TRPC channel-mediated neuroprotection by PDGF involves Pyk2/ERK/CREB pathway (original) (raw)
Main
Human immunodeficiency virus (HIV) infection selectively targets the basal ganglia region of the brain resulting in loss of dopaminergic neurons, that have been implicated in the clinical manifestation of HIV-1-associated dementia (HAD).1, 2 Accordingly, mounting evidence on the involvement of dopamine impairment in HAD comes from case studies on pathological specimens of HIV-infected patients demonstrating loss of dopaminergic neurons.1 Furthermore, experimental studies have also indicated that nigrostriatal neurons in the basal ganglia are susceptible to HIV-1 protein-mediated toxicity.3 As there is no evidence of direct infection of neurons by HIV-1, it is hypothesized that neuronal cell death is a consequence of the toxic effects of viral and cellular neurotoxins that are released from virus-infected and/or activated cells.4 One of the potent viral toxins implicated in neuronal injury/death is the virus transactivator protein, HIV-1 Tat that can be secreted from infected cells and can be taken up by the neighboring non-infected cells, including neurons.5 Indeed, HIV-1 Tat is known to inhibit tyrosine hydroxylase (TH) gene expression in dopaminergic neuronal cells thereby contributing to motor abnormalities observed in HAD patients.3, 6
Neuronal homeostasis is maintained by the fine balance between neurotrophic versus neurotoxic factors. Various neurotrophic factors, such as brain-derived neurotrophic factor, nerve growth factor, and glial cell line-derived neurotrophic factor have been implicated in the protection of neurons against neurotoxins.7, 8, 9 In this study, we explored the role of yet another neurotrophic factor, platelet-derived growth factor (PDGF) that has been documented to be critical for the development of brains of postnatal rats.10 PDGF belongs to a family of five dimeric ligands (PDGF-AA, -AB, -BB, -CC, and -DD) assembled from four gene products (PDGF-A-D) that act via two classical receptors tyrosine kinases (RTKs), PDGF-α receptor (PDGF-_α_R), and PDGF-β receptor (PDGF-_β_R). PDGF-_β_R has been implicated in neuroprotection following ischemic events in the rat.11 Members of the PDGF family have multiple roles during embryogenesis and in a variety of pathological situations in the adult.12 Among these members, PDGF has been shown to protect primary hippocampal neurons against glutamate-induced neuronal damage.13 In our previous findings we have reported that PDGF can exert neuroprotection against the HIV envelope protein-mediated toxicity in SH-SY5Y cells.14, 15 PDGF-_β_R is known to be expressed in 90% of TH-positive neurons and has been shown to be involved in TH gene expression and neuronal survival.16 However, whether PDGF can regulate dopaminergic neurons against HIV protein toxicity remains to be explored.
Neurotrophic family of growth factors is known to be essential for the survival of neurons.7 The mechanism of action of the neurotrophins on neuron survival is mediated by the extracellular signal-regulated protein kinase (ERK)/cAMP-response element-binding protein (CREB) and phosphoinositide-3-kinase (PI3K)/Akt pathways. In addition, receptors for the neurotrophins, RTKs, that can stimulate PLC-γ to activate transient receptor potential canonical channels (TRPC), have also been shown to have a role in neuroprotection.17 Recent studies have revealed these TRPC channels to be Ca2+-permeable, nonselective cation channels.17 Transient receptor potential (TRP) channels are formed by homomeric or heteromeric complexes of TRP proteins that constitute at least six subfamilies: TRPC (TRP-canonical), TRP-vanilloid, TRPM (TRP-melastatin), TRP-mucolipins, TRPPs (TRP-polycystins), and TRPA1 (TRP-ankyrin transmembrane protien1).18, 19, 20 These channels are important in various physiological processes ranging from sensation to male fertility. PDGF receptor belongs to RTKs, and the mammalian TRPC channels can be activated by RTKs.18, 21, 22 We thus sought to examine whether TRPC channels were involved in regulating neuronal protection mediated by PDGF and, if so, what were the downstream effectors for TRPC channel activation.
In this study we show direct evidence of the neuroprotective action of PDGF against HIV Tat toxicity through two distinct signaling pathways-TRPC/Ca2+/ERK and PI3K/Akt. Our data support the possibility that PDGF signaling in the midbrain neurons may contribute to the maintenance of neuronal survival.
Results
PDGF attenuates HIV-1 Tat cytotoxicity in rat primary midbrain neurons
To examine whether PDGF could abrogate Tat cytotoxicity, rat primary midbrain neurons were first exposed to HIV Tat protein (14 nM) for 24 h and cell viability assessed by [3(4,5-dimethylthiazol-2-yl)-2.5 diphenyltetrazolium bromide] (MTT) assay. As shown in Figure 1a, Tat exposure resulted in a decrease in cell viability (32%; P<0.001). Treatment of neuronal cells with either the heat-inactivated or mutant Tat, however, did not exert any cytotoxicity in these cells. We next sought to examine the neuroprotective effect of PDGF against Tat toxicity. Rat neurons were pre-treated with PDGF (20 ng/ml; concentration pre-determined based upon our previous work 14, 15) for 30 min followed by exposure of cells to Tat protein (14 nM). Twenty-four hours later cell viability was measured using the MTT assay. As shown in Figure 1b, pre-treatment of cells with PDGF followed by exposure to HIV Tat resulted in increase in cell viability. PDGF alone did increase cell viability, although this effect was not statistically significant compared with control. These findings were also confirmed by an immunostaining assay using the anti-microtubule-associated protein-2 (MAP-2) antibody. As shown in Figures 1c and d there was loss of neuronal processes as well as MAP-2 immunoreactivity in the Tat-treated group, an effect that was ameliorated by pre-treatment of cells with PDGF. Cells cultured in the presence of PDGF alone did not demonstrate any change in dendrite length compared with the untreated control cells.
Figure 1
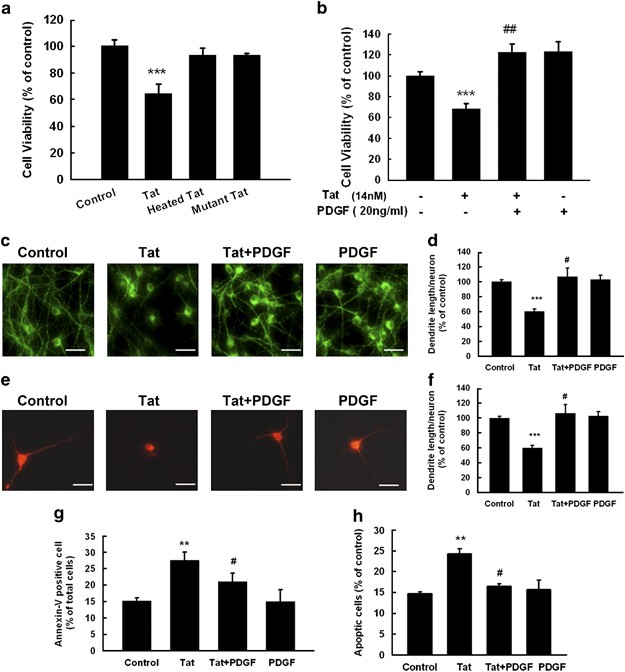
PDGF exerts neuroprotection against Tat toxicity. (a) Effect of HIV Tat on the cell survival of rat primary midbrain neurons using the MTT assay. (b) PDGF protects rat primary midbrain neurons against Tat neurotoxicity using the MTT assay. (c) Immunostaining of rat primary midbrain neurons treated with Tat and/or PDGF for 24 h with anti-MAP-2 antibody. Scale bar: 50 _μ_m. (d) Densitometric scan of neuritis (from panel c) expressed as a ratio of neurite length/neuron. (e) Immunostaining of rat primary midbrain neurons treated with Tat and/or PDGF using anti-TH-Ab. Scale bar: 50 _μ_m. (f) Densitometric scan of neuritis (from panel e) using the same method as in (d). Rat primary midbrain neurons treated with Tat and/or PDGF were stained for apoptosis marker Annexin V-FITC using flow cytometric analysis. (g) Hoechst 33342 staining monitored by fluorescence microscopy. (h) Quantification of Annexin V-positive (g) and Hoechst-positive (h) cells as the percentage of total cells. All the data in these figures are presented as mean±S.E.M. of four individual experiments. **P<0.01; ***P<0.001 versus control group; #P<0.05; ##P<0.01 versus Tat (14 nM) group
In addition to MAP-2 staining the neuroprotective effects of PDGF were further corroborated by morphological analysis of dopaminergic neurons following immunocytochemical staining of the cells with TH, a characteristic marker of these neurons. As shown in Figure 1e and f, treatment of neuronal cells with HIV-1 Tat resulted in ∼40% decrease in the total dendrite length per TH-immunoreactive neuron, pre-treatment of neuronal cultures with PDGF resulted in enhanced TH-immunoreactivity (red fluorescence).
