New findings from genetic association studies of schizophrenia (original) (raw)
Abstract
In the past 20 years, association studies of schizophrenia have evolved from analyses in lesser than 100 subjects of one or two markers in candidate genes to systematic analyses of association at a genome-wide level in samples of thousands of subjects. During this process, much of the emergent literature has been difficult to interpret and definitive findings that have met with universal acceptance have been elusive, largely because studies have been underpowered for such conclusions to be drawn. Nevertheless, in the course of the past few years, a few promising candidate genes have been reported for which the evidence is positive across multiple studies, and more recently, genome-wide association studies have yielded findings of a compelling nature. It is clear that genetic studies in schizophrenia have borne fruit, a process that can be expected to accelerate in the next few years, and that these findings are providing new avenues for research into the pathophysiology of this poorly understood disorder.
Similar content being viewed by others
Introduction
Schizophrenia is a severe psychiatric disorder with a lifetime risk of ∼1%. It is characterized by psychotic symptoms (delusions and hallucinations), apathy, altered emotional reactivity and disorganized behavior.1 Subtle cognitive and behavioral signs are often present from childhood onwards, but the characteristic features generally have their onset in the late teens and early 20s. Outcomes vary, but the typical course is one of relapses followed by only partial remission and a marked reduction in social and occupational functions.
Schizophrenia has been the subject of an extensive series of family, twin and adoption studies (Figure 1), which clearly show variance in population risk is largely the result of genetic factors, with heritability being ⩾80%. Patterns of recurrence in families do not suggest that the generality of schizophrenia can be attributed to a collection of single major genes, and are most compatible with a complex multi-locus model.3, 4 Similar to other disorders with complex inheritance,5 it has been increasingly postulated that most of the genetic variance will be attributable to weak genetic effects as proposed many years ago,6 a hypothesis compatible with meta-analyses of the linkage data.7 Those hypotheses and data do not, however, preclude the existence of highly penetrant mutations in occasional families; indeed, there is now compelling evidence for several such events (St Clair et al.8 and see below).
Figure 1
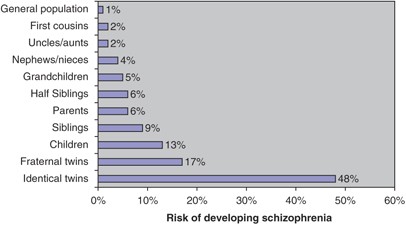
Graph shows the risk of developing schizophrenia in the relatives of a schizophrenic proband. The data are based on the review of studies compiled by Gottesman.2
Given the increasing emphasis by most researchers on small genetic effects, it has been proposed that pure association approaches independent of genetic linkage offer the greatest potential in schizophrenia and other complex disorders,9 as they are better powered to detect such effects. However, the requirement to genotype hundreds of thousands of markers to attain even a modest degree of coverage of common human genomic variation has until recently restricted the application of pure association approaches to studies of functional candidate genes. Until now, the results of such studies have been disappointing, and in general, approaches that have been based on position rather than on functional hypothesis appear to have met with greater success. In this review, our focus is on the results emerging from new approaches rather than going over old ground. Nevertheless, it should be noted that several candidate susceptibility genes have been identified on the basis of an initial report of linkage, and for some of these, for example dysbindin (DTNBP1),10 neuregulin 1 (NRG1)11 and D -amino oxidase activator (DAOA),12 although the evidence is not decisive, there is a substantial amount of support from follow-up studies.13 Moreover, the gene encoding Disrupted in Schizophrenia 1 (DISC1) was also identified based on positional data,15 which was disrupted by a balanced translocation that co-segregates with schizophrenia and other major mental disorders in a single family.8 Although subsequent population studies have not provided much evidence for additional associations with common variation in DISC1, DISC1 seems likely to be a true schizophrenia susceptibility gene, which is a perfect illustration of the desirability of retaining both linkage and association approaches as tools for gene discovery. We will not consider the classic positional candidate genes further in this review, although we note that many potentially important insights and new hypotheses concerning the pathophysiology of schizophrenia have been the gained through their study.13, 14, 16, 17 However, whether these are relevant to the disorder is yet to be confirmed based on evidence supporting their status as susceptibility genes, failing which a successful therapeutic intervention based on these insights is required.
