Functional Neural Development from Human Embryonic Stem Cells: Accelerated Synaptic Activity via Astrocyte Coculture (original) (raw)
Abstract
How a naive human neuroepithelial cell becomes an electrophysiologically active neuron remains unknown. Here, we describe the early physiological development of neurons differentiating from naive human embryonic stem (hES) cells. We found that differentiating neuronal cells progressively decrease their resting membrane potential, gain characteristic Na+ and K+ currents, and fire mature action potentials by 7 weeks of differentiation. This is similar to the maturation pattern observed in animals, albeit on a greatly expanded time scale. An additional 3 weeks of differentiation resulted in neurons that could fire repetitive trains of action potentials in response to depolarizing current pulses. The onset of spontaneous synaptic activity also occurred after 7 weeks of differentiation, in association with the differentiation of astrocytes within the culture. Cocultures of hES cell-derived neuroepithelial cells with exogenous astrocytes significantly accelerated the onset of synaptic currents but did not alter action potential generation. These findings suggest that the development of membrane characteristics and action potentials depend on the intrinsic maturation of Na+ and K+ currents, whereas synaptic transmission is enhanced by astrocytes, which may be achieved independently of the maturation of action potentials. Furthermore, we found that although astrocyte-conditioned medium accelerated synaptic protein localization, it did not increase synaptic activity, suggesting a contact-dependant mechanism by which astrocytes augment synaptic activity. These results lay the foundation for future studies examining the functional development of human neurons and provide support for the potential application of human cells in restorative neuronal therapies.
Keywords: electrophysiology, action potential, forebrain, glia, neuronal progenitor cell, synaptic communication
Introduction
During mouse cortical development, few physiological changes occur at late embryonic stages when newborn neurons generated in the ventricular zone migrate through the intermediate zone. The majority of these migrating neurons express immature sodium (Na+) and potassium (K+) currents (_I_Na and _I_K) characterized by small amplitudes relative to adult cortical neurons. In contrast, after reaching the cortical plate, a rapid maturation occurs in which _I_K, _I_Na, and action potential (AP) amplitudes increase in parallel with a decrease in AP duration. Simultaneously, early postnatal neurons gain the capacity to fire repetitive trains of APs (Annis et al., 1993; Picken Bahrey and Moody, 2003). These characteristics, together with synaptic activity, comprise the hallmarks of a mature functional neuron.
Neurons produced from mouse embryonic stem (mES) cells have been shown to follow a similar pattern of maturation in vitro (Bain et al., 1995; Strubing et al., 1995; Finley et al., 1996; Benninger et al., 2003). mES cell-derived neurons exhibit an early expression of _I_K, _I_Na, and I Ca within 6 d of differentiation, and spontaneous and stimulated APs can be seen by 1 week (Strubing et al., 1995). mES cells plated onto organotypic hippocampal mouse slices have demonstrated that similar types of ionic currents are expressed within the first 5 d of culture and increase over the next 2 weeks, in correlation with an increase in AP amplitude and a decrease in AP duration (Benninger et al., 2003). Despite these advances, the majority of physiological studies using mES cell-derived neurons have focused on the maturation of a heterogeneous population of neurons with multiple regional characteristics (Bain et al., 1995; Strubing et al., 1995; Finley et al., 1996; Benninger et al., 2003; Du and Zhang, 2004). This has made direct comparisons between developmental studies in brain slices and mES cell-derived neurons difficult to interpret.
The most detailed accounts of human neural development come from anatomical studies (O'Rahilly, 1999) that outline a complex process by which neuroepithelial tissue forms the major CNS structures during neurogenesis, but they do not provide insights into functional neuronal maturation. For this, primary fetal cultures (Kato et al., 1985; Erkman et al., 1989; Kerkovich et al., 1999), expanded neural progenitors derived from fetal brain tissues (Chalmers-Redman et al., 1997; Svendsen et al., 1997, 1998; Carpenter et al., 1999; Piper et al., 2001), and immortalized neuronal cell lines (Li et al., 2000) have been the primary models for monitoring human neuronal development. These methods have provided viable neurons capable of physiological maturation, but the temporal development of these neurons varies with tissue age, in vitro expansion, and culturing method (Piper et al., 2001). In contrast, human embryonic stem (hES) cells, capable of differentiating into all cell types (Thomson et al., 1998; Reubinoff et al., 2000), allow the systematic functional evaluation of neural development under highly reproducible conditions. Neuroepithelial cells can be efficiently generated from hES cells and patterned to regionalized neural progenitors (Zhang et al., 2001; Perrier et al., 2004; Li et al., 2005; Singh Roy et al., 2005; Yan et al., 2005). Interestingly, the early stages of neural development from hES cells correspond to early anatomical brain development (for review, see Zhang, 2006). This cellular model of differentiation forms a foundation to explore functional neuronal development and decipher how these physiological traits are regulated.
Here, we report that hES cell-derived neuroepithelial cells uniformly expressing forebrain markers progressively acquire fundamental electrophysiological properties in a time-dependent manner from 3 to 10 weeks of differentiation. We also found that synaptic activity in hES cell-derived neuronal cultures temporally correlates with astrocyte differentiation and exogenous astrocytes can promote synaptic transmission before AP maturation.
Materials and Methods
Cell culture.
hES cells [H9 line, passage day 27 (P27) to P35] were maintained and differentiated according to previously published methods (Thomson et al., 1998; Zhang et al., 2001) with the following modifications. hES cells were lifted from the mouse fibroblast feeder layer and maintained in suspension for 3 d in hES cell medium. Cells were allowed to grow for an additional 3 d in neural medium containing DMEM/F-12 (Invitrogen, San Diego, CA) supplemented with N2 (1:100; Invitrogen), MEM nonessential amino acids (1:100; Invitrogen), heparin (2 μg/ml; Sigma, St. Louis, MO), and fibroblast growth factor 2 (20 ng/ml; R & D Systems, Minneapolis, MN). The resulting hES cell aggregates were then plated onto tissue culture wells and allowed to develop into neuroepithelial “rosette” cells. After 7 d of rosette formation, cells were enzymatically lifted from the culture wells with dispase (1 mg/ml; Roche Diagnostics, Indianapolis, IN) and suspended in neural medium for another 7 d. Neuroepithelial aggregates were then plated onto polyornithine–laminin-coated glass coverslips (100 μg/ml polyornithine, 10 μg/ml laminin; Sigma) in a neuronal differentiation medium (NDM) containing Neurobasal medium (Invitrogen), N2 (Invitrogen), MEM nonessential amino acids (Invitrogen), supplemented with BDNF (10 ng/ml; PeproTech, Rocky Hill, NJ), glial-derived neurotrophic factor (10 ng/ml; R & D Systems), cAMP (1 μm; Sigma), ascorbic acid (200 μm; Sigma), and laminin (20 μg/ml; Sigma), with medium changes occurring every other day.
For astrocyte coculture, cortical astrocytes were isolated from embryonic day 14 (E14) mouse embryos using previously described methods (Cole and de Vellis, 2001). Cells were allowed to grow to confluency in DMEM (Invitrogen) and 10% fetal bovine serum (FBS; Invitrogen), enzymatically dissociated with trypsin–EDTA (0.05%; Invitrogen), and passaged three times to obtain a nearly pure astrocytic culture. Astrocytes were then plated at a density of 25,000 cells per 10 mm of coverslip. After 4 d, DMEM and 10% FBS were removed, and the hES cell-derived neural cell aggregate (after 3 weeks of hES cell differentiation) was allowed to attach to the astrocyte monolayer for 2 h. Cocultures were then fed with 1 ml of NDM with growth factors (see above). Exchange of half of the medium occurred every other day. To examine the effects of secreted astrocyte factors, astrocytes were plated at a density of 10,000 cells per transwell culture insert (Corning, Corning, NY) in DMEM and 10% FBS. Cells were fed for 4 d with DMEM and 10% FBS before being transferred to newly plated neuroepithelial sphere cultures for differentiation. Astrocytes were monitored visually to ensure survival for the duration of the experiment.
