Viral and cellular microRNAs as determinants of viral pathogenesis and immunity (original) (raw)
. Author manuscript; available in PMC: 2011 Apr 19.
Published in final edited form as: Cell Host Microbe. 2008 Jun 12;3(6):375–387. doi: 10.1016/j.chom.2008.05.002
Summary
MicroRNAs have recently emerged as key post-transcriptional regulators of gene expression in multicellular eukaryotes. It is increasingly clear that microRNAs of both viral and cellular origin can positively or negatively influence viral replication. Viral microRNAs can directly alter host physiology, including components of the immune system, and host microRNAs can directly alter the virus life cycle. Here, we discuss what is known about how viral and cellular microRNAs influence viral replication and pathogenic potential through their regulation of viral mRNAs or by reshaping cellular gene expression.
Introduction
MicroRNAs (miRNAs) are a class of ~22 nt long non-coding RNAs transcribed from the genomes of all multicellular organisms and some viruses (Bartel, 2004; Cullen, 2004). Over 400 different miRNAs have been identified in humans (Griffiths-Jones et al., 2006; Griffiths-Jones et al., 2008; Landgraf et al., 2007). miRNA biogenesis begins with the transcription of a pri-miRNA precursor, typically by RNA polymerase II (Pol II), containing the mature miRNA sequence within the stem of an imperfect ~80 nt RNA hairpin. The nuclear RNase III enzyme Drosha, acting in concert with the double-stranded (ds) RNA binding protein DGCR8, excises the ~60 nt pre-miRNA hairpin, which is exported to the cytoplasm by Exportin 5. The cytoplasmic RNase III enzyme Dicer then cleaves the pre-miRNA to give an imperfect, ~22 nt long RNA duplex, featuring 2 nt 3′-overhangs on each strand. The miRNA strand, which generally exhibits weaker 5′ base pairing, is preferentially incorporated into miRNA effector complexes, called RNA induced silencing complexes (RISCs), consisting minimally of the miRNA and one of the four Argonaute (Ago) proteins. The unincorporated strand, called the passenger or miRNA* strand, is degraded. If 5′ base pairing is similar at both ends of the duplex, each strand may be incorporated into RISC as a functional miRNA. The non-canonical generation of pre-miRNAs from short introns (“mirtrons”) by splicing and debranching, instead of Drosha-processing, has been described (Berezikov et al., 2007; Okamura et al., 2007; Ruby et al., 2007). These pre-miRNAs or introns are also recognized by Exportin5 and then processed by Dicer.
In animal cells, Ago2-containing RISCs can cleave mRNAs that are highly complementary to the miRNA. This process is indistinguishable from RNA interference (RNAi) induced by artificial small interfering RNAs (siRNAs)(Zeng et al., 2003). However, miRNA-directed cleavage of mRNAs is rare and the only example thus far in mammalian cells is miR-196, which exhibits a perfect match with its mRNA target (Yekta et al., 2004). Instead, RISC-bound miRNAs guide the recognition of partial matches within the 3′ untranslated region (3′ UTR) of target mRNAs and induce the translational inhibition of these mRNAs, potentially leading to modest mRNA destabilization. Perfect base pairing of the miRNA “seed region” (nt 2–7 from the miRNA 5′ end) to the 3′ UTR of an mRNA target is often, but not always, required for miRNA-mediated repression (Lewis et al., 2005). In many cases, miRNAs and their 3′UTR binding sites are evolutionary conserved, facilitating the computational identification of authentic miRNA targets (Rajewsky, 2006). Even though many miRNAs have been identified, and many mRNA targets have been computationally predicted, the biological significance of miRNA-mediated regulation is understood in only a few instances. Nevertheless, specific miRNAs have already been implicated in diverse biological processes, including development, cellular differentiation, proliferation, apoptosis, and oncogenesis (Bushati and Cohen, 2007). The computational prediction that individual miRNAs may regulate several hundred genes, and that more than 30% of animal genes may be subject to regulation by miRNAs, underscores the emerging importance of miRNA-mediated gene regulation (Krek et al., 2005; Lewis et al., 2005).
miRNAs are attractive candidates as virally encoded regulators of viral and host cell gene expression due to their small size, their lack of immunogenicity and their remarkable functional flexibility. It is therefore not surprising that several DNA viruses have been found to express viral miRNAs. Several scenarios for the role of miRNAs in regulating viral replication and pathogenesis are conceivable. Viral miRNAs may directly regulate viral and/or host cell gene expression to benefit the virus (Figure 1). Viruses may use cellular miRNAs for their replication, and in some cases the expression of cellular miRNAs may be induced or inhibited to reshape the cellular gene expression environment to the benefit of the virus (Figure 2). Finally, the expression of certain cellular miRNAs may be disadvantageous to the virus, through their interaction with viral mRNAs or because of their cellular functions, and may therefore either limit virus replication or be downregulated by the virus. Regardless of whether interactions with cellular miRNAs enhance or limit virus replication, cellular miRNAs may have exerted a significant effect on viral genome evolution and certainly have the potential to regulate the tissue tropism of viruses in vivo. Known examples of each scenario will be discussed in this review. This review will discuss examples of the many roles of miRNAs in interactions between viruses and their hosts.
Figure 1. Potential mechanisms of viral miRNA function.

(A) dsDNA virus-derived miRNAs may target viral transcripts that are transcribed antisense to the miRNA precursor for degradation by RISC (green circle). (B) Viral miRNAs may inhibit translation of viral transcripts carrying imperfect matches to the miRNA. (C) Viral miRNAs may engage in novel interactions with host mRNAs (red) or function as orthologs of cellular miRNAs and therefore inhibit the normal mRNA targets of the orthologous host cell miRNA (purple).
Figure 2. Potential effect of cellular miRNAs on virus replication.
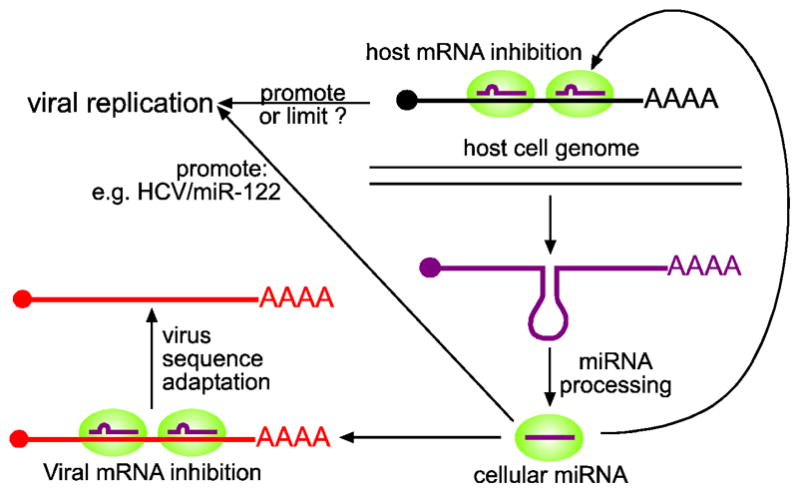
Cellular miRNAs (purple) may bind viral RNAs and thereby directly promote viral replication, as in the case of miR-122, which promotes HCV RNA replication. Cellular miRNAs may mediate the recognition of viral mRNAs (red) by RISC (green circle) and thereby limit virus replication. In this case, viral mRNAs would likely be under evolutionary pressure to avoid matches to cellular miRNAs abundantly expressed in their physiological host cells. Finally, cellular miRNAs might indirectly promote or limit virus replication through regulation of their endogenous mRNA targets. In this case, viruses could benefit from re-shaping the cellular miRNA environment.
Virally encoded miRNAs
Known viral miRNAs exclusively derive from dsDNA viruses, mainly of the herpesvirus family, but also from simian polyomaviruses and human adenovirus (Table 1). These miRNAs have been identified by cDNA cloning of small RNAs from virally infected cells or based on computational prediction and subsequent validation of their expression. Using an algorithm to identify potential viral pre-miRNA stem-loop structures, Pfeffer et al. (2005) successfully predicted some of the subsequently validated miRNAs encoded by the virus families listed in Table 1. This report also predicted pre-miRNA-like hairpin structures in members of the poxvirus family of dsDNA viruses, but the existence of poxviral miRNAs has not yet been experimentally validated. Poxviruses replicate exclusively in the cytoplasm and therefore do not have ready access to the nuclear stages of the classical miRNA biogenesis pathway. It remains possible, however, that poxviruses have evolved unconventional mechanisms of miRNA processing. For other virus families, the probability of encoding miRNAs was predicted to be low (Pfeffer et al., 2005). Consistent with this hypothesis, miRNA cloning from cells infected with the retroviruses human immunodeficiency virus type-1 (HIV-1) and human T-cell leukemia virus type 1 (HTLV-1), with the dsDNA virus human papillomavirus 31 (HPV31), and with two positive-strand RNA viruses, yellow fever virus (YFV) and hepatitis C virus (HCV), did not result in the identification of any viral miRNAs (Cai et al., 2006a; Lin and Cullen, 2007; Pfeffer et al., 2005; Randall et al., 2007). This is perhaps surprising for HPV, a nuclear dsDNA virus, but is expected for viruses with RNA genomes as excision of an encoded miRNA would result in the degradation of the genomic RNA. Nevertheless, others have reported the detection of HIV-1 miRNAs (Omoto et al., 2004; Ouellet et al., 2008), and the question of whether HIV-1 miRNAs exist remains controversial.