Functional candidate genes
The pathophysiology of schizophrenia is largely unknown but many hypotheses have been proposed. The choice of putative functional candidate genes is therefore almost unbounded by membership of any functional category. The largest number of studies relate to targets that encode proteins involved in neurotransmission, particularly dopaminergic and serotonergic, but it is difficult to think of any neurotransmitter system that has no plausibility, or that has not been the topic of investigation at one time or the other. Reflecting the widely held neuro-developmental hypothesis of schizophrenia, the other popular category of candidate genes is that relating to brain development. Regarding other disorders, the quality of the functional candidate gene literature reflects the evolution of technology and genomic resources of the times, with the vast majority of studies comprising tests of one or two variants in small samples. Even studies based on systematic mutation scanning have focused on subsets of the functional elements of genes, typically exons and promoter regions. Given these considerations, particularly the small genetic effect sizes (odds ratio (OR) <1.1) now thought typical of all but a small proportion of common susceptibility variants (Zeggini et al.18), in retrospect, it would have been more surprising had the results been more reproducible than they have been. Negative findings abound, as do positive findings that have either not been replicated or have seemed insufficiently convincing because there have been few, if any, attempts to replicate them. The failure of candidate gene studies does not, however, invalidate the approach. As better information about the pathophysiology of schizophrenia emerges from the application of large samples to genome-wide association approaches, the application of those same large samples to detailed studies of candidate genes may meet with greater levels of success.
Meanwhile, to what extent any of the functional candidate data represent true positives is sufficiently debatable for there to be a wide spectrum of opinion concerning their interpretation. Nevertheless, there are a number of comments that can be made with considerable confidence. First, in schizophrenia nothing has emerged with support on a par with, for example, the evidence for CFH in macular degeneration,19 APOE in Alzheimer's disease20 or human leukocyte antigen in type I diabetes.21 Second, while most of the reported positives may not withstand testing in adequately powered samples, no functional candidate has yet been excluded from containing common risk alleles, let alone rare low penetrance alleles. Third, if as may be the case (see below), schizophrenia is a highly polygenic disorder, a substantial number of the hundreds of positive findings that are currently not well supported are likely to be true positives.
In the presence of a vast and inconsistent literature, a new approach to schizophrenia research has been the establishment of the SzGene database,22 the goal of which is to collate association data and, where possible, perform systematic meta-analyses. The resource is online and is free to access (http://www.schizophreniaforum.org/res/sczgene/default.asp). On the basis of the April 2007 freeze, which contained data from 1179 publications relating to 3608 variants in 516 genes, the curators of the database performed meta-analyses where data were available from at least four case–control samples. Of 118 variants in 52 genes that met this criterion, 24 variants were nominally (_P_⩽0.05) associated with schizophrenia. Only two of these were associated at a P<0.001, one was in the dopamine DRD2 receptor while the other was one in the gene TPH1 which is one of two enzymes that encodes tryptophan hydroxylase, the rate limiting enzyme in the synthesis of serotonin, a neurotransmitter whose function is targeted by several second generation antipsychotics.
It may be relevant that both were among those single nucleotide polymorphisms (SNPs) that had been tested in the fewest number of subjects (473 and 879 cases, respectively). Nevertheless, in particular, the variant in DRD2, rs6277, has a strong functional plausibility, in that although it is synonymous, it appears to alter mRNA stability and translational efficiency,23 with the associated allele conferring higher mRNA stability and translational efficiency. The gene itself is also a particularly appealing candidate because the hypothesis that psychotic symptoms arise from excess dopamine is one of the more convincing hypotheses relating to schizophrenia, and this specifically relates to dopamine activity at DRD2 as there is a strong correlation between the therapeutic efficacy of anti-psychotic drugs and their affinity for blocking this receptor.24 Although the most recent data in the SzGene database (Figure 2) continue to support the finding at DRD2 as does a recent meta-analysis of TPH1,25 both genes require additional support in larger samples before the findings can be viewed with confidence.
Figure 2
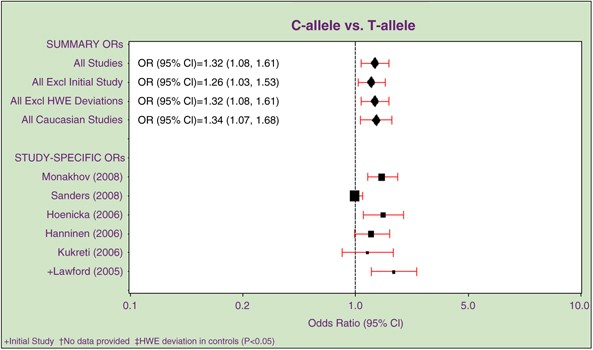
Example screen-shot to show the graphical output from SzGene website. In this example, the graph shows the pooled odds ratio estimates of all published association studies for polymorphism rs6277 in DRD2.
