MicroRNAs in Cardiac Development (original) (raw)
Abstract
The transcriptional regulation of cardiovascular development requires precise spatiotemporal control of gene expression, and heterozygous mutations of transcription factors have frequently been implicated in human cardiovascular malformations. A novel mechanism involving post-transcriptional regulation by small, noncoding microRNAs (miRNAs) has emerged as a central regulator of many cardiogenic processes. We are beginning to understand the functions that miRNAs play during essential biologic processes, such as cell proliferation, differentiation, apoptosis, stress response, and tumorigenesis. The identification of miRNAs expressed in specific cardiac and vascular cell types has led to the discovery of important regulatory roles for these small RNAs during cardiomyocyte differentiation, cell cycle, conduction, and vessel formation. Here, we overview the recent findings on miRNA regulation in cardiovascular development. Further analysis of miRNA function during cardiovascular development will allow us to determine the potential for novel miRNA-based therapeutic strategies.
Similar content being viewed by others
Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.
During the last decade, animal studies and advances in human genetics have highlighted the need for precise regulation of key molecular pathways during embryonic development. This is particularly true for the cardiovascular system, in which haploinsufficiency typically causes human disease [59]. The dosage of cardiogenic pathways can be controlled at numerous levels, some of which have been well studied. In particular, the transcriptional regulation of cardiomyocyte differentiation and cardiac morphogenesis is highly conserved across species, and heterozygous mutations of transcription factors have frequently been implicated in human cardiac malformations [46]. Recently, post-transcriptional regulation by small noncoding RNAs, such as microRNAs (miRNAs), has emerged as a central regulator of many cardiogenic processes.
miRNAs are a large class of evolutionarily conserved, small, noncoding RNAs, typically 20–26 nucleotides (nt) in length, that primarily function post-transcriptionally by interacting with the 3′ untranslated region (UTR) of specific target mRNAs in a sequence-specific manner (reviewed in Zhao and Srivastava [57]) The first animal miRNA was described in 1993 as a regulator of developmental timing in Caenorhabditis elegans [27, 52], but it was not until 2001 that miRNAs were recognized to be widespread in all eukaryotes, including vertebrates [26, 37, 39]. More than 650 miRNAs are encoded in the human genome, and each is assumed to target >100 mRNAs, resulting in mRNA degradation or translational inhibition. Interactions between miRNAs and mRNAs are thought to require sequence homology in the 5′ end of the miRNA; however, significant variance in the degree of complementation in the remaining sequence allows a single miRNA to target a wide range of mRNAs, often regulating multiple genes within a common pathway. As a result, more than one third of mRNAs in the mammalian genome are believed to be regulated by one or more miRNAs [8].
Despite advances in miRNA discovery, the role of miRNAs in physiologic and pathophysiologic processes is just emerging. It has become clear that miRNAs play diverse roles in fundamental biologic processes, such as cell proliferation, differentiation, apoptosis, stress response, and tumorigenesis. Identification of miRNAs expressed in specific cardiac cell types has led to the discovery of important regulatory roles for these small RNAs during cardiomyocyte differentiation, cell cycle, and conduction, as well as during stages of cardiac hypertrophy in adults, indicating that miRNAs may be as important as transcription factors in controlling cardiac gene expression.
Here, we review the basic mechanisms by which miRNAs function, with a focus on the role of miRNAs during development of the heart and vessels. It appears that a network of miRNAs can be superimposed on well-described signaling and transcriptional networks with considerable intersection between the two. Ultimately, knowledge of the function and regulation of specific miRNAs and their mRNA targets in the heart will lead to a deeper understanding of cardiac cell-fate decisions and morphogenesis and ultimately could result in the development of novel therapeutic or preventive approaches for heart disease.
miRNA Organization, Biogenesis, and Target Recognition
miRNAs regulate gene expression at the post-transcriptional level through mRNA degradation, translational repression, or miRNA-mediated mRNA decay. Mature miRNAs are formed in a multistep biologic process involving critical endonucleases (Fig. 1). miRNAs are initially transcribed from the genome into long (several kilobases) 5′ capped, polyadenylated (poly(A)) primary transcripts (primiRNAs) by RNA polymerase II [7]. Some miRNAs interspersed among repetitive DNA elements, such as Alu repeats (5′ AG/CT 3′), can also be transcribed by RNA polymerase III [5]. The miRNA-encoding portion of the pri-miRNA forms a hairpin structure that is recognized and cleaved in the nucleus by a microprocessing complex. This complex consists of the double-stranded RNA-specific nuclease DROSHA and its cofactor, DiGeorge syndrome critical region 8 (DGCR8) [25]. The resulting approximately 70-nt hairpin precursor miRNA (pre-miRNA) is exported to the cytoplasm by the RAN-GTP–dependent nuclear transport receptor, exportin-5, which acts by recognizing a 2- to 3-base pair overhang of the pre-miRNA stem-loop structure [4, 56]. A complex of the RNAse III-like ribonuclease, Dicer, and the transactivator RNA-binding protein then cleaves the pre-miRNA to release the mature miRNA duplex.