Table 1.
Summary of known virus-encoded miRNAs.
| Virus Family | Subfamily/Genus | Name | Host | Number of validated pre-miRNAs | Names | References |
|---|---|---|---|---|---|---|
| Herpesviruses | α/Simplexvirus | HSV-1 | human | 1 | miR-H1 | Cui et al., 2006; |
| α/Mardivirus | MDV1 | chicken | 12 | miR-M1 to miR-M12 | Burnside et al., 2006; Yao et al., 2008; | |
| MDV2 | chicken | 17 | miR-M14 to miR-M30 | Yao et al., 2007 | ||
| β/Cytomegalovirus | hCMV | human | 11 | miR-UL22A, miR-UL36, miR-UL70, miR- UL112, miR-UL148D, miR-US4, miR-US5-1, miR-US5-2, miR-US25-1, miR-US25-2, miR- US33 | Dunn et al., 2005; Grey et al., 2005; Landgraf et al., 2007; Peffer et al., 2005 | |
| mCMV | murine | 18 | miR-M23-1, miR-M23-2, miR-M44-1, miR- M55-1, miR-M87-1, miR-M95-1, miR-m01-1, miR-m01-2, miR-m01-3, miR-m01-4, miR- m21-1, miR-m22-1, miR-m59-1, miR-m59-2, miR-m88-1, miR-m107-1, miR-m108-1, miR- m108-2 | Buck et al., 2007; Dölken et al., 2007; | ||
| γ1/Lymphocryptoviruses | EBV | human | 23 | miR-BART1 to miR-BART20; miR-BHRF1-1 to miR-BHRF1-3 | Cai et al., 2006b; Grundhoff et al., 2006; Pfeffer et al., 2004 | |
| rLCV | simian | 16 | miR-rL1-1 to miR-rL1-16 | Cai et al., 2006b | ||
| γ2/Rhadinoviruses | KSHV | human | 12 | miR-K12-1 to miR-K12-12 | Cai et al., 2005; Grundhoff et al., 2006; Pfeffer et al., 2005; Samols et al., 2005 | |
| RRV | simian | 7 | miR-rR1-1 to miR-rR1-7 | Schäfer et al., 2007 | ||
| MHV68 | murine | 9 | miR-M1-1 to miR-M1-9 | Landgraf et al., 2007; Pfeffer et al., 2005 | ||
| Polyomaviruses | SV40 | simian | 1 | miR-S1 | Sullivan et al., 2005 | |
| SA12 | simian | 1 | unnamed | Cantalupo et al., 2005 | ||
| Adenoviruses | hAV | human | 1 | unnamed | Andersson et al., 2005; Aparicio et al., 2005; Sano et al., 2006 |
Inspection of Table 1 shows that herpesviruses are currently unique in expressing multiple viral miRNAs. Herpesviruses are large (~125–240 kb) dsDNA viruses that cause persistent infections of their host, invariably associated with the establishment of a reservoir of latently infected cells (Pellett and Roizman, 2007). Viral miRNA expression has been demonstrated for several human pathogenic herpesviruses, including Epstein-Barr virus (EBV), Kaposi’s Sarcoma associated herpesvirus (KSHV), human cytomegalovirus (hCMV) and herpes simplex virus type 1 (HSV-1). Of note, EBV and KSHV are tumorigenic γ-herpesviruses, which establish latency in B lymphocytes and can cause B-cell lymphomas, such as Burkitt’s or Hodgkin’s lymphoma (by EBV) or primary effusion lymphoma (PEL, can be caused by KSHV) as well as a range of tumors in other cell types, including nasopharyngeal carcinoma (by EBV) and Kaposi’s sarcoma (by KSHV) (Ganem, 2007; Kieff and Rickinson, 2007). The finding that cellular miRNAs—so-called oncomirs—can play a role in cellular transformation raises the possibility that viral miRNAs could also display oncogenic potential (Gottwein et al., 2007; He et al., 2005; Skalsky et al., 2007).
Expression of viral miRNAs
Herpesviral miRNAs derived from non-coding RNAs, from non-coding regions and open reading frames of protein-coding mRNAs, as well as from intronic regions, have all been identified. To date, no viral “mirtrons” have been reported. Like cellular miRNAs, herpesvirus miRNAs are processed from Pol II transcripts, with the exception of the murine gammaherpesvirus 68 (MHV68) miRNAs, which appear to be derived from RNA polymerase III (Pol III) transcripts (Pfeffer et al., 2005). Known adenoviral miRNAs derive from the viral VA1 non-coding RNA (Andersson et al., 2005; Aparicio et al., 2006; Sano et al., 2006), a short Pol III transcript that is expressed at very high levels in infected cells and functions as an inhibitor of cellular protein kinase R (PKR), an intracellular sensor of dsRNA (Mathews and Shenk, 1991; Thimmappaya et al., 1982). This allows adenovirus to evade inhibitory cellular responses induced by viral dsRNAs. A very minor portion of the basal stem of VA1 (<1%) is processed by Dicer into functional RISC-associated small RNAs (Andersson et al., 2005; Aparicio et al., 2006; Sano et al., 2006). The transfection of synthetic 2′-O-methyl-modified oligonucleotides antisense to these VA1 miRNAs resulted in a reduction of adenovirus titer (Aparicio et al., 2006). However, due to their high affinity for RNA, these oligonucleotides might also disrupt VA1 folding and thereby lead to the loss of other functions of VA1, such as inhibition of PKR. Therefore, further experiments are required to establish whether VA1-derived RISC-associated small RNAs are an irrelevant byproduct of VA1 overexpression or whether they indeed function in the adenoviral life cycle.
The expression of the γ-herpesvirus miRNAs identified to date, with the exception of the rhesus Rhadinovirus (RRV) miRNAs, is primarily associated with viral latency (Cai et al., 2005; Cai et al., 2006b; Grundhoff et al., 2006; Landgraf et al., 2007; Pfeffer et al., 2005; Pfeffer et al., 2004; Samols et al., 2005; Schäfer et al., 2007). In contrast, the hCMV, murine cytomegalovirus (mCMV) and HSV-1 miRNAs identified to date are all expressed during productive virus replication (Buck et al., 2007; Cui et al., 2006; Dölken et al., 2007; Dunn et al., 2005; Grey et al., 2005). A report suggesting that the HSV-1 latency associated transcript (LAT), which is the only HSV-1 transcript expressed during HSV-1 latency, gives rise to a miRNA (miR-LAT) was subsequently retracted (Gupta et al., 2006, 2008). Since no LAT-derived proteins have been found, it remains an attractive hypothesis that one or more LAT derived miRNAs exist and are important mediators of HSV-1 latency.
In several cases, viral miRNAs are expressed from polycistronic transcripts, thereby ensuring their co-regulation. EBV miRNAs are grouped into two clusters, which are differentially expressed in different forms of EBV latency (Cai et al., 2006b; Grundhoff et al., 2006; Pfeffer et al., 2004). The BART miRNA cluster contains 20 miRNA precursors within the introns of the BamA rightward transcripts (BARTs). The BHRF1 miRNA cluster, found adjacent to BHRF1 (BamH1 fragment H rightward open reading frame 1), contains three miRNA precursors (Cai et al., 2006b; Grundhoff et al., 2006; Pfeffer et al., 2004). Expression of the full repertoire of EBV latent proteins, consisting of six EBV nuclear antigens (EBNAs), three membrane proteins (latent membrane proteins, LMPs), two small RNAs (EBERs) and the BARTs, is referred to as the latency III program (Kieff and Rickinson, 2007). The latency III expression pattern is observed in lymphoblastoid cell lines, resulting from the transformation of primary B cells by EBV in vitro, as well as in B cell lymphomas arising in immunocompromised individuals in vivo. More restricted, and therefore less immunogenic, patterns of latent EBV protein expression (latency I and II programs) have been described in vitro and in vivo (Kieff and Rickinson, 2007). The expression of the BHRF1 miRNA cluster is associated with the latency III replication program (Cai et al., 2006b; Xing and Kieff, 2007). In contrast, the BART miRNA cluster is expressed at very low levels in EBV-infected B-cells, but expressed at high levels in EBV-infected epithelial cells undergoing type II latency (Cai et al., 2006b; Kim do et al., 2007; Lo et al., 2007).