As HIV Tat mediates neurotoxicity by the programmed cell death pathway, we next sought to examine whether PDGF-mediated protection of neurons involved inhibition of Tat-induced neuronal apoptosis using Annexin-V binding and propidium iodide uptake. As shown in Figure 1g, there was an increase in Annexin-V-positive cells (27.5 versus 15.2%) in Tat-treated cultures compared with control cells as evidenced by flow cytometry. In contrast, PDGF pre-treatment before Tat exposure resulted in decreased numbers of apoptotic cells (27.5 versus 20.9% – Tat treated versus PDGF/Tat). The above findings were further confirmed by staining cells with Hoechst 33342, a nuclear dye that stains nuclei. Pre-treatment of neurons with PDGF reduced Tat-induced neuronal apoptosis as quantified in Figure 1h.
PDGF receptor is necessary for PDGF-mediated neuroprotection
As PDGF mediates signaling through its cognate receptor PDGF-_β_R, we next sought to examine the expression and phosphorylation pattern of this receptor in rat primary neurons. As shown in Figure 2a, rat primary midbrain neurons expressed PDGF-_β_R, which was rapidly phosphorylated by treatment of neurons with PDGF as evidenced by western blot analysis. Furthermore, pre-treatment of cells with the PDGF receptor antagonist STI-571 (1 μ M) for 1 h abolished this effect.
Figure 2
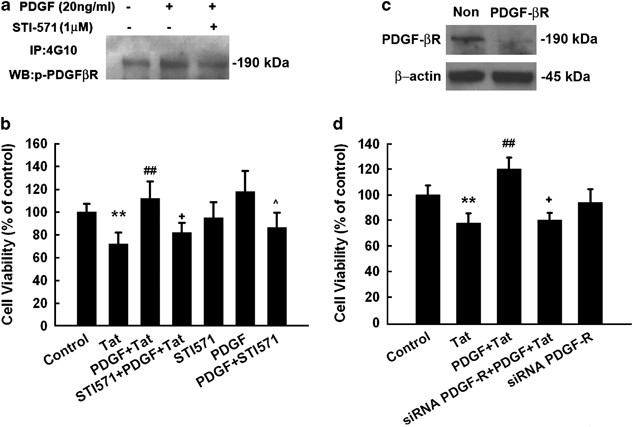
PDGF-_β_R is critical for PDGF-mediated neuroprotection. PDGF receptor antagonist blocked PDGF-β_R phosphorylation induced by PDGF as determined by coimmunoprecipitation (a) and also significantly inhibited PDGF-mediated neuroprotection (b). The data are presented as mean±S.E.M. of four individual experiments. **P<0.01 versus control group; ##P< 0.01 versus Tat-treated group; +P<0.05 versus both PDGF and Tat-treated group; ^_P<0.05 versus both PDGF alone-treated group. (c) Western blot analysis of whole cell lysates from rat primary midbrain neurons transfected with siRNAs against PDGF-_β_R or nonsense (Non) siRNA, using antibodies specific for PDGF-_β_R. Data are representative of three independent experiments. (d) Transfection of rat primary midbrain neurons with siRNAs specific for PDGF-_β_R or nonsense (Non) siRNA abolished PDGF-mediated neuroprotection. The data are presented as mean±S.E.M. of four individual experiments. **P<0.01 versus control group; ##P< 0.01 versus Tat-treated group; +P<0.05 versus both PDGF and Tat-treated group
We next assessed whether the protective effects of PDGF were mediated via its binding to its cognate PDGF-_β_R. As shown in Figure 2b receptor tyrosine kinase antagonist STI-571 was able to inhibit PDGF-mediated neuroprotection in rat primary neurons, although not to that of levels with HIV Tat alone. Exposure of neurons to PDGF alone did increase cell viability, although this effect was not statistically significant. In the presence of STI-571, PDGF-mediated increase in cell viability was significantly inhibited. We acknowledge the fact that STI-571 is not a specific antagonist for PDGF-_β_R, because it is also known to inhibit, PDGF-_α_R, as well as other kinase activities. Hence as an alternative approach we next sought to knock down PDGF-_β_R expression in neurons using the short-interfering (si) RNA strategy. As shown in Figure 2c, PDGF-_β_R short-interfering RNAs (siRNA) abrogated the expression of PDGF-_β_R in neurons, whereas also blocking the neuroprotection mediated by PDGF (Figure 2d), thus confirming the role of PDGF-_β_R in this process.
TRPC channels are required for PDGF-mediated neuroprotection
Because Ca2+ is known to mediate cell survival and TRPC is a member of TRP superfamily that functions as Ca2+ influx channels, it is possible that Ca2+ influx through TRPC channels is required for the neuroprotection mediated by PDGF in primary midbrain neurons. To test this hypothesis, cells were pre-treated with TRP channel blocker and assessed for PDGF-mediated protection against Tat toxicity. As shown in Figure 3a, in neurons pre-treated with the SKF96365 (20 _μ_M), a nonspecific inhibitor of store-operated Ca2+ entry and TRP channels,23, 24 PDGF-mediated neuroprotection against Tat toxicity was inhibited compared with cells not treated with the inhibitor.
Figure 3
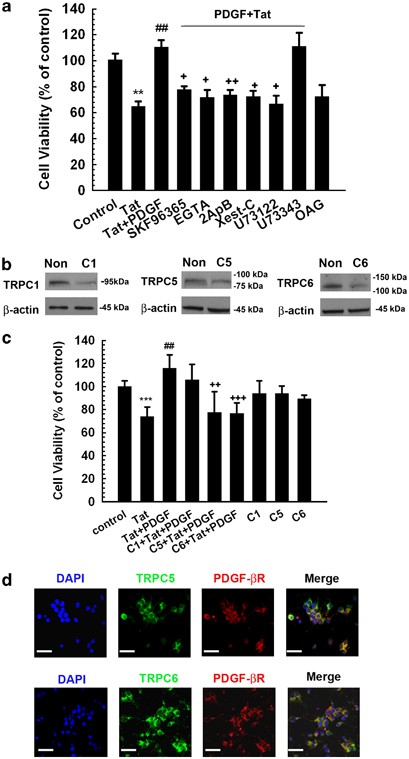
TRPC channels are required for PDGF-mediated neuroprotection. (a) Cell viability of rat primary neurons exposed to Tat and/or PDGF in the absence or presence of indicated drugs. (SKF96365: 20 μ M; EGTA: 2 mM; 2ApB: 100 μ M; Xest-C: 1 μ M; U73122: 1 μ M; U73343: 1 μ M; OAG: 50 μ M). All the data in these figures are presented as mean±S.E.M. of four individual experiments. **P<0.01 versus control group; ##P<0.01 versus Tat-treated group; +P<0.05; ++P<0.01 versus both PDGF and Tat-treated group. (b) Western blot analysis of whole cell lysates from rat primary midbrain neurons transfected with siRNAs against TRPC1, 5, 6 or nonsense (Non) siRNA using antibodies specific for either TRPC1, 5 or 6. Data are representative of three independent experiments. (c) Rat primary neurons transfected with TRPC1, 5 or 6 siRNAs were monitored for PDGF-mediated cell viability. The data are presented as mean±S.E.M. of four individual experiments. ***P<0.001 versus control group; ##P< 0.01 versus Tat-treated group; ++P<0.01, +++P<0.001 versus both PDGF and Tat-treated group. (d) Co-localization of TRPC5/6 and PDGF-_β_R in rat primary midbrain neuronal cultures. Midbrain neurons grown for 10 days were double-stained for TRPC5/6 (green) and PDGF-_β_R (red). DAPI was the nuclei counter stain (blue). Confocal images were taken at a magnification of × 60 (objective lens) with an Eclipse C1 Plus confocal microscope (Nikon). Scale bar: 50 _μ_m
In our previous study we have demonstrated that rat primary midbrain neurons express TRPC1, 5 and 6 protein.25 To ascertain the TRPC subtype(s) critical for PDGF-mediated neuroprotection, each of the TRPC subtypes (1, 5 or 6) was individually downregulated using the specific siRNAs, followed by assessment of PDGF-induced neuroprotection. As shown in Figure 3b siRNA against TRPC1, 5 and 6 suppressed expression of TRPC 1, 5 and 6, respectively. Interestingly however, TRPC 5 and 6 siRNAs, but not TRPC 1 siRNA alleviated PDGF-mediated neuroprotection (Figure 3c). These findings thus underpin the roles of TRPC 5 & 6 in neuroprotection exerted by PDGF. To further examine the role of TRPC 5 and 6 in PDGF-mediated neuroprotection, we next sought to determine whether TRPC5 and TRPC 6 co-localized with PDGF-_β_R in primary midbrain neurons. As shown in Figure 3d, both the TRPC channels co-localized with the PDGF- _β_R in these cells, thus further validating the role of these channels.
