Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus (original) (raw)
Main
The maintenance of epithelial barrier function at mucosal sites such as the intestine and respiratory tract is critical for limiting exposure to environmental stimuli, commensal bacteria and invading pathogens1,2,3. Studies have highlighted many roles for innate lymphoid cells (ILCs) in regulating immunity and/or inflammation at the intestinal barrier4,5. ILCs are a diverse family of cells of the immune system that are heterogeneous in their tissue location, cytokine production and effector functions4,5. Although the lineage relationships among these heterogeneous ILC populations remain poorly understood, they are hypothesized to originate from a common progenitor cell dependent on the transcription factor Id2 (refs. 4,6). On the basis of differences in their expression of the transcription factor RORγt, mouse ILCs can be functionally categorized into at least two populations. RORγt+ ILCs include CD4+ lymphoid tissue–inducer cells (LTi cells), NKp46+ ILCs and a population of CD4−NKp46− ILCs, all of which express interleukin 17A (IL-17A) and/or IL-22 and can promote intestinal immunity and/or inflammation4,7,8,9,10. A second group of RORγt− ILCs expresses the T helper type 2 (TH2) cell–associated cytokines IL-4, IL-5 and IL-13 and is composed of nuocytes, natural helper cells (NHCs), innate helper type 2 cells and multipotent progenitor type 2 cells. These cells are activated in response to the epithelial cell–derived cytokines IL-25 and/or IL-33 and can promote TH2 cytokine–dependent protective immunity to helminth parasites11,12,13,14.
Although the phenotypically distinct ILC populations described above have been identified in intestinal and lymphoid tissue compartments of mice, whether ILCs are present at barrier surfaces in humans and whether they influence immune responses or tissue homeostasis at extraintestinal sites remain unclear. One published study has identified a population of ILCs in the lungs of mice that resembles NHCs and nuocytes in phenotype and cytokine-expression profile15. After exposure to a high dose of influenza virus subtype H3N1, these lung ILCs promote airway hyper-reactivity early after infection via an IL-13-dependent mechanism. However, the potential influence of lung ILCs on other aspects of immunity, inflammation or tissue repair and remodeling in the respiratory tract remains unknown.
The repair and remodeling of damaged or inflamed tissue is a complex process that involves many factors, including cytokines, chemokines, growth factors and extracellular matrix proteins that restore tissue homeostasis after injury16,17. Tissue remodeling after acute injury requires a balance between promoting beneficial repair responses that drive cell proliferation and acting to limit such responses once the tissue has been adequately remodeled16,17. Failure to either appropriately initiate or resolve these repair responses can have detrimental effects, including loss of tissue integrity or function and promotion of chronic inflammation or tissue fibrosis16,17. The cellular and molecular regulators of tissue remodeling after injury or infection at mucosal tissues such as the lung are not well understood.
In this study, we used infection with the H1N1 influenza virus A/Puerto Rico/8/34 strain of influenza virus (recombinant virus expressing the lymphocytic choriomeningitis virus epitope gp33; called 'PR8' here) and identified a previously unrecognized role for ILCs in promoting the restoration of tissue homeostasis in the lungs. In mice, lung-resident ILCs lacked expression of lineage markers (Lin−) and expressed cell surface markers associated with NHC populations, including CD90, CD25, CD127 and T1-ST2, and produced IL-5 and IL-13 in response to stimulation with IL-33. An analogous population of Lin− lung ILCs was also present in the bronchoalveolar lavage (BAL) fluid and lung parenchyma of humans. ILCs accumulated in the lungs of wild-type mice or mice deficient in recombination-activating gene 1 (_Rag1_−/− mice) after experimental infection with influenza virus, and depletion of CD90+ ILCs or blockade of signaling via the IL-33 receptor (IL-33R) in influenza virus–infected mice resulted in diminished lung function, loss of airway epithelial integrity and impaired respiratory tissue remodeling. Genome-wide transcriptional profiling of lung ILCs identified enrichment for genes encoding molecules that regulate wound-healing processes, including the epidermal growth factor family member amphiregulin. Amphiregulin restored lung function and promoted tissue remodeling in mice depleted of ILCs and infected with influenza virus. Collectively, our data identified the presence of ILCs in the lung of both humans and mice and demonstrated a crucial role for mouse lung ILCs in regulating airway epithelial integrity and orchestrating pulmonary tissue homeostasis after experimental infection with influenza virus.