KSHV expresses at least 12 distinct miRNAs during latency from polycistronic transcripts that also encode the KSHV latent proteins (Cai and Cullen, 2006). The expression of two KSHV miRNAs, miR-K12-10 and miR-K12-12, is substantially enhanced upon experimental induction of lytic replication by 12-_O_-tetradecanoyl phorbol-13-acetate (TPA) (Gottwein et al., 2006; Pfeffer et al., 2005). The biological significance of the observed TPA responsiveness of KSHV miR-K12-10 and miR-K12-12, as well as some EBV miRNAs, is unknown. In contrast, the hCMV and mCMV miRNAs are scattered throughout the viral genome and are expressed in lytically infected fibroblasts (Buck et al., 2007; Dölken et al., 2007; Dunn et al., 2005; Grey et al., 2005; Pfeffer et al., 2005). In most cases, expression of these miRNAs occurs during the early phase of the viral life cycle, as indicated by the loss of miRNA expression upon treatment of cells with cycloheximide, an inhibitor of protein synthesis, which blocks the production of the immediate early viral proteins required for early gene expression (Buck et al., 2007; Dölken et al., 2007; Grey et al., 2005). In the case of hCMV and mCMV, as well as HSV-1, the examination of viral miRNA expression during latency is hampered by the lack of appropriate cell culture systems. Taken together, accumulating evidence suggests that different viral miRNAs may have roles during both viral latency and productive infection. Herpesviral latency is characterized by highly restricted viral protein expression, thereby reducing the visibility of the virus to the host immune system. The expression of viral miRNAs during latency may therefore provide herpesviruses with a non-immunogenic way to stably modify their cellular environment. miRNAs expressed exclusively during productive virus replication may serve different purposes than those expressed during viral latency. Since miRNAs act at the mRNA level, and do not directly affect the expression of pre-existing proteins, miRNAs expressed during the relatively brief productive replication cycle may be more likely to target viral transcripts, cellular mRNAs for proteins induced by viral replication or proteins with a short half-life.
Evolutionary conservation of viral miRNAs
The sequences of cellular miRNAs, and of their mRNA target sites, are frequently highly conserved (Lewis et al., 2005). In contrast, herpesviral miRNA sequences are generally poorly conserved (Cai et al., 2005; Cai et al., 2006b; Schäfer et al., 2007). However, in closely related viruses, such as EBV and rhesus lymphocryptovirus (rLCV), KSHV and RRV, and Marek’s disease virus type 1 (MDV-1) and MDV-2, the location of miRNA loci are conserved (Burnside et al., 2006; Cai et al., 2005; Cai et al., 2006b; Schäfer et al., 2007; Yao et al., 2007; Yao et al., 2008). rLCV, which diverged from EBV at least 13 million years ago, encodes miRNAs that are arranged in a similar fashion and in the same genomic location as in EBV (Cai et al., 2006b). Of the 23 validated EBV miRNAs and 16 validated rLCV miRNAs, 8 are conserved at the level of the mature miRNA sequence (Cai et al., 2006b). RRV represents the closest KSHV relative for which miRNA expression has been examined so far. While the known RRV miRNAs are encoded in the same genomic region as in KSHV, they do not exhibit sequence homology (Schäfer et al., 2007). The chicken α-herpesviruses MDV-1 and MDV-2 diverged at least 26 million years ago (Yao et al., 2007). The miRNAs encoded by these viruses are clustered in homologous regions of the viral genomes, which are transcribed during viral latency, but again are not homologous in sequence (Burnside et al., 2006; Yao et al., 2007; Yao et al., 2008). In contrast to the clustered miRNAs seen in γ-herpesviruses, the β-herpesviruses hCMV and mCMV encode miRNAs that are dispersed throughout the virus genomes, at mostly different locations, and exhibit no evident sequence homology (Buck et al., 2007; Dölken et al., 2007; Dunn et al., 2005; Grey et al., 2005; Pfeffer et al., 2005). While several of the hCMV pre-miRNA stem-loops and mature miRNA sequences are predicted to be conserved in chimpanzee CMV, these miRNAs have not been validated experimentally (Grey et al., 2005; Pfeffer et al., 2005).
The lack of conservation of viral miRNA sequences does not necessarily indicate that these miRNAs do not have important or even, in cases with similar expression patterns, similar functions. Since herpesviruses are highly adapted to their specific host, there should not be strong evolutionary pressure to target regions of cellular transcripts that are highly conserved and any given transcript could conceivably be targeted through diverse 3′ UTR sites. In addition, miRNA-mediated repression of cellular genes required for different steps in a single pathway might have similar biological consequences. Finally, for viral miRNAs targeting viral transcripts, the miRNA and the target may have co-evolved. Consequently, viral miRNAs may have very similar functions yet show no sequence identity. On the other hand, even a single point mutation in the seed region can lead to a dramatic shift in miRNA function due to the loss or acquisition of a large number of cellular or viral mRNA targets. Therefore, it is also possible that viral miRNA function exhibits greater evolutionary plasticity than viral protein function.
Regulation of viral gene expression by viral miRNAs
Viral miRNAs may regulate viral transcripts and thereby generate or fine-tune patterns of viral protein expression (Figure 1A and B). Known examples of viral mRNA targets include transcripts that are transcribed antisense to the viral miRNA precursor (Barth et al., 2008; Pfeffer et al., 2004; Sullivan et al., 2005). Binding to the perfectly matched miRNA can therefore target these transcripts for degradation by Ago2-containing RISC complexes, if they are co-expressed during the virus life cycle. More recently, other viral transcripts have been shown to be recognized by viral miRNAs through imperfect 3′ UTR matches (Grey et al., 2007; Lo et al., 2007; Murphy et al., 2008).
Viral miRNAs can induce degradation of viral transcripts
EBV miR-BART2 was among the first viral miRNAs to be identified and exhibits perfect complementarity to the 3′ UTR of the mRNA for the EBV DNA polymerase BALF5, which is transcribed antisense to miR-BART2 (Pfeffer et al., 2004). Initial evidence for cleavage of BALF5 mRNA by miR-BART2 came from the detection of an unusual BALF5 transcript, the 3′ end of which precisely matched the predicted miR-BART2 cleavage site, upon induction of productive EBV replication (Furnari et al., 1993). Subsequently, BART2-programmed RISC was shown to indeed cleave BALF5 mRNA (Barth et al., 2008). Since BALF5 is a lytic gene product, it is unclear whether latently expressed miR-BART2 specifically evolved to target BALF5 and if this interaction is relevant for the maintenance of EBV latency.
Sullivan et al. (2005) identified one miRNA stem-loop precursor in the genome of the polyoma virus SV40 which gives rise to two miRNAs (miR-S1-5p and miR-S1-3p) expressed late during viral infection. Both SV40 derived miRNAs are perfectly complementary to early mRNAs transcribed antisense to the pre-miRNA precursor and direct the cleavage of these early transcripts, coding for the large and small T antigens, by RISC (Figure 1A). SV40 lacking miRNA expression, due to the selective disruption of the pre-miRNA structure, expressed higher levels of early mRNAs and, consequently, higher levels of large and small T antigen (Sullivan et al., 2005). While the loss of miRNA-mediated downmodulation of T antigen was insignificant for virus replication, it increased cellular visibility to SV40-specific cytotoxic T lymphocytes (CTLs), resulting in a higher susceptibility of SV40 miRNA mutant infected cells to CTL killing (Sullivan et al., 2005). The finding that the polyomavirus simian agent 12 (SA12) expresses a homologous miRNA (Cantalupo et al., 2005), and that the pre-miRNA hairpin is also conserved in the related BK virus and JC virus, suggests that polyomavirus miRNAs may provide a significant advantage to these viruses in vivo (Sullivan et al., 2005).
In addition to the EBV BALF5 and SV40 T antigen mRNA, several other viral transcripts are transcribed antisense to herpesviral miRNAs and may therefore be regulated similarly (Burnside et al., 2006; Pfeffer et al., 2005; Yao et al., 2007; Yao et al., 2008). These include the hCMV UL114 transcript, which is transcribed antisense to miR-UL112 (Pfeffer et al., 2005). However, co-expression of UL114 with miR-UL112 may not result in UL114 cleavage (Grey and Nelson, 2008), suggesting that viral mRNAs may have evolved to escape regulation by miRNAs with perfect complementarity, presumably because RNA folding blocks accessibility to the complementary region. This is consistent with the finding that target RNA folding is a key determinant of the efficacy of designed siRNAs (Brown et al., 2005). Sequences transcribed antisense to known miRNA stem-loop structures may have an increased propensity to fold into stem-loop structures themselves. Consequently, possible regulatory relationships between miRNAs and their antisense mRNA transcripts need to be individually verified.
Viral miRNAs can regulate viral transcripts with partial homology
The identification of viral targets of (herpes)viral miRNAs that are regulated by imperfect matches is more difficult than for targets with perfectly complementary matches (Figure 1B). Algorithms used for miRNA target prediction typically detect conserved matches to the miRNAs (Rajewsky, 2006). Viral miRNAs are not widely conserved, however, and 3′ UTR sequence conservation between closely related viral species could be due to other reasons, such as an overlap of transcripts and regulatory regions. Finally, herpesviral 3′ UTRs are not well annotated. In the absence of miRNA sequence conservation, the computational prediction of viral mRNA targets may lead to false positives due to the fact that the predicted target sites may be inaccessible.