PDGF activates its receptor and stimulates phospholipase C (PLC), resulting in inositol triphosphate (IP3)-dependent release of Ca2+ from intracellular stores and Ca2+ influx from extracellular sources.26 Interference of the PLC-IP3 pathway thus ought to suppress the protection exerted by PDGF. As expected, pre-treatment of neurons with the PLC inhibitor U73122 (1 μ M) did indeed abolish the neuroprotective action of PDGF against Tat toxicity, whereas its inactive analog U73343 (1 μ M) did not exert this effect. The neuroprotective effect of PDGF also depended on IP3R activation, because exposure of neurons to antagonists specific for IP3 receptor, 2-ApB (100 μ M) as well as Xest-C (1 μ M), resulted in suppression of PDGF-mediated neuroprotection. Treatment of neurons with OAG (50 μ M), a membrane permeable analog of diacylglycerol (DAG); however, did not protect rat primary midbrain neurons against Tat toxicity, suggesting thereby that PLC–DAG pathway was not involved in PDGF-mediated neuroprotection. Taken together, these findings confirmed the role of PLC–IP3R pathway in PDGF-mediated neuronal protection against Tat toxicity.
TRPC channels contribute to PDGF-induced intracellular Ca2+ elevations
To study the effects of PDGF on the intracellular Ca2+ transients (i) in rat primary midbrain neurons, we first measured PDGF-induced intracellular [Ca2+]i release using Fluo-4/AM imaging. As shown in Figure 4a and b, exposure of neurons to PDGF triggered a rapid and substantial [Ca2+]i increase in primary midbrain neurons. This response was inhibited by the PDGF receptor antagonist STI-571 (Figure 4c). Furthermore, this elevation in [Ca2+]i-induced by PDGF was suppressed by culturing cells in Ca2+-free medium in the presence of EGTA (2 mM), suggesting thereby that PDGF-induced [Ca2+]i increase depended on the extracellular Ca2+ influx (Figure 4c).
Figure 4
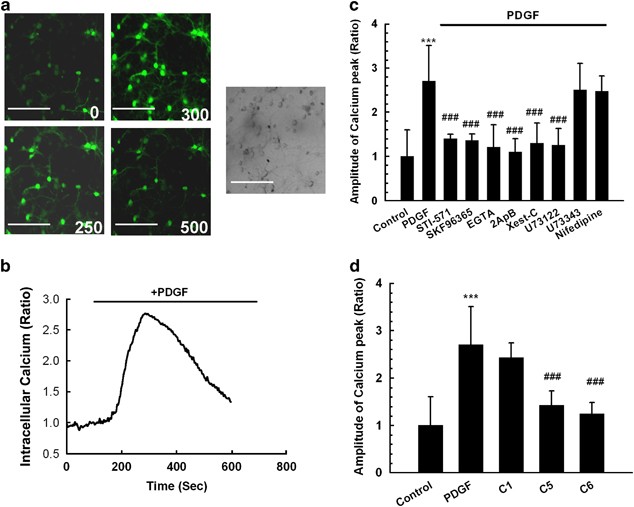
TRPC channels contribute to PDGF-induced intracellular Ca2+ elevations. (a) Rat primary neurons loaded with Fluo-4 [Ca2+]i-sensitive fluorophores before and after PDGF treatment were recorded within a single field using a Fluoview 300 confocal microscope (left panel, numbers in the panels indicate time in seconds) and differential interference contrast (right panel). Scale bar: 100 _μ_m. (b) Changes in intracellular [Ca2+]i levels in neurons following PDGF treatment were measured using the Fluo-4/Fura Red ratio, and the change in ratio is illustrated from a typical neuron. (c) Changes in fluorescence amplitude (Fluo-4/Fura Red) in rat primary neurons exposed to PDGF in the absence or presence of the indicated drugs (STI-571:1 μ M; SKF96365: 20 μ M; EGTA: 2 mM; 2ApB: 100 μ M; Xest-C: 1 μ M; U73122: 1 μ M; U73343: 1 μ M; Nifedipine: 10 μ M). All the data in these figures are presented as mean±S.E.M. of four individual experiments. ***P<0.001 versus control group; ###P<0.001 versus Tat-treated group. (d) PDGF-mediated calcium influx in rat primary neurons transfected with either TRPC1, 5 or 6 siRNA. The data are presented as mean±S.E.M. of four individual experiments. ***P<0.001 versus control group; ###P< 0.001 versus PDGF-treated group
As TRPC channels are Ca2+-permeable cation channels, we next wanted to assess whether PDGF-induced [Ca2+]i elevations in primary midbrain neurons were mediated by TRPC. To confirm the role of TRPC in PDGF-induced [Ca2+]i elevations, rat midbrain neurons were pre-treated with the SKF96365 (20 μ M) before treatment of the cells with PDGF. As shown in Figure 4c cells pre-treated with the SKF96365 showed marked reduction of PDGF-induced elevation of [Ca2+]i, thus underscoring the role of TPRC in this process. In contrast, the L-type voltage-gated calcium channel blocker nifedipine (10 μ M) did not affect PDGF-induced [Ca2+]i elevation. Having determined the role of specific TRPC proteins in neuroprotection-mediated by PDGF, we next sought to dissect the role of specific TRPC channels in PDGF-induced Ca2+ influx. Similar to our findings of the role of TRPC 5 and 6 in PDGF-mediated neuroprotection, TRPC5 & 6 siRNA, but not TRPC 1 siRNA attenuated Ca2+-influx induced by PDGF (Figure 4d).
As PDGF/PDGF-_β_R activation stimulates PLC, the next set of experiments was conducted using the PLC inhibitor U73122 (1 μ M). Similar to findings with the SKF96365, neurons pre-treated with U73122, failed to show PDGF-induced [Ca2+]i elevation (Figure 4c). Interestingly, pre-treatment of neurons with the inactive analog of U73122 (U73343) had no effect on PDGF-induced Ca2+ influx. Furthermore, pre-treatment of neurons with the IP3R antagonists 2-ApB and Xest-C resulted in inhibition of PDGF-induced [Ca2+]i elevation. In summary, PDGF-induced [Ca2+]i elevation depends on the PLC–IP3R pathway.
PDGF-mediated activation of TRPC channels involves amplification of the ERK signal via the Pyk2 pathway
ERK/mitogen-activated protein kinase (MAPK) pathway has been shown to have a crucial role in antiapoptotic mechanisms. It was therefore of interest to examine the effect of PDGF on ERK activation in rat primary neurons. As shown in Figure 5a, exposure of neurons to PDGF resulted in a sustained and time-dependent activation of ERK, which was partially inhibited by pre-treating cells with the PDGF-_β_R antagonist STI-571. PDGF-mediated ERK activation was completely abrogated by the MAPK/ERK kinase (MEK) inhibitor U0126 (20 μ M), but not by the PI3K inhibitor LY294002 (20 μ M) (Figure 5b). The functional role of PDGF-induced ERK activation in mediating neuroprotection was also corroborated using cell viability assays, wherein PDGF failed to exert its protective effect in cells pre-treated with MEK inhibitor, thereby underscoring the role of this pathway in PDGF-mediated neuroprotection (Figure 5c).
Figure 5
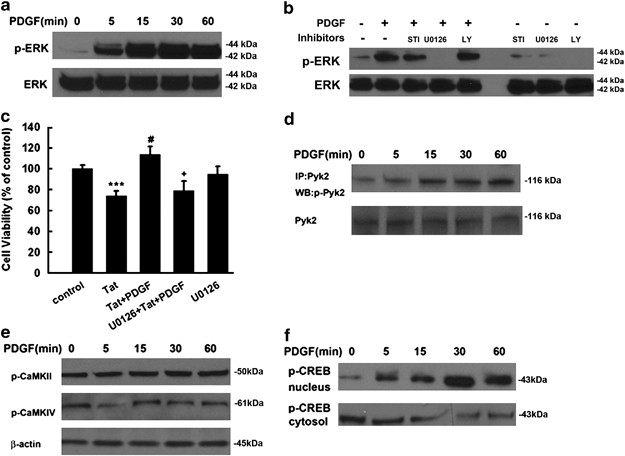
PDGF-induced neuroprotection involves activation of ERK. (a) PDGF-induced time-dependent and sustained phosphorylation of ERK. (b) PDGF maintained increased phosphorylation of ERK for at least 1 h, and this effect was abolished by PDGF receptor antagonist STI-571, MEK inhibitor U0126, but not by PI3-K inhibitor LY294002; STI: STI-571: LY: LY294002. (c) Pre-treatment of neurons with MEK inhibitor U0126 (20 μ M) for 1 h significantly attenuated the protective effect of PDGF. Data are presented as mean±S.E.M. of four individual experiments. ***P<0.001 versus control group; #P<0.05 versus Tat-treated group; +P<0.05 versus both PDGF and Tat-treated group. PDGF exposure resulted in increased time-dependent phosphorylation of Pyk2 (d), but not of CaMKII and CaMKIV (e) in rat primary neurons. PDGF exposure resulted in increased time-dependent phosphorylation of CREB in the nuclear fraction with a concomitant decrease in the cytosolic fraction in rat primary neurons (f)
Activation of the ERK pathway is known to be mediated by an upstream signaling molecule – the Ca2+-sensitive, proline-rich tyrosine kinase 2 (Pyk2). In our findings we demonstrated PDGF-mediated induction of Pyk2 as early as 5 mins following stimulation of the cells (Figure 5d). In addition to Pyk2, CaMKs, another class of Ca2+-sensitive signaling molecules are also known to have critical roles in cell survival. We thus sought to examine whether PDGF-mediated neuroprotection also involved activation of CaMKs. As shown in Figure 5e PDGF failed to activate either CaMK II or CaMKIV, thereby ruling out the role of these molecules in PDGF-mediated neuroprotection.