Fig. 1
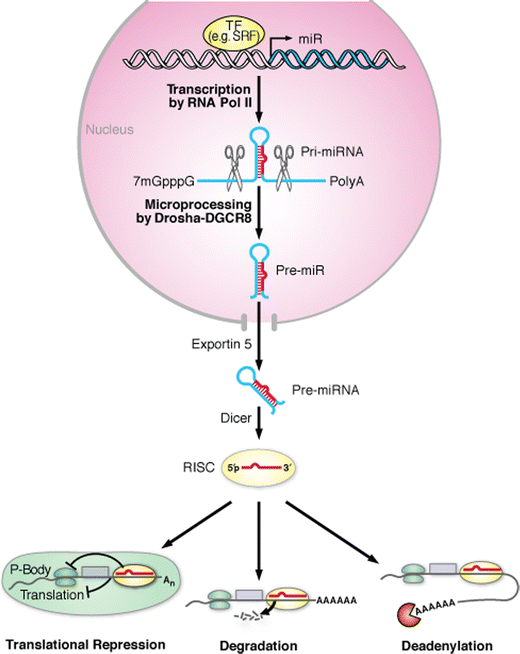
Schematic representation of miRNA biogenesis and function. Transcription of miRNA genes is typically mediated by RNA polymerase II (pol II) and can be controlled by various transcription factors (TF). The initial transcripts, termed “primiRNAs,” can range from a few hundred nucleotides to several kilobases long. The primiRNA has a characteristic stem-loop structure that can be recognized and cleaved by the RNase III endonuclease Drosha, along with its partner DGCR8 (DiGeorge syndrome critical region 8 gene; also known as Pasha). The cleavage product, an approximately 70-nt stem-loop pre-miRNA, is exported from the nucleus by Exportin 5. In the cytoplasm, another RNase III enzyme, Dicer, further cleaves the pre-miRNA into a double-stranded mature miRNA (approximately 21 nt), which is incorporated into the RISC, thus allowing preferential strand separation of the mature miRNA to repress mRNA translation or destabilize mRNA transcripts through cleavage or deadenylation (adapted from Zhao and Srivastava [57])
An asymmetry in the relative thermodynamic stability of the 5′ ends of the miRNA duplex results in preferential loading of the less stable approximately 22-nt strand into the RNA-induced silencing complex (RISC); the other strand is degraded, although in some cases both strands are incorporated into the RISC [22, 40, 43]. The RISC helps mediate miRNA–mRNA interactions and subsequent mRNA repression or destabilization [19]. miRNAs typically bind to the 3′ UTRs of their mRNA targets with imprecise complementarity. Typically, the degree of Watson–Crick base-pairing between bases 2 and 7 (the “seed region”) at the 5′ end of the miRNA is critical for binding mRNA targets [38, 48] and mediating repression. RISC-bound miRNAs may also be sequestered away from translational machinery in processing bodies, which act by recruiting poly(A) nucleases to help modulate deadenylation of mRNA and thereby prevent translation [16, 23, 29].
miRNAs can be found in exons or introns of noncoding transcripts with independent enhancer regulation and in the introns and 3′ UTRs of protein-coding transcripts. They can also overlap with either an exon or an intron, depending on the alternative splicing pattern. In flies and worms, some miRNAs in intronic regions bypass Drosha processing and enter the miRNA biogenesis pathway as pre-miRNAs [41]. In many cases, miRNAs are clustered near other miRNAs, and they are transcriptionally coregulated and share cooperative regulatory roles.