Immunochemical staining.
Immunolabeling of hES cell-derived neurons was performed according to previously established methods (Zhang et al., 2001; Li et al., 2005). The primary antibodies used were as follows: polyclonal βIII-tubulin (1:5000; Covance Research Products, Princeton, NJ), monoclonal βIII-tubulin (1:1000; Sigma), monoclonal microtubule-associated protein 2a/b (MAP2a/b) (1:3000; Sigma), polyclonal GFAP (1:1000; Dako, Carpenteria, CA), monoclonal S100β (1:1000; AbcamCambridge, MA), polyclonal synapsin-1 (1:1000; Calbiochem, La Jolla, CA), goat Otx-2 (1:2000; R & D Systems), Bf-1 (1:1000; a gift from Dr. Y. Sasai, University of Tokyo, Tokyo, Japan). The secondary antibodies used were Alexa-Fluor donkey anti-rabbit 488, Alexa-Fluor donkey anti-mouse 594, and Alexa-Fluor donkey anti-goat 594 (Invitrogen), which were all used at a concentration of 1:1000.
For synaptic punctate analysis, independent MAP2, synapsin-1, and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) images were taken and loaded into MetaMorph software (Molecular Devices, Sunnyvale, CA). Single neurons that were not part of a cluster of cells were identified by MAP2 and DAPI staining. Synapsin-1 puncta were identified by eye and confirmed by applying equivalent thresholds to synapsin-1 images across treatment groups. For punctate density analysis, regions of interest (ROIs) were outlined by tracing 50 μm segments of randomly selected dendrites from MAP2 images and transferred to synapsin-1 images for counting. To ensure accurate counts, MetaMorph software was used to count thresholded events within the same ROI. Results obtained by eye and software analysis were similar.
Electrophysiology.
Coverslips were placed in a bath solution containing the following (in mm): 127 NaCl, 1.2 KH2PO4, 1.9 KCl, 26 NaHCO3, 2.2 CaCl2, 1.4 MgSO4, and 10 glucose at 295 mOsm. The bath solution was continuously bubbled with 95% O2/5% CO2 to maintain pH. Precise application of drugs was achieved using a gravity-fed drug barrel system. All reagents were diluted in extracellular solution and continuously bubbled with 95% O2/5% CO2 during the experiment. Tetrodotoxin (TTX; 1 μm), 4-aminopyridine (4-AP; 1 mm), bicuculline (20 μm), and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 20 μm) were obtained from Sigma. Tetraethylammonium (TEA; 500 μm) was obtained from Fluka (Neu-Ulm, Germany). Recording pipettes with resistances of 4–7 MΩ were filled with an intracellular recording solution containing the following (in mm): 140 K-gluconate, 10 Na+-HEPES, 10 BAPTA, and 4 Mg2+-ATP, pH 7.2, 290 mOsm. For recording postsynaptic currents (PSCs), the pipette solution contained the following (in mm): 20 KCl, 121 K-gluconate, 10 Na+-HEPES, 10 BAPTA, and 4 Mg2+-ATP, pH 7.2, 290 mOsm. All chemicals were obtained from Sigma. Neurons were visualized using an Olympus Optical (Tokyo, Japan) BX51WI microscope with differential interference contrast optics at 40×. Voltage-clamp and current-clamp recordings were obtained using a MultiClamp 700B amplifier (Molecular Devices). Signals were filtered at 4 kHz and sampled at 100 kHz using a Digidata 1322A analog-to-digital converter (Molecular Devices). All data were stored on a computer hard disk and analyzed with pClamp 9.0 (Molecular Devices) and MiniAnalysis (Synaptosoft, Fort Lee, NJ) software. Capacitance and series resistance were compensated (typically 50–80%). The liquid junction potential was adjusted according to previously published methods (Barry, 1994) (JPCalc in Clampex; Molecular Devices). Voltage recordings were appropriately adjusted. All recordings were performed at 21–23°C.
AP amplitude was measured from the threshold to the peak of the voltage deflection. AP half-width was measured as the duration of the AP at half of the maximum amplitude. To hold the resting membrane potential (RMP) between −45 and −50 mV for measurements of AP amplitude and half-width, current injection was manually set at the beginning of the experiment and monitored/adjusted throughout. Na+ and K+ currents were elicited by current injections from a holding potential of −70 or −80 mV. Peak Na+ currents and peak K+ currents were measured using pClamp 9.0 (Molecular Devices). Sustained K+ currents were measured at the end of a 1000 ms current injection. Transient K+ current amplitudes were obtained by subtracting the sustained current from the peak.
Results
hES cell-derived neural progenitors express forebrain markers and sequentially generate neurons and astrocytes
Neuroepithelial cells were differentiated and enriched from hES cells using minimal morphogen treatment (see Materials and Methods) for 3 weeks (Fig. 1A), after which neuroepithelial spheres were plated to allow for neuronal differentiation. Twenty-four hours after plating, βIII-tubulin+ neurons with unipolar and bipolar morphology appeared in the periphery of the cluster (Fig. 1B). The majority of these βIII-tubulin+ neurons also began to express the dendritic marker MAP2 (89.2 ± 2.9%, 252 cells counted from four cultures) mainly in the cell body and the leading neurite (Fig. 1B). With time in culture, particularly after 6–7 weeks of differentiation (3–4 weeks after plating), many cells displayed more complex dendritic arborizations (Fig. 1C,D).
Figure 1.
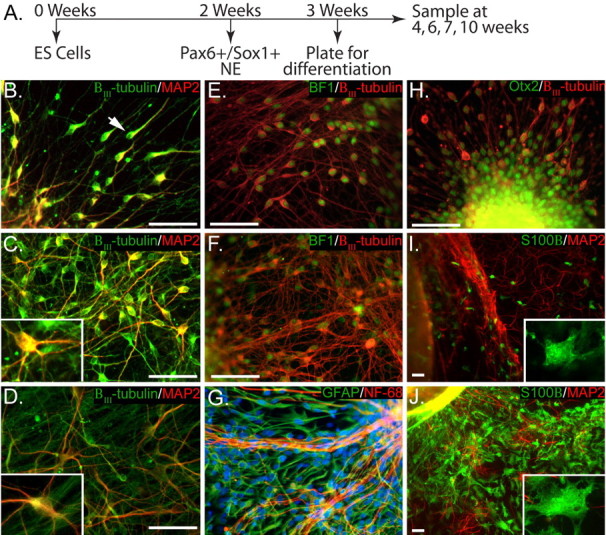
hES cell-derived neuroepithelia express forebrain markers and sequentially generate neurons and astrocytes. A, Timeline for neural and glial differentiation; H9 hES cells were differentiated into Pax6+/Sox1+ neuroepithelial cells (NE) within 2 weeks, enriched in suspension culture for an additional week, and plated on a laminin–polyornithine substrate for up to 7 weeks. B, Immature, unipolar (arrow), and bipolar neurons expressing βIII-tubulin were observed migrating away from the neuroepithelial cluster within 24 h after plating. C, D, After 4–10 weeks of differentiation (1–7 weeks after plating), neurons expressed both βIII-tubulin and MAP2 and exhibited increasingly complex multipolar morphology. Insets provide greater magnification of cells to clearly show morphological changes. E, F, H, The vast majority of immature neurons (βIII-tubulin+) were positive for Otx2 (H) and Bf-1 (E), the expression of which was retained in mature cultures (F). I, Astrocytes (S100β+) began to appear after 7 weeks of differentiation (4 weeks after plating), initially exhibiting simple morphology with few processes (inset). G, J, After 10 weeks of differentiation (7 weeks after plating), astrocytes were much larger, displayed more complex multipolar morphology (inset), and stained for GFAP (G) and S100β (J). NF-68, Neurofilament-68. Scale bars, 50 μm.