Results
Lung-resident ILCs resemble NHCs
To determine whether ILCs are present at extraintestinal mucosal sites, we used flow cytometry to characterize cells isolated from the lung tissues of naive wild-type C57BL/6 mice or _Rag1_−/− mice. We identified a population of Lin− cells that lacked expression of lineage markers associated with T cells (CD3, CD5, TCRβ and CD27), B cells (B220), macrophages (CD11b), dendritic cells (CD11c) or natural killer (NK) cells (NK1.1). These Lin− cells expressed CD90 (alloantigen Thy-1), CD25 (α-chain of the receptor for IL-2) and CD127 (α-chain of the receptor for IL-7; Fig. 1a), a pattern of surface marker expression consistent with ILCs4,5. Further examination of these Lin−CD90+CD25+ lung ILCs showed a lack of expression of either the coreceptor CD4 or the activating receptor NKp46 (Fig. 1b), which distinguish lung-resident ILCs from conventional CD4+ LTi cells or NKp46+ ILCs4. However, lung ILCs did express the activation markers CD44 and ICOS as well as the lineage marker Sca-1 and cell surface marker c-Kit (Fig. 1b), similar to the phenotype of nuocytes and NHCs12,13. Furthermore, CD90+CD25+ lung ILCs expressed the mouse IL-33R subunit T1-ST2 (Fig. 1b). This Lin− population of ILCs was a relatively rare population, with a total of 2 × 104 to 3 × 104 cells in naive mice, which represented 0.4–1% of total live cells in the lung (Fig. 1c). Collectively, these data indicate that lung-resident ILCs most closely resembled nuocytes and NHCs, originally reported to be present in secondary lymphoid tissue and fat-associated lymphoid clusters12,13.
Figure 1: ILCs in the lung resemble nuocytes and NHCs in phenotype and cytokine profile.
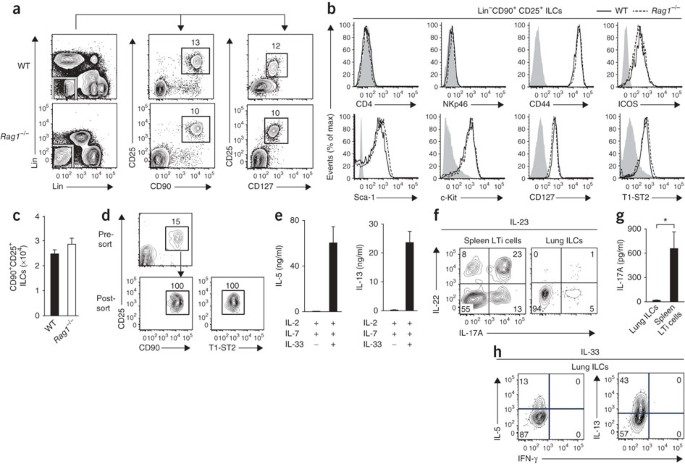
(a) Identification of lung ILCs in C57BL/6 wild-type (WT) and _Rag1_−/− mice by flow cytometry as CD90+CD25+CD127+Lin− cells that lack expression of CD3, CD5, NK1.1, CD27 and TCRβ (vertical axes) and B220, CD11b and CD11c (horizontal axes). Numbers adjacent to outlined areas indicate percent cells in each throughout. (b) Expression of cell surface markers on Lin−CD90+CD25+ lung ILCs in C57BL/6 wild-type and _Rag1_−/− mice. Gray shading, isotype-matched control antibody. (c) Absolute number of CD90+CD25+ ILCs in naive wild-type or _Rag1_−/− lungs. (d) Flow cytometry analysis of the purity of CD90+CD25+T1-ST2+ wild-type lung ILCs before and after sorting, gated on live Lin− cells. (e) Enzyme-linked immunosorbent assay of IL-5 and IL-13 in supernatants of CD90+CD25+T1-ST2+ lung ILCs sorted by flow cytometry and cultured for 4 d with IL-2 plus IL-7, with or without IL-33. (f) Intracellular cytokine staining for IL-22 and IL-17A in Lin−CD90+CD25+ lung ILCs or Lin−CD90+CD4+ splenic LTi cells obtained from wild-type mice and stimulated for 12 h with recombinant IL-23 (50 ng/ml) and for 4 h with the phorbol ester PMA and ionomycin plus