As CREB is a transcriptional factor that lies downstream of ERK and has an important role in cell survival, we next examined CREB phosphorylation following PDGF treatment using western blot analysis. As shown in Figure 5f, treatment of neurons with PDGF resulted in increased nuclear CREB phosphorylation that correlated well with a concomitant decrease in cytosolic CREB.
Having determined the role of TRPC channels in PDGF-mediated neuroprotection we next wanted to dissect the role of these channels in PDGF-mediated ERK activation pathway. Pre-treatment of neurons with either the TRP channels blocker SKF96365 (20 μ M) or EGTA (2 mM) markedly attenuated PDGF-induced phosphorylation of Pyk2, ERK and CREB (Figure 6a–c). These findings suggested that PDGF-mediated neuroprotection via the TRPC/Ca2+ pathway involves phosphorylation of Pyk2, ERK and CREB-signaling molecules.
Figure 6
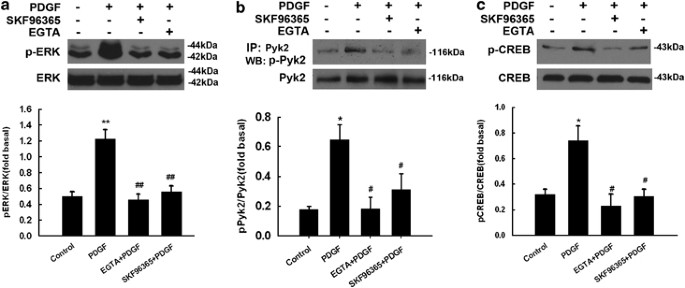
TRPC channels are critical for PDGF-induced ERK/CREB activation. Representative immunoblot of PDGF-mediated ERK, Pyk2 and CREB activation in rat primary neurons exposed to PDGF in the presence of TRPC blocker and EGTA (a–c; upper panels). Densitometric analysis of pERK/ERK, pPyk2/Pyk2 and pCREB/CREB from four individual experiments. (a–c; lower panels). *P<0.05;**P<0.01 versus control group; #P<0.05,##P<0.01 versus PDGF group
To further confirm the role of specific TRPC protein(s) in PDGF-induced ERK, Pyk2 and CREB phosphorylation, each of the TRPC subtypes (1, 5 or 6) was individually downregulated using specific siRNA followed by assessment of PDGF-induced ERK, Pyk2 and CREB phosphorylation. Interestingly, siRNA for both TRPC 5 and 6 decreased PDGF-mediated Pyk2, ERK and CREB phosphorylation (Figure 7a–c), whereas TRPC 1 siRNA exerted no such effect. These findings thus underpinned the roles of TRPC 5 & 6 in PDGF-mediated ERK, Pyk2 and CREB phosphorylation.
Figure 7
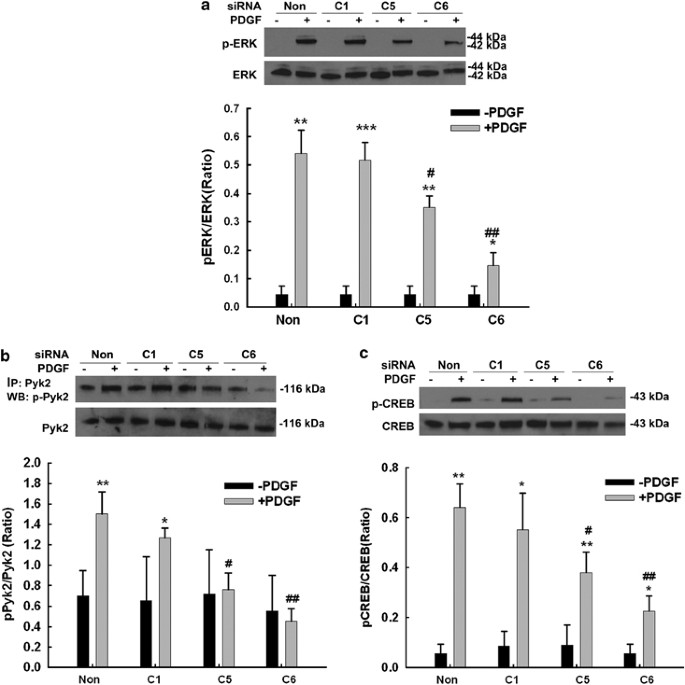
TRPC5 and 6 are essential for PDGF-mediated phosphorylation of Pyk2, ERK and CREB. Representative immunoblot of PDGF-mediated phosphorylation of ERK, Pyk2 and CREB in rat primary neurons in the presence or absence of siRNAs TRPC1, 5 or 6 (a–c; upper panels). Densitometric analyses of PDGF-mediated phosphorylation of ERK, Pyk2 and CREB in the presence or absence of siRNAs from three individual experiments. (a–c; lower panels). *P<0.05; **P<0.01; ***P<0.001 versus control group; #P<0.05, ##P<0.01 versus PDGF with nonsense siRNA group
TRPC channels are not required for PDGF-induced Akt activation
In addition to the MEK/ERK pathway, PI3K–Akt pathway also has a critical role in cell survival. We thus examined the role of the latter pathway in PDGF-mediated neuroprotection. As shown in Figure 8a, following exposure of neurons to PDGF there was an enhanced activation of Akt, which was abrogated by both the PDGF-_β_R antagonist STI-571 (1 μ M) as well as the PI3 K inhibitor LY294002 (20 μ M) (Figure 8b). We next wanted to assess the functional role of PI3K–Akt in PDGF-mediated neuroprotection against Tat. As shown in Figure 8c, PDGF pre-treatment resulted in the inhibition of Tat neurotoxicity and this effect was reversed in the presence of PI3K inhibitor LY294002. These results suggested the involvement of PI3K/Akt pathway in PDGF-mediated neuroprotection against HIV Tat. Further confirmation of the involvement of Akt pathway in PDGF-mediated neuroprotection was shown with the use of adenovirus vector constructs encoding a dominant-interfering form of Akt. As shown in Figure 8d, in neurons infected with the dominant-interfering form of Akt, PDGF failed to protect against Tat toxicity. Infection of neurons with wild type Akt, on the other hand, resulted in PDGF-mediated neuroprotection as expected.
Figure 8
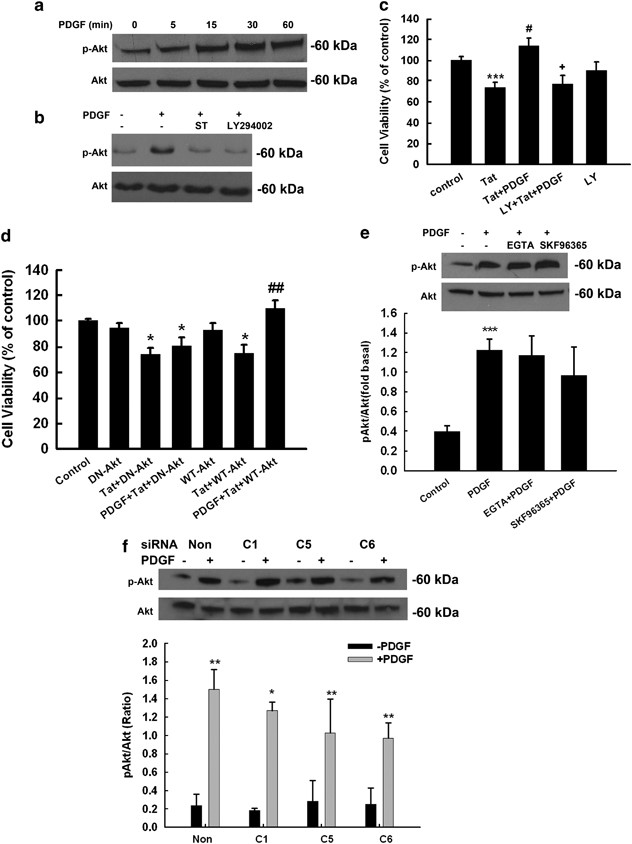
TRPC channels are not critical for PDGF-induced Akt activation (a) PDGF-induced time-dependent activation of Akt. (b) PDGF maintained sustained phosphorylation of Akt for at least 1 h and this effect was attenuated by both the PDGF receptor antagonist STI-571 and PI3-K inhibitor LY294002. (c) Pre-treatment of neurons with LY294002 (20 μ M) for 1 h resulted in inhibition of PDGF-mediated protection using MTT analysis. Data are presented as mean±S.E.M. of four individual experiments. ***P<0.001 versus control group; #P<0.05 versus Tat-treated group; +P<0.05 versus both PDGF and Tat-treated group. (d) Infection of neurons with the dominant-interfering Akt (DN-Akt) resulted in abrogation of PDGF-mediated neuroprotection. Infection with WT-Akt (WT-Akt) construct had no effect on cell survival. Data are presented as mean±S.E.M. of four individual experiments. *P< 0.05 versus adenovirus alone group; ##P< 0.01 versus Tat-treated group. (e) Representative immunoblot of PDGF-mediated Akt activation in rat primary neurons exposed to PDGF in the presence or absence of TRP blocker and EGTA (upper panel). Densitometric analyses of pAkt/Akt from the three individual experiments is presented (lower panel). ***P<0.001 versus control group. (f) Representative immunoblot of PDGF-mediated Akt phosphorylation in rat primary neurons transfected with TRPC1, 5 or 6 siRNAs (upper panel). Densitometric analyses of PDGF-mediated phosphorylation of Akt in rat neurons depleted of TRPC1, 5 or 6 from three individual western blots (lower panel). *P<0.05; **P<0.01 versus control group
Having determined that TRPC channels were involved in regulation of ERK/CREB activation, we next wanted to dissect the role of these channels in PDGF-mediated Akt activation pathway. As shown in Figure 8e, pre-treatment of neurons with both the TRP channel blocker SKF96365 as well as EGTA did not attenuate PDGF-induced Akt activation. Furthermore, siRNA for TRPC (s) did not significantly decrease PDGF-mediated Akt phosphorylation (Figure 8f). These findings thus suggested that PDGF-mediated Akt phosphorylation is independent of TRPC.
