-term effects of Akt on mTORC1 signalling, it cannot explain the immediate effects of Akt activation on mTORC1, which are blocked by Akt phosphorylation-site mutants of TSC2.
Another enticing mechanism is the possibility that phosphorylation of TSC2 alters its subcellular localization, such that it can no longer act as a GAP for Rheb. One study supporting such a mechanism separated crude membrane from cytosolic fractions and found that growth-factor stimulation led to an increase in the levels of TSC2 within the cytosolic fraction [84]. This effect was PI3K-dependent, could also be stimulated by activated Akt and required both Ser939 and Ser981 on TSC2. In that study, both TSC1 and Rheb were found exclusively in the membrane fraction, and unlike TSC2, did not show an increase in the cytosolic fraction following growth-factor stimulation. From these findings it was concluded that Akt-mediated phosphorylation of TSC2 on Ser939 or Ser981 inhibits the TSC1–TSC2 complex by triggering release of TSC2 from TSC1 at an intracellular membrane also occupied by Rheb. A caveat with this model is that it requires significant and rapid dissociation of TSC2 from TSC1 upon phosphorylation – something that has not been detected in the majority of studies to date. In addition, two previous studies found that the intact TSC1–TSC2 complex is present in both membrane and cytosolic fractions of cell and tissue homegenates [87,88]. However, these latter studies did not examine the effects of growth factors on the fractionation properties of TSC1 and TSC2.
Interestingly, phosphorylation of Ser939 and/or Ser981 creates a binding site on TSC2 for 14-3-3 proteins [84], a mechanism of regulation shared by several other Akt substrates (reviewed in [81]). Examining interactions between endogenous 14-3-3 proteins and TSC2, another study found that Ser939 and Thr1462 were both required for 14-3-3 binding to TSC2 downstream of PI3K signalling [89]. Therefore it is likely that 14-3-3 binding to some combination of phosphorylated Ser939, Ser981 and Thr1462 contributes to Akt-mediated inhibition of TSC2. However, in 14-3-3 pull-down experiments, both TSC1 and TSC2 were found to bind, and 14-3-3 did not affect the association between TSC1 and TSC2 [90,91]. It will be important to determine whether 14-3-3-bound TSC2 is hindered in its in vitro GAP activity towards Rheb, thereby suggesting a more direct Akt-dependent inhibitory mechanism on the TSC1–TSC2 complex.
ERK–RSK (extracellular-signal-regulated kinase–p90 ribosomal protein S6 kinase)
In addition to the PI3K–Akt pathway, activation of ERK signalling can also stimulate mTORC1 activity. This is most readily seen with growth stimuli that predominantly activate ERK, but not PI3K, such as the phorbol ester PMA. This ERK-specific activation of mTORC1 involves phosphorylation of the TSC1–TSC2 complex by RSK [also known as p90RSK or MAPKAP kinase1 (mitogen-activated protein kinase-activated protein) kinase-1] [92,93], which is activated by ERK and has overlapping substrate specificity with Akt [94]. RSK was found to phosphorylate a novel site at the C-terminus of TSC2 (Ser1798) and, to a lesser extent, the two conserved Akt sites (Ser939 and Thr1462) [93]. Phosphorylation-site mutants of TSC2 lacking these three sites impaired PMA-induced mTORC1 signalling to S6K1. A number of additional sites on TSC2 were found by tandem-MS analyses to be weakly induced by PMA [83], including an ERK consensus site (Ser664). This site, and a second site on TSC2 (Ser540), were independently found to be directly phosphorylated by ERK and to contribute to ERK-mediated activation of mTORC1 signalling [95]. Strikingly, phosphorylation of Ser540 and/or Ser664 by ERK was found to disrupt the association between TSC1 and TSC2. This effect was also detected following phosphorylation of the TSC1–TSC2 complex in vitro, suggesting that it is direct and does not require other proteins. Collectively, these data suggest that ERK signalling activates mTORC1 through multisite phosphorylation of TSC2 by both ERK and its downstream target RSK.
Cytokine signalling
The activation of mTORC1 downstream of most cytokines is likely to occur through the Akt and ERK signalling mechanisms described above. However, recently the pro-inflammatory cytokine TNF_α_ (tumour necrosis factor α) has been found to stimulate mTORC1 signalling through IKK β (inhibitory κB kinase β)-mediated phosphorylation of TSC1 [96]. IKK β can associate with TSC1 and appears to phosphorylate both Ser487 and Ser511. The Ser511 site is a typical IKK β phosphorylation site and is conserved amongst vertebrate TSC1 orthologues, whereas Ser487 is a non-canonical site that is found only in mammalian TSC1. Tsc1_−/_− mouse embryo fibroblasts expressing TSC1 mutants lacking these sites lose their responsiveness to TNF α for activation of mTORC1, whereas phosphomimetic mutation lead to a basal increase in mTORC1 signalling. Those authors proposed a mechanism involving rapid dissociation of the complex and increased degradation of TSC1. However, the results suggest minimal effects on the stability of the TSC1–TSC2 complex, and the precise mechanism of acute complex inhibition by phosphorylation of these sites is not known.