brefeldin A. Numbers in quandrants indicate percent cells in each throughout. (g) Enzyme-linked immunosorbent assay of IL-17A in supernatants of CD90+CD25+T1-ST2+ lung ILCs or Lin−CD90+CD4+ LTi spleen cells sorted by flow cytometry and cultured for 4 d with IL-2 plus IL-7, with IL-23. (h) Intracellular cytokine staining of Lin−CD90+CD25+ lung ILCs obtained from wild-type mice treated for 7 d in vivo with 500 ng recombinant IL-33 and stimulated for 4 h ex vivo with PMA and ionomycin plus brefeldin A. *P < 0.05 (unpaired Student's _t_-test). Data are representative of more than three experiments with at least four mice per genotype (a–c; mean and s.e.m. in c), three independent experiments (d,e; mean and s.e.m. of three replicates in e, each consisting of ILCs sorted from five pooled lungs), two or more experiments with three to four mice each (f,h) or two independent experiments (g; mean and s.e.m. of three replicates, each consisting of ILCs sorted from five pooled lungs or spleens).
Nuocytes and NHCs can be activated by IL-33 alone or in combination with IL-7 to produce IL-5 and IL-13 (refs. 12,13). Although stimulation of sort-purified CD90+CD25+T1-ST2+ lung ILCs with IL-2 and IL-7 had no apparent effect on the secretion of IL-5 or IL-13, the addition of IL-33 resulted in more production of both IL-5 and IL-13 (Fig. 1d,e). Unlike CD4+ splenic LTi cells (Fig. 1f) or NKp46+ ILCs7,8,9, CD90+CD25+ lung ILCs had minimal expression of IL-22 or IL-17A in response to stimulation with IL-23 (Fig. 1f,g). Consistent with the results obtained by in vitro stimulation with IL-33, the administration of recombinant IL-33 in vivo resulted in a greater frequency of lung-resident ILCs that produced the TH2 cell–associated cytokines IL-5 and IL-13 but not the TH1 cell–associated cytokine interferon-γ (Fig. 1h). Together these results indicated that ILCs in the respiratory tract shared a cell-surface phenotype and cytokine profile that most closely resembled that of nuocytes and NHCs.
Lung ILC development requires Id2 but not commensals
Members of the ILC family, including LTi cells, NKp46+ ILCs and NHCs, share a developmental requirement for the transcriptional regulator Id2 (refs. 12,18,19,20). To assess the influence of Id2 on the development of lung-resident ILCs, we measured Id2 expression in sort-purified lung CD90+CD25+ ILCs. Quantitative analysis of Id2 mRNA expression showed similar abundance of Id2 mRNA in lung-resident ILCs and conventional splenic CD4+ LTi cells (Fig. 2a), a population known to be Id2 dependent18,19,20. Consistent with a developmental requirement for Id2, lung-resident CD90+CD25+ ILCs were not detectable in chimeric mice deficient in Id2 (Fig. 2b), thereby developmentally linking this cell population to the Id2-dependent ILC family.
Figure 2: The development of lung ILCs requires Id2 but is independent of microbial signals.