PDGF-mediated protection of dopaminergic neurons against HIV Tat in vivo
To investigate the relevance of PDGF-mediated protection in vivo, the neuroprotective effect of PDGF on dopaminergic neuronal survival was determined in adult mice injected intrastriatally with either saline or PDGF followed by microinjection with HIV-1 Tat. Seven days later mice brains were examined for TH-positive neurons in the substantia nigra. As shown in Figure 9a, there was increased neuronal loss in Tat-injected mice as evidenced by decreased TH staining. This effect was ameliorated in mice pre-treated with PDGF as quantified in Figure 9b, indicating that PDGF protected dopaminergic neurons against HIV Tat. Similar to saline-injected mice, exposure to PDGF alone had no significant effect on neuronal survival. Furthermore, to confirm the role of TRPC in PDGF-mediated neuroprotection against Tat, we pre-treated mice with the TRP channels blocker SKF96365 before treatment with PDGF and HIV Tat. Pre-treatment of mice with SKF96365 (0.2 _μ_mol) followed by exposure to PDGF and Tat resulted in attenuation of PDGF-mediated neuroprotection. Injection of the mice with the TRP channel blocker, SKF96365 alone did not exert any effect on the dopaminergic neurons.
Figure 9
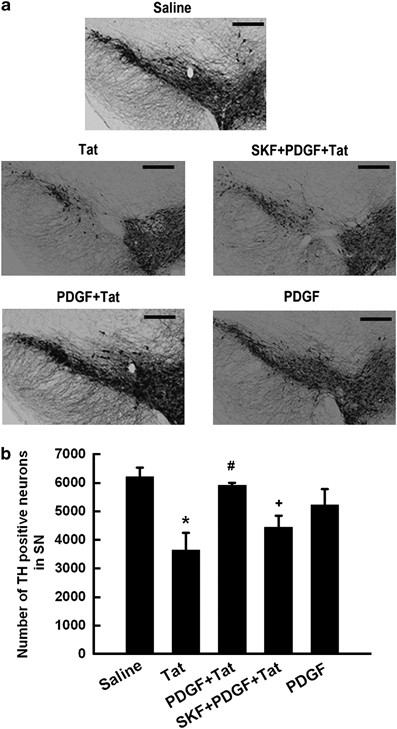
PDGF protects dopaminergic neurons against Tat-induced in the substantia nigra. (a) Representative mesencephalic sections from different groups of mice treated with PDGF and/or HIV Tat in the presence or absence of TRPC blocker SKF96365 were examined for TH-positive neurons counted by stereology. (b) There was increased loss of TH-positive neurons in the substantia nigra of Tat alone-treated group of mice compared with the saline controls. Pre-treatment with PDGF resulted in amelioration of Tat toxicity in the substantia nigra neurons, and this effect was significantly attenuated in mice pre-treated with SKF96365 (0.2 _μ_mol). *P<0.05 versus saline group; #P<0.05 versus Tat; +P<0.05 versus PDGF+Tat. SN: Substantia Nigra. Scale bar: 300 _μ_m. SKF/SKF96365 (0.2 _μ_mol)
Discussion
In this study, we show that in vivo PDGF was able to rescue dopaminergic neurons from HIV Tat neurotoxicity. Confirmation of the neuroprotective role of PDGF against Tat toxicity was also corroborated in vitro in primary cultures of rat midbrain neurons. We showed a novel role of the Ca2+-permeable channel TRPC in PDGF-mediated neuroprotection in rat neurons, indicating that exogenous PDGF activated TRPC resulting in amplification of downstream ERK signaling via the Pyk2 pathway, followed by nuclear translocation of CREB and ultimately culminating in neuronal survival.
Nigrostriatal neurons are susceptible to HIV-1 protein-mediated toxicity and HIV Tat is known to inhibit TH gene expression in dopaminergic neurons contributing to motor abnormalities in HAD patients.3 Expression of PDGF-_β_R in the dopaminergic neurons and its role in neuronal communication and possibly in dopaminergic neurotransmission circuitry has been shown by Othberg et.al.16 In our studies, we detected the presence and activation of PDGF-_β_R in rat primary cultured midbrain neurons. In addition, using the pharmacological and siRNA approaches we also showed that PDGF-mediated neuroprotection involved activation of PDGF-_β_R.
A novel finding of this study is the role of TRPC in PDGF-mediated neuroprotection against HIV Tat, thereby lending credence to previous reports indicating the involvement of TRPC signaling in neuroprotection.17, 27 The mammalian TRPC channel family consists of seven members, TRPC1-7, that appear to function as receptor-operated channels, analogous to the TRP channels involved in Drosophilia phototransduction.28 With the exception of TRPC2, these channels are widely distributed in the mammalian brain. Consistent with the previous reports showing the co-localization of TRPC (1,5 and 6) with the dopaminergic neurons in substantia nigra,29, 30, 31 our findings also provide evidence that TRPC5/6 were colocalized with PDGF-_β_R in primary midbrain neurons. The mammalian TRPC channels can be activated by G-protein-coupled receptors and RTKs.18, 21, 22 PDGF-_β_R belongs to the RTK family and is known to activate PLC, leading to hydrolysis of phosphatidylinositol-4,5-biphosphate into membrane-bound DAG and soluble IP3. Generation of IP3 results in IP3 receptor-mediated release of Ca2+ from intracellular stores as well as Ca2+ influx from outside the cell.26
PDGF triggers [Ca2+]i transients in neuronal precursor cells,32 however, how PDGF regulates [Ca2+]i remains unclear. Here, we report that PDGF induces [Ca2+]i elevation through engagement of the PDGF-_β_R because PDGF receptor antagonist was able to abolish PDGF-mediated [Ca2+]i elevations. Furthermore, the increase of [Ca2+]i induced by PDGF was primarily due to Ca2+ increase through the PLC/IP3-induced Ca2+ release and the TRPC channels. These findings were supported by a pharmacological approach using inhibitors specific for PLC (U73122), IP3R (2ApB and Xest-C) and TRP channels (SKF96365), all of which effectively blocked PDGF-mediated [Ca2+]i elevations. PDGF-mediated calcium influx from extracellular source was confirmed by EGTA, an extracellular Ca2+ chelator, which also blocked PDGF-mediated elevation of [Ca2+]i.
Influx of Ca2+ is known to regulate numerous physiological processes through a wide range of target proteins such as ERK, Pyk2 and CREB,33, 34, 35 which are essential for neuronal survival. In fact, autophosphorylation of Pyk2 on the conserved tyrosine residues Y 40236, 37 has been shown to result in downstream activation of ERK. In this study, we showed that [Ca2+]i elevations triggered by PDGF through the TRPC channels activated CREB through the Pyk2/ERK pathway. Consistent with the previous reports that ERK and its downstream effector CREB have a role in neuroprotection,38, 39, 40 PDGF-mediated neuroprotection also involved activation of the ERK pathway.
The seven TRPC channels are divided into two groups based upon their homology and mechanisms of activation. This study showed that TRPC 5 and 6, but not TRPC1 were required for PDGF-mediated neuroprotection against Tat toxicity. Previous findings have shown the protective role of TRPC1 in MPTP-induced toxicity in SH-SY5Y cells.31 Recent studies have shed light on the mechanisms underlying the activation of TRPC channels by a growth factor such as brain-derived neurotrophic factor.17 In this study, activation of TRPC3 and TRPC6 stimulated two signaling pathways: Ca2+/Ras/MEK/ERK and Ca2+/CaM/CaMK that converged on CREB activation.17
Our findings also unravel the role of TRPC activation by yet another neurorprotective growth factor PDGF. The mechanism of protection by PDGF however, involved action of ERK as well as the PI3K/Akt pathways, an effect that is consistent with the previous reports showing that PDGF stimulates PI3K/Akt pathways in various other cell types.38, 39, 40 ERK pathway can be activated via the PI3K activation or independent of it, depending on the cell type.41 In this study, PDGF-mediated neuroprotection via ERK activation was independent of PI3K pathway, because PI3K inhibitor failed to inhibit PDGF-mediated ERK phosphorylation. Role of Akt pathway in the neuroprotective effects of PDGF was further confirmed using a dominant-interfering form of Akt. Unlike the TRPC-mediated activation of Pyk2/ERK/CREB, PDGF-mediated activation of Akt was independent of TRPC channel activation.
