Nocturnal Frontal Lobe Epilepsy (original) (raw)
…
14k Accesses
71 Citations
11 Altmetric
1 Mention
Abstract
Nocturnal frontal lobe epilepsy (NFLE) is a syndrome of heterogeneous etiology, characterized by the occurrence of sleep-related seizures with different complexity and duration. Genetic, lesional, and cryptogenetic NFLE forms have been described. NFLE is generally considered a benign clinical entity, although severe, drug-resistant forms do exist. A significant proportion of sleep-related complex motor seizures, hardly distinguishable from NFLE, originate outside the frontal lobe. Moreover, the distinction of NFLE from the non-rapid eye movement arousal parasomnias may be challenging. A correct diagnosis of NFLE should be based on a diagnostic approach that includes the anamnestic, video–polysomnographic, morphological, and genetic aspects. Studies on the relationships between genes, arousal regulatory mechanisms, and epileptogenesis, using both clinical and experimental models of NFLE might provide key insights in the interrelationship between sleep and epilepsy.
Similar content being viewed by others
Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.
Introduction
Seizures originating from the frontal lobe mainly occur during sleep [1, 2]. The definition of nocturnal frontal lobe epilepsy (NFLE) has been introduced in order to identify patients affected by frontal lobe epilepsy whose seizures are almost exclusively sleep related. In the last few decades, the spread and refinement of video–polysomnographic (V-PSG) technology has allowed NFLE to be fully defined and described, so that it is now considered a distinct epileptic syndrome; nevertheless, some NFLE case can be hardly distinguishable from non-rapid eye movement (NREM) parasomnias [3–7] and from sleep-related epileptic seizures originating outside the frontal lobe [8–10].
It is now recognized that NFLE is not a homogeneous disease as familial, idiopathic, sporadic, cryptogenetic, or symptomatic forms do exist [4, 11–13]. In addition, a genetic heterogeneity is also evident within the familial type [14].
NFLE is usually considered a benign clinical epileptic syndrome because seizures occur almost exclusively during sleep, and in the majority of patients the pharmacological treatment is effective; however, severe and drug-resistant forms, occasionally associated with mental retardation, have been described [12, 13, 15, 16••]. All the aforementioned issues will be reviewed and discussed in the following sections in order to give a comprehensive overview of the pathophysiological and clinical aspects of NFLE.
Epidemiology of NFLE
No epidemiologic data are available on NFLE prevalence. The frequency of pure sleep-related seizures, defined as “seizures occurring exclusively or predominantly (>90 %) from sleep” clusters around 12 % of people with epilepsy [17, 18], the majority of patients being affected by focal epilepsy [19, 20]. NFLE is probably not rare, accounting for 13 % of polysomnographic recordings for nocturnal motor disorders in a retrospective study performed at a tertiary centre in Italy [12] and for 6.3 % of a large series of focal drug-resistant epileptic patients (unpublished data). Because many cases of NFLE are misdiagnosed as parasomnias, the clinical relevance of NFLE is underestimated, especially in children [4].
Genetic Forms of NFLE
A family history for epilepsy is frequently observed in NFLE patients [12]. After the report of several families showing a clear autosomal dominant inheritance, autosomal dominant NFLE (ADNFLE) was introduced as a distinct clinical entity with marked intrafamilial variability in disease severity [4, 11, 21]. The genetic origin of the disease was confirmed by a linkage study in Australian kindred. More specifically, a locus was identified at chromosome 20q13.2-q13.3 [22] and two different mutations were detected in the gene coding the α4 subunit of the neuronal nicotinic acetylcholine receptor (nAChR) (CHRNA4) [23, 24]. Both mutations were shown to have major effects on receptor function in vitro [23–26]. With the identification of these mutations, subsequently confirmed in families of different origins [27–29], ADNFLE became the first epilepsy in which genetic bases were detected, apparently showing features of a monogenic disease. Three de novo or inherited CHRNA4 mutations, occasionally associated with mild-to-moderate mental retardation were later reported [15, 30–37]. ADNFLE was quickly recognized as a genetically heterogeneous disorder as most of the described families did not show mutations in the CHRNA4 gene [14, 38], and new loci and genes were reported in the following years. In particular, a second locus was identified at chromosome 15q24 in one family [38] and a third locus spanning the pericentromeric region of chromosome 1 was identified in an Italian ADNFLE family [39], the latter containing the gene coding for the β2 subunit of the nACh receptor (CHRNB2) (Table 1) [40]. Further CHRNB2 mutations were disclosed in different unrelated families and one sporadic case [41–46] (Table 1); in two families a coexistence of both epilepsy and specific deficits in tasks of verbal memory was observed [44, 45]; in two patients ictal single-photon emission computed tomography showed a significant hyperperfusion of the cingulated gyrus [45].
Table 1 Known loci and mutations associated with autosomal dominant nocturnal frontal lobe epilepsy
A fourth locus was mapped on chromosome 8p21.2-8q12 in 2006 in an Italian family and a mutation was identified in the α2 subunit of the nAChR (CHRNA2) gene (Table 1). These patients exhibited prominent fear and organized motor behaviors, which led the authors to speculate that seizures could have a fronto-limbic involvement. In vitro studies confirmed functional alterations of the mutated protein, and postmortem brain examination disclosed a high expression of CHRNA2 in thalamic structures [51]. To date, this is the only reported ADNFLE kindred carrying CHRNA2 mutations.
The findings of a genetic alteration of the cholinergic system and the in vivo documentation [52] of an alteration of nAChR density distribution in ADNFLE patients may give some insights into understanding the relationships between genes, epileptogenesis, and arousal regulatory processes. Indeed, the nAChR are extensively distributed in the brain and contribute to the regulation of sleep and arousal oscillations, at both the cortical and subcortical level [53]. Moreover, they play a role in the release of other neurotransmitters such as gamma-aminobutyric acid (GABA) and glutamate. In animal models, it has been shown that a mutation of the nicotinic receptors may create an unbalanced excitation/inhibition circuitry within the GABAergic reticular thalamic neurons, thus favouring seizures through the synchronizing effect of spontaneous thalamocortical oscillations [54]. However, other experimental studies have reported an involvement of nAChR in the regulation of arousals, sleep stability, and the activity–rest pattern [53, 55•]. We can speculate that the observed genetic alterations in NFLE create the conditions for both arousal instability and seizure generation. Indeed, it is recognized that NFLE patients may have a high number of arousals and that nocturnal motor events tend to occur during unstable sleep [56, 57, 58•]. With these assumptions in mind, we can also hypothesize that the high prevalence of parasomnias in the personal and family history of NFLE patients [59••] relies on a common alteration of the arousal regulatory system.
However, ADNFLE is not only related to mutations in the cholinergic system. Since 2005, the involvement of other genes, not belonging to the nACh receptor subunit family, has been recognized in the ADNFLE pathogenesis. In particular, Combi et al. [47] found two new putative ADNFLE loci on chromosomes 3p22-p24 and 8q11.2-q21.1; among the genes mapping in these regions, the one most probably involved in ADNFLE pathogenesis is corticotropin-releasing hormone (CRH) on chromosome 8. A gene sequencing procedure disclosed two nucleotide variations (Table 1) in the CRH promoter region in four families and in two sporadic cases [48]. An additional family was later detected, with a patient carrying both variations in compound heterozygosity [49]. The in vitro functional analysis of both variations demonstrated an altered level of protein expression [48] suggesting an interrelation between CRH concentration and neuronal excitability with a possible effect on thalamo-cortical loop dysfunctions [47, 48]. Very recently, a further CRH mutation has been found in the protein pro-sequence region of the CRH of two affected siblings of an Italian ADNFLE family [50•]. The recurrence of variants causing altered levels of the CRH secretion suggests that individuals with such an altered hormone level could be more prone to develop ADNFLE [50•].
