Sequence analysis of mutations and translocations across breast cancer subtypes (original) (raw)
- Letter
- Open access
- Published: 20 June 2012
- Kristian Cibulskis1 na1,
- Claudia Rangel-Escareno4 na1,
- Kristin K. Brown5 na1,
- Scott L. Carter1,
- Abbie M. Frederick1,
- Michael S. Lawrence1,
- Andrey Y. Sivachenko1,
- Carrie Sougnez1,
- Lihua Zou1,
- Maria L. Cortes1,
- Juan C. Fernandez-Lopez4,
- Shouyong Peng2,
- Kristin G. Ardlie1,
- Daniel Auclair1,
- Veronica Bautista-Piña6,
- Fujiko Duke1,
- Joshua Francis1,
- Joonil Jung1,
- Antonio Maffuz-Aziz6,
- Robert C. Onofrio1,
- Melissa Parkin1,
- Nam H. Pho1,
- Valeria Quintanar-Jurado4,
- Alex H. Ramos1,
- Rosa Rebollar-Vega4,
- Sergio Rodriguez-Cuevas6,
- Sandra L. Romero-Cordoba4,
- Steven E. Schumacher1,2,
- Nicolas Stransky1,
- Kristin M. Thompson1,
- Laura Uribe-Figueroa4,
- Jose Baselga3,7,
- Rameen Beroukhim1,2,3,8,
- Kornelia Polyak2,3,9,
- Dennis C. Sgroi3,10,
- Andrea L. Richardson2,3,11,
- Gerardo Jimenez-Sanchez4 nAff15,
- Eric S. Lander1,3,12,
- Stacey B. Gabriel1,
- Levi A. Garraway1,2,3,
- Todd R. Golub1,3,13,14,
- Jorge Melendez-Zajgla4,
- Alex Toker3,5,
- Gad Getz1,
- Alfredo Hidalgo-Miranda4 &
- …
- Matthew Meyerson1,2,3,8
Nature volume 486, pages 405–409 (2012)Cite this article
- 46k Accesses
- 1078 Citations
- 44 Altmetric
- Metrics details
Subjects
Abstract
Breast carcinoma is the leading cause of cancer-related mortality in women worldwide, with an estimated 1.38 million new cases and 458,000 deaths in 2008 alone1. This malignancy represents a heterogeneous group of tumours with characteristic molecular features, prognosis and responses to available therapy2,3,4. Recurrent somatic alterations in breast cancer have been described, including mutations and copy number alterations, notably ERBB2 amplifications, the first successful therapy target defined by a genomic aberration5. Previous DNA sequencing studies of breast cancer genomes have revealed additional candidate mutations and gene rearrangements6,7,8,9,10. Here we report the whole-exome sequences of DNA from 103 human breast cancers of diverse subtypes from patients in Mexico and Vietnam compared to matched-normal DNA, together with whole-genome sequences of 22 breast cancer/normal pairs. Beyond confirming recurrent somatic mutations in PIK3CA11, TP536, AKT112, GATA313 and MAP3K110, we discovered recurrent mutations in the CBFB transcription factor gene and deletions of its partner RUNX1. Furthermore, we have identified a recurrent MAGI3–AKT3 fusion enriched in triple-negative breast cancer lacking oestrogen and progesterone receptors and ERBB2 expression. The MAGI3–AKT3 fusion leads to constitutive activation of AKT kinase, which is abolished by treatment with an ATP-competitive AKT small-molecule inhibitor.
Similar content being viewed by others
Main
Breast cancers are classified according to gene-expression subtypes: luminal A, luminal B, Her2-enriched (Her2 is also known as ERBB2), and basal-like14. Luminal subtypes are associated with expression of oestrogen and progesterone receptors and differentiated luminal epithelial cell markers. The subtypes differ in genomic complexity, key genetic alterations and clinical prognosis2,3,4,15. To discover genomic alterations in breast cancers, we performed whole-genome and whole-exome sequencing of 108 primary, treatment-naive, breast carcinoma/normal DNA pairs from all major expression subtypes (Table 1 and Supplementary Tables 1–3), 17 cases by whole-exome and whole-genome sequencing, 5 cases by whole-genome sequencing alone, and 86 cases by whole-exome sequencing alone.