Energy- and oxygen-sensing pathways
In addition to amino acids, protein synthesis requires sufficiently high levels of cellular energy, in the form of ATP, and mTORC1 signalling is very sensitive to energy depletion [97]. A primary mechanism by which mTORC1 senses the status of intracellular ATP is through direct phosphorylation of TSC2 by AMPK (AMP-dependent protein kinase) [98,99]. Conditions of energy stress lead to an increase in intracellular levels of AMP, which binds to AMPK, triggering its subsequent activation by upstream kinases, including the tumour suppressor LKB1 (for a review on AMPK regulation and function, see [100]). Once active, AMPK phosphorylates downstream targets to increase energy-generating catabolic processes and decrease energy-depleting anabolic processes, such as protein synthesis. AMPK phosphorylates TSC2 on at least two residues (Ser1387 and Thr1271), of which Ser1387 better meets the consensus for an AMPK site. TSC2 mutants that cannot be phosphorylated on these sites have been shown to block the inhibitory effects of energy stress-inducing agents (e.g. 2-deoxyglucose) on mTORC1 signalling [98]. These findings suggest that AMPK inhibits mTORC1 under these poor growth conditions, at least in part, by phosphorylating and activating TSC2. However, the molecular mechanism by which phosphorylation of these residues activates the TSC1–TSC2 complex is unknown, and direct effects on its GAP activity toward Rheb have not been demonstrated. Interestingly, AMPK-dependent phosphorylation of TSC2 on Ser1387 primes TSC2 for further phosphorylation by GSK3_β_ (glycogen synthase kinase 3_β_) [101]. This appears to be specific to GSK3_β_ regulated within the Wnt pathway, rather than downstream of Akt, as Wnt signalling inhibits GSK3_β_-mediated phosphorylation of TSC2. An independent study also found that GSK3_β_ could phosphorylate TSC1 in vitro [102], but the identity of the sites and potential in vivo regulatory effects are not known. GSK3_β_ phosphorylates TSC2 on Ser1371, Ser1375, Ser1379, and Ser1383 N-terminal to the AMPK site, and this somehow contributes to TSC1–TSC2 complex activation and subsequent mTORC1 inhibition under conditions of energy depletion. Therefore Wnt signalling, which inhibits GSK3_β_, appears to activate mTORC1 by overcoming AMPK-mediated activating effects on the TSC1–TSC2 complex [101]. This represents a very interesting mode of signal integration by the TSC1–TSC2 complex. However, it is currently unclear as to what developmental state or physiological condition might dictate such cross-talk between energy-sensing pathways and Wnt signalling.
mTORC1 signalling is also inhibited under conditions of oxygen depletion (i.e. hypoxia) [103]. Hypoxia appears to block mTORC1 signalling through two mechanisms, both of which involve regulation of the TSC1–TSC2 complex. As oxygen is required for aerobic ATP production through mitochondrial oxidative phosphorylation, hypoxia causes energy stress and activates AMPK. Therefore hypoxia can inhibit mTORC1 through AMPK-mediated phosphorylation and activation of the TSC1–TSC2 complex [104]. However, AMPK-independent effects of hypoxia on mTORC1 have also been described [105]. This mechanism involves a transcriptional target of HIF_α_ (hypoxia-inducible factor α) called REDD1 (also known as RTP801, DDIT4 or Dig2). Two genes encoding the Drosophila orthologues of REDD1, scylla and charybdis, were found to be genetic suppressors of an overgrowth phenotype caused by Akt activation and were placed upstream of the TSC1–TSC2 complex genetically [106]. Subsequent studies in mammalian cells found that the induction of REDD1 expression under hypoxic conditions was required for proper attenuation of mTORC1 signalling [105], demonstrating that this mode of regulation is conserved. The REDD1 protein does not have any apparent functional domains. However, REDD1 has recently been found to bind to 14-3-3 proteins, in part, through a motif surrounding Ser137 [89]. The surprising data accompanying this finding suggests that REDD1 inhibits mTORC1 by reversing Akt-mediated inhibition of the TSC1–TSC2 complex. The proposed mechanism involves the movement of 14-3-3 proteins bound to two Akt phosphorylation sites on TSC2 (Ser939 and Thr1462) from these sites to Ser137 on REDD1, thereby activating the TSC1–TSC2 complex. As 14-3-3 proteins are thought to bind only to phosphorylated motifs, it will be important to determine whether this site on REDD1 is indeed phosphorylated and to identify the protein kinase responsible for triggering this unusual mode of regulation. It is also worth noting that this putative phosphorylation site is not conserved in the Drosophila orthologues of REDD1. Finally, REDD1 has also been found to be induced by energy stress and to play a role in AMPK-mediated TSC1–TSC2-dependent regulation of mTORC1 [107]. Although more work is needed to fully understand the molecular mechanism(s) of REDD1 function, these recent findings suggest a tight link between Akt signalling and the sensing of both cellular oxygen and energy at the level of the TSC1–TSC2 complex.
Cell-cycle regulators
How the TSC1–TSC2 complex, mTORC1 signalling and protein synthesis in general are regulated over the course of the cell cycle is poorly understood. However, there are studies suggesting a role for CDKs (cyclin-dependent kinases) in the regulation of the TSC1–TSC2 complex. Nocodazole-induced arrest of the cell cycle in G2/M-phase was found to lead to an increase in the phosphorylation of TSC1 on a proline-directed site, and this was blocked by a pharmacological inhibitor of CDKs [108]. Three consensus CDK sites on human TSC1 (Thr417, Ser584 and Thr1047) appear to be the target of this phosphorylation, and these sites are phosphorylated in vitro by CDK1–cyclin B complexes. Overexpression of TSC1 lacking these phosphorylation sites is slightly more efficient than wild-type TSC1 at suppressing S6K activation, suggesting that phosphorylation of these sites might inhibit the TSC1–TSC2 complex. However, the molecular mechanism of such an inhibition and whether mTORC1 is activated at the G2-phase/M-phase transition, by this or any other mechanism, are currently unknown. In addition, cyclin D has been found to associate with TSC2 in overexpression studies, and overexpression of CDK6–cyclin D complexes appears to increase phosphate incorporation into both TSC1 and TSC2, albeit at unknown sites [109]. However, whether CDK4/6–cyclin D directly phosphorylates the TSC1–TSC2 complex to regulate mTORCl activity at the G1-phase/S-phase transition is not known.