In 2012, a further gene on chromosome 9 encoding the sodium-activated potassium channel subunit 1 (KCNT1) was associated with ADNFLE. In particular, four variants in KCNT1 were identified in three families and in a sporadic ADNFLE case (Table 1), all showing a severe ADNFLE phenotype with early onset, high prevalence of intellectual disabilities, and psychiatric or behavioral problems, including psychosis, catatonia, and aggression. Contrary to all the previously found mutations, KCNT1 mutations showed complete penetrance [16••]. Very recently, mutations in the DEPDC5 gene were reported as responsible for different types of focal epilepsies, including ADNFLE [60••]. However, the prevalence of mutations in this gene remains to be assessed.
In summary, ADNFLE is a heterogeneous genetic syndrome that can be incidental to mutations in different genes; to date, mutations have been reported in six genes (Table 1). Mutations in these genes, however, account only for a minority of cases [61] and their mean penetrance ranges from 60 % to 80 %. Hence, further studies are needed to better characterize this heterogeneous syndrome. Given the high intrafamilial variability and the overlapping features of the clinical manifestations, ADNFLE patients do not show a clear distinction from sporadic NFLE cases, except for certain ADNFLE mutations frequently associated with specific additional neurological or psychiatric symptoms (Table 1). Hence, neuropsychological testing and psychiatric assessment are mandatory in ADNFLE patients [62•].
The attempt to define a genotype–phenotype correlation is hampered by the rarity of the disease and by difference in study designs. Available data suggest that no single feature can indicate one particular form and that no correlation between functional effects of the mutations and the severity of the disease exists [29, 62•].
Symptomatic Forms of NFLE
Studies, conducted in patients with drug-resistant epilepsy, have shown that type II focal cortical dysplasia (type II FCD; Taylor-type cortical dysplasia) [63] is the most frequent etiological substrate of NFLE [64•, 65•, 66, 67]. However, although type II FCD is more frequently found within the frontal lobe, it has been shown that, regardless of its anatomical localization, type II FCD increases the risk of sleep-related epilepsy, in particular the type IIb [64•, 68]. Type IIb FCD is characterized by the presence of dysmorphic neurons, connected through an abnormal GABAergic circuit, and the so called “balloon cells” (BC). BC are not directly involved in the genesis of paroxysmal discharges, but seem to act as “bridges” between different areas of clustered neuronal hyperexcitability, enabling rapid propagation of electrical activity via their particular rearranged gap junctions [69]. During wakefulness, type IIb FCD shows a peculiar pattern of interictal activity that is characterized by the occurrence of rhythmic and subcontinuous spike- and polyspike waves with a frequency usually between 1 and 3 Hz, alternating with short bursts of fast discharges interrupted by suppression of activity. These burst of fast discharges are relatively rare and sporadic during wakefulness, but become numerous during slow wave sleep, when they tend to recur pseudo-periodically [65•, 68, 70, 71] and often spread over surrounding nonlesional areas and develop into a seizure [65•, 68]. Type IIb FCD seems to be an interesting “in vivo” model to study the relationship between sleep mechanisms and epileptogenesis, as well as a peculiar “in vitro” model for pharmacological studies.
Clinical Features of NFLE
ADNFLE and sporadic NFLE show similar clinical and electroencephalographic features [11–13, 21]. In the high majority of NFLE patients, seizures begin before the age of 20 years, with a peak during childhood [4, 11–13, 21], although onset during adulthood has been also reported [11, 12, 21]. Seizure frequency is usually high, and patients generally experience many seizures a night [12, 13], although the frequency may diminish during adulthood [11, 21]. Of note, occasional seizures during wakefulness can occur [12, 13]. Subjective seizure manifestations, uncommon during the night, have been reported by patients during these diurnal events; however, auras generally have a poor localizing and lateralizing value [13, 72].
NFLE patients may show different sleep-related motor events of increasing complexity and duration, even during a single night. These include 1) short-lasting (2–4 s) stereotyped movements involving the limbs, the axial musculature, and/or the head [11–13, 58•, 73]; 2) paroxysmal arousals (PAs), characterized by sudden and brief arousals (5–10 s in duration) sometimes accompanied by stereotyped movements, vocalization, frightened expression, and fear [11–13, 74]; and 3) major attacks, lasting 20–30 s, characterized by asymmetric tonic or dystonic posturing, or complex movements such as pelvic thrusting, pedalling, choreo-athetoid, and ballistic movements of the limbs [11–13, 21]. Some patients may show ictal deambulatory behaviors often associated with frightened expression and fear [12, 13, 51, 75]. Owing to the recurrence of nocturnal motor events, several NFLE patients may complain of nonrestorative sleep and of daytime sleepiness [12, 13, 71, 74, 76].
The increasing complexity of NFLE ictal motor behaviors, from minor to major events, reflects a different duration and propagation of the discharge within the frontal lobe [77, 78]. It has been shown that the seizure onset in patients with asymmetric tonic or dystonic posturing is generally localized in the posterior portion of the frontal cingulated gyrus and in the posterior mesial frontal cortex with a primary involvement of the supplementary motor area [13, 65•, 79, 80]. In patients with seizures characterized by hyperkinetic automatisms and complex motor behaviors, the region of seizure onset may involve the dorso-lateral and anterior frontal regions (fronto-polar and frontal antero-mesial regions) [13, 65•, 80]; in particular, the epileptic manifestations characterized by the association of fear and more organized motor behaviors (the so-called “epileptic nocturnal wandering”) seem to correspond to the activation of anterior cingulated, orbito-polar, and temporal regions (Fig. 1) [10, 13, 51, 81]. Some of these clinical manifestations have been interpreted as a release of inhibition (provoked by the seizure) on the innate behavioral automatisms and survival behaviors, programmed in cortical and subcortical central pattern generators [82, 83].
Fig. 1
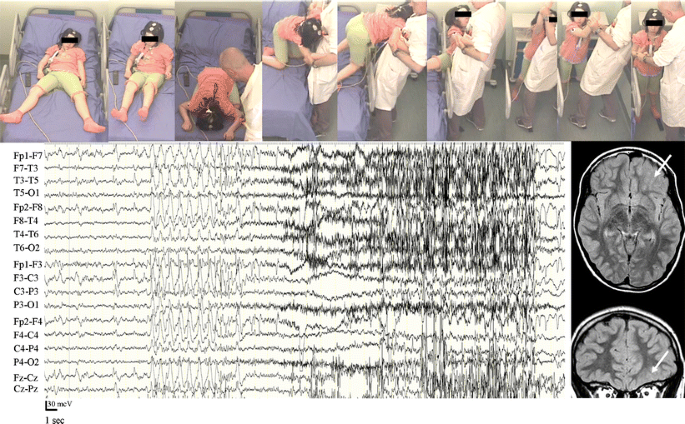
Video-electroencephalography recordings of an episode of epileptic nocturnal wandering. Magnetic resonance imaging shows the presence of a focal cortical dysplasia in the left orbital region (arrows). Postsurgical histopathologic examination revealed the presence of a type II focal cortical dysplasia (Taylor-type cortical dysplasia)
The interictal and ictal scalp electroencephalography (EEG) features of NFLE patients are often uninformative, especially in cases of seizures originating from the mesial frontal cortex [12, 78]. However, prolonged video–EEG recordings can sometimes be useful in characterizing the EEG abnormalities in selected subgroups of patients undergoing presurgical investigation [13]. Unfortunately, the reduced contribution of scalp EEG to localization and lateralization of the seizure onset zone is accompanied by a low incidence of positive findings using magnetic resonance imaging [12, 13].