Table 1 Sample collections successfully completed sequencing and analysis
In total, whole-exome sequencing was performed on 103 tumour/normal pairs, 54 from Mexico and 49 from Vietnam, targeting 189,980 exons comprising 33 megabases (Mb) of the genome and with a median of 85.1% of targeted bases covered at least 30-fold across the sample set. This analysis revealed a total of 4,985 candidate somatic substitutions (see https://confluence.broadinstitute.org/display/CGATools/MuTect for methods and data sets) and insertions/deletions (indels, see https://confluence.broadinstitute.org/display/CGATools/Indelocator for methods) in the target protein-coding regions and the adjacent splice sites, ranging from 14 to 307 putative events in individual samples (Supplementary Table 4). These mutations represented 3,153 missense, 1,157 silent, 242 nonsense, 97 splice site, 194 deletions, 110 insertions and 32 other mutations (Supplementary Table 5). The total mutation rate was 1.66 per Mb (range 0.47–10.5) with a non-silent mutation rate of 1.27 per Mb (range 0.31–8.05), similar to previous reports in breast carcinoma6,7,8,9. The mutation rate in breast cancer exceeds that of haematologic malignancies and prostate cancer, but is significantly lower than in lung cancer and melanoma10,16,17,18,19. The most common mutation events observed are C to T transition events in CpG dinucleotides (Fig. 1 and Supplementary Fig. 4).
Figure 1: Most significantly mutated genes in breast cancer as determined by whole-exome sequencing ( n = 103).
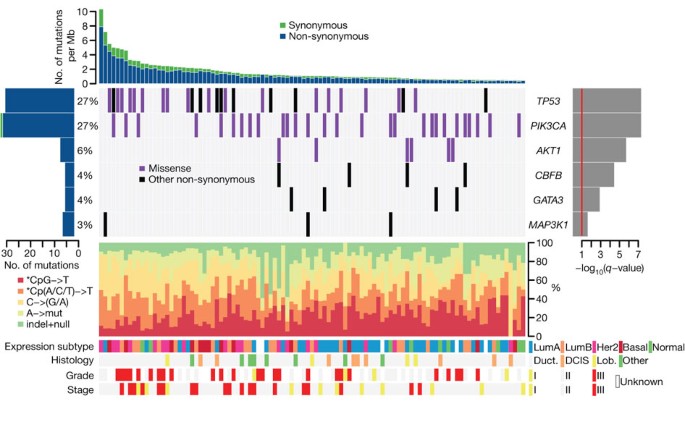
Upper histogram, rates of sample-specific mutations (substitutions and indels). Green, synonymous; blue, non-synonymous. Left histogram, number of mutations per gene and percentage of samples affected (colour coding as in upper histogram). Central heat map, distribution of significant mutations across sequenced samples (‘Other non-synonymous’ mutations: nonsense, indel and splice-site). Right histogram, −log10 score of MutSig q value. Red line at q = 0.1. Lower chart: top, rates of non-silent mutations within categories indicated by legend; bottom, key molecular features of samples in each column. DCIS, ductal carcinoma in situ; Duct., infiltrating ductal carcinoma; Lob., infiltrating lobular carcinoma; Lum, luminal.
We performed validation experiments on 494 candidate mutations (representing all significantly mutated genes and genes in significantly mutated gene sets) using a combination of mass-spectrometric genotyping, 454 pyrosequencing, Pacific Biosciences sequencing and Illumina sequencing of matched formalin-fixed paraffin-embedded tissue, and confirmed the presence of 94% of protein-altering point mutations (Supplementary Table 4 and Supplementary Fig. 5); this validation rate is consistent with previous results that 95% of point mutations can be validated with orthogonal methods16,17. Only 18 out of 39 (46%) indels among significantly mutated genes were confirmed.
Six genes were found to be mutated with significant recurrence in the 103 whole-exome sequenced samples, by analysis with the MutSig algorithm16,17 (https://confluence.broadinstitute.org/display/CGATools/MutSig) at a false discovery rate (FDR) < 0.1 after correction for multiple hypothesis testing (Supplementary Table 6a), manual review of reads, and subsequent orthogonal confirmation of somatic events (Fig. 1 and Supplementary Fig. 6). One gene, CBFB, is identified for the first time as a significantly mutated gene in breast cancer or any other epithelial cancer, to our knowledge, whereas the other five genes (TP53, PIK3CA, AKT1, GATA3 and MAP3K1) have previously been reported as mutated in breast cancer7,10,13. This significantly mutated genes list, as any list produced by a statistical method, is probably incomplete and reflects the statistical power of our cohort size—larger sample sets will provide further statistical power.