Stress signalling through p38 and MAPKAP kinase-2
TSC2 was found to be phosphorylated on Ser1254 by MAPKAP kinase-2, which is activated downstream of p38 MAPK (mitogen-activated protein kinase) [110]. Ser1254 phosphorylation was shown to create a 14-3-3-binding site on TSC2, but others have failed to observe a role for this site in 14-3-3 binding [84,89]. Furthermore, whether this phosphorylation stimulates or inhibits TSC1–TSC2 complex function is not clear. However, conditions of cellular stress, which often activate p38, would be predicted to activate the TSC1–TSC2 complex to inhibit mTORC1 signalling.
Phosphorylation sites with unknown kinases
Many additional in vivo phosphorylation sites on TSC1 and TSC2 have been identified by MS studies. Eight new sites on both TSC1 and TSC2 have been reported [83], but the regulatory inputs and kinases responsible for these 16 phosphorylation events are unknown. Although some of these sites are likely to represent novel regulatory mechanisms, it remains possible that many are unregulated sites involved in protein folding or stability. However, this analysis did reveal a novel site (Ser1364) on TSC2 as being robustly phosphorylated in response to PMA, and the use of pharmacological inhibitors suggest that phosphorylation of Ser1364 might be dependent on a protein kinase C isoform. Therefore the TSC1–TSC2 complex is phosphorylated on dozens of sites, many of which remain to be characterized, that might contribute to its signal-integrating capacity and downstream modulation of mTORC1 activity.
THE TSC1–TSC2 COMPLEX AND mTORC2
Although it is now clear that the TSC1–TSC2 complex is a critical upstream inhibitor of mTORC1, a role for this complex in the regulation of mTORC2 and its subsequent phosphorylation of Akt has been more difficult to ascertain. Much of the difficulty in this regard stems from the existence of an mTORC1-dependent feedback mechanism that, especially in cells lacking the TSC1–TSC2 complex, blocks growth-factor-stimulated phosphorylation of Akt (reviewed in [111]). This feedback mechanism affects the IRS (insulin receptor substrate) proteins and, in some cells, the PDGF (platelet-derived growth factor) receptor, and renders the PI3K–Akt pathway resistant to insulin/IGF-1 or PDGF stimulation [112–115]. This effect can be restored, at least partially, by prolonged treatment of TSC-deficient cells with rapamycin. However, Akt phosphorylation and downstream signalling appear to be attenuated more generally upon disruption of the TSC1–TSC2 complex, as seen under full serum growth conditions or in TSC-related tumours [50,115,116]. Therefore, in a recent study, we asked whether mTORC2 kinase activity was affected by loss of the TSC genes and might contribute to the decrease in Akt-Ser473 phosphorylation in these settings [117]. Indeed, mTORC2 kinase activity, assayed via Rictor immunoprecipitation kinase assays on an exogenous Akt substrate, was found to be severely blunted in a variety of cells lacking an intact TSC1–TSC2 complex. Importantly, this effect on mTORC2 activity could be separated from the regulation of both Rheb and mTORC1 by the TSC1–TSC2 complex. Finally, the TSC1–TSC2 complex was found to physically associate with mTORC2, but not with mTORC1. The molecular mechanism through which the TSC1–TSC2 complex promotes mTORC2 activation, and whether some of the pathways that regulate the complexes ability to inhibit mTORC1 also effect its activation of mTORC2, are currently unknown.
DISEASE IMPLICATIONS: TSC AND BEYOND
It is now recognized that aberrant mTORC1 activation is a common molecular event in a number of hamartoma syndromes. Given the large number of signalling inputs that regulate mTORC1 through the TSC1–TSC2 complex (Figure 4), it is not surprising that loss of the TSC tumour suppressors appears to give rise to the highest levels of constitutive mTORC1 signalling. However, loss of a number of upstream tumour suppressors, which effect specific inputs into the pathway, are likely to lead to more moderate basal increases in mTORC1. Elevated mTORC1 signalling has been detected in tumours of mouse models with mutations in the PTEN (see, e.g., [118, 119]), LKB1 [99] or NF1 (neurofibromatosis 1) [120,121] tumour suppressors, which recapitulate some clinical aspects of Cowden disease, Peutz-Jeghers syndrome and neurofibromatosis type 1 respectively. Loss of these tumour suppressors leads to either activation of pathways that inhibit the TSC1–TSC2 complex (PTEN and NF1) or inhibition of pathways that activate the TSC1–TSC2 complex (LKB1), thus leading to increased mTORC1 signalling. For instance, cells lacking PTEN or NF1 display growth-factor-independent phosphorylation of TSC2 on the inhibitory Akt sites (Ser939 and Thr1462) [21,122]. Furthermore, the resulting elevation in mTORC1 activity is essential for tumour development in mouse models of these tumour syndromes, as mTORC1 inhibitors have potent antitumour activity in these mice [118,119,121,123]. Finally, as with TSC tumours, activation of mTORC1 signalling has been detected in the tumours of patients with NF1- or PTEN-associated syndromes [120,124].
Figure 4. Signal integration by the TSC1–TSC2 complex.