Previous studies have found that hES cells can be differentiated into neurons with diverse regional identities depending on the culture conditions (Du and Zhang, 2004). To examine which regional identity our cultures adopted over time, we analyzed the expression of homeodomain proteins, including the forebrain-specific transcription factor Otx2 (Simeone et al., 1992), the telencephalic marker Bf-1 (Xuan et al., 1995; Shimamura and Rubenstein, 1997; Watanabe et al., 2005), and Hoxb4, which is expressed by the hindbrain and spinal cord. Twenty-four hours after plating, the vast majority of cells were positive for Bf-1 (97.2 ± 0.9%, 1058 cells counted from six cultures) (Fig. 1E) as well as for Otx2 (98.9 ± 0.3%, 1037 cells counted from six cultures) (Fig. 1H). None of the cells within the cluster or in the periphery were stained for HoxB4 (data not shown). The forebrain identity was retained at least until 7 weeks (4 weeks after plating), because the vast majority of neurons continued to coexpress Otx2 (96.7 ± 1.1%, 420 cells counted from six cultures) or Bf-1 (96.8 ± 1.2%, 369 cells counted from four cultures) (Fig. 1F) with βIII-tubulin.
Analysis of glial differentiation indicated that after 6 weeks of differentiation (3 weeks after plating), no cells expressed the astrocyte-specific Ca2+-binding protein S100β (Van Eldik et al., 1984) (data not shown). We used S100β, in addition to the traditional astrocytic marker GFAP, because GFAP is also expressed in neural precursor cells (Kriegstein and Gotz, 2003; Casper and McCarthy, 2006). Beginning after 7 weeks (4 weeks after plating), a small population of S100β+ cells began to appear in the periphery of the cell cluster (Fig. 1I). At this time, the astrocytes had a simple, round morphology with few short processes emanating from the cell body (Fig. 1I). By 9 weeks in culture, astrocytes labeled with GFAP or S100β were more broadly distributed, appearing throughout the neuronal network (Fig. 1G,J). At these later time points, a more complex morphology was apparent, with multiple processes protruding from the cell body (Fig. 1J, inset). From these results, we conclude that a relatively uniform population of forebrain progenitors and neurons can be differentiated from hES cells and that the temporal pattern of neurogenesis/gliogenesis from _in vitro_-produced human neural progenitors is preserved in our culture system.
Progressive changes in AP characteristics correspond to morphological and cytochemical maturation
We next characterized the electrophysiological maturation of neurons from 3 to 10 weeks in culture by measuring a range of passive and active properties, including RMP, capacitance, input resistance (_R_in), APs evoked by depolarizing current pulses, and spontaneous synaptic activity. We found that RMP became steadily more hyperpolarized, reaching an average value of −58.1 ± 3.2 mV by 10 weeks of age (7 weeks after plating). Cell capacitance, which primarily reflects cell size, increased significantly from an average of 17 ± 1.4 pF at week 3 (n = 21) to 25.6 ± 2.2 pF by week 10 (n = 11; p < 0.05). Similarly, _R_in, which is negatively correlated with levels of channel expression and activity, decreased with age in culture (Table 1). These data suggest that neurons increased in size by ∼50% and began to express significantly more ion channels at the membrane surface, potentially contributing to the observed reduction in RMP.
Table 1.
Passive membrane properties of developing hES-derived neurons
| Time (weeks) | RMP (mV) | Cap (pF) | _R_in (MΩ) |
|---|---|---|---|
| 4 | −39.2 ± 2.3 | 17.0 ± 1.4 | 2518.8 ± 280.1 |
| 6 | −45.8 ± 2.7 | 23.2 ± 1.5 | 2769.2 ± 503.1 |
| 7 | −49.8 ± 2.3 | 24.2 ± 1.6 | 1190.8 ± 158.2 |
| 10 | −58.1 ± 3.2 | 25.6 ± 2.2 | 1807.3 ± 253.6 |
Next, we tested the ability of our neurons to fire APs and measured their characteristics in response to depolarizing current pulses when cells were allowed to rest at their natural RMP. After 4 weeks (1 week after plating), 50% of neurons (n = 20) were able to fire an AP (Fig. 2B) with a clear threshold of activation that could be blocked by TTX (data not shown). The remaining 4-week-old cells displayed a transient voltage deflection (Fig. 2A, 4W) that was not blocked by TTX or CdCl2 (data not shown). The percentage of cells able to fire an AP increased to 83% by 6 weeks (n = 12), and these cells acquired a more abrupt threshold of activation (Fig. 2A, 6W, arrow). By 7 weeks, 88% of cells could fire APs (n = 9), also displaying increased amplitude and a decreased duration (Fig. 2A, 7W). By 10 weeks, 100% of neurons could fire APs (Fig. 2A, 10W) (n = 16).
Figure 2.

AP maturation occurs from 3 to 10 weeks. A, Representative voltage responses to a 30 pA current injection are shown for neurons grown for 4, 6, 7, and 10 weeks (W). APs were determined by clear evidence of threshold (arrow). Repetitive trains of APs were observed in some cells at 10 weeks (10Wi). B, Summarized data from these neurons demonstrate that the number of APs fired in response to a 30 pA current pulse increased significantly by 7 weeks of culture. C, D, AP amplitude (C) increased significantly between 6 and 7 weeks of differentiation and was mirrored by a significant decrease in half-width (D). E, When neurons were held between −55 and −60 mV, no significant difference in AP amplitude was observed between 6 and 7 weeks. F, The significant difference between 6 and 7 weeks in AP half-width remained when cells were held at negative potentials. *p < 0.05; **p < 0.01. NS, Nonsignificant. Error bars indicate SEM.
Using AP amplitude and duration (half-width) as measures of neuronal maturity, we found that AP amplitude remained unchanged from 4 to 6 weeks of differentiation (Fig. 2C) (4 weeks, n = 10; 6 weeks, n = 8; p > 0.5). However, between 6 and 7 weeks, a significant increase in amplitude occurred (Fig. 2C) (6 weeks, n = 8; 7 weeks, n = 7; p < 0.05). AP amplitude remained elevated at later time points, paralleling a significant decrease in the _R_in (Table 1). The increase in AP amplitude that occurred between 6 and 7 weeks was mirrored by a significant decrease in the AP duration, which then remained constant through 10 weeks of differentiation (Fig. 2D).
The ability of neurons to fire a train of APs in response to current injection also correlates with cell maturity (Gao and Ziskind-Conhaim, 1998; Benninger et al., 2003; Picken Bahrey and Moody, 2003). We found that after 4 weeks (1 week after plating), neurons were unable to fire multiple APs in response to a 30 pA current pulse, and some were unable to fire even a single AP, leading to a mean of <1 (Fig. 2B). Over time, the mean number of APs increased, reaching significance by 7 weeks in culture when compared with 4-week-old cells (Fig. 2B). Although many cells displayed repetitive spiking, the second and third spikes were typically diminished in amplitude and prolonged in duration (Fig. 2A, 7W and 10W). In contrast, by 10 weeks, a subset of neurons was able to fire a train of APs without diminished amplitudes or prolonged durations on subsequent spikes (Fig. 2A, 10Wi).