Consistent with the in vitro study showing the involvement of TRPC in PDGF-mediated neuroprotection, we found that pre-treatment of adult mice with PDGF also resulted in rescue of Tat-mediated dopaminergic neuronal loss in the substantia nigra. Interestingly, this effect was attenuated by injecting TRP channel blocker-SKF96365 directly into the striatum. This study for the first time shows that TRPC channels are involved in PDGF-mediated neuroprotection. These findings lend credence to the previously published reports indicating the role of TRPC channels in granule cell survival.17
In summary, activation of the PDGF/PDGF-_β_R axis resulted in stimulation of PLC/IP3 R pathway leading to activation of TRPC channels, which in turn, resulted in elevation of [Ca2+]i transients, culminating in activation of Pyk2/ERK/CREB, but not PI3K/Akt pathway (as shown in Figure 10). PDGF-mediated neuronal survival also involved the Pyk2/ERK/CREB pathway as well as the parallel PI3K/Akt pathway. Taken together our findings suggest that although the two pathways involved in PDGF-mediated neuroprotection against Tat operate independent of each other, their combined actions are necessary for the observed neuroprotective effect of PDGF. A better understanding of these molecular pathways could be critical for the development of therapeutic strategies against HAD.
Figure 10
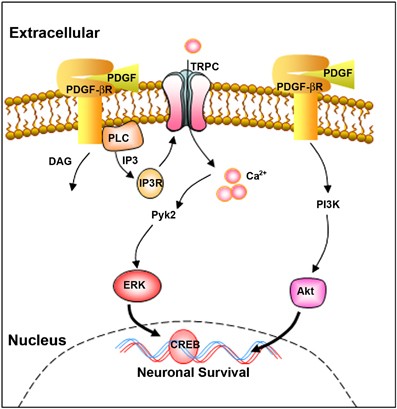
Schematic illustration showing putative-signaling pathways involved in PDGF-mediated neuroprotection in rat primary neurons. PDGF-mediated engagement of the PDGF-β receptor stimulates the PLC/IP3 R pathway, which in turn, activates TRPC channels resulting in elevation of [Ca2+]i transients. [Ca2+]i elevation in turn results in activation of Pyk2/ERK pathways leading to CREB activation and consequently neuronal survival. In addition to this, PDGF/PDGF-_β_R axis can also activate another distinct pathway – PI3K/Akt, which can also lead to potentiation of neuronal survival
Materials and Methods
Animals
C57BL/6N mice were purchased from (Charles River Laboratories Inc., Wilmington, MA, USA). All the animals were housed under conditions of constant temperature and humidity on a 12-h light, 12-h dark cycle, with lights on at 0700 h. Food and water were available ad libitum. All animal procedures were performed according to the protocols approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center.
Neuronal culture and treatment
Primary neurons were prepared as described earlier with a few modifications.42 Briefly, midbrain tissues were dissected from embryonic day 13 to 14 Sprague–Dawley rats and dissociated with a mild mechanical trituration. Dissociated cells were seeded first in DMEM/F12 supplemented with 10% fetal bovine serum. After 1 day, the cultures were supplemented with serum-free neurobasal medium containing B27 (50:1), 2 mM glutamax supplemented with antibiotics (100 U/ml penicillin and 100 _μ_g/ml streptomycin). After 7 days, the initial plating cultures consisted of 90% neuron-specific nuclear protein immunoreactive neurons of which 3% were TH-positive neurons. Based upon our earlier studies, primary midbrain neurons were treated with PDGF (R & D system, Minneapolis, MN, USA; 20 ng/ml) and HIV-1 Tat (UK College of Medicine, Lexington, KY, USA; 14 nM) at pre-determined concentrations. Treatment of neurons with pharmacological inhibitors (STI-571:1 μ M; SKF96365: 20 μ M; EGTA: 2 mM; U73122: 1 μ M; U73343: 1 μ M; 2ApB: 100 μ M; Xest-C: 1 μ M; Nifedipine: 10 μ M; U0126: 20 μ M; LY294002: 20 μ M) involved pre-treatment of cells with the respective inhibitors for 1 h followed by Tat and/or PDGF exposure.
Surgery and microinjection
Ten-week-old C57BL/6N mice were anesthetized with 2.5% isofluorane and placed in a stereotaxic apparatus for cannula implantation. Using the stereotaxic coordinates, AP+0.86 mm posterior, ML-1.8 mm lateral to midline, and DV −3.5 mm to bregma according to Franklin and Paxinos mouse brain atlas. A permanent 26-G stainless steel guide cannula (C315G; Plastics One, Roanoke, VA, USA) was implanted into the right striatum. The guide cannula was secured in place using gel adhesive and dental cement applied sequentially to the skull. A 33-G stainless steel dummy cannula was used to seal the guide cannula when not in use. After surgery the animals were housed individually to avoid damage to guide and dummy cannula.
Following striatal cannulation animals were allowed to recover for 7 days and were randomly divided into six groups (_n_=5): Saline, Tat, PDGF alone, PDGF plus Tat, and SKF96365 plus PDGF plus Tat (using two doses of the SKF96365 – see below) groups. Tat and PDGF each were injected at a concentration of 1 _μ_g per 2 _μ_l/mouse/day for 2 days. Saline control mice were injected with the same volume of sterile saline (2 _μ_l). In the PDGF plus Tat group of mice, PDGF was first microinjected for 2 days followed by HIV-1 Tat microinjection subsequently for 2 days. To determine the optimal dose of the TRP channel blocker in mice, SKF96365 freshly dissolved in 0.9% sterile saline, was injected at two different concentrations, (0.02 _μ_mol and 0.2 _μ_mol/2 _μ_l/mouse; concentrations determined from our in vitro study) for 2 consecutive days, followed by PDGF and Tat microinjections. All microinjections were performed using a 33-G stainless steel injector connected to a 10 _μ_l syringe, which was operated by an infusion pump set at the rate of 0.4 _μ_l/min. An additional minute was allowed for diffusion and prevention of backflow through the needle track before the injector was withdrawn. Histological verification of the striatum cannula was performed at the end of each experiment.
Cell survival
The percentage of surviving neurons in the presence of PDGF and /or HIV-1 Tat was estimated using the biochemical and histological analyses listed below.
MTT
Cell viability was measured by mitochondrial dehydrogenases MTT method. Primary midbrain neurons were seeded in 96-well plates at a density of 105cells per cm2 for 7 days, following which neurons were exposed to fresh medium containing various concentrations of PDGF for 20 min at 37 °C. Subsequently, HIV-1 Tat (14 nM) was added into the medium and cells were incubated for another 24 h (concentration pre-determined from our previous work).25 After incubation for up to 24 h, 20 _μ_l MTT tetrazolium salt dissolved in Hank's balanced salt solution at a final concentration of 5 mg/ml was added to each well and incubated in the CO2 incubator for 4 h. Finally, the medium was aspirated from each well and 200 _μ_l of DMSO was added to dissolve the formazan crystals and the absorbance of each well was obtained using a Dynatech MR5000 plate counter at test and reference wavelengths of 570 and 630 nm, respectively.
Hoechst staining
To quantify apoptotic neurons, neurons were fixed and stained with Hoechst 33324 (Sigma, St Louis, MO, USA). The morphological features of apoptosis (cell shrinkage, chromatin condensation, and fragmentation) were monitored by fluorescence microscopy (Olympus BX 60, Tokyo, Japan). At least 400 cells from 12 randomly selected fields per dish were counted, and each treatment was performed in triplicate.
Flow cytometry
Neuronal apoptosis was estimated using the Annexin-V Fluorescein (FITC) apoptosis detection kit (Oncogene Research Products, San Diego, CA, USA) according to the manufacturer's instructions. The cell samples were analyzed in a flow cytometry apparatus (Becton Dickinson FACSVantage SE, San Jose, CA, USA). Annexin V binds to phosphatidylserine that is translocated during apoptosis from the inner to the outer leaflet of the plasma membrane. Live cells with intact membranes are distinguished by their ability to exclude propidium iodide, which readily penetrates dead or damaged cells. Dual analysis was introduced using a quadrant dot plot, in which necrotic cells were identified as single PI-positive, early apoptotic cells were Annexin V-FITC-positive only, and cells in late apoptosis were recognized as double-positive for annexin V-FITC and propidium iodide. Cells that stained negative for both annexin V-FITC and propidium iodide were classified as live cells. Finally, the number of cells in each category was expressed as a percentage of the total number of stained cells counted.