The TSC1–TSC2 complex monitors growth conditions through multiple signalling pathways, which either inhibit or activate the complex to affect mTORC1 activity. See the text for details. Further abbreviations: Dvl, dishevelled; Frz, frizzled; MEK, MAPK/ERK kinase; TNFR, tumour-necrosis-factor receptor; VHL, von Hippel–Lindau tumour suppressor; VPS34, vacuolar protein sorting 34.
Given the variety of oncoproteins [RTKs (receptor tyrosine kinases), PI3K, Akt, Ras, Raf and Wnt] and tumour suppressors (PTEN and LKB1) that lie upstream (Figure 4) and whose genes are amplified or mutated in a large percentage of sporadic human malignancies, defects in the regulation of the TSC1–TSC2 complex are likely to contribute to tumorigenesis and cancer progression. Activation of mTORC1 has been suggested in several mechanistic studies to be required for neoplastic transformation by oncogenic Ras or Akt (see, e.g., [125–127]. Furthermore, increased phosphorylation of the TSC1–TSC2 complex on the Akt, ERK and IKK_β_ sites have been detected in human cancers with elevated mTORC1 signalling [96,128,129]. Therefore dysregulation of the TSC1–TSC2 complex and subsequent activation of mTORC1 is likely to be a common molecular event in tumorigenesis.
At first sight, the above discussion would indicate that the TSC disease should be a cancer predisposition syndrome and that mutations in the TSC genes might be common in sporadic cancers. However, TSC is predominantly a tumour syndrome of benign growths and, where examined, TSC gene mutations have not been detected in human malignancies. As discussed above, Akt signalling is blocked upon loss of the TSC1–TSC2 complex through both mTORC1-dependent feedback mechanisms [112– 114] and loss of mTORC2 activity [117]. It is likely that the limited malignancy potential of tumours lacking the TSC genes is due, at least in part, to these multiple mechanisms leading to attenuation of Akt, which is a potent oncoprotein regulating many aspects of tumour progression (reviewed in [81]). In support of this idea, Akt signalling was found to be attenuated in liver tumours from a mouse model of TSC, and re-activation of Akt, through loss of one copy of PTEN, correlated with a dramatic increase in tumour severity [50]. Therefore, although constitutive mTORC1 signalling is likely to drive tumorigenesis in the TSC disease, loss of Akt signalling is likely to distinguish these tumours from the more aggressive tumours arising from activation of upstream oncogenic pathways.
As the PI3K–Akt pathway is the critical mediator of insulin action on liver, muscle and fat cells, proper regulation of the TSC1–TSC2 complex by Akt contributes to the maintenance of metabolic homoeostasis. For instance, Akt-mediated phosphorylation of TSC2 and subsequent activation of mTORC1 is likely to play a role in stimulating the anabolic growth of muscle downstream of insulin. However, aberrantly high mTORC1 signalling has also been implicated in the development of insulin resistance (reviewed in [111]). It has been found that mTORC1 activation downstream of growth factors, cytokines or prolonged insulin leads to insulin resistance in cell-culture models. Furthermore, mice lacking the downstream target of mTORC1, S6K1, display enhanced peripheral insulin sensitivity [130]. As discussed above, these effects have been attributed to negative feedback mechanisms affecting IRS proteins [112,113], which normally relay the signal from the insulin receptor to PI3K and Akt. Therefore it is likely that conditions of mTORC1-dependent insulin resistance are, at least partially, triggered through aberrant upstream inhibition of the TSC1–TSC2 complex.
Although the advances in our understanding of the regulation and function of the TSC1–TSC2 complex have been immense, there is much we do not understand regarding how this small G-protein switch integrates a full network of upstream signals to regulate mTORC1 activity in various cell types. The study of this complex offers a unique opportunity to define molecular mechanisms by which cells perceive and integrate diverse signals to impose tight control over critical cellular processes. Finally, given the diversity of complex human diseases in which regulation of the TSC1–TSC2 complex goes awry, it is important that we understand not only how this signalling network is wired in normal cells, but also how it is rewired in disease states.
Acknowledgments
We thank Christian C. Dibble of this Department for providing critical comments on the manuscript before its submission. J.H. is supported by a National Science Scholarship from the Agency for Science, Technology and Research, Singapore. Research in B. D. M.’s laboratory on the regulation and function of the TSC1–TSC2 complex is supported-by grants from the Tuberous Sclerosis Alliance, the LAM Foundation, the American Diabetes Association and the National Institutes of Health (grants R01-CA122617 and P01-CA120964).