Somatic mutations in TP53 and PIK3CA were each present in 27% of samples, consistent with published frequencies10,20 (Fig. 1). TP53 mutations occur in samples with a higher mutation rate (_t_-test P = 0.0079 comparing samples with mutation rates greater than or less than the median 1.66 mutations per Mb) and were distributed across the gene in sites reported in COSMIC (http://www.sanger.ac.uk/genetics/CGP/cosmic/). Also, using the ABSOLUTE algorithm for determining allele-specific copy number21, we observed that 21 out of 31 TP53 mutations were homozygous (Supplementary Table 4). PIK3CA mutations were clustered in the helical (amino acids 542/545; 40%) and kinase domains (amino acid 1047; 47%)20. Six samples harboured the AKT1 E17K mutation that alters the pleckstrin-homology (PH) domain and leads to activation of the kinase12. AKT1 and PIK3CA mutations, which activate the phosphatidylinositol-3-kinase (PI3K) pathway, were mutually exclusive in our data set. MAP3K1, recently reported as mutated in oestrogen-receptor-positive breast cancers10, harboured five mutations in three patients with oestrogen-receptor-positive disease, and followed a pattern consistent with positive selection for recessive inactivation of the gene. In total, two frameshift, two nonsense and one missense mutation, combined with a homozygous deletion spanning the coding region were observed. Although the point mutations seemed to be heterozygous by copy-number analysis, two patients harboured dual mutations, consistent with compound heterozygous inactivation, although confirmatory phasing data were not available. The GATA3 transcription factor gene harboured mutations in four patients with luminal tumours, including three previously unknown frameshift mutations near the 3′-end of the coding sequence. We also identified one previously described splice-site mutation that disrupts zinc-finger domains in GATA3 required for DNA binding13.
CBFB, encoding the core-binding-factor beta subunit, was mutated in four oestrogen-receptor-positive samples, with one nonsense mutation and three truncating frameshift mutations (Fig. 2a). CBFB somatic mutations have been noted in isolated cases of breast cancer6,10. This is the first report of these mutations recurring at a significant rate above background; the sample size is not sufficient to determine whether these mutations are specific for oestrogen-receptor-positive subtypes. CBFB encodes the non-DNA-binding component of a heterodimeric protein complex, together with the DNA-binding RUNX proteins encoded by RUNX1, RUNX2 and RUNX3. Copy-number analysis, using the ABSOLUTE algorithm21, provides further evidence for loss of function of the RUNX1/CBFB complex in breast cancer: the cases with CBFB mutations seem to have hemizygous deletions of one parental allele, whereas two additional cases harbour homozygous deletions of RUNX1 (Fig. 2b, c and Supplementary Figs 7 and 8). Oncogenic rearrangements of RUNX1 or CBFB are common in acute myeloid leukaemia22,23 (including the CBFB–MYH11 translocation believed to have dominant negative function22). This is to our knowledge the first report of inactivation of this transcription factor complex in epithelial cancers.
Figure 2: CBFB mutations and RUNX1 deletions.
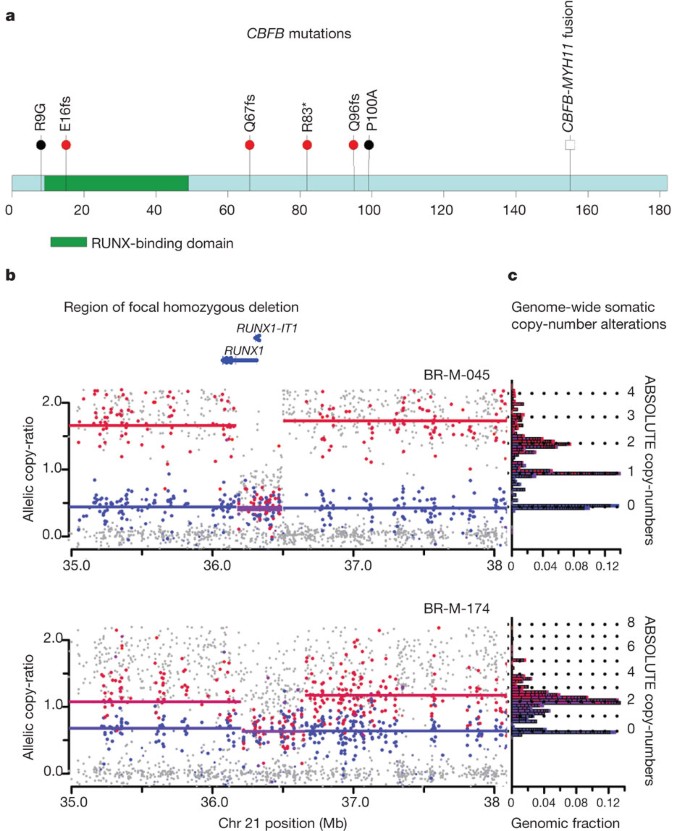
a, CBFB coding region diagram, RUNX-binding domain in green. Mutations identified in this study (red bullets), previously identified mutations6,10 (black bullets), and known CBFB–MYH11 fusion indicated. b, Allelic copy ratios for the 3-Mb region surrounding RUNX1 in samples BR-M-045 and BR-M-174. Dots indicate copy-ratios for individual SNP alleles. Red, higher copy-ratio allele for informative SNPs that are heterozygous in matched normal DNA; blue, lower-copy ratio SNPs; grey, uninformative SNPs (homozygous in matched normal). Lines indicate inferred segmental copy-ratios. Red, higher-copy segment; blue, lower-copy segment; purple, equal-copy segment. c, Histogram depicting bins of segmented copy number (y axis), with inferred integral copies shown by dotted lines; the length of each horizontal block corresponds to the fraction of the haploid genome at the copy number level, or ‘genomic fraction’ (x axis).