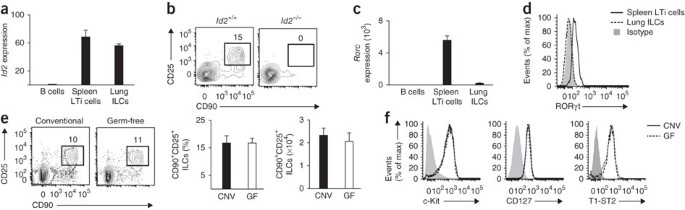
(a) Expression of Id2 mRNA in Lin−CD90+CD25+ lung ILCs and Lin− CD90+CD4+ splenic LTi cells obtained from C57BL/6 wild-type mice and purified by sorting; results are normalized to those for β-actin and are presented relative to expression in purified B220+ B cells. (b) Flow cytometry of CD90+CD25+ lung ILCs from wild-type (Id2+/+) or Id2-deficient (_Id2_−/−) bone marrow chimeras at 10 weeks after reconstitution, gated on live Lin− donor cells. (c) Expression of Rorc mRNA in cells as in a. (d) Flow cytometry analysis of RORγt expression in Lin−CD90+CD25+ lung ILCs or Lin−CD90+CD4+ LTi cells. (e) Flow cytometry (left), total frequency (middle) and absolute number (right) of Lin−CD90+CD25+ lung ILCs from conventional C57BL/6 mice (CNV) or germ-free mice (GF). (f) Cell surface expression of c-Kit, CD127 and T1-ST2 on Lin−CD90+CD25+ lung ILCs from conventional mice or germ-free mice. Gray shading (d,f), isotype-matched control antibody. Data are representative of two independent experiments (a,c; mean and s.e.m. of three replicates, each consisting of spleens or lungs pooled from five mice), three experiments with two to four chimeras per genotype (b), more than three experiments with three mice (d) or two independent experiments (e,f; mean and s.e.m. of three conventional or germ-free mice in e).
On the basis of their expression of RORγt, ILCs can be categorized into two populations, one critically dependent on RORγt for development (LTi cells and NKp46+ ILCs) and the other independent of RORγt (nuocytes and NHCs)4. In contrast to splenic LTi cells, lung ILCs did not have abundant expression of Rorc mRNA (encoding RORγt; Fig. 2c) or RORγt protein (Fig. 2d). This provided further evidence that lung ILCs were more closely related to nuocytes and NHCs than to LTi or NKp46+ ILC populations.
Signals derived from commensal bacterial communities have also been linked to the development and/or activation of several ILC subsets9,21,22. To determine whether live commensal bacteria–derived signals influenced the development of lung-resident ILCs, we examined ILCs in the lungs of conventional or germ-free C57BL/6 mice. These mice had a similar frequency, total number and surface phenotype of lung ILCs (Fig. 2e,f), which indicated that development of lung ILCs was dependent on Id2 but independent of signals derived from live commensal bacteria.
ILCs in human lung and airways
Studies have described heterogeneous populations of LTi-like cells and NKp46+ cells that express IL-17A and/or IL-22 and are located in human lymphoid tonsil and Peyer's patch tissues23,24,25,26,27. However, whether human ILC populations exist at other barrier surfaces, such as the respiratory tract, and what surface markers they express has not been examined before, to our knowledge. Similar to findings obtained with mice, we identified a Lin− population of cells in healthy human lung parenchymal tissue that lacked expression of markers for T cells (CD3 and TCRαβ), dendritic cells (CD11c), NK cells (CD56), macrophages (CD11b) or B cells (CD19) but that expressed CD127 (Fig. 3a). The Lin−CD127+ ILC population in the lung parenchyma expressed CD25 and the human IL-33R subunit ST2 (Fig. 3b), consistent with the cell-surface phenotype of the lung-resident ILC population found in mouse lung (Fig. 1a,b) and the mouse nuocytes and NHCs identified before12,13. To determine if an analogous ILC population exists in the airway, we examined BAL fluid from recipients of transplanted lungs. Similar to the population of ILCs found in lung parenchyma, we identified a Lin−CD127+ ILC population that also expressed CD25 and ST2 in BAL fluid from seven of nine recipients examined (Fig. 3c,d). Thus, these data provided evidence of NHC- and nuocyte-like ILC populations in human respiratory tissue.
Figure 3: Lin−CD127+CD25+ST2+ ILCs in human lung and airways.