Abbreviations used
AMPK
AMP-dependent protein kinase
4E-BP1
eukaryotic-translation-initiation-factor-4E-binding protein 1
CDK
cyclin-dependent kinase
eIF4
eukaryotic translation initiation factor 4
FKBP12
FK506-binding protein 12
GAP
GTPase-activating protein
GEF
guanine-nucleotide exchange factor
GSK3β
glycogen synthase kinase 3β
IGF-1
insulin-like growth factor 1
IKK_β_
inhibitory κB kinase β
InR
insulin/IGF-1 receptor
LAM
lymphangioleiomyomatosis
IRS
insulin receptor substrate
LST8
lethal with SEC13 protein 8
MAPK
mitogen-activated protein kinase
MAPKAP
mitogen-activated protein kinase-activated protein
mTORC
mammalian target of rapamycin
mTORC1
mammalian target of rapamycin complex 1
NF1
neurofibromatosis 1
PDGF
platelet-derived growth factor
PI3K
phosphoinositide 3-kinase
PTEN
phosphatase and tensin homologue deleted on chromosome 10
Rheb
Ras homologue enriched in brain
Rictor
rapamycin-insensitive companion of mTOR
RSK
p90 ribosomal protein S6 kinase
RTK
receptor tyrosine kinase
S6K
70 kDa ribosomal protein S6 kinase
TNFα
tumour necrosis factor α
TOR
target of rapamycin
TORC1
TOR complex 1
TORC2
TOR complex 2
TSC
tuberous sclerosis complex
TSC1–TSC2 complex
hamartin–tuberin complex
References
- 1.Kandt RS, Haines JL, Smith M, Northrup H, Gardner RJ, Short MP, Dumars K, Roach ES, Steingold S, Wall S, et al. Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat Genet. 1992;2:37–41. doi: 10.1038/ng0992-37. [DOI] [PubMed] [Google Scholar]
- 2.Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–1315. doi: 10.1016/0092-8674(93)90618-z. [DOI] [PubMed] [Google Scholar]
- 3.van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A, Halley D, Young J, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805–808. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- 4.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA. 2000;97:6085–6090. doi: 10.1073/pnas.97.11.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 6.Wienecke R, Konig A, DeClue JE. Identification of tuberin, the tuberous sclerosis-2 product. Tuberin possesses specific Rap1GAP activity. J Biol Chem. 1995;270:16409–16414. doi: 10.1074/jbc.270.27.16409. [DOI] [PubMed] [Google Scholar]
- 7.Xiao GH, Shoarinejad F, Jin F, Golemis EA, Yeung RS. The tuberous sclerosis 2 gene product, tuberin, functions as a Rab5 GTPase activating protein (GAP) in modulating endocytosis. J Biol Chem. 1997;272:6097–6100. doi: 10.1074/jbc.272.10.6097. [DOI] [PubMed] [Google Scholar]
- 8.Maheshwar MM, Cheadle JP, Jones AC, Myring J, Fryer AE, Harris PC, Sampson JR. The GAP-related domain of tuberin, the product of the TSC2 gene, is a target for missense mutations in tuberous sclerosis. Hum Mol Genet. 1997;6:1991–1996. doi: 10.1093/hmg/6.11.1991. [DOI] [PubMed] [Google Scholar]
- 9.Jin F, Wienecke R, Xiao GH, Maize JC, Jr, DeClue JE, Yeung RS. Suppression of tumourigenicity by the wild-type tuberous sclerosis 2 (Tsc2) gene and its C-terminal region. Proc Natl Acad Sci USA. 1996;93:9154–9159. doi: 10.1073/pnas.93.17.9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Slegtenhorst M, Nellist M, Nagelkerken B, Cheadle J, Snell R, van den Ouweland A, Reuser A, Sampson J, Halley D, van der Sluijs P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet. 1998;7:1053–1057. doi: 10.1093/hmg/7.6.1053. [DOI] [PubMed] [Google Scholar]
- 11.Plank TL, Yeung RS, Henske EP. Hamartin, the product of the tuberous sclerosis 1 (TSC1) gene, interacts with tuberin and appears to be localized to cytoplasmic vesicles. Cancer Res. 1998;58:4766–4770. [PubMed] [Google Scholar]
- 12.Hodges AK, Li S, Maynard J, Parry L, Braverman R, Cheadle JP, DeClue JE, Sampson JR. Pathological mutations in TSC1 and TSC2 disrupt the interaction between hamartin and tuberin. Hum Mol Genet. 2001;10:2899–2905. doi: 10.1093/hmg/10.25.2899. [DOI] [PubMed] [Google Scholar]
- 13.Gao X, Pan D. TSC1 and TSC2 tumour suppressors antagonize insulin signaling in cell growth. Genes Dev. 2001;15:1383–1392. doi: 10.1101/gad.901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Potter CJ, Huang H, Xu T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001;105:357–368. doi: 10.1016/s0092-8674(01)00333-6. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto S, Bandyopadhyay A, Kwiatkowski DJ, Maitra U, Matsumoto T. Role of the Tsc1–Tsc2 complex in signaling and transport across the cell membrane in the fission yeast Schizosaccharomyces pombe. Genetics. 2002;161:1053–1063. doi: 10.1093/genetics/161.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benvenuto G, Li S, Brown SJ, Braverman R, Vass WC, Cheadle JP, Halley DJ, Sampson JR, Wienecke R, DeClue JE. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene. 2000;19:6306–6316. doi: 10.1038/sj.onc.1204009. [DOI] [PubMed] [Google Scholar]
- 17.Chong-Kopera H, Inoki K, Li Y, Zhu T, Garcia-Gonzalo FR, Rosa JL, Guan KL. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J Biol Chem. 2006;281:8313–8316. doi: 10.1074/jbc.C500451200. [DOI] [PubMed] [Google Scholar]
- 18.Nellist M, Verhaaf B, Goedbloed MA, Reuser AJ, van den Ouweland AM, Halley DJ. TSC2 missense mutations inhibit tuberin phosphorylation and prevent formation of the tuberin–hamartin complex. Hum Mol Genet. 2001;10:2889–2898. doi: 10.1093/hmg/10.25.2889. [DOI] [PubMed] [Google Scholar]
- 19.Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 2001;105:345–355. doi: 10.1016/s0092-8674(01)00332-4. [DOI] [PubMed] [Google Scholar]
- 20.Stocker H, Hafen E. Genetic control of cell size. Curr Opin Genet Dev. 2000;10:529–535. doi: 10.1016/s0959-437x(00)00123-4. [DOI] [PubMed] [Google Scholar]
- 21.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumour suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 22.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 23.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 24.Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: a regulator of cell size. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- 25.Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS, Ru B, Pan D. Tsc tumour suppressor proteins antagonize amino acid–TOR signalling. Nat Cell Biol. 2002;4:699–704. doi: 10.1038/ncb847. [DOI] [PubMed] [Google Scholar]