Grey et al. (2007) computationally predicted target sites for hCMV miR-UL112 in the 3′ UTRs of hCMV genes, using an algorithm that calculates the binding energy of miRNAs to candidate matches. Thirteen candidate target 3′ UTRs, which were conserved in chimpanzee CMV, were further screened by 3′ UTR luciferase reporter assays. Three of these responded to miR-UL112, including those from IE72 (IE1) and UL120/121, two mRNAs encoded within the hCMV major immediate early region (MIE). The authors went on to demonstrate the specific regulation of an IE72-based reporter, as well as the IE72 protein itself, by miR-UL112. In contrast, expression of IE86, an immediate early transactivator expressed from the MIE that lacks a predicted 3′ UTR target, was not inhibited. Independently, Murphy et al. (2008) also computationally predicted IE72 to be a target for miR-UL112 and validated this interaction using 3′ UTR indicator assays. Since miR-UL112 is expressed early and accumulates during viral infection (Grey et al., 2005), miR-UL112 may function to inhibit IE72 expression during the later stage of viral replication. Murphy et al. (2008) constructed hCMV mutant viruses in which either miR-UL112 expression was selectively disrupted or which lacked the 7 nt seed match to miR-UL112 in the 3′ UTR of IE72. Both viruses replicated with wild type kinetics in a fibroblast cell line, but exhibited higher levels of IE72 protein synthesis late during productive infection. While the biological significance of this finding is currently unclear, the authors hypothesized that miR-UL112 may promote the transition from productive replication to latent infection. Interestingly, the ectopic expression of a synthetic miR-UL112 mimic early during infection resulted in the reduced expression of a number of hCMV immediate early proteins, including IE72, but also the non-targeted IE86 (Grey et al., 2007). This effect was accompanied by a reduced hCMV genome copy number. miR-UL112 had no effect on HSV-1 genome copy numbers, thus suggesting no potent non-specific effect on general viral replication, e.g. due to induction of an interferon response. Because this phenotype does not reflect the pattern of immediate early (IE) gene expression in an IE72 knockout virus, it is likely that suppression of IE72 expression by miR-UL112 is not the sole reason for the observed effect. Instead, other viral, or possibly cellular, mRNA targets of miR-UL112 may be important.
Another group recently used computational methods to predict candidate matches to EBV BART miRNAs in the 3′ UTR of EBV latent membrane protein 1 (LMP1) (Lo et al., 2007). LMP1 is expressed during EBV latency and functions as a constitutively active tumor necrosis factor (TNF) receptor (Kieff and Rickinson, 2007). LMP1 is essential for transformation by EBV in culture and is believed to contribute to EBV tumorigenesis (Kieff and Rickinson, 2007). Of the EBV miRNAs tested, miR-BART-1-5p, miR-BART-16, and miR-BART-17-5p downmodulated LMP1 protein expression (Lo et al., 2007). The authors further demonstrated that altered LMP1 protein levels resulted in the downmodulation of known LMP1 activities, resulting in an increase in cellular sensitivity to apoptotic stimuli, as well as a decrease in the LMP1-mediated activation of nuclear factor-kappa B (NF-κB). Therefore, these miR-BART miRNAs may buffer LMP1 expression levels, thereby balancing LMP1 growth stimulation and transforming activity with potential toxic effects of the protein. The significance of this finding in the context of EBV infections remains unclear.
Regulation of host gene expression by viral miRNAs
It is possible that some miRNAs have evolved explicitly to regulate viral genes and may largely avoid targeting cellular mRNAs. On the other hand, most viral miRNAs will likely downmodulate at least some cellular mRNAs and thereby exert physiological effects (Figure 1C). It is perhaps surprising that so far no viral miRNA has been found to be perfectly complementary to a cellular transcript, and to trigger its degradation, since this would presumably be the most effective way to downregulate a factor that limits viral replication.
Viral miRNAs are not widely conserved, and, with the notable exception of viral miRNAs with cellular orthologs (see below), there is no obvious reason to presume that they target conserved regions in cellular mRNAs. Because established algorithms for the prediction of targets for cellular miRNAs rely on conservation of seed matches (Rajewsky, 2006), they may not be optimal for the prediction of cellular mRNA targets for viral miRNAs, because they would miss miRNA binding sites that are not conserved between species. On the other hand, applying only matching criteria, without considering conservation as an additional criterion, could result in a large number of false positive predictions, since many predicted sites might not be accessible due to mRNA folding. An alternative approach to mRNA target identification is microarray analysis of cellular gene expression upon ectopic expression of individual or multiple miRNAs, or after inhibition of miRNA function using antisense reagents, to identify candidate mRNA targets for miRNAs (Bushati and Cohen, 2007; Krutzfeldt et al., 2005). The main disadvantage of this approach is that many authentic targets will be missed because mRNA levels are not reduced enough for reliable detection by microarray analysis. This approach does, however, have the potential to reveal indirect downstream consequences of miRNA-mediated regulation on cellular gene expression.
Viral miRNAs define “novel” regulatory relationships
Samols et al. (2007) performed gene expression profiling of 293 cells stably transfected with a plasmid expressing 10 KSHV miRNAs and found that 65 mRNAs were significantly downregulated, while 8 mRNAs were upregulated. 3′ UTR analysis showed that seed matches to the KSHV miRNAs were more common in the 3′ UTRs of downregulated genes than when 3′ UTRs were randomly shuffled, suggesting the presence of authentic targets for viral miRNA regulation. Three of these candidate targets, secreted phosphoprotein 1 (osteopontin, SPP1), plasticity related gene 1 (PRG1) and thrombospondin 1 (THBS1), were further validated using 3′ UTR luciferase indicator assays. While the mRNA for SPP1 was >20 fold downregulated, Samols et al. did not observe a reduction in the SSP1 protein. THBS1 is an extracellular matrix glycoprotein with diverse functions mediated through manifold interactions with matrix proteins, cytokines and cell associated proteins (Bornstein, 2001). Among other functions, THBS1 possesses anti-angiogenic and anti-tumorigenic properties and binds and activates transforming growth factor-β (TGF-β), a multifunctional cytokine (Bornstein, 2001). The authors observed a >10 fold reduction in THBS1 protein expression in 293 cells stably expressing the KSHV miRNA cluster (Samols et al., 2007). Surprisingly, robust de-repression of a THBS1 3′ UTR luciferase indicator was observed using 2′-O-methyl-RNAs specific for 9 out of 10 individual miRNAs encoded in the cluster, which would suggest that each of these 9 KSHV miRNA contributes to full inhibition of THBS1. Based on the repression of luciferase reporters for TGF-β activity in 293 cells stably expressing the KSHV miRNA cluster, Samols et al. postulated that reduced levels of THBS1 protein translate into reduced TGF-β activity. However, the authors did not show that the reduced TGF-β activity was indeed due to reduced THBS1 expression. Further studies are therefore needed to help clarify the role of KSHV miRNA-mediated THBS1 repression in physiological target cells for KSHV, and the functional implications of this proposed interaction. Given that Kaposi’s sarcoma (KS) lesions are characterized by deregulated angiogenesis (Ganem, 2007), and that KSHV infected PEL cells are resistant to TGF-β induced cell cycle arrest and apoptosis (Di Bartolo et al., 2008), the repression of THBS1 expression by KSHV miRNAs may be important for KSHV pathogenesis.
To overcome the current limitations for the computational prediction of cellular mRNA targets for viral miRNAs, Stern-Ginossar et al. (2007) developed an algorithm that depends on the occurrence of multiple binding sites for a single miRNA in cellular 3′ UTRs and/or stringent matching criteria to identify two cellular targets for hCMV miR-UL112. Stern-Ginossar et al. did not report a comprehensive validation of their algorithm and it is therefore hard to estimate the overall performance of their approach. As discussed above, it seems likely that even stringent matching criteria would lead to a large number of false positive predictions if site accessibility is not used as a further criterion. Nevertheless, this method did identify two interesting cellular targets for miR-UL112 (Stern-Ginossar et al., 2007). The authors show that miR-UL112 downregulates major histocompatibility complex class 1-related chain B (MICB) and, to a lesser extent MICA, expression. These proteins are stress-induced ligands of the natural killer (NK) cell activating receptor NKG2D. Ligation of NKG2D triggers killing of virus-infected cells by NK cells. Consequently, miR-UL112 specific downregulation of MICB translated into the reduced susceptibility of virally infected cells to killing by NK cells (Stern-Ginossar et al., 2007). Of note, several hCMV proteins, including UL16, have previously been implicated in the escape of hCMV-infected cells from NK cell killing (Wilkinson et al., 2008), providing an example of functional synergy between viral proteins and viral miRNAs. MICB is regulated exclusively at the level of translation and the interaction with miR-UL112 is apparently not dependent on an intact seed region (Stern-Ginossar et al., 2007), and would therefore have been missed using microarray analysis or target prediction algorithms that depend on seed matching. As miR-UL122 has also been shown to target the mRNA for the hCMV immediate early protein IE72 (see above) (Grey et al., 2007; Murphy et al., 2008), hCMV miR-UL112 appears to target both cellular and viral targets at the translational level.