The authors additionally ranked the associated genes using three sets of criteria, namely the amount of evidence, the consistency of the evidence and protection from bias.26 In the associated genes, four SNPs met the most stringent category for each criterion. These SNPs were in the D1 dopamine receptor (DRD1), DTNBP1, TPH1 and methylenetetrahydrofolate reductase (MTHFR). However, a recent meta-analysis that included additional data27 no longer supports the marker in MTHFR, and therefore this gene will not be considered further.
The association at DRD1 together with the data on DRD2 would appear to provide etiological credibility to the dopamine hypothesis of schizophrenia, but it is premature to consider DRD1, a confirmed susceptibility gene. Not a single positive association study has yet been reported for this gene. Although this is not incompatible with a true finding of small effect, in the meta-analysis of Allen et al.22 the statistical support was weak (_P_=0.037) and would not even be gene-wide significant allowing for other variants at this locus. Moreover, the meta-analysis was based on only 725 cases and 1000 controls. In this context, it should be noted that previously, another dopamine receptor, DRD3 showed somewhat stronger statistical support which survived several waves of meta-analysis only for the significance to disappear as the sample size was increased to >10 000 subjects.28
DTNBP1, which codes for the dystrobrevin binding protein 1 (commonly known as dysbindin), is located on 6p24-22 within a schizophrenia linkage region10 and has been mentioned briefly earlier. The protein is expressed pre- and post-synaptically with high levels of expression in the cerebellum and the hippocampus.29 After the initial report of association,10 a large number of positive studies have been reported. However, there has been little consistency in the specific associated alleles or haplotypes,30 which suggests that if the association is correct, there is considerable variation in the linkage disequilibrium structure at the locus, or that there are multiple functional variants that contribute to the association. The greater consistency evident from the SzGene meta-analysis adds weight to the hypothesis that DTNBP1 is a true susceptibility gene for schizophrenia, although caveats remain concerning the modest support (_P_=0.003) from the meta-analysis. At a functional level, several of the associated haplotypes are associated with reduced DTNBP1 expression in the post-mortem brain31 compatible with other observations of underexpression of DTNBP1 in schizophrenia.29, 32 How DTNBP1 might be involved in schizophrenia is the topic of intense investigation, but leading hypotheses are that as a component of the biogenesis of lysosome-related organelles complex 1 (BLOC-1), variation in DTNBP1 might perturb pre-synaptic trafficking of neurotransmitter vesicles, in particular, those containing glutamate and dopamine33, 34 or impair internalization of dopamine DRD2 receptors.35
The development of the SzGene database is a welcome and timely addition to the schizophrenia genetics research environment, and represents a major step in facilitating an unbiased evaluation of the extensive genetic literature. The interested reader is therefore directed to a recent review by its curators in which some of its strengths and weaknesses are more extensively discussed.36
Genome-wide association studies
Genome-wide association studies (GWASs) have been widely embraced by those researching the genetics of complex disorders because they incorporate the power to detect small effects with the huge advantage of the positional genetics design, which requires no specific knowledge of pathogenesis. For some phenotypes, GWAS approaches have transformed our knowledge of genetic susceptibility37 and the early evidence for schizophrenia suggest similar gains might accrue.
At the time of writing, there have been six published GWASs of schizophrenia, of which three have been based on DNA pooling and one has been limited to non-synonymous SNPs.38 In the first study based on individual genotyping (178 cases and 144 controls), the authors reported only the strongest result (_P_=3.7 × 10−7), which was equidistant (∼350 kb) from two genes, colony stimulating factor 2 receptor alpha (CSF2RA) and short stature homeobox isoform b (SHOX). CSF2RA is a subunit of a cytokine receptor, which regulates granulocytes and macrophages, whereas SHOX is a putative transcription factor that is not present in some people with short stature. The authors targeted CSF2RA and the neighboring interleukin 3 receptor alpha (IL3RA) in a second small sample (71 cases and 31 controls) for sequencing. A number of significant associations were observed across the two genes, but to what extent these relate to the findings from the GWAS stage is unclear. A priori, replication in such a small sample would seem extremely unlikely unless the effect sizes are much larger that at a typical complex disease locus.
The second GWAS with individual genotypes39 was larger (738 cases and 733 controls), but none of the findings achieved genome-wide significance,40 and in the absence of follow-up data it is impossible to draw conclusions regarding any specific locus. It may be relevant that the sample was ethnically heterogeneous, which may have adversely impacted upon power.