In recent years it has been shown that sleep-related complex motor attacks, similar to those occurring in NFLE patients, may also originate from the temporal lobe [8, 71, 72, 81], the insular-opercular region [9, 65•, 84••, 85, 86], and the posterior cerebral regions [65•]. A careful analysis of aura features may be particularly useful in the differential diagnosis of NFLE. Indeed, these patients may report subjective manifestations that could suggest an origin of seizure outside the frontal lobe, such as epigastric, acoustic sensations, and/or déjà vu in temporal lobe onset [72]; laryngeal and throat sensations, dysarthria, hypersalivation, unpleasant or electrical paresthesiae, diffuse or restricted to a small cutaneous area, in opercular, and/or insular onset [9, 65•, 84••, 87]; and visual hallucinations in occipital seizures [65•].
Differential Diagnosis
Many of the differences between NFLE and non-epileptic sleep-related events have now been clarified, and a number of clinical features have been categorized to help physicians in the differential diagnosis (Table 2). For instance, episodes of sleep terror and sleepwalking are isolated and rare (one episode every 1–4 months), without any stereotypic complex motor patterns such as dystonic posturing, ballistic movements, tremor, OR choreoathetosis, and tend to disappear with time. REM sleep behavior disorders (RBD) can be readily differentiated from NFLE. RBD affects predominantly elderly men (around 60 years of age), and episodes prevail in the second half of the night, unlike arousal disorders and epileptic nocturnal wandering. RBD are characterized by polymorphic behavior consistent with aggressive dream content associated with the typical polygraphic finding of REM sleep without atonia. Nocturnal panic attacks are also characterized by a sudden awakening from sleep with dramatic autonomic activation and a sensation of imminent death, but these episodes often recur only once per night and are prolonged (many minutes) [89].
Table 2 Clinical features of nocturnal frontal lobe seizures (NFLS) and the most common parasomnias (modified from [88])
Nevertheless, the differential diagnosis may remain a challenge for clinicians, and some controversial points are still a matter of debate. First, the distinction between the different disorders is complicated by the similarity of behavioral patterns encountered in non-REM parasomnias, RBD, and nocturnal frontal lobe seizures (NFLS), and by the possible coexistence of these sleep disturbances in the same individual [12]. Second, there is no gold standard objective test for parasomnias or NFLE. Finally, the diagnostic potential of the conventional work flow—clinical assessment (history taking) followed by laboratory investigations, such as interictal EEG and nocturnal V-PSG—is still hampered by the lack of internationally accepted diagnostic criteria [7, 90].
Distinguishing NFLE from non-epileptic sleep-related events, namely arousal disorders, may be challenging and sometimes impossible on the basis of history-taking alone [7, 88]. A reliable semeiological description of motor events occurring during the night is often difficult to collect from a witness or sleep partner because observers may be lacking or, if present, not fully reliable or awake when attacks occur. Moreover, subjective semeiological elements are often absent in all types of motor events during sleep. To date, the available clinical diagnostic tools for nocturnal motor episodes, that is, the standard criteria, are unreliable, as in the case of several parasomnias [91], or are still lacking, as in the case of NFLE. Recent efforts to obtain a systematic assessment of the diagnostic reliability of clinical history have devised two instruments: the Frontal Lobe Epilepsy and Parasomnias scale [92] and the Structured Interview for NFLE [93•]. Albeit clinically useful, these tools are limited by contradictory diagnostic accuracy [7, 94].
Interictal EEGs fail to disclose epileptiform abnormalities in a substantial percentage of NFLE patients [12], whereas interictal epileptiform discharges may also occur in some patients with parasomnias, especially those with RBD (up to 26 %) [95]. Sleep V-PSG is unanimously considered the “gold-standard” diagnostic test, but it is expensive, with a limited availability, and does not always capture the event in a single-night recording. Moreover, even when the nocturnal episode has been recorded, the diagnosis remains doubtful because ictal scalp EEG fails to disclose epileptiform abnormalities or because the episode captured is a minor motor event, like the so-called PA, for which the diagnosis is not reliable, even among experts [6, 7]. To make video analysis of nocturnal paroxysmal events more reliable a diagnostic algorithm focusing on the semeiological features of the arousal parasomnias and NFLS has recently been proposed [96••].
Despite the limits of V-PSG, the possibility of analyzing the video of the nocturnal attack remains an important diagnostic tool, making home video recording a useful adjunct when episodes are infrequent, even if the onset of the episode is missed [88, 96••, 97]. Indeed, it has been shown that while the behavioral aspects characterizing the onset of a paroxysmal episode may be similar in NFLE and NREM parasomnias, the analysis of the evolution and the offset of the events are more critical for discriminating between parasomnias and NFLE seizures [96••]. However, although a home-made video can be very helpful in the diagnosis of major episodes (hypermotor seizures), the lack of reliable semeiological criteria for PAs and other minor motor events diminishes its diagnostic use for less complex brief attacks [6, 73, 96••, 97]. However, in patients with a high clinical suspicion of NFLE, the presence of a high number of sleep-related minor motor events might be interpreted as an indirect marker of epileptiform activity [73, 98].
Response to therapy is not an ideal diagnostic tool, but may contribute to the diagnosis, especially when V-PSG is impracticable or has proven unhelpful. Unfortunately, even the response to antiepileptic drugs does not discriminate between NFLE and parasomnias. Although carbamazepine is effective at low doses in most NFLE patients, this treatment did not modify seizure frequency at all in 32 % of patients [12]. Moreover, long-term follow-up data are lacking for both parasomnias and NFLE.
In conclusion, no single, valid, and reliable diagnostic procedure (including V-PSG) or diagnostic algorithm has been defined for parasomnias and NFLE. We need to improve the sensitivity of the available diagnostic tools, studying the use of home-made videos before or instead of hospitalization and V-PSG. It is also important to devise common criteria to develop a single classification of motor events occurring during sleep and thereby make VPSG more reliable. Finally, we need to improve our knowledge on the long-term follow-up of sleep disorders, particularly when a definite diagnosis (i.e., epileptic or non-epileptic) has not been established. An international consensus on these issues is therefore advisable [7].
Treatment
Many and early reports documented carbamazepine as the drug of choice in NFLE/ADNFLE patients [11, 12, 39, 99], and basic studies of ADNFLE mutations suggested that mutated nAChR receptors demonstrate an increased sensitivity to carbamazepine [100].
According to the literature, about two thirds of NFLE/ADNFLE patients benefit from carbamazepine administration. This drug is usually given at low doses (200–1,000 mg at bedtime), abolishing seizures in ~20 % of cases, and giving significant relief (at least 50 % seizure reduction) in another 48 %. One third of patients are drug-resistant and most of these patients have many seizures per month [12, 101].