- 26.Radimerski T, Montagne J, Hemmings-Mieszczak M, Thomas G. Lethality of Drosophila lacking TSC tumour suppressor function rescued by reducing dS6K signaling. Genes Dev. 2002;16:2627–2632. doi: 10.1101/gad.239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–3171. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- 28.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 29.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 30.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 31.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 32.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 33.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 34.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 35.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 36.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 37.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearson RB, Dennis PB, Han JW, Williamson NA, Kozma SC, Wettenhall RE, Thomas G. The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J. 1995;14:5279–5287. doi: 10.1002/j.1460-2075.1995.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, Sonenberg N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15:2852–2864. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mothe-Satney I, Yang D, Fadden P, Haystead TA, Lawrence JC., Jr Multiple mechanisms control phosphorylation of PHAS-I in five (S/T)P sites that govern translational repression. Mol Cell Biol. 2000;20:3558–3567. doi: 10.1128/mcb.20.10.3558-3567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goncharova EA, Goncharov DA, Eszterhas A, Hunter DS, Glassberg MK, Yeung RS, Walker CL, Noonan D, Kwiatkowski DJ, Chou MM, et al. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation: a role for the TSC2 tumour suppressor gene in pulmonary lymphangioleiomyomatosis (LAM) J Biol Chem. 2002;277:30958–30967. doi: 10.1074/jbc.M202678200. [DOI] [PubMed] [Google Scholar]
- 43.Jaeschke A, Hartkamp J, Saitoh M, Roworth W, Nobukuni T, Hodges A, Sampson J, Thomas G, Lamb R. Tuberous sclerosis complex tumour suppressor-mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J Cell Biol. 2002;159:217–224. doi: 10.1083/jcb.jcb.200206108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, Onda H. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in TSC1 null cells. Hum Mol Genet. 2002;11:525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- 46.Kenerson HL, Aicher LD, True LD, Yeung RS. Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumours. Cancer Res. 2002;62:5645–5650. [PubMed] [Google Scholar]
- 47.Karbowniczek M, Yu J, Henske EP. Renal angiomyolipomas from patients with sporadic lymphangiomyomatosis contain both neoplastic and non-neoplastic vascular structures. Am J Pathol. 2003;162:491–500. doi: 10.1016/S0002-9440(10)63843-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El-Hashemite N, Zhang H, Henske EP, Kwiatkowski DJ. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet. 2003;361:1348–1349. doi: 10.1016/S0140-6736(03)13044-9. [DOI] [PubMed] [Google Scholar]
- 49.Crino PB. Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol. 2004;19:716–725. doi: 10.1177/08830738040190091301. [DOI] [PubMed] [Google Scholar]
- 50.Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumours lacking Tsc2. Genes Dev. 2005;19:1773–1778. doi: 10.1101/gad.1314605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.El-Hashemite N, Walker V, Zhang H, Kwiatkowski DJ. Loss of Tsc1 or Tsc2 induces vascular endothelial growth factor production through mammalian target of rapamycin. Cancer Res. 2003;63:5173–5177. [PubMed] [Google Scholar]
- 52.Lee L, Sudentas P, Donohue B, Asrican K, Worku A, Walker V, Sun Y, Schmidt K, Albert MS, El-Hashemite N, et al. Efficacy of a rapamycin analog (CCI-779) and IFN-γ in tuberous sclerosis mouse models. Genes Chromosomes Cancer. 2005;42:213–227. doi: 10.1002/gcc.20118. [DOI] [PubMed] [Google Scholar]
- 53.Messina MP, Rauktys A, Lee L, Dabora SL. Tuberous sclerosis preclinical studies: timing of treatment, combination of a rapamycin analog (CCI-779) and interferon-γ, and comparison of rapamycin to CCI-779. BMC Pharmacol. 2007;7:14. doi: 10.1186/1471-2210-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- 55.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salisbury S, Franz DN. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davies DM, Johnson SR, Tattersfield AE, Kingswood JC, Cox JA, McCartney DL, Doyle T, Elmslie F, Saggar A, de Vries PJ, Sampson JR. Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med. 2008;358:200–203. doi: 10.1056/NEJMc072500. [DOI] [PubMed] [Google Scholar]
- 57.Patel PH, Thapar N, Guo L, Martinez M, Maris J, Gau CL, Lengyel JA, Tamanoi F. Drosophila Rheb GTPase is required for cell cycle progression and cell growth. J Cell Sci. 2003;116:3601–3610. doi: 10.1242/jcs.00661. [DOI] [PubMed] [Google Scholar]
- 58.Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signaling network. Nat Cell Biol. 2003;5:566–571. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- 59.Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003;5:559–566. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5:578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 61.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tee AR, Manning BD, Roux PP, Cantely LC, Blenis J. Tuberous sclerosis complex gene products, tuberin and hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 63.Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. 2003;278:32493–32496. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- 64.Garami A, Zwartkruis FJT, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–1466. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- 65.Im E, vonLintig FC, Chen J, Zhuang S, Qui W, Chowdhury S, Worley PF, Boss GR, Pilz RB. Rheb is in a high activation state and inhibits B-Raf kinase in mammalian cells. Oncogene. 2002;21:6356–6365. doi: 10.1038/sj.onc.1205792. [DOI] [PubMed] [Google Scholar]