The only other GWAS published with individual genotypes41 was based on an initial GWAS of 479 UK cases and 2937 UK controls, with sequential follow-up of loci surpassing a threshold P <10−5 in up to 6829 cases and 9897 controls from Europe, the United States, Australia, Israel, Japan and China. Of 12 loci followed up, strong, replicated support (P<5 × 10−4) was observed in 3 of the loci, whereas another two were nominally significant. The distribution of test statistics for the 12 follow-up SNPs was highly improbable (_P_=9 × 10−8) under the null hypothesis that these were a random set of SNPs unrelated to disease, strongly supporting the existence of true associations. The best supported locus was in the vicinity of a putative transcription regulator (zinc finger protein 804A or ZNF804A). Although the evidence (_P_=1 × 10−7) for association fell short of genome-wide significance (_P_=5 × 10−8, Dudbridge and Gusnanto40), this threshold was surpassed when the affected phenotype was extended to include bipolar disorder (_P_=9 × 10−9), a phenotype for which there is prior evidence for shared genetic risk with schizophrenia.42 Thus, the ZNF804A locus is very likely a true susceptibility locus for schizophrenia and bipolar disorder, and the main challenge now is, under the assumption that it is a transcription factor, to understand what targets are regulated and which of these are relevant to pathogenesis.
It has been argued that as schizophrenia is associated with reduced fecundity, common variants may not be relevant to its etiology, the idea being that selection pressure would remove such variants from the population. The identification of even one common susceptibility variant for schizophrenia now allows this hypothesis to be refuted, and it seems virtually inevitable that because one common risk variant of small effect escapes purification (the OR for ZNF804A in the replication samples was 1.09), there will be many others. Power analysis of our study leads to similar conclusions. As we only had power of <0.001 to detect an effect of this size in any specific gene, it is probable this low power was offset by the existence of hundreds of genes of similar effect size, one of which we detected.
Three GWASs38, 44, 45 were based on genotyping of pooled DNA samples, whereby DNA from cases are mixed together in a single quantitative assay as are controls and the differences in allele frequency are estimated. All three identified interesting rather than definitive findings, but of particular note, the study of Shifman and colleagues provided strong evidence for a female-specific association between reelin (RELN) and schizophrenia, a finding they were able to replicate in samples from Europe and the United States (meta-analysis _P_=8.8 × 10−7, uncorrected for models and gender-specific tests). Although awaiting further replication, the association with reelin is compatible with the widely held hypothesis of schizophrenia as a neuro-developmental disorder, reelin being involved in corticogenesis and also being associated with an autosomal-recessive form of lissencephaly.46
The total combined sample sizes reported thus far for which there are GWAS data based on individual genotyping are still small, numbering fewer than 1500 cases, and power to capture small genetic effects is still limited. Nevertheless, the findings provide cause for optimism that larger scale endeavors will meet with success in identifying additional loci. One caveat with respect to these larger endeavors is that unlike almost all other medical disorders, the diagnosis of schizophrenia is unsupported by validating laboratory tests. As a result, it is at least conceivable that efforts to extract more power by combining multiple samples might be hampered by increases in the amount of heterogeneity present.47 To what extent this concern will operate in practice is a real, but open question.
Copy number variants
It is now clear that copy number variants (CNVs) are ubiquitous in the population, a conclusion that stands even though early studies appear to have overestimated the physical size of the sequences contained within each CNV.48 This has led to the proposition that, in addition to their known role in a number of fairly uncommon syndromes, CNVs might contribute to more common phenotypes. Among these, neuro-developmental disorders including autism and mental retardation (MR) have been the most prominent focus of CNV research, and the first generation of genome-wide CNV scans have shown that de novo CNV mutations of about 100 kb or more contribute to about 10% of cases of these disorders (for example, Sebat et al.49 and Autism Genome Project Consortium50).
The idea that a proportion of schizophrenia might be caused by CNVs has been around for some time, indeed until recently, this was the only class of DNA variation where a pathogenic role was virtually undisputed. Thus, the 22q11DEL gives rise to dramatic increase in the risk of schizophrenia,51, 52 along with the other variable features of the velo-cardio-facial syndrome. To date, several specific schizophrenia risk genes within the deleted region have been suggested, but as yet none are supported by multiple independent replications.