Oxcarbamazepine (at a mean dose of 30.4 ±11.7 mg/kg/day) fully controlled nocturnal seizures in a single study on eight children between the age of 4 and 16 years with NFLE (some of whom were refractory to previous antiepileptic medications) [102].
Topiramate administered as single or add-on therapy from 50 to 300 mg daily at bedtime was found to be effective in about 90 % of cases in a small series of 24 NFLE patients. In the follow-up, ranging from 6 months to 6 years, the authors found 25 % of patients to be seizure-free and 62.5 % with a reduction in seizures of at least 50 % [103].
Varadkar et al. [104] reported a reduction or complete control of nocturnal seizures in three members of an ADNFLE family with acetazolamide, 500 mg at night, as add-on therapy to carbamazepine.
Treatment with nicotine transdermal patches had a beneficial effect on seizure frequency in a single patient with ADNFLE, whose seizures were refractory to standard antiepileptic drugs [105]. The beneficial effect of nicotine on seizure frequency was later reported in 9 out of 22 patients from two Norwegian ADNFLE pedigrees with CHRNA4 mutations [106], supporting the role of a nicotine defect in the arousal pathway in NFLE/ADNFLE patients. Recently, in a case–control family study, Naldi et al. [107] failed to find any significant difference in the distribution of tobacco use among NFLE patients and their relatives compared to that in the control group.
Surgical treatment may provide excellent results for selected drug-resistant NFLE patients both for seizures and for epilepsy-related sleep disturbances, such as fragmented sleep and excessive daytime somnolence [13, 71]. An in-depth pre-surgical evaluation, often including invasive recording by stereo EEG, is mandatory for resective surgery [13]. In a population of 21 drug-resistant NFLE patients, after at least 12 months following surgery, 76 % of patients were completely seizure-free and the remainder reported significant improvements in seizure frequency and/or intensity [13]. Surgical treatment can also be effective in patients with sleep-related complex motor seizures with an extrafrontal origin [8, 10, 65•, 72, 84••, 85, 86].
In summary, the interpretation of NFLE as relatively benign clinical entity, because seizures occur only or predominantly during sleep, is not completely true. Some patients suffering from rare and brief attacks tolerate them well and choose not to undergo therapy because they do not feel incapacitated by the seizures. However, a third of NFLE/ADNFLE patients are drug-resistant, and also in patients responding to treatment, withdrawal of the antiepileptic therapy is often followed by the reappearance of the seizures [101]. Finally, discrepancies between seizure outcome reported by patient and bed partner and V-PSG recordings can be found. The subjective effectiveness of antiepileptic treatment could be mostly related to the partial reduction of the longer and more complex seizures, opening the debate to the identification of clinical and sleep measures, which can better define a more objective response to treatment [108•].
Conclusions
NFLE is a syndrome of heterogeneous etiology characterized by the occurrence of sleep-related seizures with various motor and affective clinical manifestations, depending on the involved neuronal networks within the frontal lobe. Moreover, a significant proportion of sleep-related complex motor seizures have an extra-frontal origin, predominantly temporal and insular. NFLE is generally considered a benign clinical entity, although severe, drug-resistant forms do exist. Comprehensive clinical and instrumental investigations have enabled us to shed light on the wide spectrum of paroxysmal events occurring during sleep; however, in a small percentage of cases, the differential diagnosis between NFLE and NREM parasomnias can still be challenging. It is therefore imperative to adopt a comprehensive diagnostic approach that includes the anamnestic, V-PSG, morphologic, and genetic aspects. Finally, we believe that further studies on the relationships between genes, arousal regulatory mechanisms, and epileptogenesis using both clinical and experimental models of NFLE could provide key insight in the interrelationship between sleep and epilepsy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- Crespel A, Baldy-Moulinier M, Coubes P. The relationship between sleep and epilepsy in frontal and temporal lobe epilepsies: practical and physiopathologic considerations. Epilepsia. 1998;39:150–7.
CAS PubMed Google Scholar - Herman ST, Walczak TS, Bazil CW. Distribution of partial seizures during the sleep–wake cycle: differences by seizure onset site. Neurology. 2001;56:1453–9.
CAS PubMed Google Scholar - Zucconi M, Ferini-Strambi L. NREM parasomnias: arousal disorders and differentiation from nocturnal frontal lobe epilepsy. Clin Neurophysiol. 2000;111 Suppl 2:S129–35.
PubMed Google Scholar - Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann F, et al. Autosomal dominant frontal epilepsy misdiagnosed as sleep disorder. Lancet. 1994;343:515–7.
CAS PubMed Google Scholar - Zucconi M, Oldani A, Ferini-Strambi L, Bizzozero D, Smirne S. Nocturnal paroxysmal arousals with motor behaviors during sleep: frontal lobe epilepsy or parasomnia? Clin Neurophysiol. 1997;14:513–22.
CAS Google Scholar - Vignatelli L, Bisulli F, Provini F, Naldi I, Pittau F, Zaniboni A. Interobserver reliability of video recording in the diagnosis of nocturnal frontal lobe seizures. Epilepsia. 2007;48:1506–11.
PubMed Google Scholar - Bisulli F, Vignatelli L, Provini F, Lugaresi E, Tinuper P. Parasomnias and nocturnal frontal lobe epilepsy (NFLE): lights and shadows – controversial points in the differential diagnosis. Sleep Med. 2011;12 Suppl 2:S27–32.
PubMed Google Scholar - Nobili L, Cossu M, Mai R, Tassi L, Cardinale F, Castana L, et al. Sleep-related hyperkinetic seizures of temporal lobe origin. Neurology. 2004;62:482–5.
CAS PubMed Google Scholar - Ryvlin P, Minotti L, Demarquay G, Hirsch E, Arzimanoglou A, Hoffman D, et al. Nocturnal hypermotor seizures, suggesting frontal lobe epilepsy, can originate in the insula. Epilepsia. 2006;47:755–65.
PubMed Google Scholar - Nobili L, Francione S, Cardinale F, Lo RG. Epileptic nocturnal wanderings with a temporal lobe origin: a stereoelectroencephalographic study. Sleep. 2002;25:669–71.
PubMed Google Scholar - Oldani A, Zucconi M, Asselta R, Modugno M, Bonati MT, Dalpra L, et al. Autosomal dominant nocturnal frontal lobe epilepsy. A video-polysomnographic and genetic appraisal of 40 patients and delineation of the epileptic syndrome. Brain. 1998;121:205–23.
PubMed Google Scholar - Provini F, Plazzi G, Tinuper P, Vandi S, Lugaresi E, Montagna P. Nocturnal frontal lobe epilepsy: a clinical and polygraphic overview of 100 consecutive cases. Brain. 1999;122:1017–31.
PubMed Google Scholar - Nobili L, Francione S, Mai R, Cardinale F, Castana L, Tassi L, et al. Surgical treatment of drug-resistant nocturnal frontal lobe epilepsy. Brain. 2007;130:561–73.
CAS PubMed Google Scholar - De Marco EV, Gambardella A, Annesi F, Labate A, Carrideo S, Forabosco P, et al. Further evidence of genetic heterogeneity in families with autosomal dominant nocturnal frontal lobe epilepsy. Epilepsy Res. 2007;74:70–3.