Significance analysis restricted to somatic mutations in genes reported in COSMIC revealed three significantly mutated genes, PIK3CA, TP53 and ERBB2, the latter below the significance threshold in the complete analysis (Supplementary Table 7). ERBB2 contained somatic mutations in three samples, with two being identical S310F mutations (these two samples are distinct on the basis of their germline and somatic genotypes). The S310F mutation can activate ERBB2 and is transforming in vitro (personal communication from H. Greulich). Neither sample with the S310F activating mutation has ERBB2 amplification (Supplementary Fig. 9). The two samples belong to the Her2-enriched and luminal B subtypes, which typically have ERBB2 amplification; this supports the notion that the observed mutations have a driving role in these tumours10,24.
To identify candidate genomic rearrangements, we applied the dRanger algorithm16,17 to the 22 cases with paired tumour/normal whole-genome sequencing data (Supplementary Table 8). The rate of rearrangements ranged from a median of 30 rearrangements per sample in the luminal A subtype (range 0–218) to the basal-like and Her2-enriched subtypes with a median of 237 and 246 rearrangements, respectively (Supplementary Fig. 10); the rates are similar to a recent report15. We performed polymerase chain reaction (PCR) amplification on a subset of the candidate rearrangements (Supplementary Methods) and confirmed 89 out of 165 events (54%). No rearrangement was seen in more than one sample (Supplementary Table 8). In addition, we did not identify rearrangements previously observed by DNA sequencing15 nor by complementary DNA (cDNA)-sequencing, including MAST and NOTCH family-gene fusions25.
The discovery of recurrent driver rearrangements in other epithelial cancers26,27 led to a closer examination of the list of confirmed rearrangements. In a triple-negative, basal-like subtype tumour, we observed a rearrangement between the genes MAGI3 (membrane-associated guanylate kinase, WW and PDZ domain containing 3) on chromosome 1p and AKT3 (v-akt murine thymoma viral oncogene homologue 3) on chromosome 1q, resulting in a balanced translocation from intron 9 in MAGI3 to intron 1 of AKT3 (Fig. 3a). The previously unknown fusion genes were confirmed in tumour DNA by sequencing the product of PCR amplification (Fig. 3b). The MAGI3 disruption is complemented by a hemizygous deletion of the other allele (Supplementary Fig. 11a). The expression levels of individual exons of MAGI3 and AKT3 correspond to the predicted 5′-_MAGI3–AKT3_-3′ fusion (Supplementary Fig. 11b), with this sample having the highest AKT3 expression in the data set. Expression of the fusion gene was confirmed in the tumour sample by PCR amplification of the cDNA (Fig. 3b).
Figure 3: MAGI3–AKT3 fusion gene.
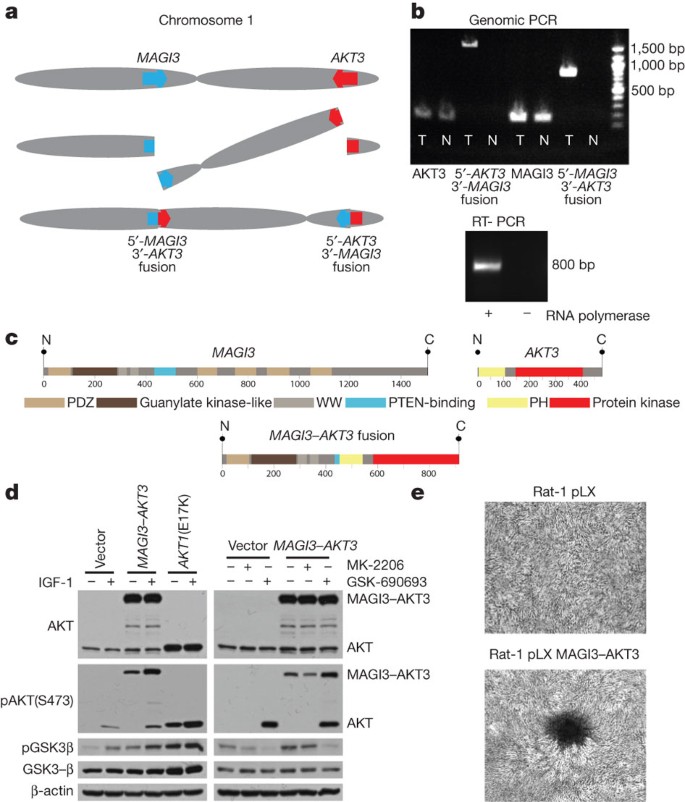
a, Diagram of balanced translocation between MAGI3 and AKT3. b, Top, genomic DNA PCR for AKT3, MAGI3 and both fusion products in tumour (T) and normal (N). Bottom, cDNA PCR of fusion gene in tumour. c, Above, MAGI3 and AKT3 protein domains; below, putative fusion protein. d, Immunoblots of lysates from ZR-75 cells transfected with vector, MAGI3–AKT3 fusion, or AKT1 E17K mutant, grown in low-serum media, for the indicated antibodies. Left, infected cells with and without insulin growth factor 1 (IGF-1) stimulation; right, treatment of vector or MAGI3–AKT3 overexpressing cells with AKT inhibitors MK-2206 and GSK-690693. e, Focus formation assays with Rat-1 cells expressing pLX control or MAGI3–AKT3, and stained with crystal violet.