Analyses of neuronal dendrites
Primary rat midbrain neurons were stained with anti-MAP-2 antibody and dopaminergic neurons were identified with the anti-TH as described previously with minor modifications.43 For the measurement of the dendrite length, images of individual MAP-2/TH-positive neurons were recorded. The length of each fiber originating from each neuronal cell body and subsequent branches was measured, and a sum of total dendrite length for each neuron was calculated using Image Pro-plus software. For each well, 50 neurons were analyzed for total dendrite length. Results were expressed as a percentage of the control cultures using a Nikon TE2000E microscope with a digital camera (Photometrics, Tucson, AZ, USA).43
Measurement of [Ca2+]i
The changes in [Ca2+]i were monitored using Fluo-4/AM dissolved in dimethyl sulfoxide. The rat midbrain neurons in 35-mm culture dishes were rinsed twice with Physiological Solution (PS, 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 0.5 mM MgCl2, 10 mM glucose, 5.5 mM HEPES, pH 7.4) followed by incubation in PS containing 5 μ M Fluo-4/AM (Molecular Probes, Eugene, OR, USA) at 37 °C for 40 min. Cells were then rinsed twice with the PS, mounted on a perfusion chamber and scanned every second using confocal microscopy (Fluoview 300; Olympus). Fluorescence excitation at 488 nm was provided by an argon laser, and the emitted light (515 nm) captured along with transmitted images. All analyses of [Ca2+]i were processed at a single-cell level and are expressed as the relative fluorescence intensity. For ratiometric analysis, cells were loaded (using procedures described above) with Fluo-4 and Fura-Red. Fluorescence values from subcellular regions were calculated as mean pixel values±S.E., and a ratio was calculated by using the Fluo-4/Fura Red combination. All of these studies were done at least four times.
Western blotting
Treated cells were lysed using the Mammalian Cell Lysis kit (Sigma) and the NE-PER Nuclear and Cytoplasmic Extraction kit (Pierce, Rockford, IL, USA). Equal amounts of the corresponding proteins were electrophoresed in a sodium dodecyl sulfate-polyacrylamide gel in reducing conditions followed by transfer to PVDF membranes. The blots were blocked with 5% non fat dry milk in phosphate-buffered saline. The western blots were then probed with antibodies recognizing the phosphorylated forms of PDGF-_β_R,ERK1/2, Pyk2, Akt, CREB (Cell Signaling, Danvers, MA, USA 1:200), and _β_-actin (Sigma, 1:4000) The secondary antibodies were alkaline phosphatase conjugated to goat anti-mouse/rabbit IgG (1:5000). Signals were detected by chemiluminescence (Pierce). All of western blot was repeated at least three times.
Coimmunoprecipitation
The procedure for immunoprecipitation was performed as described previously.15 Briefly, rat primary neurons were treated with PDGF and/ or PDGF-receptor antagonist STI-571 or siRNAs followed by lysis in RIPA buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 0.1% SDS, 1.0% NP-40 and 0.5% sodium deoxycholate) containing proteinase and phosphatase inhibitors. For each sample, 200 _μ_g of protein was used for coimmunoprecipitation. The sample protein was incubated with 2 _μ_g diluted anti-4-G-10 or Pyk2 antibody (Millipore, Billerica, MA, USA or Cell signaling) overnight at 4°C followed by incubation with 20 _μ_l of protein A-sepharose for 3 h at 4°C. The mixture was then centrifuged (at 6000 g for 30 s) and the cell pellets were rinsed twice with RIPA, followed by boiling in 2 × western blot loading buffer for 4 min. After spinning (at 6000 g for 30 s) the supernatants were subjected to western blot as described above.
Adenovirus infection
Primary midbrain neurons were infected with adenoviral constructs containing the WT or dominant-interfering forms of Akt (kind gift from Dr K Walsh). Neurons infected for 48 h with adenovirus constructs were subsequently treated with Tat and/or PDGF followed by assessment of cell survival as described above.
Short-interfering (si) RNA transfection
siRNA targeted against PDGF-_β_R were obtained from Thermo Scientific Dharmacon RNAi Technologies (Chicago, IL, USA; Accell SMART Pool, Cat no. E-091874-00). siRNAs targeted against TRPC1, 5 and 6 were obtained from Ambion (Austin, TX, USA). The sequences of these siRNAs are listed in Table 1. For siRNA transfection, we used the rat Neuron Nucleofector Kit (Amaxa, Gaithersburg, MD, USA) according to the manufacturer's instructions. Briefly, dissociated cells were re-suspended in transfection medium, mixed with the respective siRNAs (200 nM), and electroporated using the fixed program (O-03) for optimal neuronal transfection. Cells were then quickly centrifuged, resuspended and plated. The knockdown efficiency of siRNAs was determined after 7 days of transfection by western blotting.
Table 1 siRNA sequences of rat TRPC1, 5 and 6
Immunochemistry
For immunocytochemistry primary midbrain neurons were plated on cover slips. After 7 days, cells were fixed with 4% paraformaldehyde for 15 min at room temperature followed by permeabilization with 0.3% Triton X-100 in PBS. Cells were then incubated with a blocking buffer containing 10% normal goat serum in PBS for 1 h at room temperature followed by addition of rabbit anti-TRPC 5 and 6 (1:100; Alomone Lab, Jerusalem, Israel) and mouse anti-PDGF-_β_R (1:500; Cell Signalling) antibodies and incubated overnight at 4 °C. Finally, the secondary AlexaFluor 488 goat anti-rabbit IgG and AlexaFluor 569 goat anti-mouse IgG, was added at a 1:500 dilution for 2 h to detect TRPC and PDGF- _β_R. Cells were washed three times in buffer and mounted with Vectashield onto slides (Vector Laboratories, Burlingame, CA, USA).Confocal images were taken at a magnification of × 60(objective lens) with an Eclipse C1 Plus confocal microscope (Nikon).
For immunohistochemistry mice were perfused by transcardial perfusion using chilled 4% paraformaldehyde. Free-floating sections encompassing the entire midbrain were sectioned at 40 _μ_m on a cryostat. For TH immunostaining, tissue sections were incubated with primary antibodies overnight at 4 °C. Primary antibodies used in this study were as follows: rabbit anti-TH (1:4000, Sigma). Immunostaining was visualized by using 3, 3′-diaminobenzidine as the substrate. Quantitative (neuronal number) estimates of TH-positive cell bodies were performed ipsilaterally in the areas of interest using stereology method.
Statistical analysis
Data were expressed as mean±S.E.M. Significance of differences between control and samples treated with various drugs was determined by one-way ANOVA followed by post hoc least significant difference test. Values of P<0.05 were taken as statistically significant.
Abbreviations
CaMKs:
calcium/calmodulin-dependent kinases
CREB:
cAMP-response element-binding protein
DAG:
diacylglycerol
ERK:
extracellular signal-regulated protein kinase
HIV-1:
human immunodeficiency virus-1
HAD:
HIV-1-associated dementia
IP3:
inositol triphosphate
MAP-2:
microtubule-associated protein-2
MTT:
3(4,5-dimethylthiazol-2-yl)-2.5 diphenyltetrazolium bromide
TH:
tyrosine hydroxylase
MEK:
MAPK/ERK kinase
∣PI3K:
phosphoinositide-3-kinase
PDGF:
platelet-derived growth factor
PLC:
phospholipase C
Pyk2:
proline-rich tyrosine kinase 2
TRP:
transient receptor potential
TRPC:
transient receptor potential canonical channels
siRNA:
short-interfering RNAs
References
- Nath A, Anderson C, Jones M, Maragos W, Booze R, Mactutus C et al. Neurotoxicity and dysfunction of dopaminergic systems associated with AIDS dementia. J Psychopharmacol 2000; 14: 222–227.
Article CAS Google Scholar - Mocchetti I, Nosheny RL, Tanda G, Ren K, Meyer EM . Brain-derived neurotrophic factor prevents human immunodeficiency virus type 1 protein gp120 neurotoxicity in the rat nigrostriatal system. Ann N Y Acad Sci 2007; 1122: 144–154.
Article CAS Google Scholar - Zauli G, Secchiero P, Rodella L, Gibellini D, Mirandola P, Mazzoni M et al. HIV-1 Tat-mediated inhibition of the tyrosine hydroxylase gene expression in dopaminergic neuronal cells. J Biol Chem 2000; 275: 4159–4165.
Article CAS Google Scholar - Eugenin EA, D'Aversa TG, Lopez L, Calderon TM, Berman JW . MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem 2003; 85: 1299–1311.
Article CAS Google Scholar - Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE et al. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med 2000; 6: 1380–1387.
Article CAS Google Scholar - Everall IP, Hansen LA, Masliah E . The shifting patterns of HIV encephalitis neuropathology. Neurotox Res 2005; 8: 51–61.
Article CAS Google Scholar - Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Graos MM et al. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ 2005; 12: 1329–1343.
Article CAS Google Scholar - Arthur DB, Georgi S, Akassoglou K, Insel PA . Inhibition of apoptosis by P2Y2 receptor activation: novel pathways for neuronal survival. J Neurosci 2006; 26: 3798–3804.
Article CAS Google Scholar - Deierborg T, Soulet D, Roybon L, Hall V, Brundin P . Emerging restorative treatments for Parkinson's disease. Prog Neurobiol 2008; 85: 407–432.
Article CAS Google Scholar - Smits A, Kato M, Westermark B, Nister M, Heldin CH, Funa K . Neurotrophic activity of platelet-derived growth factor (PDGF): Rat neuronal cells possess functional PDGF beta-type receptors and respond to PDGF. Proc Natl Acad Sci USA 1991; 88: 8159–8163.