AP amplitude is influenced by the number of available Na+ channels, which is partially controlled by the RMP. Because we found that cells became progressively more hyperpolarized with time in culture, we asked whether hyperpolarization alone could account for the observed changes in AP amplitude. To test this hypothesis, we held cells at an RMP of approximately −55 mV (range, −55 to −60 mV), which is the mean RMP of 7-week-old cells, and applied the same current pulses as above to elicit APs. When held at −55 mV, the significant change in AP maturation between 6 and 7 weeks was eliminated (Fig. 2E). Instead, a gradual increase in AP amplitude was observed over the entire 7 week period studied, with significantly larger amplitudes found at 7 and 10 weeks compared with those observed at 4 weeks (week 4, 35.4 ± 2.2 mV; week 7, 46.0 ± 2.8 mV; week 10, 49.9 ± 3.4; p < 0.05). These data suggest that when neurons were not held at hyperpolarized potentials, there was a reduced availability of Na+ channels in 6-week-old cells (compared with 7- and 10-week-old cells). Interestingly, this occurred despite a nonsignificant difference in RMP between 6- and 7-week-old cells (Table 1). Furthermore, the significant decrease in AP half-width observed between 6- and 7-week-old cells remained when RMP was adjusted (Fig. 2F) (p < 0.01). Thus, we reasoned that there must be other ionic currents besides Na+ that were affecting AP maturation in our cells (see below).
Na+ currents increase with cellular maturation
The steady increase in AP amplitude when cells were held between −55 and −60 mV suggests that reduced RMP over time in culture was not the only factor affecting AP amplitude. We therefore examined whether an increased number of Na+ channels could also contribute to increases in AP amplitude. Large, rapidly activating inward currents were reliably induced by voltage steps from a holding potential of −70 mV (Fig. 3Ai) and were blocked completely by TTX applied to the extracellular solution (Fig. 3Aii). Notably, when we examined these currents over time, there was no significant difference between peak _I_Na at 6 and 7 weeks as expected. There was, however, a trend toward increased _I_Na between these two time points (see Discussion), and 7-week-old cells did show significantly larger _I_Na than 4-week-old neurons (Fig. 3B,C) (week 4, n = 39; week 7, n = 9; p < 0.05). Current–voltage plots for 4-week-old cells (filled bars) and 7-week-old cells (open bars) showed an ∼6 mV leftward shift in the half-maximal current in older cultures. This is consistent with previous reports suggesting that rat neocortical neurons demonstrate a significant shift in the voltage dependence of activation for _I_Na from P3 to P21 (Cummins et al., 1994).
Figure 3.

Peak _I_Na increases during development. Ai, Rapidly inactivating voltage-gated inward currents were elicited by step depolarizations from −50 to 50 mV from a holding potential of −70 mV. Aii, These inward currents were completely blocked by TTX (1 μm). B, Pooled data demonstrate that peak _I_Na increases during differentiation, reaching significance after 7 weeks (*p < 0.05). Error bars indicate SEM. C, I–V relationship for peak inward current is shown for neurons after 4 weeks (filled diamonds) and 7 weeks (open squares) of differentiation. Seven-week-old neurons display larger peak _I_Na and a distinct (∼6 mV) leftward shift in half-maximal current, whereas maximal current was always elicited at 0 mV.
K+ current changes parallel AP maturation
To determine whether alterations in K+ currents contributed to changes in the AP half-width and the ability to fire multiple APs, we analyzed outward currents in response to voltage steps from a holding potential of −80 mV. We observed two distinct K+ current components. The first displayed a large, rapidly inactivating outward current in 86% of neurons tested (Fig. 4Ai). Treatment with 4-AP blocked the transient component of the outward current (Fig. 4Aii) that could also be blocked by prepulses to more positive holding potentials (−40 mV; data not shown), indicative of _I_A potassium currents (Nerbonne and Gurney, 1989; McCobb et al., 1990; Birnbaum et al., 2004). The transient outward current could be isolated by subtracting the 4-AP-treated current from the nontreated current (Fig. 4Aiii). The remaining sustained current was present in all cells and could be reduced with 12 mm TEA (Fig. 4Aiv). The sustained current was not affected by prepulses to more positive holding potentials (data not shown), consistent with descriptions of the sustained rectifying _I_K current (Nerbonne and Gurney, 1989; McCobb et al., 1990).
Figure 4.

_I_A increases parallel AP maturation, whereas _I_K decreases throughout maturation. Ai, A representative trace showing a fast inactivating and a sustained-outward current elicited by voltage steps from a holding potential of −80 mV. Aii, 4-AP (1 mm) eliminated the fast inactivating K+ current, leaving a sustained K+ current. Aiii, Subtraction of the sustained current from total K+ current isolated the transient K+ current. Aiv, A concentration of 12 μm TEA further reduced the sustained K+ current. B, Summarized data demonstrate that transient K+ current density significantly increased between 6 and 7 weeks and remained elevated until 10 weeks of differentiation. C, Sustained K+ current density declined steadily over time in culture and was significantly reduced by 10 weeks of differentiation (*p < 0.05). Error bars indicate SEM.
To compensate for changes in cell size, we calculated the maximum K+ current density for both _I_A and _I_K. Similar to changes seen in APs, _I_A density showed a significant increase in amplitude between 6 and 7 weeks of differentiation (Fig. 4B) (week 6, n = 7; week 7, n = 8; p < 0.05), then remained unchanged until 10 weeks. Unlike _I_A, the sustained _I_K density steadily decreased during differentiation, becoming significant only by 7 weeks after plating (Fig. 4C) (week 4, n = 9; week 7, n = 7; p < 0.05). The temporal correlation between the relatively sudden change in AP characteristics between 6 and 7 weeks of development and the sudden increase in _I_A density suggests a role for development of this K+ current in AP maturation.
Spontaneous synaptic activity correlates with astrocytic outgrowth
In addition to the generation of APs, the ability of a neuron to form synaptic connections within a neural network is an integral component of functional maturity. Therefore, we examined the localization of synaptic proteins as well as the generation of spontaneous PSCs from 4 to 10 weeks of neuronal maturation. Within the first 3 weeks after plating (3–6 weeks total), the synaptic protein synapsin-1 was diffusely distributed within the cytoplasm and processes (Fig. 5Ai, arrowhead), rarely forming synaptic puncta along MAP2+ dendrites (Fig. 5Ai, arrow). However, synapsin-1 puncta changed dramatically at later time points (Fig. 5Aii), increasing from 4.4 ± 4.0% at 6 weeks (746 cells counted from three cultures) to 85.7 ± 3.0% by 9 weeks (Fig. 6B) (403 cells counted from four cultures; p < 0.05).
Figure 5.

Endogenous and exogenous astrocytes induce synapsin localization and promote synaptic activity. Ai, In neurons plated without exogenous astrocytes, synapsin-1 (green) was diffusely distributed in the cytoplasm of MAP2-positive neurons (red) after 6 weeks of differentiation (arrowhead). Representative voltage-clamp recordings from cells held at −70 mV showed no spontaneous PSCs in these cells (below). Aii, In contrast, neurons grown on a monolayer of astrocytes displayed punctate localization of synapsin-1 (arrows) and prominent PSCs (below) at 6 weeks of differentiation. Aiii, Aiv, After 9 weeks, both groups displayed punctuate localization of synapsin-1 protein (arrows) and noticeable synaptic activity. Bi, Bii, Synaptic activity was composed of AMPA receptor-mediated currents that were blocked with CNQX (100 μm), as well as GABA receptor-mediated currents that could be blocked with bicuculline (bic.) (50 μm). C, Summarized data show that significantly more neurons displayed synaptic activity from 1 to 3 weeks when grown on astrocytes (open bars) than those grown on laminin (filled bars; *p < 0.05). At later time points, there was no significant difference between the number of cells displaying synaptic activity when neurons were plated with or without astrocytes. Scale bars, 20 μm.
Figure 6.