The rearrangement produces an in-frame fusion gene with a predicted MAGI3–AKT3 fusion protein that combines MAGI3 lacking the second PDZ domain, reported to bind to PTEN and be required for the inhibitory effect of PTEN on the PI3K pathway28, together with an AKT3 region that retains an intact kinase domain but has a disruption of the pleckstrin homology domain before the glutamate at position 17 (Fig. 3c). AKT3 shares significant homology to AKT1 and is reported to be the dominant AKT family member expressed in hormone-receptor-negative breast cancers29. Together, the MAGI3–AKT3 translocation and deletion of MAGI3 could result in the combined loss of function of a tumour suppressor gene (PTEN) and activation of an oncogene (AKT3).
To evaluate oncogenic activity of the MAGI3–AKT3 fusion, we expressed the fusion gene ectopically in ZR-75 cells. The MAGI3–AKT3 fusion protein is constitutively phosphorylated at serine 473 in the AKT3 kinase domain (numbered according to the wild-type protein) in the absence of growth factors (Fig. 3d); ectopically expressed AKT1 with an engineered E17K mutation is likewise constitutively phosphorylated (Fig. 3d), as previously reported12. Constitutive activation of the MAGI3–AKT3 kinase in turn activates downstream pathways as demonstrated by phosphorylation of GSK3β, an AKT substrate (Fig. 3d). Phosphorylation of GSK3β by the MAGI3–AKT3 fusion can be inhibited with an ATP-competitive small molecule AKT inhibitor, GSK-690693, but not with an allosteric AKT inhibitor, MK-2206, that interacts with the PH domain of AKT (Fig. 3d). Overexpression of the MAGI3–AKT3 fusion gene in Rat-1 fibroblast cell lines led to loss of contact inhibition and focus formation (Fig. 3e).
We screened 235 additional breast cancer samples for the presence of the 5′-_MAGI3–AKT3_-3′ fusion event by PCR with reverse transcription (RT–PCR) of cDNA followed by Sanger sequencing of breakpoints. The fusion was present in 8 of the 235 samples, including 5 out of 72 triple-negative (oestrogen-receptor-, progesterone-receptor- and Her2-negative) samples (Supplementary Fig. 12).
The power provided by whole-genome and whole-exome sequencing of a relatively large and diverse breast cancer sample set has enabled several significant discoveries, including the identification of recurrent inactivating mutations in CBFB and of a recurrent translocation of MAGI3–AKT3. The mutations in CBFB, RUNX1 and GATA3 suggest the importance of understanding epithelial cell differentiation and its regulatory transcription factors in breast cancer pathogenesis. The recurrent genomic fusion involving AKT3 suggests that the use of ATP-competitive AKT inhibitors should be evaluated in clinical trials for the treatment of fusion-positive triple-negative breast cancers, a subtype where limited therapeutic options exist beyond systemic cytotoxic chemotherapy.
Methods Summary
All samples were obtained under institutional IRB approval and with documented informed consent. Breast cancer specimens from Mexico were paired with peripheral blood normal DNA whereas the Vietnamese samples were paired with DNA from normal adjacent breast tissue. Tumour RNA for each case was analysed on exon arrays to determine breast cancer expression subtype using the PAM50 classification method, whereas tumour/normal DNA pairs were analysed for copy number, allelic imbalance, and ancestry using single nucleotide polymorphism (SNP) arrays. A total of 108 samples, 17 both whole-genome sequencing and whole-exome sequencing, 86 whole-exome sequencing only, and 5 whole-genome sequencing only, passed initial qualification metrics, library construction, and successfully achieved desired sequencing depth (100× whole-exome sequencing; 30× whole-genome sequencing) on the Illumina sequencing platform (Supplementary Figs 1–3, Supplementary Tables 2 and 3). Tumour-specific point mutations, small insertions/deletions (indels), and rearrangements were detected by comparing tumour DNA to its paired normal DNA and using a series of algorithms to identify somatic events (Supplementary Fig. 2)16,17. Additional mutation calling was performed separately on tumour and normal DNA to identify germline mutation events that may confer susceptibility to breast carcinoma. Allele-specific copy number of each gene/mutation was determined using the HAPSEG and ABSOLUTE analysis methods. Confirmation of point mutations and indels was performed using mass-spectrometry-based genotyping and orthogonal next-generation sequencing methods, whereas putative in-frame genomic rearrangements were PCR-amplified from DNA to confirm the presence of the event.