Viral orthologs of cellular miRNAs
Most viral miRNAs do not share seed homology with cellular miRNAs and therefore likely target novel sites on cellular mRNAs, i.e. they engage in novel, virus-specific regulatory relationships. However, at least some viral miRNAs seem to have evolved to hijack existing and, presumably, highly evolved gene regulatory networks (Figures 1, 3). It was recently reported that KSHV miR-K12-11 is an ortholog of cellular miR-155 (Figure 3). miR-K12-11 and miR-155 are identical along their 5′-terminal 8 nt, i.e. including the entire seed region (Gottwein et al., 2007; Skalsky et al., 2007). Using microarray analysis of BJAB cell pools stably expressing levels of miR-K12-11 comparable to those observed in latently KSHV infected PEL cell lines, miR-K12-11 downregulated genes were specifically enriched for predicted targets for miR-155 (Gottwein et al., 2007). A number of microarray-identified candidate target genes for KSHV miR-K12-11 and a number of previously predicted targets of miR-155 were similarly regulated by both miRNAs, as determined by 3′ UTR luciferase indicator assays, real time (RT)-PCR and Western Blotting. Similar findings were reported by Skalsky et al. (2007), who showed that the 3′ UTR of BACH-1, a predicted target of miR-155, responds equally to both miR-K12-11 and miR-155.
Figure 3. Viral miRNAs with cellular orthologs.

Seed alignments of viral miRNAs with their known or candidate cellular orthologs (homologous bases are in red) are shown. KSHV miR-K12-11 functions as an ortholog of cellular miR-155 (Gottwein et al., 2007; Skalsky et al., 2007). Other potential orthologs of viral miRNAs referred to in the text are listed. Nucleotides that distinguish hsa-miR-18a from hsa-miR-18b are in black and bold. miRNAs are designated as in miRBase (http://microrna.sanger.ac.uk/). Abbreviations: hsa, homo sapiens (human); gga, gallus gallus (chicken); mmu, mus musculus (mouse); mghv, murine gammaherpesvirus 68 (MHV68).
miR-155, the product of the Bic gene (Eis et al., 2005), is transiently expressed in macrophages, T- and B-lymphocytes upon treatment with inflammatory stimuli, and after T- or B-cell receptor ligation, and loss of miR-155 expression in knockout mice revealed defects in adaptive immune responses (Haasch et al., 2002; O’Connell et al., 2007; Rodriguez et al., 2007; Thai et al., 2007). Importantly, constitutive expression of miR-155 in B cells is associated with the development of B-cell lymphomas in humans, mice and chickens, leading to the classification of miR-155 as an oncomir. Bic was first identified as a gene that is commonly activated by proviral insertion in avian leukosis virus (ALV) induced B-cell lymphomas in chicken (Tam et al., 1997). Transgenic expression of miR-155 in the B-cell compartment in mice results in the development of B-cell lymphomas and miR-155 expression is elevated in several types of human B-cell lymphomas, such as Hodgkin’s lymphoma, as well as in other cancers (Costinean et al., 2006; van den Berg et al., 2003). While the mechanism by which the constitutive expression of miR-155 in B cells causes tumorigenesis is not understood, the finding that KSHV miR-K12-11 is an ortholog of miR-155 raises the possibility that miR-K12-11 may contribute to the development of KSHV-associated lymphomas. Interestingly, the oncogenic α-herpesvirus MDV-1 encodes a miRNA, miR-M4, that also shares the same 5′-terminal 8 nt, suggesting that this viral miRNA may also function as an ortholog of miR-155 in infected chickens (Figure 3). Remarkably, MDV-2, which lacks a miR-155 ortholog, is not oncogenic in chickens (Schat and Calnek, 1978). While MDV-1 also encodes other proteins, such as the meq oncogene, that are not present in MDV-2, it is tempting to speculate that MDV-1 miR-M4 may contribute to tumorigenesis in this system. Validated targets for miR-155 include proteins with known roles in B-cell function, such as PU.1, but also a protein with known pro-apoptotic functions, tumor protein p53 inducible nuclear protein 1 (TP53INP1), which has been suggested to contribute to the development of miR-155-expressing pancreatic tumors (Gironella et al., 2007; Vigorito et al., 2007). It is interesting that both KSHV miR-K12-11 and MDV-1 miR-M4 only share the first 8 nt with cellular miR-155. Therefore, it remains possible that functional differences between these miRNAs exist.
A subset of other viral miRNAs also share sequence homology with cellular miRNAs. MHV68 miR-M1-4 shares the 5′-terminal 9 nts with murine miR-151, a miRNA of unknown function (Figure 3). Other seed homologies are more limited, and extend only over nts 2–7, corresponding to the minimal miRNA seed region. However, since 6 nt seed base pairing can be sufficient for the regulation of some targets, these miRNAs may also share targets with their cellular counterparts. The most interesting of these miRNAs are EBV miR-BART5, rLCV miR-rL1-8 and MHV68 miR-1-7-5p, all of which share perfect seed homology with cellular miR-18a and miR-18b, encoded in the miR-17-92 cluster, which also has oncomir function (Figure 3) (He et al., 2005). Since the seed homology is more limited, and does not extend to nt 8 of the miRNAs, targets that depend on base pairing to nt 8 of the miRNA may not be shared between these miRNAs. In conclusion, it has been shown that KSHV miR-K12-11 is an ortholog of cellular miR-155 and in other cases, similar relationships to cellular miRNAs are suggested by seed identity to cellular miRNAs. In these cases, known functions of the suspected cellular orthologs and computationally predicted targets of these miRNAs may provide a useful starting point for functional analysis.
Impact of host miRNAs on virus replication and pathogenesis
Given the scope of miRNA-mediated gene regulation in the mammalian system, it is expected that cellular miRNAs also directly or indirectly affect virus replication and pathogenesis (Figure 2). Viral genomic RNAs or mRNAs could have evolved to directly interact with host miRNAs to facilitate certain steps of their life cycle. In other cases, such interactions would be inhibitory and viral mRNAs may be under selective pressure to avoid binding to abundant cellular miRNAs. Finally, the cellular miRNA composition in infected cells is likely to indirectly affect viruses, because many pathways that promote or limit viral replication or the survival of infected cells are likely to be regulated by cellular miRNAs (Figure 2). In either case, viruses may therefore gain an advantage by reshaping the cellular miRNA composition. Several recent reports hint at the scope of viral interactions with host miRNAs and are discussed below.
Viruses may exploit direct interactions with host miRNAs
A striking example of a cellular miRNA required for virus replication is miR-122, which facilitates the replication of the pathogenic, positive strand RNA virus HCV (Jopling et al., 2005; Randall et al., 2007). In mammals, miR-122 expression is confined to the liver, where it is the most abundant miRNA and may help maintain liver tissue identity (Esau et al., 2006; Krutzfeldt et al., 2005). miR-122 regulates fatty acid and cholesterol biosynthesis, pathways central to liver function and the HCV life cycle (Esau et al., 2006; Krutzfeldt et al., 2005). Jopling et al. (2005) reported that inhibition of miR-122 function reduced HCV genotype 1 RNA replication, without notable effects on HCV protein translation or RNA stability. This dependence on miR-122 function required binding to a site in the 5′ UTR of the HCV genomic RNA. The importance of miR-122 for the complete HCV life cycle was subsequently also confirmed in an HCV genotype 2 cell culture system (Randall et al., 2007). Furthermore, conservation of the miR-122 binding site in all 6 HCV genotypes, which otherwise differ by ~30% at the nucleotide level, suggests that the interaction of HCV with miR-122 is relevant in vivo (Jopling et al., 2005). Finally, the liver specific expression of miR-122 may contribute to the tissue tropism of HCV.
The mechanism for this unusual function of miR-122 in HCV replication is still unknown. A simple explanation would be an indirect enhancement of HCV replication caused by the sequestration of miR-122 away from its endogenous target mRNAs. However, while the effect of HCV replication on the regulation of endogenous miR-122 targets has not been evaluated to date, the role of miR-122 in HCV RNA replication is likely direct. While a miR-122 binding-deficient, mutant HCV replicon exhibited reduced RNA replication, this mutant could be rescued by expression of a miR-122 variant bearing compensatory base changes (Jopling et al., 2005). More likely scenarios for miR-122 function in HCV replication therefore include a direct role of miR-122, or miR-122-associated proteins, in the localization and/or replication of HCV genomic RNA. Interestingly, the individual knockdown of Drosha, DGCR8, Dicer, and of the four Ago proteins was reported to result in reduced HCV replication (Randall et al., 2007). However, this finding has not been linked so far to the requirement for miR-122 function in HCV replication and further experiments are needed to delineate any general role of the miRNA pathway in HCV replication. It will also be interesting to see if other RNA viruses, especially flaviviruses, engage in similar interactions with host miRNAs.