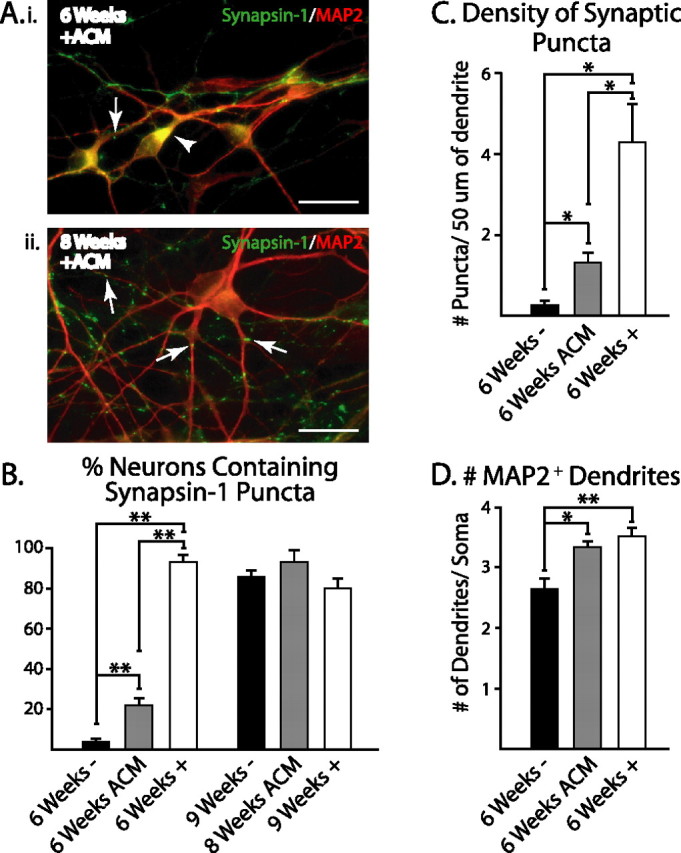
ACM induces the early onset of synapsin-1 localization. Ai, After 6 weeks of differentiation, neurons grown with ACM primarily showed diffuse synapsin-1 staining within MAP2+ neurons (arrowhead), which became punctate after 8 weeks of differentiation (Aii, arrows). B, ACM significantly increased the percentage of MAP2+ neurons with punctate synapsin-1 staining compared with untreated cultures (6 weeks −) after 6 weeks of differentiation. However, this effect was significantly smaller than the effects seen with direct astrocyte contact (6 weeks +). By 8–9 weeks, all groups showed similar percentages of punctate synapsin-1 staining. C, Similarly, the number of puncta per 50 μm of dendrite after 6 weeks of differentiation was significantly increased with ACM treatment but not to the same extent as direct astrocyte contact. D. The mean number of MAP2+ dendrites emanating from cell bodies after 6 weeks of differentiation was increased with ACM and direct contact with astrocytes. *p < 0.05; **p < 0.01. Scale bars, 20 μm. Error bars indicate SEM.
To examine whether the observed increase in synaptic protein localization correlated with the development of synaptic activity, we used whole-cell voltage-clamp recordings to monitor spontaneous synaptic currents. For the first 6 weeks in culture, cells did not display any spontaneous synaptic activity (Fig. 5Ai,C). Spontaneous synaptic activity was first seen at 7 weeks in culture in 40% of cells (Fig. 5C) (n = 5). Interestingly, this was also the time point corresponding to the outgrowth of astrocytes within the culture (Fig. 1I) and the observed punctate localization of synapsin-1 (Fig. 5Aii). Two types of spontaneous events were observed within the culture: an inward current with a rapid rise and decay that was blocked by the AMPA antagonist CNQX (Fig. 5Bi) and a more slowly decaying inward current that was blocked by the GABAA antagonist bicuculline (Fig. 5Bii). The negative deflection of the current trace (Fig. 5Bii) is consistent with a chloride-mediated current resulting from a depolarized reversal potential for chloride (−33 mV) under our recording conditions (see Materials and Methods).
Astrocytes promote synaptic transmission but not AP maturation
Because synaptogenesis and synaptic transmission correlated with the timing of astrocyte outgrowth (Figs. 1I, 5A,C), we postulated that coculture of hES cell-derived neurons with astrocytes before endogenous astrocytes develop would accelerate the development of synapses and synaptic transmission. Enhanced synaptic activity by astrocyte coculture has been consistently observed in rat retinal ganglion cells (Pfrieger and Barres, 1997; Ullian et al., 2004a) but has shown varying responses in other neuronal cell types (Ullian et al., 2004b; Steinmetz et al., 2006). We found that, in contrast to neurons grown in the absence of exogenous astrocytes (Figs. 5Ai, Aiii, 6B), 93.4 ± 3.1% of MAP2+ neurons (417 cells counted from three cultures) grown on a monolayer of mouse cortical astrocytes displayed punctate synapsin-1 staining by 6 weeks in vitro (Figs. 5Aii, 6B). In cells grown on astrocytes, we also observed synaptic activity as early as 1 week after plating. Furthermore, 27% of cells grown with astrocytes from 1 to 3 weeks in vitro displayed synaptic activity (Fig. 5Aii,C), a significantly larger proportion than those grown without astrocytes (0 of 15 without astrocytes, 6 of 22 with astrocytes; χ2 = 4.88; p < 0.05).
At later time points, there was no significant difference between the two groups in the percentage of neurons that displayed synaptic activity (Fig. 5C). Interestingly, the percentage of cells with spontaneous synaptic activity did not increase beyond 50% (Fig. 5D) for either group at any time point studied. Analysis of frequency and amplitude of spontaneous synaptic events did not differ between groups at each time point or within groups at different time points (data not shown). Together, these data suggest that exogenous astrocytes are sufficient to induce synaptic formation to the same extent as endogenous astrocytes and at significantly earlier time points. Furthermore, cells displaying synaptic activity between 4 and 6 weeks of differentiation on monolayers of astrocytes did not show increased AP generation as measured by the number of APs fired during a 30 pA, 500 ms current pulse (with astrocytes: 1.33 ± 0.21, n = 6; without astrocytes: 1.05 ± 0.26, n = 21; p > 0.05). The above findings indicate that the intrinsic firing capability of neurons is regulated separately from the formation of functional synaptic connections.
Previous studies have postulated that factors secreted from astrocytes were responsible for the increases in synaptic activity observed with astrocyte coculture, with some reports demonstrating increases (Pfrieger and Barres, 1997; Mauch et al., 2001; Steinmetz et al., 2006) and others showing no change (Christopherson et al., 2005). We examined this question using astrocytes grown on transwell membranes suspended within developing neuronal cultures. After 6 weeks of differentiation, no cells (0 of 13) displayed spontaneous synaptic activity with astrocyte-conditioned medium (ACM; data not shown). This was a time point at which direct neuron–astrocyte contact did give rise to synaptic activity (Fig. 5Aiii,C). After 8 weeks in culture, 36.4% of cells (4 of 11) displayed spontaneous synaptic activity consistent with previous data from wild-type cultures (Fig. 5Aii,C; and data not shown).
Although we did not observe an increase in the fraction of synaptically active cells with ACM treatment, significantly more MAP2+ cells (22.0 ± 3.3%; 550 cells counted from three cultures) grown with ACM displayed synaptic puncta after 6 weeks of total differentiation (Fig. 6B) compared with wild-type cultures (p < 0.05). Interestingly, almost all cells grown with astrocytes (93.4 ± 3.1%; 417 cells counted from four cultures) displayed punctate synapsin-1 localization, a significantly greater proportion than cells treated with ACM (Fig. 6B) (p < 0.01). In addition, the density of synaptic puncta varied, with cells treated with ACM having intermediate levels of puncta per 50 μm section of dendrite (Fig. 6C). To test whether these differences were because of changes in dendritic arborization, shown to be augmented by cholesterol in ACM (Goritz et al., 2005), we counted the number of primary dendrites emanating from cell bodies in each treatment group. We found that the number of dendritic protrusions in ACM-treated cultures (3.3 ± 0.1; 74 cells counted from three cultures) and astrocyte-contact cocultures (3.5 ± 0.1; 78 cells counted from four cultures) was significantly greater than wild-type cultures (2.7 ± 0.2; 62 cells counted from four cultures) (Fig. 6D) (p < 0.05). Interestingly, there was no difference between ACM-treated and astrocyte-supplemented cultures. This suggests that although astrocyte-secreted factors are able to induce synaptic protein localization and changes in dendritic structure, direct contact with astrocytes is required to accelerate the onset of synaptic activity during early neuronal maturation.