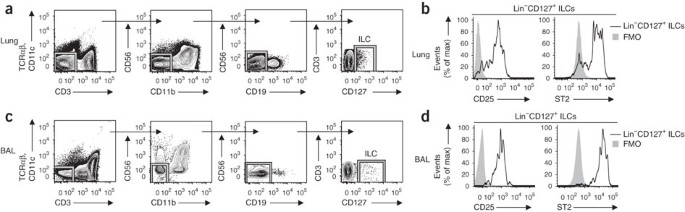
(a) Gating strategy for the identification of CD127+Lin− ILCs (CD3−TCRαβ−CD11c−CD11b−CD19−CD56−) in human lung parenchyma tissue (lower lobe; cadaver tissue) by flow cytometry, gated on live cells. (b) Expression of CD25 and ST2 on Lin−CD127+ human lung parenchyma cells. FMO, fluorescence-minus-one control. (c) Gating strategy as in a for cells in BAL fluid (from lung transplant recipients). (d) Expression of CD25 and ST2 on Lin−CD127+ cells in BAL fluid (as in b). Data are representative of one experiment per donor (among four donors (lung) or seven donors (BAL fluid)).
Influenza virus induces ILC responses in the lung
ILCs have been shown to mediate antibacterial or antihelminth immunity in the intestine7,9,11,12,13,14; however, the functional importance of ILCs in regulating immunity and/or inflammation in the lung is not well understood. To investigate whether lung ILCs influence immunity, inflammation or tissue homeostasis in the lung, we used a model of respiratory infection with mouse-adapted PR8. In response to intranasal infection with PR8, CD90+CD25+ ILCs accumulated in the lung parenchyma in both wild-type C57BL/6 and _Rag1_−/− mice (Fig. 4a,b). Wild-type mice controlled viral replication and recovered after infection with this dose of PR8 (0.5 median lethal dose (LD50) or ∼300 median tissue culture infectious dose (TCID50)), and although _Rag1_−/− mice were unable to clear infection and eventually succumbed within 12–14 d, the early ILC response and pathologic consequences of infection, including the diminished lung function and degree of lung immunopathology, were similar in immunocompetent and immunodeficient hosts (Supplementary Fig. 1). Together these data demonstrated that ILCs responded to infection with influenza virus in the lung in the presence or absence of the adaptive immune system, which suggests a potential role for lung ILCs in regulating innate immunity, inflammation and/or tissue homeostasis in the lung.
Figure 4: Depletion of CD90+ ILCs during influenza infection results in diminished lung function, compromised epithelial integrity and impaired airway remodeling.
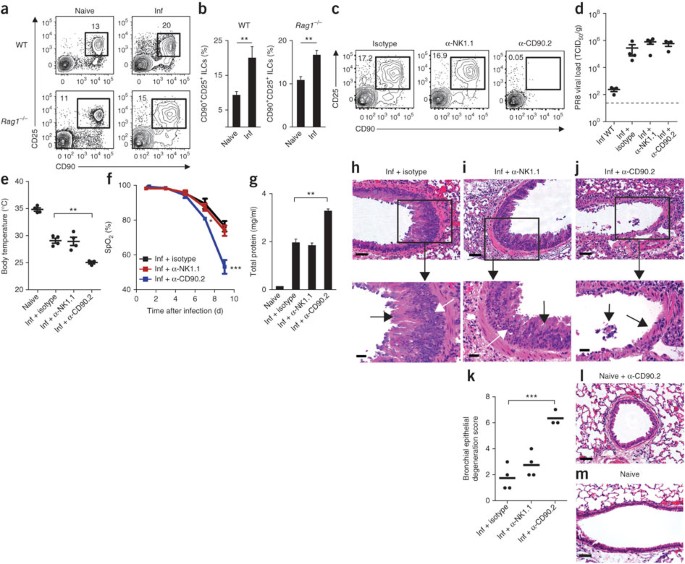
(a,b) Flow cytometry (a) and frequency (b) of Lin−CD90+CD25+ ILCs in the lung parenchyma of naive mice (Naive) or mice infected intranasally with 0.5 LD50 PR8 (Inf), assessed 16 d (wild type) or 10 d (_Rag1_−/−) after infection. (c) Flow cytometry of Lin−CD90+CD25+ ILCs from the lungs of _Rag1_−/− mice infected intranasally with 0.5 LD50 PR8 on day 0 and treated intraperitoneally with 200 μg isotype, mAb to NK1.1 (α-NK1.1) or mAb to CD90.2 (α-CD90.2) the day before infection (day –1) and on days 2, 5 and 8 after infection, assessed 10 d after infection. (d) Quantitative PCR analysis of the viral load of PR8 in wild-type mice infected as in c and left untreated (Inf WT) or in _Rag1_−/− mice infected and treated with isotype (Inf + isotype), mAb to NK1.1 (Inf + α-NK1.1) or mAb to CD90.2 (Inf + α-CD90.2) as in c, assessed 10 d after infection and presented as TCID50 