In a recent report, Huang et al. (2007) proposed that HIV-1 exploits miRNA mediated regulation to maintain HIV-1 latency in resting CD4+ T lymphocytes. In acquired immunodeficiency syndrome (AIDS) patients, a small number of HIV-1 infected activated CD4+ lymphocytes return to a quiescent state to become CD4+ memory T cells. This process is associated with the transcriptional silencing of the integrated HIV-1 genome, due to the limited activity in these cells of factors required for HIV-1 transcription (Lassen et al., 2004). These authors propose that the few HIV-1 transcripts produced in these cells are subject to silencing by several cellular miRNAs, specifically miR-150, miR-28, miR-125b, miR-223, and miR-382, which are overexpressed in resting CD4+ T lymphocytes compared to their activated counterparts (Huang et al., 2007). The presence of matches to these miRNAs in the shared 3′ portion of HIV-1 transcripts conferred miRNA mediated regulation to luciferase reporters carrying HIV-1 genome fragments. The inhibition of all of these miRNAs, using a combination of 2′-O-methyl-oligonucleotides, resulted in increased HIV-1 protein expression and virus production from resting CD4+ T cells transfected with infectious HIV-1 clones or from patient-derived resting CD4+ T cells. Since a mutational analysis of the putative miRNA binding sites has not been reported, it remains possible that the observed effects are indirect and that these miRNAs regulate CD4+ T cells properties required for HIV-1 latency. While HIV-1 latency in resting CD4+ T cells is a major obstacle to curing HIV-1 by available anti-retroviral drugs, which invariably target replicating virus, there is currently no evidence that HIV-1 is under evolutionary pressure to establish or maintain a latent reservoir and therefore the physiological relevance of the proposed interactions is not yet clear.
Antiviral miRNAs?
Another possible scenario is that cellular miRNAs interact with viral genomic RNAs or mRNAs and thereby inhibit virus replication. Several viruses, including retroviruses, are accessible to inhibition by experimentally introduced siRNAs, making it likely that they are also accessible to inhibition by miRNA-programmed RISC (Gitlin and Andino, 2003). Viruses may be under selective pressure to avoid functional binding sites for miRNAs, which are abundantly expressed in their normal target cells, and viral genomes are likely optimized for replication in that environment. This hypothesis suggests that the fortuitous repression of viral mRNAs by cellular miRNAs present in non-target tissues could pose a significant hurdle to successful infection and thereby might serve as a key determinant of viral tissue tropism. This hypothesis is supported by the finding that engineered miRNA target sites can regulate the tissue tropism of lentiviral vectors in vivo (Brown et al., 2006). Similarly, the introduction of miRNA target sites for two muscle-specific miRNAs into the genome of the oncolytic picornavirus Coxsackievirus A21 (CVA21) effectively abolished CVA21 replication in murine muscle cells and, consequently, CVA21 induced myositis. In contrast, the oncolytic properties of CVA21 in tumor cells lacking these muscle-specific miRNAs were fully preserved (E. J. Kelly & S. J. Russell, personal communication).
Several recent reports suggest that replication of wild-type viruses can also be inhibited by endogenous cellular miRNAs, raising the question of whether specific cellular miRNAs may have specifically evolved to limit virus replication. Lecellier et al. showed that endogenous cellular miR-32, expressed in HeLa and BHK21 cells, can limit the replication of the retrovirus primate foamy virus (PFV) in cell culture through an interaction with a poorly conserved region in the 3′ portion of the PFV genome (Lecellier et al., 2005). It is currently unclear if such an interaction would occur in the natural context and thereby limit PFV replication. Similarly, Otsuka et al. observed that Dicer knockout mice are hypersensitive to infection by vesicular stomatitis virus (VSV), a negative-strand RNA virus (Otsuka et al., 2007). This effect was at least partially due to the loss of miR-24 and miR-93 expression in these mice. Both miRNAs were shown to downregulate viral mRNAs and mutant VSV, in which miR-24 and miR-93 binding was abolished, replicated with higher efficiency in wild-type, but not Dicer knockout, mice. While this result demonstrates that VSV could be targeted by miRNAs, there is currently no evidence that these cellular miRNAs restrict VSV replication in its natural hosts. Finally, Pedersen et al. (2007) demonstrated that interferon treatment of hepatocytes causes the upregulation of several cellular miRNAs, including miR-1, miR-30, miR-128, miR-196, miR-296, miR-351, miR-431, and miR-448, some of which have seed matches in the HCV genome. In the case of miR-196 and miR-1, the kinetics of miRNA expression were analyzed and showed a transient upregulation of these miRNAs by 3- and 10-fold, respectively, in interferon-β treated cells compared to untreated cells. miRNA levels returned to the uninduced status within 24 hours after interferon treatment. The transfection of HCV replicon-containing hepatocytes with synthetic miRNA mimics for miR-196, miR-296, miR-351, miR-431, and miR-448, resulted in a 2- to 5-fold reduction in HCV RNA accumulation. At least in the case of miR-196 and miR-448, the antiviral effect of the transfected miRNA mimics was correlated with the presence of matches to these miRNAs in the viral genome. The levels and expression kinetics of transfected miRNA mimics are likely to be different from the transient expression of the endogenous miRNAs after interferon induction, and therefore it is currently unclear if this inhibitory effect would also exist with endogenously expressed miRNAs. The treatment of HCV replicon-containing hepatocytes with interferon reduces HCV RNA content by about 10-fold, whereas HCV RNA levels in interferon-treated cells were only marginally increased when these miRNAs were inhibited, suggesting that if these miRNAs indeed limit HCV replication, they must act in concert with other antiviral mechanisms (Pedersen et al., 2007). Interestingly, miR-122 was downregulated by the treatment of hepatocytes with interferon. However, rescued miR-122 expression alone was also not able to rescue HCV replication in interferon treated cells (Pedersen et al., 2007). Therefore, the relevance of the interferon-induction or repression of cellular miRNAs to HCV replication remains unclear. The fact that many of the binding sites for the interferon-induced miRNAs are not broadly conserved between HCV genotypes also suggests that interferons limit HCV replication primarily through miRNA-independent mechanisms.
It is possible that reported interactions between viruses and cellular miRNA that result in restriction of viral replication may have resulted from fortuitous binding in settings that are irrelevant to the normal cellular targets of the virus in vivo, or that the reported interactions are not likely to be limiting to viral replication. This hypothesis is underscored by the fact that most of the miRNAs that were reported to have antiviral functions are conserved in a range of organisms that is far broader than the species tropism of the inhibited viruses, suggesting that host cell miRNA sequence conservation is due to unrelated selection pressures. Moreover, viruses should also be able to easily escape such inhibitory interactions by mutation and, indeed, rapid viral escape from experimentally introduced siRNAs has been reported for a range of viruses (Westerhout et al., 2005). These considerations suggest that future claims for inhibition of viral replication by endogenous cellular miRNAs will need to carefully address the physiological relevance of the proposed inhibitory interactions.
Indirect effects of host miRNAs
In addition to direct roles of cellular miRNAs in modulating virus replication, the expression pattern of cellular miRNAs may also indirectly promote or limit virus replication and/or pathogenicity by repressing cellular factors that influence these processes. It is likely that viruses have evolved ways to selectively upregulate supportive miRNAs or to downregulate limiting miRNAs, and thereby optimize the cellular environment for their replication.
Triboulet et al. (2007) reported that HIV-1 replication in T lymphocytes is enhanced in the context of reduced Dicer and Drosha expression, suggesting that cellular miRNAs may limit HIV-1 replication. They further showed that miRNA expression from the cellular miR-17-92 cluster, comprising miR-17-5p, miR-18, miR-19a, miR-20a, miR-19b-1, and miR-92-1, is downregulated in HIV-1 infected T lymphocytes by an unknown mechanism. Ectopic expression and/or inhibition of endogenous miRNAs suggested that miR-17-5p and miR-20a limit HIV-1 replication by reducing PCAF expression. PCAF, a histone acetyl transferase, has been proposed to promote HIV-1 transcriptional elongation (Kiernan et al., 1999). Enhanced PCAF expression does not fully account for the phenotype induced by Dicer or Drosha knockdown, however, since the knockdown of Dicer or Drosha, in the context of a PCAF knockdown, still increased reported gene expression from an HIV-1 based indicator plasmid (Triboulet et al., 2007).
While viruses may have evolved to downregulate the expression of certain cellular miRNAs, they may also upregulate cellular miRNAs that benefit the virus. Several miRNAs have been reported to be upregulated by HIV-1 infection, with unknown consequences (Triboulet et al., 2007; Yeung et al., 2005). Similarly, EBV infection results in the upregulation of several cellular miRNAs, including miR-146 and miR-155, possibly due to B-cell activation by EBV (Cameron et al., 2008; Jiang et al., 2006; Yin et al., 2008). This hypothesis is supported by the finding that miR-146 induction is a consequence of the activation of NF-κB by the EBV LMP1 protein (Cameron et al., 2008). The significance of the upregulation of these miRNAs for EBV latency, and their possible role in the induction of EBV-associated tumors, is currently unclear. Interestingly, the tumorigenic herpesviruses KSHV, and possibly MDV-1, encode viral orthologs of miR-155 (Burnside et al., 2006; Gottwein et al., 2007; Skalsky et al., 2007; Yao et al., 2008). The benefit of miR-155 expression to these viruses remains to be elucidated, but the expression of miR-155, or its functional orthologs, may contribute to viral oncogenesis in all three systems, with KSHV and MDV-1 encoding a viral miR-155 ortholog while EBV instead induces expression of endogenous miR-155.