There is now compelling evidence that additional CNVs contribute to schizophrenia. Kirov and colleagues43, 44 detected a single de novo event after scanning 93 subjects; a duplication of 1.4 Mb of chromosome 15. They also observed a rare transmitted deletion that affects part of neurexin 1 (NRXN1). Although not de novo, it was also carried by an affected sibling and was of particular interest as deletions of NRXN1 had been observed in both autism and Mental Retardation (MR) (for example, Autism Genome Project Consortium50). Moreover, an NRXN1 CNV has been independently observed in a schizophrenic proband,53 and a large consortium has subsequently shown an increased rate of CNVs spanning exons of NRXN1 in schizophrenia.54 Thus, there is now strong evidence that reduced function of NRXN1 increases risk of a number of neuro-developmental disorders, including schizophrenia. Interestingly, the de novo duplication reported by Kirov et al.43, 44 also included the gene encoding amyloid-β A4 precursor protein-binding (APBA2), a protein with which NRXN1 interacts. Together, NRXN1 and APBA2 play a role in synaptic development and function, processes that are likely fundamental to schizophrenia etiology.
De novo CNV events were also uncommon in a study of childhood onset schizophrenia53 in stark contrast to another study where a comparison of the de novo rate in cases and controls revealed a rate of about 10% in cases and <2% in controls.55 The design of the study of Kirov and colleagues is very likely to underestimate the de novo rate, but it is not evident why the other two studies differ.53, 55 Which is the more representative remains to be elucidated.
Although the rate of de novo CNVs in schizophrenia is unresolved, three studies have now shown that the total burden (number of events or number of genes spanned by those events) of rare CNVs is increased in the disorder.53, 56, 57 Moreover, two of these have been large enough to identify association with specific rare loci as well as with total burden, and here the findings are in striking agreement.56, 57 The same two deletions were associated with schizophrenia in both studies, one at 1q21.1 (position 142.5–145.5 Mb), the other at 15q13.2 (28–31 Mb). The CNVs were rare and had strong effects on disease risk, with ORs of 6.6 and 17.9, respectively. This is now becoming a familiar pattern, and each CNV locus has also been associated with other phenotypes—in particular, the 1q21.1 locus is associated with microcephaly, various physical abnormalities and neuro-developmental abnormalities, including MR, autism and attention deficit hyperactivity disorder (ADHD).58 Each CNV spans multiple genes that are clearly high-priority candidate genes for the disorder, although it remains to be proven that more subtle variation within genes mapping to these regions contributes to disease susceptibility.
Despite the strong evidence for the involvement of specific CNV loci, it is clear that none of them is sufficient to cause disease, as each has been observed in individuals with no history of psychosis. Thus, although at least some of the loci are of fairly substantial effect, expression of disorder might still require environmental contributions or additional risk variants. Similar uncertainties surround the interpretation of the excess burden analysis. Those results could, in principle, be attributed to a number of rare highly penetrant CNVs or to more subtle effects. Finally, it is important to appreciate that we are as yet uncertain to what extent CNVs contribute to disorder. Currently, the specific associated CNVs are involved in only about 1% of cases, although this figure will probably increase as better-resolution platforms are deployed. These uncertainties aside, the early work shows CNV analysis has the potential to pinpoint novel pathogenic mechanisms and provides a strong rationale for further work in this area as a complementary approach to more traditional SNP-based analyses.
Conclusions
Association studies in schizophrenia have evolved in parallel with genome analysis technology, and mirror developments in complex disease genetics as a whole. Until recently, although there have been a number of findings particularly based on positional approaches for which the evidence is credible, and which are very likely to include true susceptibility genes, there have been no findings where the support is unequivocal and which point to specific alleles or haplotypes. Some of the more robust findings for other disorders doubtless reflect the fact that for some loci, the effect sizes are at the larger end of the spectrum seen in common disease, but most of the success is recent and has come from GWAS applied to large patient and control samples and, of vital importance, follow-up analyses in even larger samples. Such approaches are currently being applied to schizophrenia and early results are promising. The application of genome-wide approaches has also revealed that sub-microscopic chromosomal abnormalities play a role in at least some cases, and this figure will probably increase with the use of technologies with higher resolution.48 The foregoing suggests that efforts in the genetics of schizophrenia are bearing fruit, and there are strong grounds for being optimistic that research in these disorders is poised to benefit further from the same advances in genomics and post-genomics that are also being successfully applied to other common non-psychiatric disorders. The success of this work will depend among other things on the assembly of large, well-phenotyped patient samples, effective collaboration and sharing of patient resources, and the ability to handle and analyze increasingly large and complex data sets. Moreover, as more risk loci are identified, there will be an increasing imperative to focus on translating genetic findings into clinical benefit through a greater understanding of pathogenic mechanisms, improved classification and the development of new interventions.