Several algorithmic databases have been designed for miRNA target prediction that rely, for the most part, on the following criteria: (1) conservation across species, (2) complementarity of the 5′ miRNA “seed match” to the 3′ UTR (approximately 7 nt) [28, 38, 59], (3) G:U wobbles in the seed [6], (4) the thermodynamic context of target mRNA-binding sites (i.e., mRNA targets located in regions of high free energy and unstable secondary structure are favored) [58, 59], and (5) multiple miRNA-binding sites in 3′ UTR [14]. These computational programs are continuously updated to integrate new knowledge from validated miRNA–mRNA interactions (reviewed in Sethupathy [44]).
One approach that has showed the importance of miRNAs during vertebrate development has been to create mutations in Dicer, the enzyme required to process miRNAs into their active form. Dicer is encoded by a single locus in vertebrates. Zebrafish lacking maternal and endogenous Dicer die from defects in gastrulation, brain morphogenesis, somitogenesis, and heart development [18, 51]. In mice, targeted deletion of Dicer causes lethality at embryonic day 7.5 (E7.5), before body axis formation [2]. Cardiac-specific deletion of Dicer using Cre recombinase expressed under the control of the endogenous Nkx2.5 regulatory elements resulted in embryonic lethality at E12.5 [59]. The Nkx2.5-Cre is active from E8.5 during heart patterning and differentiation but only after initial commitment of cardiac progenitors [34]. It will be important to determine whether Dicer is required for earlier steps of cardiogenesis (before E8.5), such as cardiac lineage specification, because Dicer is required for embryonic stem cell differentiation [21, 35]. Deletions of Dicer in specific heart populations will show the importance of miRNA function in distinct aspects of heart development.
Organization and Regulation of miR-1 and miR-133
Two widely conserved miRNAs that display cardiac muscle- and skeletal muscle-specific expression during development and in the adult human are miR-1 and miR-133a (Fig. 2), which are derived from a common precursor transcript (bicistronic) [9, 58]. Two highly conserved loci encode the mature miR-1 (miR-1-1 and miR-1-2) and miR-133a (miR-133a-1, miR-133a-2) transcripts, which appear to be genetically redundant [24]. The mature forms of miR-1 derived from the distinct loci are identical, as are the miR-133a forms.
Fig. 2
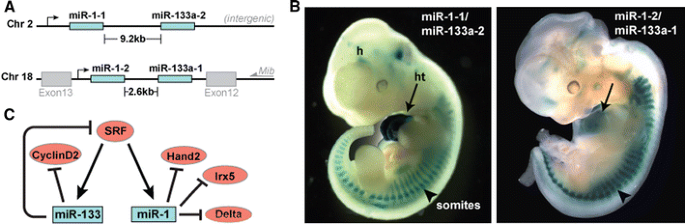
Summary of miR-1 and miR-133 genomic organization, regulation, and expression during mouse cardiogenesis. a Chromosomal locations of mouse miR-1 and miR-133a. The miR-1/133a clusters are transcribed as bicistronic transcripts. b LacZ directed by an upstream enhancer of the miR-1-2/miR-133a-2 and miR-1-1 miR-133a-1 clusters, respectively, shows expression in the heart (ht) and somites (arrowhead) at mouse embryonic day 11.5. c Cardiac expression of miR-1 and miR-133 is regulated by SRF. Targets of miR-1 and miR-133 in cardiac muscle are shown
Cardiac transcription of miR-1/miR-133 bicistronic precursors is directly regulated by myocyte enhancer factor-2 (Mef2) and serum response factor (SRF) (Fig. 2a) [58]. SRF binds to CArG motifs in promoters and enhancers of muscle-specific genes that regulate hundreds of miRNAs [36], although only a limited number have been assigned target mRNAs. In the heart, SRF binds and activates the enhancer regions of miR-1/miR-133 in vitro and in vivo through a serum-response element conserved from fly to human [58]. Concordant with their common cis- and trans-regulation, both miR-1 and miR-133 are coexpressed in cardiac and skeletal muscle throughout mouse development (Fig. 2b) and are robustly expressed in the adult human [9, 30, 58].
Function of miR-1 and miR-133a During Cardiogenesis
miR-1 expression is detectable by E8.5 in mouse and increases throughout development. Overexpression of miR-1 under the control of the β-MHC promoter diminishes the pool of proliferating ventricular myocytes by inducing a premature exit from the cell cycle. This negatively regulates cardiac growth, in part by inhibiting translation of the heart and neural crest derivative-2 protein, Hand2 [58], a basic helix-loop-helix protein involved in ventricular myocyte expansion. In mice, Hand2 is initially expressed throughout the linear heart tube and then becomes restricted to the developing atrial and ventricular myocardium with highest expression in the right ventricle. Mice that lack Hand2 die at E10.5 from right ventricular hypoplasia and decreased trabeculation in the left ventricle [47, 49, 55]. In mice overexpressing miR-1, trabeculation is also decreased, consistent with the Hand2 mutant phenotype, corroborating Hand2 as a direct target of miR-1 [58]. Mice lacking miR-1 have an increase in Hand2 protein, providing yet further evidence of Hand2 as a direct target of miR-1.