PubMed Google Scholar - Cho YW, Motamedi GK, Laufenberg I, Sohn SI, Lim JG, Lee H, et al. A Korean kindred with autosomal dominant nocturnal frontal lobe epilepsy and mental retardation. Arch Neurol. 2003;60:1625–32.
PubMed Google Scholar - Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012;44:1188–90. This study identified a new gene, encoding a sodium-gated potassium channel subunit, causing severe autosomal dominant nocturnal frontal lobe epilepsy. The mutations were identified in families and in a sporadic case. Patients with this mutation were not only affected by severe nocturnal focal seizures, but also showed intellectual and or psychiatric disability. The identification of this new gene has important implications for genetic counselling and for the understanding of the spectrum of epilepsy disorders..
CAS PubMed Google Scholar - Thomas RH, King WH, Johnston JA, Smith PE. Awake seizures after pure sleep-related epilepsy: a systematic review and implications for driving law. J Neurol Neurosurg Psychiatry. 2010;81:130–5.
CAS PubMed Google Scholar - Derry CP, Duncan S. Sleep and epilepsy. Epilepsy Behav. 2013;26:394–404.
PubMed Google Scholar - Yaqub BA, Waheed G, Kabiraj MM. Nocturnal epilepsies in adults. Seizure. 1997;6:145–9.
CAS PubMed Google Scholar - Fernandez LB, Salas-Puig J. Pure sleep seizures: risk of seizures while awake. Epileptic Disord. 2007;9:65–70.
PubMed Google Scholar - Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann E, et al. Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain. 1995;118:61–73.
PubMed Google Scholar - Phillips HA, Scheffer IE, Berkovic SF, Hollway GE, Sutherland GR, Mulley JC. Localization of a gene for autosomal dominant nocturnal frontal lobe epilepsy to chromosome 20q 13.2. Nat Genet. 1995;10:117–8.
CAS PubMed Google Scholar - Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201–3.
CAS PubMed Google Scholar - Steinlein OK, Magnusson A, Stoodt J, Bertrand S, Weiland S, Berkovic SF, et al. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum Mol Genet. 1997;6:943–7.
CAS PubMed Google Scholar - Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J. Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+ permeability, conductance, and gating of human alpha4beta2 nicotinic acetylcholine receptors. J Neurosci. 1997;17:9035–47.
CAS PubMed Google Scholar - Weiland S, Witzemann V, Villarroel A, Propping P, Steinlein O. An amino acid exchange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett. 1996;398:91–6.
CAS PubMed Google Scholar - Saenz A, Galan J, Caloustian C, Lorenzo F, Marquez C, Rodriguez N, et al. Autosomal dominant nocturnal frontal lobe epilepsy in a Spanish family with a Ser252Phe mutation in the CHRNA4 gene. Arch Neurol. 1999;56:1004–9.
CAS PubMed Google Scholar - Steinlein OK, Stoodt J, Mulley J, Berkovic S, Scheffer IE, Brodtkorb E. Independent occurrence of the CHRNA4 Ser248Phe mutation in a Norwegian family with nocturnal frontal lobe epilepsy. Epilepsia. 2000;41:529–35.
CAS PubMed Google Scholar - McLellan A, Phillips HA, Rittey C, Kirkpatrick M, Mulley JC, Goudie D, et al. Phenotypic comparison of two Scottish families with mutations in different genes causing autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia. 2003;44:613–7.
PubMed Google Scholar - Hirose S, Iwata H, Akiyoshi H, Kobayashi K, Ito M, Wada K, et al. A novel mutation of CHRNA4 responsible for autosomal dominant nocturnal frontal lobe epilepsy. Neurology. 1999;53:1749–53.
CAS PubMed Google Scholar - Phillips HA, Marini C, Scheffer IE, Sutherland GR, Mulley JC, Berkovic SF. A de novo mutation in sporadic nocturnal frontal lobe epilepsy. Ann Neurol. 2000;48:264–7.
CAS PubMed Google Scholar - Sansoni V, Nobili L, Proserpio P, Ferini-Strambi L, Combi R. A de novo mutation in an Italian sporadic patient affected by nocturnal frontal lobe epilepsy. J Sleep Res. 2012;21:352–3.
PubMed Google Scholar - Rozycka A, Skorupska E, Kostyrko A, Trzeciak WH. Evidence for S284L mutation of the CHRNA4 in a white family with autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia. 2003;44:1113–7.
CAS PubMed Google Scholar - Ito M, Kobayashi K, Fujii T, Okuno T, Hirose S, Iwata H, et al. Electroclinical picture of autosomal dominant nocturnal frontal lobe epilepsy in a Japanese family. Epilepsia. 2000;41:52–8.
CAS PubMed Google Scholar - Leniger T, Kananura C, Hufnagel A, Bertrand S, Bertrand D, Steinlein OK. A new Chrna4 mutation with low penetrance in nocturnal frontal lobe epilepsy. Epilepsia. 2003;44:981–5.
CAS PubMed Google Scholar - Chen Y, Wu L, Fang Y, He Z, Peng B, Shen Y, et al. A novel mutation of the nicotinic acetylcholine receptor gene CHRNA4 in sporadic nocturnal frontal lobe epilepsy. Epilepsy Res. 2009;83:152–6.
CAS PubMed Google Scholar - Magnusson A, Stordal E, Brodtkorb E, Steinlein O. Schizophrenia, psychotic illness and other psychiatric symptoms in families with autosomal dominant nocturnal frontal lobe epilepsy caused by different mutations. Psychiatr Genet. 2003;13:91–5.
PubMed Google Scholar - Phillips HA, Scheffer IE, Crossland KM, Bhatia KP, Fish DR, Marsden CD, et al. Autosomal dominant nocturnal frontal-lobe epilepsy: genetic heterogeneity and evidence for a second locus at 15q24. Am J Hum Genet. 1998;63:1108–16.
CAS PubMed Central PubMed Google Scholar - Gambardella A, Annesi G, De Fusco M, Patrignani A, Aguglia U, Annesi F, et al. A new locus for autosomal dominant nocturnal frontal lobe epilepsy maps to chromosome 1. Neurology. 2000;55:1467–71.
CAS PubMed Google Scholar - De Fusco M, Becchetti A, Patrignani A, Annesi G, Gambardella A, Quattrone A, et al. The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet. 2000;26:275–6.
PubMed Google Scholar - Hoda JC, Gu W, Friedli M, Phillips HA, Bertrand S, Antonarakis SE, et al. Human nocturnal frontal lobe epilepsy: pharmocogenomic profiles of pathogenic nicotinic acetylcholine receptor beta-subunit mutations outside the ion channel pore. Mol Pharmacol. 2008;74:379–91.
CAS PubMed Google Scholar - Phillips HA, Favre I, Kirkpatrick M, Zuberi SM, Goudie D, Heron SE, et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. 2001;68:225–31.
CAS PubMed Central PubMed Google Scholar - Diaz-Otero F, Quesada M, Morales-Corraliza J, Martinez-Parra C, Gomez-Garre P, Serratosa JM. Autosomal dominant nocturnal frontal lobe epilepsy with a mutation in the CHRNB2 gene. Epilepsia. 2008;49:516–20.
CAS PubMed Google Scholar - Bertrand D, Elmslie F, Hughes E, Trounce J, Sander T, Bertrand S, et al. The CHRNB2 mutation I312M is associated with epilepsy and distinct memory deficits. Neurobiol Dis. 2005;20:799–804.