Summary and conclusions
Viral miRNAs were discovered only recently and functional relationships between viruses and viral or cellular miRNAs are only now beginning to be elucidated. Recent reports suggest that at least some viral miRNAs evolved to regulate viral gene expression. Other viral miRNAs affect cellular gene expression by engaging in novel regulatory relationships or by mimicking cellular miRNAs and thereby utilizing predefined cellular regulatory networks. Conversely, cellular miRNAs can alter the viral life cycle, strikingly exemplified by the fact that host miR-122 facilitates HCV replication.
Some functions of cellular or viral miRNAs in infected cells will undoubtedly turn out to be important for virus replication in vivo and may therefore become targets for therapeutic intervention. On the other hand, the emerging interface between viruses and miRNAs may uncover novel pathways that promote or limit virus replication, and this knowledge could also help to guide drug design. Antisense inhibitors of miRNA function which are bio-available in vivo, such as antagomirs, could represent a good starting point for the development of miRNA inhibitory drugs (Gottwein et al., 2007; Krutzfeldt et al., 2005). Antisense inhibitors targeting cellular miRNAs would be predicted to have side effects, depending on the function of the inhibited miRNA, since these inhibitors would also disrupt the cellular functions of these miRNAs. An alternative strategy could be to disrupt the interaction of cellular or viral miRNAs with only one or few of their cellular or viral target sequences. As for other antiviral drugs, there is a chance of the virus becoming resistant, e.g. by mutation of the viral miRNAs or viral binding sites for cellular miRNAs. This is a particular concern for viruses with high mutation rates, such as HIV-1 or HCV. In this case, miRNA-based therapeutics, like other antiviral drugs, would likely be most effective when used in combination.
MicroRNAs represent a class of molecule produced by both viruses and their hosts that can benefit either the virus or the host, depending on the particular interaction. Such interactions are likely to play key roles in viral pathogenesis, determining the degree to which hosts restrict viral infections, and being modified over evolutionary time as the genomes of both host and pathogen evolve.
Acknowledgments
The research on miRNA function performed by the authors is supported by grants GM071408 and AI067968 from the NIH.
References
- Andersson MG, Haasnoot PC, Xu N, Berenjian S, Berkhout B, Akusjarvi G. Suppression of RNA interference by adenovirus virus-associated RNA. J Virol. 2005;79:9556–9565. doi: 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio O, Razquin N, Zaratiegui M, Narvaiza I, Fortes P. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J Virol. 2006;80:1376–1384. doi: 10.1128/JVI.80.3.1376-1384.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Barth S, Pfuhl T, Mamiani A, Ehses C, Roemer K, Kremmer E, Jaker C, Hock J, Meister G, Grasser FA. Epstein-Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic acids research. 2008;36:666–675. doi: 10.1093/nar/gkm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC. Mammalian mirtron genes. Molecular cell. 2007;28:328–336. doi: 10.1016/j.molcel.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BD, Venneri MA, Zingale A, Sergi Sergi L, Naldini L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nature medicine. 2006;12:585–591. doi: 10.1038/nm1398. [DOI] [PubMed] [Google Scholar]
- Brown KM, Chu CY, Rana TM. Target accessibility dictates the potency of human RISC. Nat Struct Mol Biol. 2005;12:469–470. doi: 10.1038/nsmb931. [DOI] [PubMed] [Google Scholar]
- Buck AH, Santoyo-Lopez J, Robertson KA, Kumar DS, Reczko M, Ghazal P. Discrete clusters of virus-encoded micrornas are associated with complementary strands of the genome and the 7.2-kilobase stable intron in murine cytomegalovirus. J Virol. 2007;81:13761–13770. doi: 10.1128/JVI.01290-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnside J, Bernberg E, Anderson A, Lu C, Meyers BC, Green PJ, Jain N, Isaacs G, Morgan RW. Marek’s disease virus encodes MicroRNAs that map to meq and the latency-associated transcript. J Virol. 2006;80:8778–8786. doi: 10.1128/JVI.00831-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- Cai X, Cullen BR. Transcriptional origin of Kaposi’s sarcoma-associated herpesvirus microRNAs. J Virol. 2006;80:2234–2242. doi: 10.1128/JVI.80.5.2234-2242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Li G, Laimins LA, Cullen BR. Human papillomavirus genotype 31 does not express detectable microRNA levels during latent or productive virus replication. J Virol. 2006a;80:10890–10893. doi: 10.1128/JVI.01175-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Schäfer A, Lu S, Bilello JP, Desrosiers RC, Edwards R, Raab-Traub N, Cullen BR. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS pathogens. 2006b;2:e23. doi: 10.1371/journal.ppat.0020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron JE, Yin Q, Fewell C, Lacey M, McBride J, Wang X, Lin Z, Schaefer BC, Flemington EK. Epstein-Barr virus latent membrane protein 1 induces cellular MicroRNA miR-146a, a modulator of lymphocyte signaling pathways. J Virol. 2008;82:1946–1958. doi: 10.1128/JVI.02136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantalupo P, Doering A, Sullivan CS, Pal A, Peden KW, Lewis AM, Pipas JM. Complete nucleotide sequence of polyomavirus SA12. J Virol. 2005;79:13094–13104. doi: 10.1128/JVI.79.20.13094-13104.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Griffiths A, Li G, Silva LM, Kramer MF, Gaasterland T, Wang XJ, Coen DM. Prediction and identification of herpes simplex virus 1-encoded microRNAs. J Virol. 2006;80:5499–5508. doi: 10.1128/JVI.00200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Transcription and processing of human microRNA precursors. Molecular cell. 2004;16:861–865. doi: 10.1016/j.molcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Di Bartolo DL, Cannon M, Liu YF, Renne R, Chadburn A, Boshoff C, Cesarman E. KSHV LANA inhibits TGF-β signaling through epigenetic silencing of the TGF-β type II receptor. Blood. 2008;111:4731–4740. doi: 10.1182/blood-2007-09-110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölken L, Perot J, Cognat V, Alioua A, John M, Soutschek J, Ruzsics Z, Koszinowski U, Voinnet O, Pfeffer S. Mouse cytomegalovirus microRNAs dominate the cellular small RNA profile during lytic infection and show features of posttranscriptional regulation. J Virol. 2007;81:13771–13782. doi: 10.1128/JVI.01313-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn W, Trang P, Zhong Q, Yang E, van Belle C, Liu F. Human cytomegalovirus expresses novel microRNAs during productive viral infection. Cell Microbiol. 2005;7:1684–1695. doi: 10.1111/j.1462-5822.2005.00598.x. [DOI] [PubMed] [Google Scholar]
- Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Furnari FB, Adams MD, Pagano JS. Unconventional processing of the 3′ termini of the Epstein-Barr virus DNA polymerase mRNA. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:378–382. doi: 10.1073/pnas.90.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem D. Kaposi’s Sarcoma-associated herpesvirus. In: Knipe DM, Howley PM, editors. Fields Virology. 2007. pp. 2847–2888. [Google Scholar]
- Gironella M, Seux M, Xie MJ, Cano C, Tomasini R, Gommeaux J, Garcia S, Nowak J, Yeung ML, Jeang KT, et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16170–16175. doi: 10.1073/pnas.0703942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L, Andino R. Nucleic acid-based immune system: the antiviral potential of mammalian RNA silencing. J Virol. 2003;77:7159–7165. doi: 10.1128/JVI.77.13.7159-7165.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwein E, Cai X, Cullen BR. Expression and function of microRNAs encoded by Kaposi’s sarcoma-associated herpesvirus. Cold Spring Harbor symposia on quantitative biology. 2006;71:357–364. doi: 10.1101/sqb.2006.71.004. [DOI] [PubMed] [Google Scholar]
- Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi J-tA, Braich R, Manoharan M, Soutschek J, Ohler U, et al. A viral microRNA functions as an ortholog of cellular miR-155. Nature. 2007;450:1096–1099. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey F, Antoniewicz A, Allen E, Saugstad J, McShea A, Carrington JC, Nelson J. Identification and characterization of human cytomegalovirus-encoded microRNAs. J Virol. 2005;79:12095–12099. doi: 10.1128/JVI.79.18.12095-12099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey F, Meyers H, White EA, Spector DH, Nelson J. A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS pathogens. 2007;3:e163. doi: 10.1371/journal.ppat.0030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey F, Nelson J. Identification and function of human cytomegalovirus microRNAs. J Clin Virol. 2008;41:186–191. doi: 10.1016/j.jcv.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic acids research. 2006;34:D140–144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic acids research. 2008;36:D154–158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundhoff A, Sullivan CS, Ganem D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA. 2006;12:733–750. doi: 10.1261/rna.2326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Gartner JJ, Sethupathy P, Hatzigeorgiou AG, Fraser NW. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature. 2006;442:82–85. doi: 10.1038/nature04836. [DOI] [PubMed] [Google Scholar]