Discussion
In the current study, we used an established system that efficiently directs the differentiation of hES cells to neuroepithelial cells (Zhang et al., 2001) to create neurons with a nearly uniform forebrain identity. By systematically tracing the functional maturation of neurons originating from hES cell-derived neuroepithelia, we present evidence that differentiating human neurons progressively acquire fundamental electrophysiological properties in a time-dependent manner, including the coordinated maturation of the RMP, characteristic _I_K and _I_Na, and spontaneous synaptic currents. We also found that synaptic transmission in hES cell-derived neuronal cultures temporally correlates with astrocyte differentiation. Interestingly, we found that exogenous astrocytes can significantly accelerate the onset of synaptic activity, although they do not significantly affect the development of APs. These findings lay the groundwork for future studies on the functional maturation of human neurons.
Morphological and histological studies demonstrate that mES cell- and hES cell-derived neurons display temporally different developmental progressions that correlate well with anatomical studies. In mES cells, rosette-like structures mimicking the neural tube are evident within 5 d (equivalent days of gestation, 8.5) and reach maturity within 7 d of differentiation [equivalent days of gestation, 10.5 (Ying et al., 2003)]. This timing is similar to mouse neural tube formation in vivo. Furthermore, mouse neurogenesis begins at E11 within the cortex and continues after birth (Bayer, 1991), much like the outgrowth of postmitotic neurons from mES cells, as reported by a number of laboratories (Bain et al., 1995; Lang et al., 2004; Miles et al., 2004) (S. C. Zhang, unpublished observations). We have previously shown that hES cells form neural tube-like rosettes after 2 weeks of differentiation (Zhang et al., 2001), which is approximately equivalent to the third week of gestation considering that hES cells are derived from 1-week-old embryos. Rosette formation temporally correlates with the formation of the neural groove by 18 d in vivo and its subsequent rostrocaudal delineation by 20 d (O'Rahilly, 1999). In developing human embryos, recent evidence has demonstrated that there is a population of migratory postmitotic neurons by 31 d, distinct from Cajal-Retzius cells, which are evident within the forebrain (Bystron et al., 2006) and now thought to be the earliest born neurons. This is in close accord with the outgrowth of neurons within our culture system at 21 d (equivalent days of gestation, 27) from ES cells.
Strikingly, some of the electrophysiological characteristics we describe for human neuronal maturation mirror those observed in animal studies and can be predicted from the temporal differences between animal and human gestation (Clancy et al., 2001). In slice cultures of the developing mouse cortex from E14 to P0, the percentage of neurons in the cortical plate able to fire APs increases from 31 to 80%. Simultaneously, AP duration in these cells decreases from 8.8 ms at E14, reaching a minimum of 3.7 ms at P7 (Picken Bahrey and Moody, 2003). Our differentiating human neurons require 4 weeks for AP duration to decrease from 6.9 to 2.3 ms, and repetitive trains of APs are not observed until 10 weeks in vitro (compared with 9 d in mouse ES cell-derived neurons). Functionally, differentiated neurons begin firing APs at 7 weeks, which coincides with the earliest signs of spontaneous twitching within the developing human fetus at 7–8 weeks (Birnholz et al., 1978). Together, these data suggest an intrinsic temporal pattern of maturation that is preserved in vitro (Zhang et al., 2001, Zhang, 2006). Thus, in addition to their possible therapeutic potential, hES cells also provide us with the first available system to study functional human neural development at its earliest stages.
Although traditional roles for sodium and potassium currents are maintained in hES cell-derived neurons, increases in _I_A seem to play a dominant role in AP repolarization. _I_Na was found to increase gradually over the first 7 weeks of differentiation, similar to changes seen in slice recordings of rodent neurons, mES cell-derived neurons, and in limited studies with human neurons (Gao and Ziskind-Conhaim, 1998; Piper et al., 2001; Benninger et al., 2003, Picken Bahrey and Moody, 2003). We also demonstrate that _I_Na, in combination with RMP, seemed to play a significant role in AP amplitude, consistent with previous reports (Hodgkin and Huxley, 1952). Although _I_K density was always greater in magnitude than _I_Na, a decrease in AP half-width occurred in parallel with a significant increase in _I_A, indicating that _I_A was a major contributor to AP maturation (Fig. 1E,F). The relative proportion of each of these currents to total _I_K was not wholly unexpected, because changes in potassium currents during development differ greatly across brain region and neuronal subtype (Ahmed, 1988; Westbrook and Kenyon, 1990; Beck et al., 1992; Spigelman et al., 1992; Raucher and Dryer, 1994; Gao and Ziskind-Conhaim, 1998; Reiff and Guenther, 1999; Nakamura et al., 2001; Deng et al., 2004).
Interestingly, some of the currents we observed may be modulated by culture conditions. A recent report by Goetz et al. (2006) demonstrated that laminin (used in this study) and fibronectin substrates can differentially affect _I_Na as well as repetitive AP firing, which we and others attribute to _I_A currents (Connor and Stevens, 1971; Neher, 1971; Storm, 1987; Kang et al., 2000). They go on to show that substrates can alter the composition of neuronal subtypes within a culture, leading to the intriguing idea that the differentiation of varying neuronal subtypes from ES cells may produce neurons with different (and appropriate) physiological characteristics. Studies using genetic manipulation to identify specific neuronal subtypes for electrophysiological characterization are currently being pursued.
Although the basic electrophysiological properties of the neuron are acquired before astrocyte development, the onset of synaptic activity is associated with the differentiation of astrocytes from our hES cells. This has striking similarity to the paralleled appearance of astrocytes and first wave of synaptogenesis in developing rodents (Slezak and Pfrieger, 2003). In vitro studies using multiple neuronal cell types have shown that the cocultures of neurons with astrocytes enhance synaptogenesis (Pfrieger and Barres, 1997; Ullian et al., 2001, 2004a; Christopherson et al., 2005; Steinmetz et al., 2006). Here we show that endogenous astrocyte differentiation is associated with the localization of synaptic proteins and the onset of PSCs. Furthermore, exogenous astrocytes increase early synaptic protein localization and the onset of PSCs without altering firing properties of the developing neurons. Interestingly, ACM has been shown to have differential effects on the incidence and frequency of spontaneous synaptic currents ranging from no change (Christopherson et al., 2005) to 23-fold increases in miniature EPSC frequency (Pfrieger and Barres, 1997). Within our developing neuronal cultures, ACM showed a modest response, significantly increasing synaptic protein localization while having no effect on the incidence of PSCs. One component of ACM, cholesterol, has been shown to increase synaptogenesis in retinal ganglion cell cultures (Mauch et al., 2001), an effect caused by increases in dendritic arborization (Goritz et al., 2005). We found that ACM increased dendritic arborization to the same extent as direct astrocyte contact. Therefore, although astrocyte-secreted factors may affect synaptogenesis in our cells, we believe that a nonsecreted factor plays a major role in increasing synaptic protein localization and synaptic activity in developing hES cell-derived neurons. The disparity between these results could be attributable to species differences or by discrepancies in developmental stages between studies.