In Drosophila, miR-1 functions to pattern the dorsal vessel (i.e., aorta/heart tube). Moreover, deletion of the single miR-1 gene (dmiR-1) results in a muscle-differentiation defect [24, 45]. In a subset of _dmiR-_1-null flies, muscle progenitors are arrested in a proliferative state and accumulate ectopically. Drosophila hand does not seem to be a target of miR-1 because the fly hand ortholog lacks miR-1-binding sites in its 3′ UTR, suggesting that miRNA–mRNA interactions may differ somewhat between species. Instead, dmiR-1 targets transcripts encoding the Notch ligand, Delta, which regulates the expansion of cardiac and muscle progenitor cells [24], suggesting that miR-1 promotes muscle differentiation through downregulation of the Notch signaling pathway. This is consistent with the known function of the Notch/Delta–signaling pathway during developmental cell-fate decisions, including those involving cardiac specification [1].
In cultured myoblasts, miR-1 promotes myoblast differentiation, whereas miR-133 stimulates myoblast proliferation [9]. Chen et al. showed in myoblast culture that miR-1 targets the histone deacetylase 4 (HDAC4) mRNA, a transcriptional repressor of Mef2-dependent activation of muscle-specific gene expression, suggesting that translational repression of HDAC4 by miR-1 enhances gene activation of Mef2-dependent promoters. They also showed that miR-133 targets SRF, which is important in muscle proliferation, differentiation, and activation of the miR-1/miR-133 transcript, thus creating a negative-feedback regulatory loop (Fig. 2c). When rat ventricular cells are subjected to oxidative stress, miR-1 and miR-133 have opposing effects on apoptosis: miR-1 targets the anti-apoptotic heat-shock proteins HSP60 and HSP70 and is apoptotic, whereas miR-133 represses caspase-9, a regulator of mitochondria-mediated apoptosis [54], and is anti-apoptotic.
Targeted deletion in mice will be invaluable for investigating the functional role of individual miRNAs. Surprisingly, disruption of just one of the two miR-1 family members, miR-1-2, results in a range of abnormalities, including cell-cycle dysregulation, heart malformations, and postnatal electrophysiological defects. Heterozygous _miR-1-2_-null mice survive to reproduce, but 50% of miR-1-2 homozygous null mice die between E15.5 and birth from ventricular septal defects and cardiac dysfunction. These defects can occur from dysregulation of a multitude of events during cardiogenesis, and it is likely that miR-1-2 regulates numerous genes during this process. Precise dosage of Hand2 is crucial for normal cardiomyocyte proliferation and development, and increased levels of Hand2 may contribute to the ventricular septal defects (Fig. 2c) [59].
_miR-1-_2-null mice that survive until birth often die suddenly [59]. Electrophysiologic testing showed a spectrum of cardiac arrhythmias in mutant mice, which may be caused in part by increased levels of the transcription factor Iroquois homeobox 5 (Irx5). Irx5 regulates the cardiac ventricular repolarization gradient by negatively regulating the expression of potassium channel genes, such as Kcnd2 [13]. The 3′ UTR of Irx5 contains a well-conserved _miR-_1–binding site and is a direct target of miR-1 (Fig. 2c) [59]. In the miR-1-2–null hearts, Irx5 transcripts were upregulated, and its target gene, Kcnd2, was correspondingly downregulated.
Postnatal mouse cardiomyocytes terminally exit the cell cycle after the first 10 days of life. However, _miR-1-_2–null adult mice have an increase in mitotic cardiac myocytes along with cardiac hyperplasia. These abnormalities could reflect the effect of miR-1 on Notch signaling and the derepression of Hand2 (Fig. 2c), which promotes myocyte expansion. In addition, genome-wide profiling of miR-1-2 mutant adult mouse hearts suggests a broad upregulation of positive regulators of the cell cycle and downregulation of tumor suppressors, indicating a shift in the “threshold” of cells to re-enter the cell cycle [59]. Whether this change promotes cardiac repair after injury remains to be determined.