A complete description of the materials and methods is provided in the Supplementary Information. Access to the data and computational algorithms used in this study can be found at https://confluence.broadinstitute.org/display/CGATools/Home.
Accession codes
Primary accessions
Protein Data Bank
Data deposits
Sequence data have been deposited in the dbGaP repository (http://www.ncbi.nlm.nih.gov/gap) under accession number phs000369.v1.p1.
References
- Jemal, A. et al. Global cancer statistics. CA Cancer J. Clin. 61, 69–90 (2011)
Article Google Scholar - Sørlie, T. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl Acad. Sci. USA 98, 10869–10874 (2001)
Article ADS Google Scholar - Chin, K. et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 10, 529–541 (2006)
Article CAS Google Scholar - Gatza, M. L. et al. A pathway-based classification of human breast cancer. Proc. Natl Acad. Sci. USA 107, 6994–6999 (2010)
Article ADS CAS Google Scholar - King, C. R., Kraus, M. H. & Aaronson, S. A. Amplification of a novel v-_erb_B-related gene in a human mammary carcinoma. Science 229, 974–976 (1985)
Article ADS CAS Google Scholar - Sjöblom, T. et al. The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 (2006)
Article ADS Google Scholar - Wood, L. D. et al. The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113 (2007)
Article ADS CAS Google Scholar - Shah, S. P. et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461, 809–813 (2009)
Article ADS CAS Google Scholar - Ding, L. et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 (2010)
Article ADS CAS Google Scholar - Kan, Z. et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873 (2010)
Article ADS CAS Google Scholar - Samuels, Y. et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554 (2004)
Article CAS Google Scholar - Carpten, J. D. et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444 (2007)
Article ADS CAS Google Scholar - Usary, J. et al. Mutation of GATA3 in human breast tumors. Oncogene 23, 7669–7678 (2004)
Article CAS Google Scholar - Sorlie, T. et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl Acad. Sci. USA 100, 8418–8423 (2003)
Article ADS CAS Google Scholar - Stephens, P. J. et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462, 1005–1010 (2009)
Article ADS CAS Google Scholar - Berger, M. F. et al. The genomic complexity of primary human prostate cancer. Nature 470, 214–220 (2011)
Article ADS CAS Google Scholar - Chapman, M. A. et al. Initial genome sequencing and analysis of multiple myeloma. Nature 471, 467–472 (2011)
Article ADS CAS Google Scholar - Pleasance, E. D. et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463, 191–196 (2010)
Article ADS CAS Google Scholar - Pleasance, E. D. et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463, 184–190 (2010)
Article ADS CAS Google Scholar - Bachman, K. E. et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol. Ther. 3, 772–775 (2004)
Article CAS Google Scholar - Carter, S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nature Biotechnol. 10.1038/nbt.2203 (29 April 2012)
- Cameron, E. R. & Neil, J. C. The Runx genes: lineage-specific oncogenes and tumor suppressors. Oncogene 23, 4308–4314 (2004)
Article CAS Google Scholar - Shigesada, K., van de Sluis, B. & Liu, P. P. Mechanism of leukemogenesis by the inv(16) chimeric gene CBFB/_PEBP2B_-MHY11 . Oncogene 23, 4297–4307 (2004)
Article CAS Google Scholar - Stephens, P. et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431, 525–526 (2004)
Article ADS CAS Google Scholar - Robinson, D. R. et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nature Med. 17, 1646–1651 (2011)
Article CAS Google Scholar - Soda, M. et al. Identification of the transforming EML4_–_ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566 (2007)
Article ADS CAS Google Scholar - Tomlins, S. A. et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310, 644–648 (2005)
Article ADS CAS Google Scholar - Wu, Y. Interaction of the tumor Suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. J. Biol. Chem. 275, 21477–21485 (2000)
Article CAS Google Scholar - Nakatani, K. et al. Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. J. Biol. Chem. 274, 21528–21532 (1999)
Article CAS Google Scholar
Acknowledgements
We would like to thank all patients who contributed samples to this study. This study was a collaboration of the Broad Institute in Cambridge, Massachusetts, USA, and the National Institute of Genomic Medicine (INMEGEN) in Mexico City, Mexico. The work was conducted as part of the Slim Initiative for Genomic Medicine, a project funded by the Carlos Slim Health Institute in Mexico. This work is part of a global effort in collaboration with the International Cancer Genome Consortium (ICGC). The authors would also like to acknowledge J. Barretina and H. Greulich for their critical review of the manuscript. In addition, we would like to acknowledge the technical expertise and data generation efforts of The Broad Institute Biological Samples, Genome Sequencing, and Genetic Analysis Platforms. S.B. has received fellowship support co-sponsored by CancerCare Manitoba and the University of Manitoba. K.K.B. is a recipient of the John Gavin Post-doctoral Fellowship, Genesis Oncology Trust of New Zealand. R.R.-V. and S.L.R.-C. received a scholarship from the Mexican Council of Science and Technology (CONACyT). R.B. is a V Foundation Scholar. A.T. is funded by NIH grant CA122099. This work was partially supported by the Dana-Farber/Harvard SPORE in breast cancer under NCI grant reference CA089393.