References
- Cardno, A. G. & Gottesman, I. I. Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. Am. J. Med. Genet. 97, 12–17 (2000).
Article CAS PubMed Google Scholar - Gottesman, I. I. Schizophrenia Genesis: The Origins of Madness, Henry Holt & Company Inc.: New York, NY, USA, 296pp (1991).
Google Scholar - McGue, M. & Gottesman, I. I. A single dominant gene still cannot account for the transmission of schizophrenia. Arch. Gen. Psychiatry 46, 478–480 (1990).
Article Google Scholar - Risch, N. Linkage strategies for genetically complex traits. I. Multilocus models. Am. J. Hum. Genet. 46, 222–228 (1990).
CAS PubMed PubMed Central Google Scholar - Wellcome Trust Case Control Consortium. Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 447, 661–678 (2007).
Article Google Scholar - Gottesman, I. I. & Shields, J. A polygenic theory of schizophrenia. Proc. Natl Acad. Sci. USA 58, 199–205 (1967).
Article CAS PubMed PubMed Central Google Scholar - Lewis, C. M., Levinson, D. F., Wise, L. H., DeLisi, L. E., Straub, R. E., Hovatta, I. et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: schizophrenia. Am. J. Hum. Genet. 73, 34–48 (2003).
Article CAS PubMed PubMed Central Google Scholar - St Clair, D., Blackwood, D., Muir, W., Carothers, A., Walker, M., Spowart, G. et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet 336, 13–16 (1990).
Article CAS PubMed Google Scholar - Risch, N. & Merikangas, K. The future of genetic studies of complex human diseases. Science 273, 1516–1517 (1996).
CAS PubMed Google Scholar - Straub, R. E., Jiang, Y., MacLean, C. J., Ma, Y., Webb, B. T., Myakishev, M. V. et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am. J. Hum. Genet. 71, 337–348 (2002).
Article CAS PubMed PubMed Central Google Scholar - Stefansson, H., Sigurdsson, E., Steinthorsdottir, V., Bjornsdottir, S., Sigmundsson, T., Ghosh, S. et al. Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 71, 877–892 (2002).
Article PubMed PubMed Central Google Scholar - Chumakov, I., Blumenfeld, M., Guerassimenko, O., Cavarec, L., Palicio, M., Abderrahim, H. et al. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc. Natl Acad. Sci. USA 99, 13675–13680 (2002).
Article CAS PubMed PubMed Central Google Scholar - Owen, M. J., Craddock, N. & O’Donovan, M. C. Schizophrenia: genes at last? Trends Genet. 21, 518–525 (2005).
Article CAS PubMed Google Scholar - Owen, M. J., O’Donovan, M. C. & Harrison, P. J. Schizophrenia: a genetic disorder of the synapse? BMJ 330, 158–159 (2005).
Article PubMed PubMed Central Google Scholar - Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A. et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423 (2000).
Article CAS PubMed Google Scholar - Harrison, P. J. & Weinberger, D. R. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 10, 40–68 (2005); image 5.
Article CAS PubMed Google Scholar - Ross, C. A., Margolis, R. L., Reading, S. A., Pletnikov, M. & Coyle, J. T. Neurobiology of schizophrenia. Neuron 52, 139–153 (2006).
Article CAS PubMed Google Scholar - Zeggini, E., Scott, L. J., Saxena, R., Voight, B. F., Marchini, J. L., Hu, T. et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 40, 638–645 (2008).
Article CAS PubMed PubMed Central Google Scholar - Klein, R. J., Zeiss, C., Chew, E. Y., Tsai, J. Y., Sackler, R. S., Haynes, C. et al. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 (2005).
Article CAS PubMed PubMed Central Google Scholar - Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S. et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl Acad. Sci. USA 90, 1977–1981 (1993).
Article CAS PubMed PubMed Central Google Scholar - Todd, J. A., Bell, J. I. & McDevitt, H. O. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature 329, 599–604 (1987).
Article CAS PubMed Google Scholar - Allen, N. C., Bagade, S., McQueen, M. B., Ioannidis, J. P., Kavvoura, F. K., Khoury, M. J. et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat. Genet. 40, 827–834 (2008).
Article CAS PubMed Google Scholar - Duan, J., Wainwright, M. S., Comeron, J. M., Saitou, N., Sanders, A. R., Gelernter, J. et al. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum. Mol. Genet. 12, 205–216 (2003).