Similarly, targeted deletion of miR-133a in mice has helped to elucidate its role during cardiac development [31]. Unlike miR-1, single mutations of either miR-133a-1 or miR-133a-2 caused no overt phenotype. However, combined deletion of both miRNAs led to partially penetrant ventricular septal defects and decreased survival at birth. Adult compound mutant mice developed dilated cardiomyopathy and lethal heart failure. miR-133a was found to suppress smooth-muscle gene expression and cardiomyocyte proliferation, in part by way of repression of SRF and cyclin D2 (Fig. 2c).
During early cell fate decisions of mouse and human embryonic stem (ES) cells, miR-1, and miR-133 are expressed just as mesoderm emerges and function in concert to promote mesoderm induction while suppressing differentiation into the ectodermal or endodermal lineages (Fig. 3) [20]. However, miR-1 and miR-133 have antagonistic effects on further adoption of muscle lineages: miR-1 promotes differentiation of mouse and human ES cells toward a cardiac fate, whereas miR-133 inhibits differentiation into cardiac muscle. miR-1 appears to exert this effect, in part, by translationally repressing the mammalian orthologue of delta, Delta-like-1 (Dll-1), similar to the repression seen in the fly (Fig. 2c) [20]. Thus, the bicistronic miR-1/miR-133 transcript encodes distinct mature miRNAs that likely share common targets yet complement each other by balancing the differentiation and proliferation of cardiac and skeletal muscle lineages.
Fig. 3
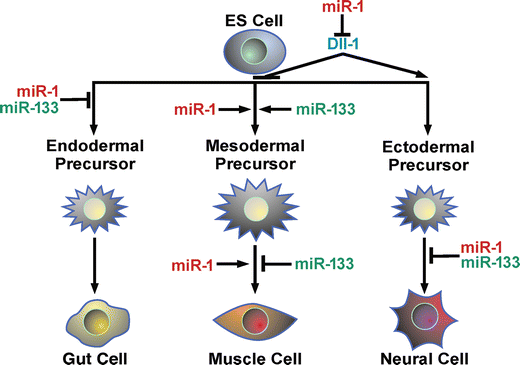
Model of miR-1/miR-133 effects during embryonic stem-cell differentiation. miR-1 and miR-133 promote differentiation of mesoderm and inhibit endoderm and ectoderm differentiation at specific stages as indicated and have opposing effects in later steps of muscle differentiation. miR-1 inhibition of Dll-1 translation, along with yet unknown targets, likely contribute to the observed effects of miR-1 (from Ivey et al. [20])
miR-143/145 Regulate Smooth-Muscle Plasticity
Vascular smooth muscle cells have the unique capacity to oscillate between a contractile phenotype and a less differentiated, more synthetic state in response to external queues. This phenotypic modulation is a major component of the vascular repair process and certain diseases, such as atherosclerosis. Not surprisingly, it involves dramatic changes in the smooth-muscle gene program, which are heavily influenced by particular miRNAs.
Among the most highly enriched microRNAs in the vascular smooth muscle are miR-143 and miR-145. These two microRNAs are cotranscribed from a single locus under the transcriptional control of SRF, myocardin, and Nkx2.5 [12]. Interestingly, their expression is strongly downregulated in injured or atherosclerotic vessels, which are comprised of synthetic smooth muscle cells [10, 12, 15, 53]. Indeed, miR-143 and miR-145 target a network of transcription factors, including Klf4, Klf5, myocardin, MRTFB, and Elk-1, which is consistent with a role for these miRNAs in regulating the quiescent versus proliferative phenotype of smooth muscle (Fig. 4) [12, 53]. Further, targeted deletion of these miRNAs in mice showed their importance in achieving vascular tone and responding to vascular injury [3, 15, 53] and led to the identification of the angiotensin-converting enzyme as a target of miR-145 [3]. miR-145 was also found to be necessary for smooth-muscle conversion from fibroblasts and sufficient to differentiate smooth muscle cells from multipotent neural crest stem cells, further implicating microRNAs as regulators of cell-fate decisions [12]. Similarly, miR-221 and miR-222 regulate smooth-muscle cell proliferation and modulate the deleterious vascular response after angioplasty [32]. Therefore, these miRNAs represent potential targets for the diagnosis and therapy of vascular disease.