- Gupta A, Gartner JJ, Sethupathy P, Hatzigeorgiou AG, Fraser NW. Retraction of Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature. 2008;451:600. doi: 10.1038/nature06621. [DOI] [PubMed] [Google Scholar]
- Haasch D, Chen YW, Reilly RM, Chiou XG, Koterski S, Smith ML, Kroeger P, McWeeny K, Halbert DN, Mollison KW, et al. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto-oncogene, BIC. Cell Immunol. 2002;217:78–86. doi: 10.1016/s0008-8749(02)00506-3. [DOI] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nature medicine. 2007;13:1241–1247. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- Jiang J, Lee EJ, Schmittgen TD. Increased expression of microRNA-155 in Epstein-Barr virus transformed lymphoblastoid cell lines. Genes Chromosomes Cancer. 2006;45:103–106. doi: 10.1002/gcc.20264. [DOI] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- Kieff E, Rickinson A. Epstein-Barr Virus and Its Replication. In: Knipe DM, Howley PM, editors. Fields Virology. 2007. pp. 2603–2654. [Google Scholar]
- Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, Maudoux F, Calomme C, Burny A, Nakatani Y, Jeang KT, et al. HIV-1 tat transcriptional activity is regulated by acetylation. The EMBO journal. 1999;18:6106–6118. doi: 10.1093/emboj/18.21.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim do N, Chae HS, Oh ST, Kang JH, Park CH, Park WS, Takada K, Lee JM, Lee WK, Lee SK. Expression of viral microRNAs in Epstein-Barr virus-associated gastric carcinoma. J Virol. 2007;81:1033–1036. doi: 10.1128/JVI.02271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. Combinatorial microRNA target predictions. Nature genetics. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen K, Han Y, Zhou Y, Siliciano J, Siliciano RF. The multifactorial nature of HIV-1 latency. Trends Mol Med. 2004;10:525–531. doi: 10.1016/j.molmed.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, Saib A, Voinnet O. A cellular microRNA mediates antiviral defense in human cells. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Lin J, Cullen BR. Analysis of the interaction of primate retroviruses with the human RNA interference machinery. J Virol. 2007;81:12218–12226. doi: 10.1128/JVI.01390-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo AK, To KF, Lo KW, Lung RW, Hui JW, Liao G, Hayward SD. Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16164–16169. doi: 10.1073/pnas.0702896104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews MB, Shenk T. Adenovirus virus-associated RNA and translation control. J Virol. 1991;65:5657–5662. doi: 10.1128/jvi.65.11.5657-5662.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:5453–5458. doi: 10.1073/pnas.0711910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007;130:89–100. doi: 10.1016/j.cell.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoto S, Ito M, Tsutsumi Y, Ichikawa Y, Okuyama H, Brisibe EA, Saksena NK, Fujii YR. HIV-1 nef suppression by virally encoded microRNA. Retrovirology. 2004;1:44. doi: 10.1186/1742-4690-1-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka M, Jing Q, Georgel P, New L, Chen J, Mols J, Kang YJ, Jiang Z, Du X, Cook R, et al. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity. 2007;27:123–134. doi: 10.1016/j.immuni.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Ouellet DL, Plante I, Landry P, Barat C, Janelle ME, Flamand L, Tremblay MJ, Provost P. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucleic acids research. 2008;36:2353–2365. doi: 10.1093/nar/gkn076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellett PE, Roizman B. The Family Herpesviridae, A Brief Introduction. In: Knipe DM, Howley PM, editors. Fields Virology. 2007. pp. 2479–2497. [Google Scholar]
- Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, et al. Identification of microRNAs of the herpesvirus family. Nature methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Zavolan M, Grasser FA, Chien M, Russo JJ, Ju J, John B, Enright AJ, Marks D, Sander C, et al. Identification of virus-encoded microRNAs. Science. 2004;304:734–736. doi: 10.1126/science.1096781. [DOI] [PubMed] [Google Scholar]
- Rajewsky N. microRNA target predictions in animals. Nature genetics. 2006;38(Suppl):S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samols MA, Hu J, Skalsky RL, Renne R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2005;79:9301–9305. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samols MA, Skalsky RL, Maldonado AM, Riva A, Lopez MC, Baker HV, Renne R. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS pathogens. 2007;3:e65. doi: 10.1371/journal.ppat.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Kato Y, Taira K. Sequence-specific interference by small RNAs derived from adenovirus VAI RNA. FEBS Lett. 2006;580:1553–1564. doi: 10.1016/j.febslet.2006.01.085. [DOI] [PubMed] [Google Scholar]
- Schäfer A, Cai X, Bilello JP, Desrosiers RC, Cullen BR. Cloning and analysis of microRNAs encoded by the primate gamma-herpesvirus rhesus monkey rhadinovirus. Virology. 2007;364:21–27. doi: 10.1016/j.virol.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schat KA, Calnek BW. Protection against Marek’s disease-derived tumor transplants by the nononcogenic SB-1 strain of Marek’s disease virus. Infect Immun. 1978;22:225–232. doi: 10.1128/iai.22.1.225-232.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, Lopez MC, Baker HV, Renne R. Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol. 2007;81:12836–12845. doi: 10.1128/JVI.01804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, Horwitz E, Prokocimer Z, Prichard M, Hahn G, et al. Host immune system gene targeting by a viral miRNA. Science. 2007;317:376–381. doi: 10.1126/science.1140956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature. 2005;435:682–686. doi: 10.1038/nature03576. [DOI] [PubMed] [Google Scholar]
- Tam W, Ben-Yehuda D, Hayward WS. bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Molecular and cellular biology. 1997;17:1490–1502. doi: 10.1128/mcb.17.3.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- Thimmappaya B, Weinberger C, Schneider RJ, Shenk T. Adenovirus VAI RNA is required for efficient translation of viral mRNAs at late times after infection. Cell. 1982;31:543–551. doi: 10.1016/0092-8674(82)90310-5. [DOI] [PubMed] [Google Scholar]
- Triboulet R, Mari B, Lin YL, Chable-Bessia C, Bennasser Y, Lebrigand K, Cardinaud B, Maurin T, Barbry P, Baillat V, et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science. 2007;315:1579–1582. doi: 10.1126/science.1136319. [DOI] [PubMed] [Google Scholar]
- van den Berg A, Kroesen BJ, Kooistra K, de Jong D, Briggs J, Blokzijl T, Jacobs S, Kluiver J, Diepstra A, Maggio E, et al. High expression of B-cell receptor inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes Chromosomes Cancer. 2003;37:20–28. doi: 10.1002/gcc.10186. [DOI] [PubMed] [Google Scholar]
- Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A, Bradley A, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27:847–859. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhout EM, Ooms M, Vink M, Das AT, Berkhout B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic acids research. 2005;33:796–804. doi: 10.1093/nar/gki220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson GW, Tomasec P, Stanton RJ, Armstrong M, Prod’homme V, Aicheler R, McSharry BP, Rickards CR, Cochrane D, Llewellyn-Lacey S, et al. Modulation of natural killer cells by human cytomegalovirus. J Clin Virol. 2008;41:206–212. doi: 10.1016/j.jcv.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, Kieff E. Epstein-Barr virus BHRF1 micro- and stable RNAs during latency III and after induction of replication. J Virol. 2007;81:9967–9975. doi: 10.1128/JVI.02244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Zhao Y, Xu H, Smith LP, Lawrie CH, Sewer A, Zavolan M, Nair V. Marek’s disease virus type 2 (MDV-2)-encoded microRNAs show no sequence conservation with those encoded by MDV-1. J Virol. 2007;81:7164–7170. doi: 10.1128/JVI.00112-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Zhao Y, Xu H, Smith LP, Lawrie CH, Watson M, Nair V. MicroRNA profile of Marek’s disease virus-transformed T-cell line MSB-1: Predominance of virus-encoded microRNAs. J Virol. 2008;82:4007–4015. doi: 10.1128/JVI.02659-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–596. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- Yeung ML, Bennasser Y, Myers TG, Jiang G, Benkirane M, Jeang KT. Changes in microRNA expression profiles in HIV-1-transfected human cells. Retrovirology. 2005;2:81. doi: 10.1186/1742-4690-2-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q, McBride J, Fewell C, Lacey M, Wang X, Lin Z, Cameron J, Flemington EK. microRNA-155 is an Epstein-Barr Virus induced gene that modulates Epstein Barr virus regulated gene expression pathways. J Virol. 2008 doi: 10.1128/JVI.02380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9779–9784. doi: 10.1073/pnas.1630797100. [DOI] [PMC free article] [PubMed] [Google Scholar]