Author information
Author notes
- Shantanu Banerji & Gerardo Jimenez-Sanchez
Present address: Present addresses: Department of Medical Oncology, CancerCare Manitoba, Winnipeg, Manitoba R3E 0V9, Canada (S.B.); Global Biotech Consulting Group, Mexico City 01900, Mexico (G.J.-S.)., - Shantanu Banerji, Kristian Cibulskis, Claudia Rangel-Escareno and Kristin K. Brown: These authors contributed equally to this work.
Authors and Affiliations
- The Broad Institute of MIT and Harvard, Cambridge, 02142, Massachusetts, USA
Shantanu Banerji, Kristian Cibulskis, Scott L. Carter, Abbie M. Frederick, Michael S. Lawrence, Andrey Y. Sivachenko, Carrie Sougnez, Lihua Zou, Maria L. Cortes, Kristin G. Ardlie, Daniel Auclair, Fujiko Duke, Joshua Francis, Joonil Jung, Robert C. Onofrio, Melissa Parkin, Nam H. Pho, Alex H. Ramos, Steven E. Schumacher, Nicolas Stransky, Kristin M. Thompson, Rameen Beroukhim, Eric S. Lander, Stacey B. Gabriel, Levi A. Garraway, Todd R. Golub, Gad Getz & Matthew Meyerson - Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, 02215, Massachusetts, USA
Shantanu Banerji, Shouyong Peng, Steven E. Schumacher, Rameen Beroukhim, Kornelia Polyak, Andrea L. Richardson, Levi A. Garraway & Matthew Meyerson - Harvard Medical School, Boston, 02115, Massachusetts, USA
Shantanu Banerji, Jose Baselga, Rameen Beroukhim, Kornelia Polyak, Dennis C. Sgroi, Andrea L. Richardson, Eric S. Lander, Levi A. Garraway, Todd R. Golub, Alex Toker & Matthew Meyerson - Instituto Nacional de Medicina Genómica, Mexico City, 01900, Mexico
Claudia Rangel-Escareno, Juan C. Fernandez-Lopez, Valeria Quintanar-Jurado, Rosa Rebollar-Vega, Sandra L. Romero-Cordoba, Laura Uribe-Figueroa, Gerardo Jimenez-Sanchez, Jorge Melendez-Zajgla & Alfredo Hidalgo-Miranda - Department of Pathology, Beth Israel Deaconess Medical Center, 330 Brookline Avenue, Boston, Massachusetts 02215,
Kristin K. Brown & Alex Toker - Instituto de Enfermedades de la Mama FUCAM, Mexico City, 04980, Mexico
Veronica Bautista-Piña, Antonio Maffuz-Aziz & Sergio Rodriguez-Cuevas - Division of Hematology and Oncology, Massachusetts General Hospital, Boston, 02114, Massachusetts, USA
Jose Baselga - Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, 02215, Massachusetts, USA
Rameen Beroukhim & Matthew Meyerson - Department of Medicine, Brigham and Women’s Hospital, Boston, 02115, Massachusetts, USA
Kornelia Polyak - Depertment of Pathology, Massachusetts General Hospital, Boston, 02114, Massachusetts, USA
Dennis C. Sgroi - Department of Pathology, Brigham and Women's Hospital, Boston, 02115, Massachusetts, USA
Andrea L. Richardson - Massachusetts Institute of Technology, Cambridge, 02139, Massachusetts, USA
Eric S. Lander - Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, 02215, Massachusetts, USA
Todd R. Golub - Howard Hughes Medical Institute, Chevy Chase, 20815, Maryland, USA
Todd R. Golub
Authors
- Shantanu Banerji
- Kristian Cibulskis
- Claudia Rangel-Escareno
- Kristin K. Brown
- Scott L. Carter
- Abbie M. Frederick
- Michael S. Lawrence
- Andrey Y. Sivachenko
- Carrie Sougnez
- Lihua Zou
- Maria L. Cortes
- Juan C. Fernandez-Lopez
- Shouyong Peng
- Kristin G. Ardlie
- Daniel Auclair
- Veronica Bautista-Piña
- Fujiko Duke
- Joshua Francis
- Joonil Jung
- Antonio Maffuz-Aziz
- Robert C. Onofrio
- Melissa Parkin
- Nam H. Pho
- Valeria Quintanar-Jurado
- Alex H. Ramos
- Rosa Rebollar-Vega
- Sergio Rodriguez-Cuevas
- Sandra L. Romero-Cordoba
- Steven E. Schumacher
- Nicolas Stransky
- Kristin M. Thompson
- Laura Uribe-Figueroa
- Jose Baselga
- Rameen Beroukhim
- Kornelia Polyak
- Dennis C. Sgroi
- Andrea L. Richardson
- Gerardo Jimenez-Sanchez
- Eric S. Lander
- Stacey B. Gabriel
- Levi A. Garraway
- Todd R. Golub
- Jorge Melendez-Zajgla