per gram of lung tissue; dashed line indicates the limit of detection. (e) Body temperature of naive _Rag1_−/− mice and _Rag1_−/− mice infected and treated as in c, assessed 10 d after infection. (f) Blood oxygen saturation (SpO2) over the course of infection as in c. (g) Total protein in BAL fluid 10 d after infection as in c. (h–j) Hematoxylin-and-eosin staining of lung tissue from _Rag1_−/− mice infected with PR8 and treated with isotype (h), anti-NK1.1 (i) or anti-CD90.2 (j) as in c, assessed 10 d after infection. Black arrows indicate goblet cell hyperplasia (h,i) or regions of epithelial shedding or necrosis in the bronchioles (j); white arrows indicate epithelial cell hyperplasia (h,i). Bottom row, enlargement of regions outlined above. Scale bars, 50 μm. (k) Histological scores of bronchial epithelial degeneration in lung sections from _Rag1_−/− mice infected and treated as in c, obtained 10 d after infection and stained with hematoxylin and eosin. (l,m) Hematoxylin-and-eosin staining of lung tissue from a naive mouse treated with anti-CD90.2 (l) and an untreated naive mouse (m). Scale bars, 50 μm. In d,e,k, each symbol represents an individual mouse; small horizontal lines indicate the mean (k) or mean and s.e.m. (d,e). *P < 0.05, **P < 0.01 and ***P < 0.001 (unpaired Student's _t_-test). Data are representative of three or more independent experiments with three to four mice per group (mean and s.e.m. in b,f,g).
Depletion of ILCs impairs lung function and tissue repair
Although both cellular and humoral arms of the adaptive immune system have been shown to be crucial for immunity to respiratory viruses, the influence of cells of the innate immune system on antiviral immunity in the lung remains poorly understood28. To examine the influence of lung ILCs on innate immunity to influenza virus, we administered isotype-matched control antibody (isotype) or depleting monoclonal antibody (mAb) to CD90.2 in vivo to PR8-infected _Rag1_−/− mice. As some NK cells can also express CD90.2 after activation, we treated an additional group of mice with mAb to NK1.1 to allow direct comparison of depletion of ILCs mediated by antibody to CD90.2 (anti-CD90.2) versus depletion of NK cells mediated by anti-NK1.1. The administration of anti-CD90.2 effectively depleted the lungs of the CD90+CD25+ population of ILCs (Fig. 4c). Depletion of CD90.2+ ILCs or NK1.1+ NK cells did not affect viral loads in the lung relative to those of isotype-treated mice (Fig. 4d), which suggested that neither NK cells nor ILCs directly contributed to innate control of viral replication. However, mice that received anti-CD90.2 had a much lower mean body temperature at day 10 after infection (Fig. 4e), which indicated exaggerated thermodysregulation and morbidity in the absence of CD90.2+ ILCs. Additionally, although neutrophil populations were unaffected by treatment with anti-NK1.1 or anti-CD90.2, the BAL fluid of mice treated with anti-CD90.2 had less eosinophilia, probably a result of lower IL-5 concentrations in the absence of ILCs (Supplementary Fig. 2a,b).