Fig. 4
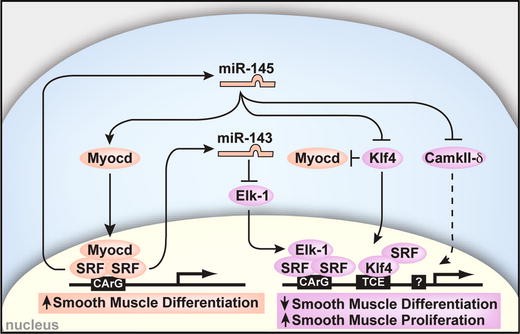
miR-143 and miR-145 are transcriptionally regulated by SRF and repress multiple factors that normally promote the synthetic smooth-muscle phenotype (lavender). These include Klf4, which interacts with SRF and also represses Myocd. miR-145 has a positive effect on Myocd activity to concurrently promote the contractile smooth-muscle phenotype (peach), thereby also reinforcing miR-145 and miR-143 expression (from Cordes et al. [12])
miR-138 Regulation of Cardiac Patterning
Intricate transcriptional networks establish chamber-specific gene expression, and these patterning events are highly conserved across species, from zebrafish to human [46]. Zebrafish are useful models to study cardiac patterning events because of their simple two-chambered heart, which consists of a single atrium and ventricle separated by the atrioventricular canal (AVC). The atrial and ventricular chambers express unique myosin genes, whereas the AVC expresses distinct genes, such as cspg2, encoding versican, notch1b, and tbx2 [11, 42]. miR-138 is a highly conserved miRNA found in many parts of the embryo, but within the zebrafish heart it is specifically expressed in the ventricular chamber [33]. Disruption of miR-138 function led to expansion of AVC gene expression into the ventricle and failure of ventricular cardiomyoctyes to fully mature. miR-138 normally restricts AVC gene expression by directly repressing cspg2 in the ventricle. This event is reinforced by ventricular repression of retinoic acid dehydrogenase, resulting in decreased retinoic acid, which is a positive regulator of cspg2 [33]. It is likely that other region-specific miRNAs will reinforce known signaling and transcriptional networks that establish patterns of gene expression throughout the developing heart tube.
miR-126 Regulation of Angiogenesis
In addition to miRNA regulation of cardiomyocytes and smooth muscle, two recent reports regarding the endothelial-specific miRNA, miR-126, illustrate what will likely be a broader function of tissue-specific miRNAs during cardiovascular development [17, 50]. miR-126, which is located in the intron of an endothelial-specific gene, Egfl7, regulates endothelial cell migration and integrity of capillary tubes in vitro [17]. In zebrafish embryos, miR-126 was similarly required for vascular integrity with vascular collapse and extensive hemorrhage observed on disruption of miR-126 function [17]. This function was conserved in mice, in which targeted deletion of miR-126 resulted in cranial hemorrhages and defects in angiogenesis [50]. miR-126 appears to function in part to promote vascular endothelial growth factor (VEGF) signaling by directly targeting multiple repressors of VEGF signaling, including Spred1 [17, 50] and Pik3r2 [17]. Mice lacking miR-126 had an impaired ability to generate new blood vessels in response to cardiac injury. Thus, miR-126-mediated regulation of angiogenesis may be a valuable therapeutic target to promote new blood vessel formation in ischemic conditions as well to inhibit angiogenesis during tumor growth.
Summary
The function of miRNAs in cardiovascular development reviewed here likely foreshadows a much broader role of dozens of miRNAs in regulating most aspects of cardiovascular development. Through their ability to post-transcriptionally regulate mRNA levels, and thus manage protein dosage, miRNAs provide finer regulation within the complex molecular networks that regulate cardiogenesis. The importance of this fine regulation is highlighted by the recognition that most known genetic causes of heart malformations in humans result from haploinsufficiency or heterozygous point mutations. The field of miRNA biology is growing rapidly, and new tools and mechanisms are becoming available. A critical hurdle will be to efficiently identify direct miRNA targets and to integrate cardiovascular enriched targets with previously described regulatory networks. With further characterization, elucidating the function of cardiac-enriched miRNAs may provide us with new diagnostic, prognostic, and therapeutic targets for many forms of cardiovascular disease.