- Alex Toker
- Gad Getz
- Alfredo Hidalgo-Miranda
- Matthew Meyerson
Contributions
S.B., K.C., C.R.E., L.Z., M.S.L., A.Y.S, A.H.R., N.S., G.G., A.H.M. and M.M. performed sequencing data analysis. S.B., A.M.F. and J.F. planned and performed fusion gene PCR and cloning. K.K.B. and A.T. planned and performed fusion gene biochemical and transformation experiments. F.D. and J.J. performed quantitative PCR experiments. C.R.-E. and J.C.F.-L. performed ancestry analysis. C.R.-E., J.C.F.-L., L.Z. and S.P. performed expression analysis. S.L.C. and G.G. performed ABSOLUTE analysis. R.R.-V., L.U.-F., S.E.S. and R.B. performed experimental work on SNP arrays and DNA copy number analysis. K.C., R.C.O. and M.P. performed mutation and rearrangement validation analysis. C.S., M.L.C., K.G.A., D.A., V.B.-P., V.Q.-J. and S.L.R.-C. collected data, processed and analysed genetic material, and provided data management. S.B., K.G.A. D.A., R.R.-V., S.L.R.-C., A.M.-A., S.R.-C., K.M.T., D.C.S., A.L.R. and A.H.-M. collected patient materials. S.B., K.C., C.R.-E., S.L.C., L.Z., A.M.F., J.C.F.-L., N.H.P., and M.M. contributed text and figures including Supplementary Information. All authors contributed to the final manuscript. K.P., D.C.S., A.L.R. and E.S.L. provided critical review of the manuscript. J.B., G.J.-S., E.S.L., S.B.G., L.A.G., T.R.G., J.M.-Z., G.G., A.H.-M. and M.M. provided leadership for the project.
Corresponding authors
Correspondence toAlex Toker, Gad Getz, Alfredo Hidalgo-Miranda or Matthew Meyerson.
Ethics declarations
Competing interests
E.S.L., L.A.G., T.R.G. and M.M. have financial interests in Foundation Medicine, which operates in the field of cancer diagnosis, but has no connection or rights to the work described in this study. They wish to declare this interest, although it does not appear to be a competing interest.
Supplementary information
Supplementary Information
This fie contains Supplementary Figures 1-12, Supplementary Methods, Supplementary Tables 1-3, 5 and 7 (see separate files for Supplementary Tables 4, 6 and 8) a Supplementary Discussion and additional references. (PDF 4315 kb)
Supplementary Table 4
Excel spreadsheet containing details of all mutations identified using whole exome sequencing. (XLS 1328 kb)
Supplementary Table 6
Excel spreadsheet with multiple tabs detailing significantly mutated genes in indicated breast cancer subtypes (XLS 173 kb)
Supplementary Table 8
Excel spreadsheet detailing all rearrangements identified using dRanger and associated PCR validation. (XLS 93 kb)
PowerPoint slides
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/).
About this article
Cite this article
Banerji, S., Cibulskis, K., Rangel-Escareno, C. et al. Sequence analysis of mutations and translocations across breast cancer subtypes.Nature 486, 405–409 (2012). https://doi.org/10.1038/nature11154
- Received: 16 June 2011
- Accepted: 20 April 2012
- Published: 20 June 2012
- Issue Date: 21 June 2012
- DOI: https://doi.org/10.1038/nature11154
This article is cited by
Editorial Summary
Mutations and translocations in breast cancer
This paper reports one of the largest whole-exome sequencing efforts in human breast cancers so far, complemented by whole-genome sequences of 22 breast cancer/normal pairs. The authors analysed diverse subtypes from patients in Mexico and Vietnam and identified recurrent mutations in the CBFB transcription factor gene and deletions of its partner RUNX1, as well as a recurrent MAGI3–AKT3 fusion enriched in triple-negative breast cancers (those lacking oestrogen and progesterone receptors and ERBB2 expression). The fusion leads to constitutive activation of AKT kinase, which can be counteracted by treatment with a small-molecule inhibitor.