The current study builds on previous evidence demonstrating that the cellular and functional differentiation of hES cells to neural lineages in vitro mirrors anatomical studies using human fetal tissues. Furthermore, these data are in close agreement with functional analyses of animal models, suggesting that neural differentiation of hES cells is a realistic tool for the study of early human neural development. Using this model, we show that fundamental properties of a neuron, such as membrane potential, AP characteristics, and ionic currents, are most likely intrinsically regulated once a cell chooses a neuronal fate, whereas synaptic transmission can be significantly influenced by astrocytes. The finding that astrocytes promote synaptic transmission may have practical relevance in developing optimal methods for transplanting immature neurons into the adult brain environment.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grant R01 NS045926 and in part by a core grant to the Waisman Center from the National Institute of Child Health and Human Development (P30 HD03352).
References
- Ahmed Z. Expression of membrane currents in rat neocortical neurons in serum-free culture. II. Outward currents. Brain Res. 1988;468:297–305. doi: 10.1016/0165-3806(88)90142-3. [DOI] [PubMed] [Google Scholar]
- Annis CM, Robertson RT, O'Dowd DK. Aspects of early postnatal development of cortical neurons that proceed independently of normally present extrinsic influences. J Neurobiol. 1993;24:1460–1480. doi: 10.1002/neu.480241103. [DOI] [PubMed] [Google Scholar]
- Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI. Embryonic stem cells express neuronal properties in vitro. Dev Biol. 1995;168:342–357. doi: 10.1006/dbio.1995.1085. [DOI] [PubMed] [Google Scholar]
- Barry PH. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J Neurosci Methods. 1994;51:107–116. doi: 10.1016/0165-0270(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Bayer SA. Neocortical development. New York: Raven; 1991. [Google Scholar]
- Beck H, Ficker E, Heinemann U. Properties of two voltage-activated potassium currents in acutely isolated juvenile rat dentate gyrus granule cells. J Neurophysiol. 1992;68:2086–2099. doi: 10.1152/jn.1992.68.6.2086. [DOI] [PubMed] [Google Scholar]
- Benninger F, Beck H, Wernig M, Tucker KL, Brustle O, Scheffler B. Functional integration of embryonic stem cell-derived neurons in hippocampal slice cultures. J Neurosci. 2003;23:7075–7083. doi: 10.1523/JNEUROSCI.23-18-07075.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Birnholz JC, Stephens JC, Faria M. Fetal movement patterns: a possible means of defining neurologic developmental milestones in utero. AJR Am J Roentgenol. 1978;130:537–540. doi: 10.2214/ajr.130.3.537. [DOI] [PubMed] [Google Scholar]
- Bystron I, Rakic P, Molnar Z, Blakemore C. The first neurons of the human cerebral cortex. Nat Neurosci. 2006;9:880–886. doi: 10.1038/nn1726. [DOI] [PubMed] [Google Scholar]
- Carpenter MK, Cui X, Hu ZY, Jackson J, Sherman S, Seiger A, Wahlberg LU. In vitro expansion of a multipotent population of human neural progenitor cells. Exp Neurol. 1999;158:265–278. doi: 10.1006/exnr.1999.7098. [DOI] [PubMed] [Google Scholar]
- Casper KB, McCarthy KD. GFAP-positive progenitor cells produce neurons and oligodendrocytes throughout the CNS. Mol Cell Neurosci. 2006;31:676–684. doi: 10.1016/j.mcn.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Chalmers-Redman RM, Priestley T, Kemp JA, Fine A. In vitro propagation and inducible differentiation of multipotential progenitor cells from human fetal brain. Neuroscience. 1997;76:1121–1128. doi: 10.1016/s0306-4522(96)00386-7. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105:7–17. doi: 10.1016/s0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- Cole R, de Vellis J. Preparation of astroycte, oligodendrocyte, and microglia cultures from primary rat cerebral cultures. In: Federoff S, Richardson A, editors. Protocols for neural cell culture, Chap 9. Ed 3. Totowa, NJ: Humana; 2001. pp. 117–127. [Google Scholar]
- Connor JA, Stevens CF. Voltage clamp studies of a transient outward membrane current in gastropod neural somata. J Physiol (Lond) 1971;213:21–30. doi: 10.1113/jphysiol.1971.sp009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Xia Y, Haddad GG. Functional properties of rat and human neocortical voltage-sensitive sodium currents. J Neurophysiol. 1994;71:1052–1064. doi: 10.1152/jn.1994.71.3.1052. [DOI] [PubMed] [Google Scholar]
- Deng P, Pang Z, Zhang Y, Xu ZC. Developmental changes of transient potassium currents in large aspiny neurons in the neostriatum. Brain Res Dev Brain Res. 2004;153:97–107. doi: 10.1016/j.devbrainres.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Du ZW, Zhang SC. Neural differentiation from embryonic stem cells: which way? Stem Cells Dev. 2004;13:372–381. doi: 10.1089/scd.2004.13.372. [DOI] [PubMed] [Google Scholar]
- Erkman L, Touzeau G, Bertrand D, Bader CR, Kato AC. Characterization of dissociated monolayer cultures of human spinal cord. Brain Res Bull. 1989;22:57–65. doi: 10.1016/0361-9230(89)90128-7. [DOI] [PubMed] [Google Scholar]
- Finley MF, Kulkarni N, Huettner JE. Synapse formation and establishment of neuronal polarity by P19 embryonic carcinoma cells and embryonic stem cells. J Neurosci. 1996;16:1056–1065. doi: 10.1523/JNEUROSCI.16-03-01056.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao BX, Ziskind-Conhaim L. Development of ionic currents underlying changes in action potential waveforms in rat spinal motoneurons. J Neurophysiol. 1998;80:3047–3061. doi: 10.1152/jn.1998.80.6.3047. [DOI] [PubMed] [Google Scholar]
- Goetz AK, Scheffler B, Chen HX, Wang A, Suslov O, Xiang H, Brustle O, Roper SN, Steindler DA. Temporally restricted substrate interactions direct fate and specification of neural precursors derived from embryonic stem cells. Proc Natl Acad Sci USA. 2006;103:11063–11068. doi: 10.1073/pnas.0510926103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29:190–201. doi: 10.1016/j.mcn.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of loligo. J Physiol (Lond) 1952;116:449–472. doi: 10.1113/jphysiol.1952.sp004717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Huguenard JR, Prince DA. Voltage-gated potassium channels activated during action potentials in layer V neocortical pyramidal neurons. J Neurophysiol. 2000;83:70–80. doi: 10.1152/jn.2000.83.1.70. [DOI] [PubMed] [Google Scholar]
- Kato AC, Touzeau G, Bertrand D, Bader CR. Human spinal cord neurons in dissociated monolayer cultures: morphological, biochemical, and electrophysiological properties. J Neurosci. 1985;5:2750–2761. doi: 10.1523/JNEUROSCI.05-10-02750.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkovich DM, Sapp D, Weidenheim K, Brosnan CF, Pfeiffer SE, Yeh HH, Busciglio J. Fetal human cortical neurons grown in culture: morphological differentiation, biochemical correlates and development of electrical activity. Int J Dev Neurosci. 1999;17:347–356. doi: 10.1016/s0736-5748(99)00036-2. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Gotz M. Radial glia diversity: a matter of cell fate. Glia. 2003;43:37–43. doi: 10.1002/glia.10250. [DOI] [PubMed] [Google Scholar]
- Lang RJ, Haynes JM, Kelly J, Johnson J, Greenhalgh J, O'brien C, Mulholland EM, Baker L, Munsie M, Pouton CW. Electrical and neurotransmitter activity of mature neurons derived from mouse embryonic stem cells by sox-1 lineage selection and directed differentiation. Eur J Neurosci. 2004;20:3209–3221. doi: 10.1111/j.1460-9568.2004.03782.x. [DOI] [PubMed] [Google Scholar]