CAS PubMed Google Scholar - Cho YW, Yi SD, Lim JG, Kim DK, Motamedi GK. Autosomal dominant nocturnal frontal lobe epilepsy and mild memory impairment associated with CHRNB2 mutation I312M in the neuronal nicotinic acetylcholine receptor. Epilepsy Behav. 2008;13:361–5.
PubMed Google Scholar - Liu H, Lu C, Li Z, Zhou S, Li X, Ji L, et al. The identification of a novel mutation of nicotinic acetylcholine receptor gene CHRNB2 in a Chinese patient: Its possible implication in non-familial nocturnal frontal lobe epilepsy. Epilepsy Res. 2011;95:94–9.
CAS PubMed Google Scholar - Combi R, Ferini-Strambi L, Montruccoli A, Bianchi V, Malcovati M, Zucconi M, et al. Two new putative susceptibility loci for ADNFLE. Brain Res Bull. 2005;67:257–63.
CAS PubMed Google Scholar - Combi R, Dalpra L, Ferini-Strambi L, Tenchini ML. Frontal lobe epilepsy and mutations of the corticotropin-releasing hormone gene. Ann Neurol. 2005;58:899–904.
CAS PubMed Google Scholar - Combi R, Ferini-Strambi L, Tenchini ML. Compound heterozygosity with dominance in the Corticotropin Releasing Hormone (CRH) promoter in a case of nocturnal frontal lobe epilepsy. J Sleep Res. 2008;17:361–2.
PubMed Google Scholar - Sansoni V, Forcella M, Mozzi A, Fusi P, Ambrosini R, Ferini-Strambi L, et al. Functional characterization of a CRH missense mutation identified in an ADNFLE family. PLoS One. 2013;8:e61306. This is the first mutation described in the coding region of the CRH gene associated with ADNFLE. Previous studies have shown that CRH promotes wakefulness and impairs sleep in a dose-dependent way, and may increase seizures susceptibility. Individuals with an altered CRH level could be more prone to developing the disease..
CAS PubMed Central PubMed Google Scholar - Aridon P, Marini C, Di Resta C, Brilli E, De Fusco M, Politi F, et al. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am J Hum Genet. 2006;79:342–50.
CAS PubMed Central PubMed Google Scholar - Picard F, Bruel D, Servent D, Saba W, Fruchart-Gaillard C, Schollhorn-Peyronneau MA, et al. Alteration of the in vivo nicotinic receptor density in ADNFLE patients: a PET study. Brain. 2006;129:2047–60.
CAS PubMed Google Scholar - Lena C, Popa D, Grailhe R, Escourrou P, Changeux JP, Adrien J. Beta2-containing nicotinic receptors contribute to the organization of sleep and regulate putative micro-arousals in mice. J Neurosci. 2004;24:5711–8.
CAS PubMed Google Scholar - Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A. 2006;103:19152–7.
CAS PubMed Central PubMed Google Scholar - Xu J, Cohen BN, Zhu Y, Dziewczapolski G, Panda S, Lester HA, et al. Altered activity-rest patterns in mice with a human autosomal-dominant nocturnal frontal lobe epilepsy mutation in the beta2 nicotinic receptor. Mol Psychiatry. 2011;16:1048–61. This experimental study shows that mice with a mutation in the beta2 nicotinic receptor may have an alteration of their sleep pattern, independent of the presence of epileptic activity. The study underlines the role of nicotinic receptors in the sleep regulation and has important implications for the interpretation of the pathophysiological mechanisms of ADNFLE..
CAS PubMed Central PubMed Google Scholar - Parrino L, De Paolis F, Milioli G, Gioi G, Grassi A, Riccardi S, et al. Distinctive polysomnographic traits in nocturnal frontal lobe epilepsy. Epilepsia. 2012;53:1178–84.
PubMed Google Scholar - Terzaghi M, Sartori I, Mai R, Tassi L, Francione S, Cardinale F, et al. Coupling of minor motor events and epileptiform discharges with arousal fluctuations in NFLE. Epilepsia. 2008;49:670–6.
PubMed Google Scholar - Halasz P, Kelemen A, Szucs A. The role of NREM sleep micro-arousals in absence epilepsy and in nocturnal frontal lobe epilepsy. Epilepsy Res. 2013;107:9–19. This is a systematic review on the role of NREM sleep-related phasic events, micro-arousals, in two major idiopathic epilepsies. Taking into account both experimental and clinical findings, the authors discuss the role of arousal fluctuations in NFLE and the relationships between cholinergic alterations, thalamocortical oscillations, and seizures..
PubMed Google Scholar - Bisulli F, Vignatelli L, Naldi I, Licchetta L, Provini F, Plazzi G, et al. Increased frequency of arousal parasomnias in families with nocturnal frontal lobe epilepsy: a common mechanism? Epilepsia. 2010;51:1852–60. This details a case-control family study that recruited 33 NFLE probands, 200 relatives of probands, 31 controls, and 194 control relatives. The authors found an increased prevalence of arousal disorders (odds ratio 4.7) and nightmares (odds ratio 2.6) among NFLE proband relatives. The study suggests a possible common pathophysiological mechanism (based on an alteration of the cholinergic system) in parasomnias and NFLE..
PubMed Google Scholar - Ishida S, Picard F, Rudolf G, Noe E, Achaz G, Thomas P, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet. 2013;45:552–5. This study shows that loss-of-function mutations in DEPDC5 are a major cause of a broad spectrum of autosomal dominant focal epilepsies with different brain localization and electroclinical expression, including ADNFLE, familial temporal lobe epilepsy, and familial focal epilepsy with variable foci. The study indicates that the seizure initiation sites may be dissociated from underlying genetic mechanisms..
CAS PubMed Google Scholar - Heron SE, Scheffer IE, Berkovic SF, Dibbens LM, Mulley JC. Channelopathies in idiopathic epilepsy. Neurotherapeutics. 2007;4:295–304.
CAS PubMed Google Scholar - Steinlein OK, Hoda JC, Bertrand S, Bertrand D. Mutations in familial nocturnal frontal lobe epilepsy might be associated with distinct neurological phenotypes. Seizure. 2012;21:118–23. This study shows that certain ADNFLE mutations might be associated with an increased risk of cognitive deficits, mental retardation, and psychiatric symptoms. These results suggest that a neuropsychological evaluation should be included in the clinical assessment of patients with ADNFLE..
PubMed Google Scholar - Blumcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52:158–74.
PubMed Central PubMed Google Scholar - Nobili L, Cardinale F, Magliola U, Cicolin A, Didato G, Bramerio M, et al. Taylor's focal cortical dysplasia increases the risk of sleep-related epilepsy. Epilepsia. 2009;50:2599–604. This study evaluated the relative roles of the topography of the epileptogenic zone and of the etiologic substrate as risk factors for sleep-related focal epilepsy. The analysis of the clinical and histopathologic features of 303 drug-resistant patients seizure-free since surgery revealed that one of every two patients with a Taylor-type cortical dysplasia, independent of its location, had a sleep-related focal epilepsy. The presence of Taylor-type cortical dysplasia increases the risk of sleep-related focal epilepsy 14-fold..