References
- Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284:770–776
Article CAS PubMed Google Scholar - Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ et al (2003) Dicer is essential for mouse development. Nat Genet 35:215–217
Article CAS PubMed Google Scholar - Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L et al (2009) Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119:2634–2647
Article CAS PubMed Google Scholar - Bohnsack MT, Czaplinski K, Gorlich D (2004) Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 10:185–191
Article CAS PubMed Google Scholar - Borchert GM, Lanier W, Davidson BL (2006) RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 13:1097–1101
Article CAS PubMed Google Scholar - Brennecke J, Stark A, Russell RB, Cohen SM (2005) Principles of microRNA-target recognition. PLoS Biol 3:404–418
Article CAS Google Scholar - Cai X, Hagedorn CH, Cullen BR (2004) Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10:1957–1966
Article CAS PubMed Google Scholar - Chaudhuri K, Chatterjee R (2007) MicroRNA detection and target prediction: integration of computational and experimental approaches. DNA Cell Biol 26:321–337
Article CAS PubMed Google Scholar - Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM et al (2006) The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38:228–233
Article CAS PubMed Google Scholar - Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q et al (2009) MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res 105:158–166
Article CAS PubMed Google Scholar - Chi NC, Shaw RM, De Val S, Kang G, Jan LY, Black BL et al (2008) Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev 22:734–739
Article CAS PubMed Google Scholar - Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH et al (2009) miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460:705–710
CAS PubMed Google Scholar - Costantini DL, Arruda EP, Agarwal P, Kim KH, Zhu Y, Zhu W et al (2005) The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell 123:347–358
Article CAS PubMed Google Scholar - Doench JG, Sharp PA (2004) Specificity of microRNA target selection in translational repression. Genes Dev 18:504–511
Article CAS PubMed Google Scholar - Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV et al (2009) The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ 16:1590–1598
Article CAS PubMed Google Scholar - Ezzeddine N, Chang TC, Zhu W, Yamashita A, Chen CY, Zhong Z et al (2007) Human TOB, an antiproliferative transcription factor, is a poly(A)-binding protein-dependent positive regulator of cytoplasmic mRNA deadenylation. Mol Cell Biol 27:7791–7801
Article CAS PubMed Google Scholar - Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD et al (2008) miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell 15:272–284
Article CAS PubMed Google Scholar - Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S et al (2005) MicroRNAs regulate brain morphogenesis in zebrafish. Science 308:833–888
Article CAS PubMed Google Scholar - Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R (2005) Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 123:631–640
Article CAS PubMed Google Scholar - Ivey KN, Muth A, Arnold J, King FW, Yeh R, Fish JE et al (2008) MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell 2:219–229
Article CAS PubMed Google Scholar - Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T et al (2005) Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19:489–501
Article CAS PubMed Google Scholar - Khvorova A, Reynolds A, Jayasena SD (2003) Functional siRNAs and miRNAs exhibit strand bias. Cell 115:209–216
Article CAS PubMed Google Scholar - Kim SK, Nam JW, Rhee JK, Lee WJ, Zhang BT (2006) miTarget: microRNA target gene prediction using a support vector machine. BMC Bioinform 7:411–422
Article Google Scholar - Kwon C, Han Z, Olson EN, Srivastava D (2005) MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci USA 102:18986–18991
Article CAS PubMed Google Scholar - Landthaler M, Yalcin A, Tuschl T (2004) The human DiGeorge syndrome critical region gene 8 and its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol 14:2162–2167
Article CAS PubMed Google Scholar - Lee RC, Ambros V (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science 294:862–864
Article CAS PubMed Google Scholar - Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854
Article CAS PubMed Google Scholar - Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20
Article CAS PubMed Google Scholar - Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R (2005) MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol 7:719–723
Article CAS PubMed Google Scholar - Liu N, Williams AH, Kim Y, McAnally J, Bezprozvannaya S, Sutherland LB et al (2007) An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc Natl Acad Sci USA 104:20844–20849
Article CAS PubMed Google Scholar - Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R et al (2008) MicroRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev 22:3242–3254
Article CAS PubMed Google Scholar - Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C (2009) A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res 104:476–487
Article CAS PubMed Google Scholar - Morton SU, Scherz PJ, Cordes KR, Ivey KN, Stainier DY, Srivastava D (2008) MicroRNA-138 modulates cardiac patterning during embryonic development. Proc Natl Acad Sci USA 105:17830–17835