Article CAS PubMed Google Scholar - Seeman, P. & Lee, T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 188, 1217–1219 (1975).
Article CAS PubMed Google Scholar - Watanabe, Y., Nunokawa, A., Kaneko, N. & Someya, T. The tryptophan hydroxylase 1 (TPH1) gene and risk of schizophrenia: a moderate-scale case–control study and meta-analysis. Neurosci. Res. 59, 322–326 (2007).
Article CAS PubMed Google Scholar - Ioannidis, J. P., Boffetta, P., Little, J., O’Brien, T. R., Uitterlinden, A. G., Vineis, P. et al. Assessment of cumulative evidence on genetic associations: interim guidelines. Int. J. Epidemiol. 37, 120–132 (2008).
Article PubMed Google Scholar - Jonsson, E. G., Larsson, K., Vares, M., Hansen, T., Wang, A. G., Djurovic, S. et al. Two methylenetetrahydrofolate reductase gene (MTHFR) polymorphisms, schizophrenia and bipolar disorder: an association study. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 147B, 976–982 (2008).
Article PubMed Google Scholar - Ma, G., He, Z., Fang, W., Tang, W., Huang, K., Li, Z. et al. The Ser9Gly polymorphism of the dopamine D3 receptor gene and risk of schizophrenia: an association study and a large meta-analysis. Schizophr. Res. 101, 26–35 (2008).
Article PubMed Google Scholar - Talbot, K., Eidem, W. L., Tinsley, C. L., Benson, M. A., Thompson, E. W., Smith, R. J. et al. Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J. Clin. Invest. 113, 1353–1363 (2004).
Article CAS PubMed PubMed Central Google Scholar - Williams, N. M., O’Donovan, M. C. & Owen, M. J. Is the dysbindin gene (DTNBP1) a susceptibility gene for schizophrenia? Schizophr. Bull. 31, 800–805 (2005).
Article PubMed Google Scholar - Bray, N. J., Preece, A., Williams, N. M., Moskvina, V., Buckland, P. R., Owen, M. J. et al. Haplotypes at the dystrobrevin binding protein 1 (DTNBP1) gene locus mediate risk for schizophrenia through reduced DTNBP1 expression. Hum. Mol. Genet. 14, 1947–1954 (2005).
Article CAS PubMed Google Scholar - Weickert, C. S., Straub, R. E., McClintock, B. W., Matsumoto, M., Hashimoto, R., Hyde, T. M. et al. Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch. Gen. Psychiatry 61, 544–655 (2004).
Article CAS PubMed Google Scholar - Kumamoto, N., Matsuzaki, S., Inoue, K., Hattori, T., Shimizu, S., Hashimoto, R. et al. Hyperactivation of midbrain dopaminergic system in schizophrenia could be attributed to the down-regulation of dysbindin. Biochem. Biophys. Res. Commun. 345, 904–909 (2006).
Article CAS PubMed Google Scholar - Numakawa, T., Yagasaki, Y., Ishimoto, T., Okada, T., Suzuki, T., Iwata, N. et al. Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum. Mol. Genet. 13, 2699–2708 (2004).
Article CAS PubMed Google Scholar - Lizuka, Y., Sei, Y., Weinberger, D. R. & Straub, R. E. Evidence that the BLOC-1 protein dysbindin modulates dopamine D2 receptor internalization and signaling but not D1 internalization. J. Neurosci. 27, 12390–12395 (2007).
Article Google Scholar - Bertram, L. Genetic research in schizophrenia: new tools and future perspectives. Schizophr. Bull. 34, 806–812 (2008).
Article PubMed PubMed Central Google Scholar - Manolio, T. A., Brooks, L. D. & Collins, F. S. A HapMap harvest of insights into the genetics of common disease. J. Clin. Invest. 118, 1590–1605 (2008).
Article CAS PubMed PubMed Central Google Scholar - Mah, S., Nelson, M. R., Delisi, L. E., Reneland, R. H., Markward, N., James, M. R. et al. Identification of the semaphorin receptor PLXNA2 as a candidate for susceptibility to schizophrenia. Mol. Psychiatry 11, 471–478 (2006).
Article CAS PubMed Google Scholar - Sullivan, P. F., Lin, D., Tzeng, J. Y., van den Oord, E., Perkins, D., Stroup, T. S. et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol. Psychiatry 13, 570–584 (2008).
Article CAS PubMed PubMed Central Google Scholar - Dudbridge, F. & Gusnanto, A. Estimation of significance thresholds for genomewide association scans. Genet. Epidemiol. 32, 227–234 (2008).