PubMed Google Scholar - Proserpio P, Cossu M, Francione S, Gozzo F, Lo Russo G, Mai R, et al. Epileptic motor behaviors during sleep: anatomo-electro-clinical features. Sleep Med. 2011;12 Suppl 2:S33–8. This study included 40 patients with drug-resistant sleep-related focal epilepsy submitted to Stereo-Electroencephalography investigation and seizure-free after surgical treatment. The study points out that a significant proportion (30%) of sleep-related complex motor seizures may originate outside the frontal lobe. These results are clinically relevant when considering drug-resistant epileptic patients in whom a surgical approach could be an effective treatment..
PubMed Google Scholar - Chassoux F, Landre E, Mellerio C, Turak B, Mann MW, Daumas-Duport C, et al. Type II focal cortical dysplasia: electroclinical phenotype and surgical outcome related to imaging. Epilepsia. 2012;53:349–58.
PubMed Google Scholar - Noli D, Bartuluchi M, Gonzalez FS, Kaltenmeier MC, Cersosimo R, Rugilo C, et al. Type II focal cortical dysplasia: electroclinical study and surgical outcome in 31 pediatric patients. Childs Nerv Syst. 2013;29:2079–87.
PubMed Google Scholar - Tassi L, Garbelli R, Colombo N, Bramerio M, Russo GL, Mai R, et al. Electroclinical, MRI and surgical outcomes in 100 epileptic patients with type II FCD. Epileptic Disord. 2012;14:257–66.
PubMed Google Scholar - Garbelli R, Frassoni C, Condorelli DF, Trovato Salinaro A, Musso N, Medici V, et al. Expression of connexin 43 in the human epileptic and drug-resistant cerebral cortex. Neurology. 2011;76:895–902.
CAS PubMed Google Scholar - Chassoux F, Devaux B, Landre E, Turak B, Nataf F, Varlet P, et al. Stereoelectroencephalography in focal cortical dysplasia: a 3D approach to delineating the dysplastic cortex. Brain. 2000;123:1733–51.
PubMed Google Scholar - Nobili L, Sartori I, Terzaghi M, Stefano F, Mai R, Tassi L, et al. Relationship of epileptic discharges to arousal instability and periodic leg movements in a case of nocturnal frontal lobe epilepsy: a stereo-EEG study. Sleep. 2006;29:701–4.
PubMed Google Scholar - Mai R, Sartori I, Francione S, Tassi L, Castana L, Cardinale F, et al. Sleep-related hyperkinetic seizures: always a frontal onset? Neurol Sci. 2005;26 Suppl 3:s220–4.
PubMed Google Scholar - Terzaghi M, Sartori I, Mai R, Tassi L, Francione S, Cardinale F, et al. Sleep-related minor motor events in nocturnal frontal lobe epilepsy. Epilepsia. 2007;48:335–41.
PubMed Google Scholar - Peled R, Lavie P. Paroxysmal awakenings from sleep associated with excessive daytime somnolence: a form of nocturnal epilepsy. Neurology. 1986;36:95–8.
CAS PubMed Google Scholar - Plazzi G, Tinuper P, Montagna P, Provini F, Lugaresi E. Epileptic nocturnal wanderings. Sleep. 1995;18:749–56.
CAS PubMed Google Scholar - Zucconi M, Oldani A, Smirne S, Ferini-Strambi L. The macrostructure and microstructure of sleep in patients with autosomal dominant nocturnal frontal lobe epilepsy. J Clin Neurophysiol. 2000;17:77–86.
CAS PubMed Google Scholar - Montagna P. Nocturnal paroxysmal dystonia and nocturnal wandering. Neurology. 1992;42(7 Suppl 6):61–7.
CAS PubMed Google Scholar - Nobili L, Francione S, Mai R, Tassi L, Cardinale F, Castana L, et al. Nocturnal frontal lobe epilepsy: intracerebral recordings of paroxysmal motor attacks with increasing complexity. Sleep. 2003;26:883–6.
PubMed Google Scholar - Morris 3rd HH, Dinner DS, Luders H, Wyllie E, Kramer R. Supplementary motor seizures: clinical and electroencephalographic findings. Neurology. 1988;38:1075–82.
PubMed Google Scholar - Rheims S, Ryvlin P, Scherer C, Minotti L, Hoffmann D, Guenot M, et al. Analysis of clinical patterns and underlying epileptogenic zones of hypermotor seizures. Epilepsia. 2008;49:2030–40.
PubMed Google Scholar - Vaugier L, Aubert S, McGonigal A, Trebuchon A, Guye M, Gavaret M, et al. Neural networks underlying hyperkinetic seizures of "temporal lobe" origin. Epilepsy Res. 2009;86:200–8.
CAS PubMed Google Scholar - Tassinari CA, Rubboli G, Gardella E, Cantalupo G, Calandra-Buonaura G, Vedovello M, et al. Central pattern generators for a common semiology in fronto-limbic seizures and in parasomnias. A neuroethologic approach. Neurol Sci. 2005;26 Suppl 3:s225–32.
PubMed Google Scholar - Tassinari CA, Cantalupo G, Hogl B, Cortelli P, Tassi L, Francione S, et al. Neuroethological approach to frontolimbic epileptic seizures and parasomnias: the same central pattern generators for the same behaviours. Rev Neurol (Paris). 2009;165:762–8.
CAS Google Scholar - Proserpio P, Cossu M, Francione S, Tassi L, Mai R, Didato G, et al. Insular-opercular seizures manifesting with sleep-related paroxysmal motor behaviors: a stereo-EEG study. Epilepsia. 2011;52:1781–91. These data reinforce the concept that sleep-related complex motor seizures can originate outside the frontal lobe and give helpful electroclinical information to distinguish patients with sleep-related insular–opercular seizures. Moreover, this study indicates that intracranial EEG in the insula can be performed safely, and surgical treatment may represent a highly effective treatment option in these drug-resistant patients..
PubMed Google Scholar - Kaido T, Otsuki T, Nakama H, Kaneko Y, Kubota Y, Sugai K, et al. Complex behavioral automatism arising from insular cortex. Epilepsy Behav. 2006;8:315–9.
PubMed Google Scholar - Dobesberger J, Ortler M, Unterberger I, Walser G, Falkenstetter T, Bodner T, et al. Successful surgical treatment of insular epilepsy with nocturnal hypermotor seizures. Epilepsia. 2008;49:159–62.
PubMed Google Scholar - Isnard J, Guenot M, Sindou M, Mauguiere F. Clinical manifestations of insular lobe seizures: a stereo-electroencephalographic study. Epilepsia. 2004;45:1079–90.
PubMed Google Scholar - Tinuper P, Provini F, Bisulli F, Vignatelli L, Plazzi G, Vetrugno R, et al. Movement disorders in sleep: guidelines for differentiating epileptic from non-epileptic motor phenomena arising from sleep. Sleep Med Rev. 2007;11:255–67.
PubMed Google Scholar - Plazzi G, Montagna P, Provini F, Tinuper P, Lugaresi E. Sudden arousals from slow-wave sleep and panic disorder. Sleep. 1998;21:548.
CAS PubMed Google Scholar - ASDA. American Academy of Sleep Medicine: The international classification of sleep disorders: diagnostic and coding manual. 2nd ed. Westchester: American Academy of Sleep Medicine; 2005.
Google Scholar - Vignatelli L, Bisulli F, Zaniboni A, Naldi I, Fares JE, Provini F, et al. Interobserver reliability of ICSD-R minimal diagnostic criteria for the parasomnias. J Neurol. 2005;252:712–7.