- 66.Tabancay AP, Jr, Gau CL, Machado IM, Uhlmann EJ, Gutmann DH, Guo L, Tamanoi F. Identification of dominant negative mutants of Rheb GTPase and their use to implicate the involvement of human Rheb in the activation of p70S6K. J Biol Chem. 2003;278:39921–39930. doi: 10.1074/jbc.M306553200. [DOI] [PubMed] [Google Scholar]
- 67.Tee AR, Blenis J, Proud CG. Analysis of mTOR signaling by the small G-proteins, Rheb and RhebL1. FEBS Lett. 2005;579:4763–4768. doi: 10.1016/j.febslet.2005.07.054. [DOI] [PubMed] [Google Scholar]
- 68.Hsu YC, Chern JJ, Cai Y, Liu M, Choi KW. Drosophila TCTP is essential for growth and proliferation through regulation of dRheb GTPase. Nature. 2007;445:785–788. doi: 10.1038/nature05528. [DOI] [PubMed] [Google Scholar]
- 69.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 Is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 70.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 71.Urano J, Comiso MJ, Guo L, Aspuria PJ, Deniskin R, Tabancay AP, Jr, Kato-Stankiewicz J, Tamanoi F. Identification of novel single amino acid changes that result in hyperactivation of the unique GTPase, Rheb, in fission yeast. Mol Microbiol. 2005;58:1074–1086. doi: 10.1111/j.1365-2958.2005.04877.x. [DOI] [PubMed] [Google Scholar]
- 72.Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, Jiang Y. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–980. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- 73.Rosner M, Hofer K, Kubista M, Hengstschlager M. Cell size regulation by the human TSC tumour suppressor proteins depends on PI3K and FKBP38. Oncogene. 2003;22:4786–4798. doi: 10.1038/sj.onc.1206776. [DOI] [PubMed] [Google Scholar]
- 74.Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- 75.Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG. the tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J Biol Chem. 2005;280:18717–18727. doi: 10.1074/jbc.M414499200. [DOI] [PubMed] [Google Scholar]
- 76.Long X, Ortiz-Vega S, Lin Y, Avruch J. Rheb binding to mTOR is regulated by amino acid sufficiency. J Biol Chem. 2005;280:23433–23436. doi: 10.1074/jbc.C500169200. [DOI] [PubMed] [Google Scholar]
- 77.Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci USA. 2005;102:14238–14243. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roccio M, Bos JL, Zwartkruis FJ. Regulation of the small GTPase Rheb by amino acids. Oncogene. 2006;25:657–664. doi: 10.1038/sj.onc.1209106. [DOI] [PubMed] [Google Scholar]
- 79.Byfield MP, Murray JT, Backer JM. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem. 2005;280:33076–33082. doi: 10.1074/jbc.M507201200. [DOI] [PubMed] [Google Scholar]
- 80.Findlay GM, Yan L, Procter J, Mieulet V, Lamb RF. A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem J. 2007;403:13–20. doi: 10.1042/BJ20061881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dan HC, Sun M, Yang L, Sui RM, Yeung RS, Halley DJJ, Nicosia SV, Pledger WJ, Cheng JQ. PI3K/Akt pathway regulates TSC tumour suppressor complex by phosphorylation of tuberin. J Biol Chem. 2002;277:35364–35370. doi: 10.1074/jbc.M205838200. [DOI] [PubMed] [Google Scholar]
- 83.Ballif BA, Roux PP, Gerber SA, MacKeigan JP, Blenis J, Gygi SP. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumour suppressors. Proc Natl Acad Sci USA. 2005;102:667–672. doi: 10.1073/pnas.0409143102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J, Guo R, Johnson CL, Kiguchi K, Walker CL. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J Cell Biol. 2006;173:279–289. doi: 10.1083/jcb.200507119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong J, Pan D. Tsc2 is not a critical target of Akt during normal Drosophila development. Genes Dev. 2004;18:2479–2484. doi: 10.1101/gad.1240504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem. 2003;278:12361–12366. doi: 10.1074/jbc.M213069200. [DOI] [PubMed] [Google Scholar]
- 87.Nellist M, van Slegtenhorst MA, Goedbloed M, van den Ouweland AMW, Halley DJJ, van der Sluijs P. Characterization of the cytosolic tuberin–hamartin complex. J Biol Chem. 1999;274:35647–35652. doi: 10.1074/jbc.274.50.35647. [DOI] [PubMed] [Google Scholar]
- 88.Yamamoto Y, Jones KA, Mak BC, Muehlenbachs A, Yeung RS. Multicompartmental distribution of the tuberous sclerosis gene products, hamartin and tuberin. Arch Biochem Biophys. 2002;404:210–217. doi: 10.1016/s0003-9861(02)00300-4. [DOI] [PubMed] [Google Scholar]
- 89.Deyoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2 mTOR signaling and tumour suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nellist M, Goedbloed MA, de Winter C, Verhaaf B, Jankie A, Reuser AJJ, van den Ouweland AMW, van der Sluijs P, Halley DJJ. Identification and characterization of the interaction between tuberin and 14-3-3ζ. J Biol Chem. 2002;277:39417–39424. doi: 10.1074/jbc.M204802200. [DOI] [PubMed] [Google Scholar]
- 91.Shumway SD, Li Y, Xiong Y. 14-3-3β binds to and negatively regulates the tuberous sclerosis complex 2 (TSC2) tumour suppressor gene product, tuberin. J Biol Chem. 2003;278:2089–2092. doi: 10.1074/jbc.C200499200. [DOI] [PubMed] [Google Scholar]
- 92.Tee AR, Anjum R, Blenis J. Inactivation of the tuberous sclerosis complex-1 and -2 gene products occurs by phosphoinositide 3-kinase/Akt-dependent and -independent phosphorylation of tuberin. J Biol Chem. 2003;278:37288–37296. doi: 10.1074/jbc.M303257200. [DOI] [PubMed] [Google Scholar]