Article CAS PubMed Google Scholar - Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ (2001) Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 31:176–180
Article CAS PubMed Google Scholar - Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ (2005) Characterization of Dicer-deficient murine embryonic stem cells. Proc Natl Acad Sci USA 102:12135–12140
Article CAS PubMed Google Scholar - Niu Z, Iyer D, Conway SJ, Martin JF, Ivey K, Srivastava D et al (2008) Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc Natl Acad Sci USA 105:17824–17829
Article CAS PubMed Google Scholar - Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B et al (2000) Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408:86–89
Article CAS PubMed Google Scholar - Rajewsky N (2006) MicroRNA target predictions in animals. Nat Genet 38(Suppl):S8–S13
Article CAS PubMed Google Scholar - Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE et al (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403:901–906
Article CAS PubMed Google Scholar - Ro S, Park C, Young D, Sanders KM, Yan W (2007) Tissue-dependent paired expression of miRNAs. Nucleic Acids Res 35:5944–5953
Article CAS PubMed Google Scholar - Ruby JG, Jan CH, Bartel DP (2007) Intronic microRNA precursors that bypass Drosha processing. Nature 448:83–86
Article CAS PubMed Google Scholar - Rutenberg JB, Fischer A, Jia H, Gessler M, Zhong TP, Mercola M (2006) Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related transcription factors. Development 133:4381–4390
Article CAS PubMed Google Scholar - Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD (2003) Asymmetry in the assembly of the RNAi enzyme complex. Cell 115:199–208
Article CAS PubMed Google Scholar - Sethupathy P, Megraw M, Hatzigeorgiou AG (2006) A guide through present computational approaches for the identification of mammalian microRNA targets. Nat Methods 3:881–886
Article CAS PubMed Google Scholar - Sokol NS, Ambros V (2005) Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev 19:2343–2354
Article CAS PubMed Google Scholar - Srivastava D (2006) Making or breaking the heart: From lineage determination to morphogenesis. Cell 126:1037–1048
Article CAS PubMed Google Scholar - Srivastava D, Thomas T, Lin Q, Kirby ML, Brown D, Olson EN (1997) Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nat Genet 16:154–160
Article CAS PubMed Google Scholar - Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM (2005) Animal microRNAs confer robustness to gene expression and have a significant impact on 3′ UTR evolution. Cell 123:1133–1146
Article CAS PubMed Google Scholar - Thomas T, Yamagishi H, Overbeek PA, Olson EN, Srivastava D (1998) The bHLH factors, dHAND and eHAND, specify pulmonary and systemic cardiac ventricles independent of left-right sidedness. Dev Biol 196:228–236
Article CAS PubMed Google Scholar - Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA et al (2008) The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell 15:261–271
Article PubMed Google Scholar - Wienholds E, Koudijs MJ, van Eeden FJ, Cuppen E, Plasterk RH (2003) The microRNA-producing enzyme Dicer1 is essential for zebrafish development. Nat Genet 35:217–218
Article CAS PubMed Google Scholar - Wightman B, Ha I, Ruvkun G (1993) Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75:855–862
Article CAS PubMed Google Scholar - Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF et al (2009) MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev 23:2166–2178
Article CAS PubMed Google Scholar - Xu C, Lu Y, Pan Z, Chu W, Luo X, Lin H et al (2007) The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci 120:3045–3052
Article CAS PubMed Google Scholar - Yamagishi H, Yamagishi C, Nakagawa O, Harvey RP, Olson EN, Srivastava D (2001) The combinatorial activities of Nkx2.5 and dHAND are essential for cardiac ventricle formation. Dev Biol 239:190–203
Article CAS PubMed Google Scholar - Zeng Y, Cullen BR (2004) Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res 32:4776–4785
Article CAS PubMed Google Scholar - Zhao Y, Srivastava D (2007) A developmental view of microRNA function. Trends Biochem Sci 32:189–197
Article CAS PubMed Google Scholar - Zhao Y, Samal E, Srivastava D (2005) Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 436:214–220
Article CAS PubMed Google Scholar - Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN et al (2007) Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 129:303–317
Article CAS PubMed Google Scholar
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
- Gladstone Institute of Cardiovascular Disease and Departments of Pediatrics and Biochemistry and Biophysics, University of California, San Francisco, CA, 94158, USA
Kimberly R. Cordes, Deepak Srivastava & Kathryn N. Ivey
Authors
- Kimberly R. Cordes
- Deepak Srivastava
- Kathryn N. Ivey
Corresponding author
Correspondence toKathryn N. Ivey.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cordes, K.R., Srivastava, D. & Ivey, K.N. MicroRNAs in Cardiac Development.Pediatr Cardiol 31, 349–356 (2010). https://doi.org/10.1007/s00246-010-9639-3
- Received: 16 December 2009
- Accepted: 17 January 2010
- Published: 07 February 2010
- Issue Date: April 2010
- DOI: https://doi.org/10.1007/s00246-010-9639-3