Article CAS Google Scholar - Egawa-Tsuzuki T, Ohno M, Tanaka N, Takeuchi Y, Uramoto H, Faigle R et al. The PDGF B-chain is involved in the ontogenic susceptibility of the developing rat brain to NMDA toxicity. Exp Neurol 2004; 186: 89–98.
Article CAS Google Scholar - Andrae J, Gallini R, Betsholtz C . Role of platelet-derived growth factors in physiology and medicine. Genes Dev 2008; 22: 1276–1312.
Article CAS Google Scholar - Beazely MA, Lim A, Li H, Trepanier C, Chen X, Sidhu BR et al. Platelet-derived growth factor selectively inhibits NR2B-containing NMDA receptors in ca1 hippocampal neurons. J Biol Chem 2008.
- Peng F, Dhillon N, Callen S, Yao H, Bokhari S, Zhu X et al. Platelet-derived growth factor protects neurons against gp120-mediated toxicity. J Neurovirol 2008; 14: 62–72.
Article CAS Google Scholar - Peng F, Dhillon NK, Yao H, Zhu X, Williams R, Buch S . Mechanisms of platelet-derived growth factor-mediated neuroprotection--implications in HIV dementia. Eur J Neurosci 2008; 28: 1255–1264.
Article Google Scholar - Othberg A, Odin P, Ballagi A, Ahgren A, Funa K, Lindvall O . Specific effects of platelet derived growth factor (PDGF) on fetal rat and human dopaminergic neurons in vitro. Exp Brain Res 1995; 105: 111–122.
Article CAS Google Scholar - Jia Y, Zhou J, Tai Y, Wang Y . TRPC channels promote cerebellar granule neuron survival. Nat Neurosci 2007; 10: 559–567.
Article CAS Google Scholar - Clapham DE . TRP channels as cellular sensors. Nature 2003; 426: 517–524.
Article CAS Google Scholar - Montell C, Birnbaumer L, Flockerzi V . The TRP channels, a remarkably functional family. Cell 2002; 108: 595–598.
Article CAS Google Scholar - Clapham DE, Julius D, Montell C, Schultz G . International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev 2005; 57: 427–450.
Article CAS Google Scholar - Ambudkar IS, Ong HL . Organization and function of TRPC channelosomes. Pflugers Arch 2007; 455: 187–200.
Article CAS Google Scholar - Ramsey IS, Delling M, Clapham DE . An introduction to TRP channels. Annu Rev Physiol 2006; 68: 619–647.
Article CAS Google Scholar - Merritt JE, Armstrong WP, Benham CD, Hallam TJ, Jacob R, Jaxa-Chamiec A et al. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem J 1990; 271: 515–522.
Article CAS Google Scholar - Zhu X, Jiang M, Birnbaumer L . Receptor-activated Ca2+ influx via human Trp3 stably expressed in human embryonic kidney (HEK)293 cells. Evidence for a non-capacitative Ca2+ entry. J Biol Chem 1998; 273: 133–142.
Article CAS Google Scholar - Yao H, Peng F, Dhillon N, Callen S, Bokhari S, Stehno-Bittel L et al. Involvement of TRPC channels in CCL2-mediated neuroprotection against tat toxicity. J Neurosci 2009; 29: 1657–1669.
Article CAS Google Scholar - White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M et al. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci USA 2005; 102: 14092–14097.
Article CAS Google Scholar - Bollimuntha S, Singh BB, Shavali S, Sharma SK, Ebadi M . TRPC1-mediated inhibition of 1-methyl-4-phenylpyridinium ion neurotoxicity in human SH-SY5Y neuroblastoma cells. J Biol Chem 2005; 280: 2132–2140.
Article CAS Google Scholar - Clapham DE, Runnels LW, Strubing C . The TRP ion channel family. Nat Rev Neurosci 2001; 2: 387–396.
Article CAS Google Scholar - De March Z, Giampa C, Patassini S, Bernardi G, Fusco FR . Cellular localization of TRPC5 in the substantia nigra of rat. Neurosci Lett 2006; 402: 35–39.
Article CAS Google Scholar - Martorana A, Giampa C, DeMarch Z, Viscomi MT, Patassini S, Sancesario G et al. Distribution of TRPC1 receptors in dendrites of rat substantia nigra: a confocal and electron microscopy study. Eur J Neurosci 2006; 24: 732–738.
Article Google Scholar - Bollimuntha S, Ebadi M, Singh BB . TRPC1 protects human SH-SY5Y cells against salsolinol-induced cytotoxicity by inhibiting apoptosis. Brain Res 2006; 1099: 141–149.
Article CAS Google Scholar - Cuddon P, Bootman MD, Richards GR, Smith AJ, Simpson PB, Roderick HL . Methacholine and PDGF activate store-operated calcium entry in neuronal precursor cells via distinct calcium entry channels. Biol Res 2008; 41: 183–195.
Article Google Scholar - Agell N, Bachs O, Rocamora N, Villalonga P . Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal 2002; 14: 649–654.
Article CAS Google Scholar - Fukuchi M, Tabuchi A, Tsuda M . Transcriptional regulation of neuronal genes and its effect on neural functions: cumulative mRNA expression of PACAP and BDNF genes controlled by calcium and cAMP signals in neurons. J Pharmacol Sci 2005; 98: 212–218.
Article CAS Google Scholar - Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 2008; 14: 738–747.
Article CAS Google Scholar - Ivankovic-Dikic I, Gronroos E, Blaukat A, Barth BU, Dikic I . Pyk2 and FAK regulate neurite outgrowth induced by growth factors and integrins. Nat Cell Biol 2000; 2: 574–581.
Article CAS Google Scholar - Girault JA, Costa A, Derkinderen P, Studler JM, Toutant M . FAK and PYK2/CAKbeta in the nervous system: a link between neuronal activity, plasticity and survival? Trends Neurosci 1999; 22: 257–263.
Article CAS Google Scholar - Kim SY, Bae JC, Kim JY, Lee HL, Lee KM, Kim DS et al. Activation of p38 MAP kinase in the rat dorsal root ganglia and spinal cord following peripheral inflammation and nerve injury. Neuroreport 2002; 13: 2483–2486.
Article CAS Google Scholar - Sodhi A, Biswas SK . Monocyte chemoattractant protein-1-induced activation of p42/44 MAPK and c-Jun in murine peritoneal macrophages: a potential pathway for macrophage activation. J Interferon Cytokine Res 2002; 22: 517–526.
Article CAS Google Scholar - Jimenez-Sainz MC, Fast B, Mayor Jr F, Aragay AM . Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal regulated kinase activation. Mol Pharmacol 2003; 64: 773–782.
Article CAS Google Scholar - Heo JS, Han HJ . ATP stimulates mouse embryonic stem cell proliferation via protein kinase C, phosphatidylinositol 3-kinase/Akt, and mitogen-activated protein kinase signaling pathways. Stem Cells 2006; 24: 2637–2648.
Article CAS Google Scholar - Yao HH, Ding JH, Zhou F, Wang F, Hu LF, Sun T et al. Enhancement of glutamate uptake mediates the neuroprotection exerted by activating group II or III metabotropic glutamate receptors on astrocytes. J Neurochem 2005; 92: 948–961.
Article CAS Google Scholar - Zhou F, Yao HH, Wu JY, Yang YJ, Ding JH, Zhang J et al. Activation of Group II/III metabotropic glutamate receptors attenuates LPS-induced astroglial neurotoxicity via promoting glutamate uptake. J Neurosci Res 2006; 84: 268–277.
Article CAS Google Scholar
Acknowledgements
We thank Dr K Walsh (Tufts University School of Medicine) for providing adenoviral constructs for the WT and dominant-interfering forms of Akt. We thank Dr Y Jia (Jackson Laboratory) for technical support with siRNA TRPC. This work was supported by Grants MH-068212, DA020392, DA023397 and DA024442 from the National Institutes of Health (SB).
Author information
Authors and Affiliations
- Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE, USA
H Yao, F Peng, X Zhu & S J Buch - Department of Pharmacology, Jiangsu Key Laboratory of Neurodegeneration, Nanjing Medical University, Nanjing, Jiangsu, China
Y Fan & G Hu
Authors
- H Yao
You can also search for this author inPubMed Google Scholar - F Peng
You can also search for this author inPubMed Google Scholar - Y Fan
You can also search for this author inPubMed Google Scholar - X Zhu
You can also search for this author inPubMed Google Scholar - G Hu
You can also search for this author inPubMed Google Scholar - S J Buch
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toS J Buch.
Additional information
Edited by RA Knight
Rights and permissions
About this article
Cite this article
Yao, H., Peng, F., Fan, Y. et al. TRPC channel-mediated neuroprotection by PDGF involves Pyk2/ERK/CREB pathway.Cell Death Differ 16, 1681–1693 (2009). https://doi.org/10.1038/cdd.2009.108
- Received: 29 January 2009
- Revised: 26 June 2009
- Accepted: 02 July 2009
- Published: 14 August 2009
- Issue Date: December 2009
- DOI: https://doi.org/10.1038/cdd.2009.108