Article PubMed PubMed Central Google Scholar - O’Donovan, M. C., Craddock, N., Norton, N., Williams, H., Peirce, T., Moskvina, V. et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 40, 1053–1055 (2008).
Article PubMed Google Scholar - Craddock, N., O’Donovan, M. C. & Owen, M. J. Genes for schizophrenia and bipolar disorder? Implications for psychiatric nosology. Schizophr. Bull. 32, 9–16 (2006).
Article PubMed Google Scholar - Kirov, G., Gumus, D., Chen, W., Norton, N., Georgieva, L., Sari, M. et al. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum. Mol. Genet. 17, 458–465 (2008).
Article CAS PubMed Google Scholar - Kirov, G., Zaharieva, I., Georgieva, L., Moskvina, V., Nikolov, I., Cichon, S. et al. A genome-wide association study in 574 schizophrenia trios using DNA pooling. Mol. Psychiatry (2008); e-pub ahead of print 11 March 2008.
- Shifman, S., Johannesson, M., Bronstein, M., Chen, S. X., Collier, D. A., Craddock, N. J. et al. Genome-wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women. PLoS Genet. 4, e28 (2008).
Article PubMed PubMed Central Google Scholar - Hong, S. E., Shugart, Y. Y., Huang, D. T., Shahwan, S. A., Grant, P. E., Hourihane, J. O. et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 26, 93–96 (2000).
Article CAS PubMed Google Scholar - Craddock, N., O’Donovan, M. C. & Owen, M. J. Genome-wide association studies in psychiatry: lessons from early studies of non-psychiatric and psychiatric phenotypes. Mol. Psychiatry 13, 649–653 (2008).
Article CAS PubMed Google Scholar - McCarroll, S. A., Kuruvilla, F. G., Korn, J. M., Cawley, S., Nemesh, J., Wysoker, A. et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 40, 1166–1174 (2008).
Article CAS PubMed Google Scholar - Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T. et al. Strong association of de novo copy number mutations with autism. Science 316, 445–449 (2007).
Article CAS PubMed PubMed Central Google Scholar - Autism Genome Project Consortium. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 39, 319–328 (2007).
Article Google Scholar - Lindsay, E. A., Greenberg, F., Shaffer, L. G., Shapira, S. K., Scambler, P. J. & Baldini, A. Submicroscopic deletions at 22q11.2: variability of the clinical picture and delineation of a commonly deleted region. Am. J. Med. Genet. 56, 191–197 (1995).
Article CAS PubMed Google Scholar - Murphy, K. C., Jones, L. A. & Owen, M. J. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch. Gen. Psychiatry 56, 940–945 (1999).
Article CAS PubMed Google Scholar - Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M. et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543 (2008).
Article CAS PubMed Google Scholar - Rujescu, D., Ingason, A., Cichon, S., Pietilainen, O. P., Barnes, M. R., Toulopoulou, T. et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. (2008); e-pub ahead of print 22 October 2008.
- Xu, B., Roos, J. L., Levy, S., van Rensburg, E. J., Gogos, J. A. & Karayiorgou, M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 40, 880–885 (2008).
Article CAS PubMed Google Scholar - International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241 (2008).
Article Google Scholar - Stefansson, H., Rujescu, D., Cichon, S., Pietilainen, O. P., Ingason, A., Steinberg, S. et al. Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–236 (2008).
Article CAS PubMed PubMed Central Google Scholar - Mefford, H. C., Sharp, A. J., Baker, C., Itsara, A., Jiang, Z., Buysse, K. et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 359, 1685–1699 (2008).
Article CAS PubMed PubMed Central Google Scholar
Author information
Authors and Affiliations
- Department of Psychological Medicine, School of Medicine, Cardiff Universty, Heath Park, Cardiff, UK
Hywel J Williams, Michael J Owen & Michael C O'Donovan
Authors
- Hywel J Williams
You can also search for this author inPubMed Google Scholar - Michael J Owen
You can also search for this author inPubMed Google Scholar - Michael C O'Donovan
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toMichael C O'Donovan.
Rights and permissions
About this article
Cite this article
Williams, H., Owen, M. & O'Donovan, M. New findings from genetic association studies of schizophrenia.J Hum Genet 54, 9–14 (2009). https://doi.org/10.1038/jhg.2008.7
- Received: 31 October 2008
- Accepted: 06 November 2008
- Published: 09 January 2009
- Issue Date: January 2009
- DOI: https://doi.org/10.1038/jhg.2008.7