- 93.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumour-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumour suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA. 2004;101:13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alessi DR, Caudwell FB, Andejelkovic M, Hemmings BA, Cohen P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 1996;399:333–338. doi: 10.1016/s0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- 95.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk: implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 96.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, et al. IKKβ suppression of TSC1 links inflammation and tumour angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 97.Dennis PB, Jaeshke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science. 2001;294:1102–1105. doi: 10.1126/science.1063518. [DOI] [PubMed] [Google Scholar]
- 98.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 99.Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumour suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 100.Hardie DG. New roles for the LKB1 → AMPK pathway. Curr Opin Cell Biol. 2005;17:167–173. doi: 10.1016/j.ceb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 101.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 102.Mak BC, Kenerson HL, Aicher LD, Barnes EA, Yeung RS. Aberrant β-catenin signaling in tuberous sclerosis. Am J Pathol. 2005;167:107–116. doi: 10.1016/s0002-9440(10)62958-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278:29655–29660. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 104.Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumour suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Reiling JH, Hafen E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004;18:2879–2892. doi: 10.1101/gad.322704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005;25:5834–5845. doi: 10.1128/MCB.25.14.5834-5845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Astrinidis A, Senapedis W, Coleman TR, Henske EP. Cell cycle-regulated phosphorylation of hamartin, the product of the tuberous sclerosis complex 1 gene, by cyclin-dependent kinase 1/cyclin B. J Biol Chem. 2003;278:51372–51379. doi: 10.1074/jbc.M303956200. [DOI] [PubMed] [Google Scholar]
- 109.Zacharek SJ, Xiong Y, Shumway SD. Negative regulation of TSC1–TSC2 by mammalian D-type cyclins. Cancer Res. 2005;65:11354–11360. doi: 10.1158/0008-5472.CAN-05-2236. [DOI] [PubMed] [Google Scholar]
- 110.Li Y, Inoki K, Vacratsis P, Guan KL. The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14-3-3. J Biol Chem. 2003;278:13663–13671. doi: 10.1074/jbc.M300862200. [DOI] [PubMed] [Google Scholar]
- 111.Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumourigenesis. J Cell Biol. 2004;167:399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1–2 tumour suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival defects. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 114.Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, Griffin JD, Kwiatkowski DJ. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–738. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K–Akt signaling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang HH, Lipovsky AI, Dibble CC, Sahin M, Manning BD. S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol Cell. 2006;24:185–197. doi: 10.1016/j.molcel.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1–TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008 doi: 10.1128/MCB.00289–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL. Enhanced sensitivity of PTEN-deficient tumours to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA. 2001;98:10314–10319. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Podsypanina K, Lee RT, Politis C, Hennessy I, Crane A, Puc J, Neshat M, Wang H, Yang L, Gibbons J, et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in PTEN+/− mice. Proc Natl Acad Sci USA. 2001;98:10320–10325. doi: 10.1073/pnas.171060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumours. Cancer Res. 2005;65:2755–2760. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- 121.Johannessen CM, Johnson BW, Williams SM, Chan AW, Reczek EE, Lynch RC, Rioth MJ, McClatchey A, Ryeom S, Cichowski K. TORC1 is essential for NF1 -associated malignancies. Curr Biol. 2008;18:56–62. doi: 10.1016/j.cub.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 122.Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumour suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 2005;102:8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wei C, Amos CI, Zhang N, Wang X, Rashid A, Walker CL, Behringer RR, Frazier ML. Suppression of Peutz–Jeghers polyposis by targeting mammalian target of rapamycin signaling. Clin Cancer Res. 2008;14:1167–1171. doi: 10.1158/1078-0432.CCR-07-4007. [DOI] [PubMed] [Google Scholar]
- 124.Abel TW, Baker SJ, Fraser MM, Tihan T, Nelson JS, Yachnis AT, Bouffard JP, Mena H, Burger PC, Eberhart CG. Lhermitte–Duclos disease: a report of 31 cases with immunohistochemical analysis of the PTEN/AKT/mTOR pathway. J Neuropathol Exp Neurol. 2005;64:341–349. doi: 10.1093/jnen/64.4.341. [DOI] [PubMed] [Google Scholar]
- 125.Aoki M, Blazek E, Vogt PK. A role of the kinase mTOR in cellular transformation induced by the oncoproteins P3k and Akt. Proc Natl Acad Sci USA. 2001;98:136–141. doi: 10.1073/pnas.011528498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 127.Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, Hahn-Windgassen A, Kiyokawa H, Hay N. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. 2006;10:269–280. doi: 10.1016/j.ccr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 128.Riemenschneider MJ, Betensky RA, Pasedag SM, Louis DN. AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling. Cancer Res. 2006;66:5618–5623. doi: 10.1158/0008-5472.CAN-06-0364. [DOI] [PubMed] [Google Scholar]
- 129.Ma L, Teruya-Feldstein J, Bonner P, Bernardi R, Franz DN, Witte D, Cordon-Cardo C, Pandolfi PP. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 2007;67:7106–7112. doi: 10.1158/0008-5472.CAN-06-4798. [DOI] [PubMed] [Google Scholar]
- 130.Urn SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]