PubMed Google Scholar - Derry CP, Davey M, Johns M, Kron K, Glencross D, Marini C, et al. Distinguishing sleep disorders from seizures: diagnosing bumps in the night. Arch Neurol. 2006;63:705–9.
PubMed Google Scholar - Bisulli F, Vignatelli L, Naldi I, Pittau F, Provini F, Plazzi G, et al. Diagnostic accuracy of a structured interview for nocturnal frontal lobe epilepsy (SINFLE): a proposal for developing diagnostic criteria. Sleep Med. 2012;13:81–7. The objective of this investigation was to measure the accuracy of anamnestic features collected during clinical history for the diagnosis of NFLE. The authors conducted a a case-control diagnostic study including a case group of NFLE and a control group of people with sleep disorders potentially confounding for NFLE. The study disclosed two major anamnestic patterns (dystonic posturing and hyperkinetic behaviors) and four minor features (duration < 2 mins, unstructured vocalization during the episode, experience of an aura preceding the motor attack, and a history of tonic–clonic seizures during sleep) with unsatisfactory sensitivity, but high specificity..
PubMed Google Scholar - Manni R, Terzaghi M, Repetto A. The FLEP scale in diagnosing nocturnal frontal lobe epilepsy, NREM and REM parasomnias: data from a tertiary sleep and epilepsy unit. Epilepsia. 2008;49:1581–5.
PubMed Google Scholar - Manni R, Terzaghi M, Zambrelli E. REM sleep behavior disorder and epileptic phenomena: clinical aspects of the comorbidity. Epilepsia. 2006;47 Suppl 5:78–81.
PubMed Google Scholar - Derry CP, Harvey AS, Walker MC, Duncan JS, Berkovic SF. NREM arousal parasomnias and their distinction from nocturnal frontal lobe epilepsy: a video EEG analysis. Sleep. 2009;32:1637–44. This study analyzed a video–EEG monitoring series focusing on the semiological features of the arousal parasomnias and NFLS with the aim of making video analysis of nocturnal paroxysmal events more reliable. Dystonic posturing and hyperkinetic automatisms (e.g., kicking, rocking, or cycling movements) were found to be specific for NFLS. In the absence of these major features the presence of verbal interaction and failure to achieve full arousal after the event strongly suggest a diagnosis of parasomnia. However, other motor patterns, mainly characterized by brief motor manifestations or complex fearful behaviors, like sitting, standing, or walking, did not discriminate between the two conditions..
PubMed Google Scholar - Nobili L. Can homemade video recording become more than a screening tool? Sleep. 2009;32:1544–5.
PubMed Google Scholar - Nobili L. Nocturnal frontal lobe epilepsy and non-rapid eye movement sleep parasomnias: differences and similarities. Sleep Med Rev. 2007;11:251–4.
PubMed Google Scholar - Lugaresi E, Cirignotta F. Hypnogenic paroxysmal dystonia: epileptic seizure or a new syndrome? Sleep. 1981;4:129–38.
CAS PubMed Google Scholar - Picard F, Bertrand S, Steinlein OK, Bertrand D. Mutated nicotinic receptors responsible for autosomal dominant nocturnal frontal lobe epilepsy are more sensitive to carbamazepine. Epilepsia. 1999;40:1198–209.
CAS PubMed Google Scholar - Provini F, Plazzi G, Montagna P, Lugaresi E. The wide clinical spectrum of nocturnal frontal lobe epilepsy. Sleep Med Rev. 2000;4:375–86.
PubMed Google Scholar - Raju GP, Sarco DP, Poduri A, Riviello JJ, Bergin AM, Takeoka M. Oxcarbazepine in children with nocturnal frontal-lobe epilepsy. Pediatr Neurol. 2007;37:345–9.
PubMed Google Scholar - Oldani A, Manconi M, Zucconi M, Martinelli C, Ferini-Strambi L. Topiramate treatment for nocturnal frontal lobe epilepsy. Seizure. 2006;15:649–52.
PubMed Google Scholar - Varadkar S, Duncan JS, Cross JH. Acetazolamide and autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia. 2003;44:986–7.
PubMed Google Scholar - Willoughby JO, Pope KJ, Eaton V. Nicotine as an antiepileptic agent in ADNFLE: an N-of-one study. Epilepsia. 2003;44:1238–40.
CAS PubMed Google Scholar - Brodtkorb E, Picard F. Tobacco habits modulate autosomal dominant nocturnal frontal lobe epilepsy. Epilepsy Behav. 2006;9:515–20.
PubMed Google Scholar - Naldi I, Bisulli F, Vignatelli L, Licchetta L, Pittau F, Di Vito L, et al. Tobacco habits in nocturnal frontal lobe epilepsy. Epilepsy Behav. 2013;26:114–7.
PubMed Google Scholar - De Paolis F, Colizzi E, Milioli G, Grassi A, Riccardi S, Puligheddu M, et al. Effects of antiepileptic treatment on sleep and seizures in nocturnal frontal lobe epilepsy. Sleep Med. 2013;14:597–604. In this study the authors report the effects of antiepileptic drug treatment on both sleep macro- and microstructure, and on video-PSG-recorded seizures in a group of NFLE patients. A significant reduction of major seizures was observed; however, the authors also report the persistence of a certain number of minor seizures and a high sleep instability. The authors suggest a peculiar sleep microstructure instability as a possible trait marker of NFLE..
PubMed Google Scholar
Acknowledgments
We thank Dr. Steve Gibbs for insightful discussions.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Lino Nobili, Paola Proserpio, Romina Combi, Federica Provini, Laura Tassi, and Paolo Tinuper declare that they have no conflict of interest.
Giuseppe Plazzi has received financial compensation for board membership for UCB Pharma and Jazz Pharmaceuticals.
Francesca Bisulli has received grants from the Italian Ministry of Health.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
- Centre for Epilepsy Surgery, Niguarda Hospital, Piazza Ospedale Maggiore 3, 20154, Milan, Italy
Lino Nobili, Paola Proserpio & Laura Tassi - Centre of Sleep Medicine, Niguarda Hospital, Milan, Italy
Lino Nobili & Paola Proserpio - Institute of Bioimaging and Molecular Physiology, National Research Council, Milan, Italy
Lino Nobili - Department of Biotechnology and Biosciences, University of Milano-Bicocca, Milan, Italy
Romina Combi - IRCCS Institute of Neurological Sciences of Bologna, Bologna, Italy
Federica Provini, Giuseppe Plazzi, Francesca Bisulli & Paolo Tinuper - Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy
Federica Provini, Giuseppe Plazzi, Francesca Bisulli & Paolo Tinuper
Authors
- Lino Nobili
- Paola Proserpio
- Romina Combi
- Federica Provini
- Giuseppe Plazzi
- Francesca Bisulli
- Laura Tassi
- Paolo Tinuper
Corresponding author
Correspondence toLino Nobili.
Additional information
This article is part of the Topical Collection on Sleep
Rights and permissions
About this article
Cite this article
Nobili, L., Proserpio, P., Combi, R. et al. Nocturnal Frontal Lobe Epilepsy.Curr Neurol Neurosci Rep 14, 424 (2014). https://doi.org/10.1007/s11910-013-0424-6
- Published: 07 January 2014
- DOI: https://doi.org/10.1007/s11910-013-0424-6