- Li R, Thode S, Zhou J, Richard N, Pardinas J, Rao MS, Sah DW. Motoneuron differentiation of immortalized human spinal cord cell lines. J Neurosci Res. 2000;59:342–352. doi: 10.1002/(sici)1097-4547(20000201)59:3<342::aid-jnr7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Li XJ, Du ZW, Zarnowska ED, Pankratz M, Hansen LO, Pearce RA, Zhang SC. Specification of motoneurons from human embryonic stem cells. Nat Biotechnol. 2005;23:215–221. doi: 10.1038/nbt1063. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- McCobb DP, Best PM, Beam KG. The differentiation of excitability in embryonic chick limb motoneurons. J Neurosci. 1990;10:2974–2984. doi: 10.1523/JNEUROSCI.10-09-02974.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles GB, Yohn DC, Wichterle H, Jessell TM, Rafuse VF, Brownstone RM. Functional properties of motoneurons derived from mouse embryonic stem cells. J Neurosci. 2004;24:7848–7858. doi: 10.1523/JNEUROSCI.1972-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura F, Morihata H, Matsuura S, Kuno M. Post-cultured development of basic electrophysiological properties of spinal neurons obtained from rat embryo. Brain Res. 2001;905:245–249. doi: 10.1016/s0006-8993(01)02530-6. [DOI] [PubMed] [Google Scholar]
- Neher E. Two fast transient current components during voltage clamp on snail neurons. J Gen Physiol. 1971;58:36–53. doi: 10.1085/jgp.58.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, Gurney AM. Development of excitable membrane properties in mammalian sympathetic neurons. J Neurosci. 1989;9:3272–3286. doi: 10.1523/JNEUROSCI.09-09-03272.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rahilly R. The embryonic human brain: an atlas of developmental stages. New York: Wiley; 1999. [Google Scholar]
- Perrier AL, Tabar V, Barberi T, Rubio ME, Bruses J, Topf N, Harrison NL, Studer L. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci USA. 2004;101:12543–12548. doi: 10.1073/pnas.0404700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Picken Bahrey HL, Moody WJ. Early development of voltage-gated ion currents and firing properties in neurons of the mouse cerebral cortex. J Neurophysiol. 2003;89:1761–1773. doi: 10.1152/jn.00972.2002. [DOI] [PubMed] [Google Scholar]
- Piper DR, Mujtaba T, Keyoung H, Roy NS, Goldman SA, Rao MS, Lucero MT. Identification and characterization of neuronal precursors and their progeny from human fetal tissue. J Neurosci Res. 2001;66:356–368. doi: 10.1002/jnr.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucher S, Dryer SE. Functional expression of A-currents in embryonic chick sympathetic neurones during development in situ and in vitro. J Physiol (Lond) 1994;479:77–93. doi: 10.1113/jphysiol.1994.sp020279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff DF, Guenther E. Developmental changes in voltage-activated potassium currents of rat retinal ganglion cells. Neuroscience. 1999;92:1103–1117. doi: 10.1016/s0306-4522(99)00044-5. [DOI] [PubMed] [Google Scholar]
- Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol. 2000;18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- Shimamura K, Rubenstein JL. Inductive interactions direct early regionalization of the mouse forebrain. Development. 1997;124:2709–2718. doi: 10.1242/dev.124.14.2709. [DOI] [PubMed] [Google Scholar]
- Simeone A, Acampora D, Gulisano M, Stornaiuolo A, Boncinelli E. Nested expression domains of four homeobox genes in developing rostral brain. Nature. 1992;358:687–690. doi: 10.1038/358687a0. [DOI] [PubMed] [Google Scholar]
- Singh Roy N, Nakano T, Xuing L, Kang J, Nedergaard M, Goldman SA. Enhancer-specified GFP-based FACS purification of human spinal motor neurons from embryonic stem cells. Exp Neurol. 2005;196:224–234. doi: 10.1016/j.expneurol.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Slezak M, Pfrieger FW. New roles for astrocytes: regulation of CNS synaptogenesis. Trends Neurosci. 2003;26:531–535. doi: 10.1016/j.tins.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Spigelman I, Zhang L, Carlen PL. Patch-clamp study of postnatal development of CA1 neurons in rat hippocampal slices: membrane excitability and K+ currents. J Neurophysiol. 1992;68:55–69. doi: 10.1152/jn.1992.68.1.55. [DOI] [PubMed] [Google Scholar]
- Steinmetz CC, Buard I, Claudepiere T, Nägler K, Pfrieger FW. Regional variation in the glial influence of synapse development in the mouse CNS. J Physiol (Lond) 2006;577:249–261. doi: 10.1113/jphysiol.2006.117358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol (Lond) 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strubing C, Ahnert-Hilger G, Shan J, Wiedenmann B, Hescheler J, Wobus AM. Differentiation of pluripotent embryonic stem cells into the neuronal lineage in vitro gives rise to mature inhibitory and excitatory neurons. Mech Dev. 1995;53:275–287. doi: 10.1016/0925-4773(95)00446-8. [DOI] [PubMed] [Google Scholar]
- Svendsen CN, Caldwell MA, Shen J, ter Borg MG, Rosser AE, Tyers P, Karmiol S, Dunnett SB. Long-term survival of human central nervous system progenitor cells transplanted into a rat model of Parkinson's disease. Exp Neurol. 1997;148:135–146. doi: 10.1006/exnr.1997.6634. [DOI] [PubMed] [Google Scholar]
- Svendsen CN, ter Borg MG, Armstrong RJ, Rosser AE, Chandran S, Ostenfeld T, Caldwell MA. A new method for the rapid and long term growth of human neural precursor cells. J Neurosci Methods. 1998;85:141–152. doi: 10.1016/s0165-0270(98)00126-5. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Christopherson KS, Barres BA. Role for glia in synaptogenesis. Glia. 2004a;47:209–216. doi: 10.1002/glia.20082. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Harris BT, Wu A, Chan JR, Barres BA. Schwann cells and astrocytes induce synapse formation by spinal motor neurons in culture. Mol Cell Neurosci. 2004b;25:241–251. doi: 10.1016/j.mcn.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Van Eldik LJ, Ehrenfried B, Jensen RA. Production and characterization of monoclonal antibodies with specificity for the S100 beta polypeptide of brain S100 fractions. Proc Natl Acad Sci USA. 1984;81:6034–6038. doi: 10.1073/pnas.81.19.6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Kamiya D, Nishiyama A, Katayama T, Nozaki S, Kawasaki H, Watanabe Y, Mizuseki K, Sasai Y. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci. 2005;8:288–296. doi: 10.1038/nn1402. [DOI] [PubMed] [Google Scholar]
- Westbrook CD, Kenyon JL. Potassium currents in chick sensory neurons change with development. Brain Res Dev Brain Res. 1990;54:1–10. doi: 10.1016/0165-3806(90)90059-8. [DOI] [PubMed] [Google Scholar]
- Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E. Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron. 1995;14:1141–1152. doi: 10.1016/0896-6273(95)90262-7. [DOI] [PubMed] [Google Scholar]
- Yan Y, Yang D, Zarnowska ED, Du Z, Werbel B, Valliere C, Pearce RA, Thomson JA, Zhang SC. Directed differentiation of dopaminergic neuronal subtypes from human embryonic stem cells. Stem Cells. 2005;23:781–790. doi: 10.1634/stemcells.2004-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Stavridis M, Griffiths D, Li M, Smith A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol. 2003;21:183–186. doi: 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
- Zhang SC. Neural subtype specification from embryonic stem cells. Brain Pathol. 2006;16:132–142. doi: 10.1111/j.1750-3639.2006.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]