Notably, depletion of CD90.2+ ILCs resulted in significantly lower lung function, as measured by pulse oximetry, which reached an average blood oxygen saturation below 54% in mice treated with anti-CD90.2 at day 9 after infection compared with 76% in isotype-treated control mice (Fig. 4f). Furthermore, depletion of CD90.2+ ILCs significantly impaired epithelial integrity, as measured by the higher total protein concentration in the BAL fluid of mice treated with anti-CD90.2 than that in BAL fluid of mice treated with isotype or anti-NK1.1 (Fig. 4g). Influenza is a cytopathic virus that replicates mainly in airway epithelial cells and causes severe acute injury to the respiratory epithelium28. Although _Rag1_−/− mice treated with isotype or depleted of NK cells eventually succumbed to infection between day 13 and day 14 because of the absence of an adaptive immune response (data not shown), they generated hyperplastic epithelial and goblet cell responses essential for repair of the damaged airway epithelial barrier (Fig. 4h,i). In contrast to treatment with isotype or anti-NK1.1, depletion of CD90.2+ ILCs resulted in an impaired ability to generate hyperplastic epithelial cell responses (Fig. 4j) and led to substantial epithelial degeneration and necrosis (Fig. 4j), including (in many cases) complete sloughing of the bronchiolar epithelial lining (Fig. 4j,k). This epithelial necrosis was probably not due to a direct effect of treatment with anti-CD90.2 on the epithelium, as bronchioles in naive mice treated with anti-CD90.2 had normal epithelial cell lining (Fig. 4l) similar to that of untreated naive mice (Fig. 4m). Collectively, these observations indicate a critical role for CD90.2+ ILCs in promoting airway epithelial integrity and restoring tissue homeostasis in the lung after acute viral infection.
Lung ILCs are sufficient to restore airway integrity
CD90 is expressed on cells of many hematopoietic and nonhematopoietic lineages29. To address the specificity of the depletion of ILCs mediated by anti-CD90.2, we examined CD90 expression on many subsets of cells of the innate immune response in the lung. Flow cytometry of macrophages, dendritic cells, neutrophils and eosinophils showed that these cell subsets lacked expression of CD90.2 (Supplementary Fig. 3), which suggested that anti-CD90.2 probably did not have a direct effect on these cell populations. To directly test whether lung-resident ILCs were the cell population responsible for promoting tissue remodeling after infection with influenza virus, we adoptively transferred congenically disparate CD90.1+ lung ILCs into PR8-infected _Rag1_−/− mice depleted of ILCs with anti-CD90.2. This system allowed the selective depletion of endogenous CD90.2+ ILCs, whereas the transferred population of CD90.1+ lung ILCs was left unaffected. Analysis of lung tissue by flow cytometry at 10 d after infection indicated that whereas anti-CD90.2 effectively abolished host CD90.2+ ILC responses in treated mice (Fig. 5a), a population of donor CD90.1+ ILCs was detectable (Fig. 5b). Mice treated with anti-CD90.2 that received transferred CD90.1+ ILCs had a higher mean body temperature at day 10 after infection than did mice that received anti-CD90.2 alone (Fig. 5c), which suggested that adoptively transferred ILCs were able to ameliorate influenza virus–induced morbidity. Additionally, analysis of blood oxygen saturation showed that the delivery of CD90.1+ lung ILCs effectively restored lung function to a degree equivalent to that of isotype-treated mice (Fig. 5d). Similar to isotype-treated control mice (Fig. 5e) and in contrast to mice that received anti-CD90.2 alone (Fig. 5f), mice treated with anti-CD90.2 that also received CD90.1+ ILCs had regions of epithelial proliferation in the bronchioles (Fig. 5g), indicative of a beneficial tissue-remodeling response. Collectively, these data provide evidence that the lung ILC population, not another CD90-expressing cell type, was critical for promoting respiratory tissue remodeling after infection with influenza virus.
Figure 5: Adoptive transfer of lung-resident ILCs promotes tissue remodeling in mice depleted of ILCs by treatment with anti-CD90.2, but blockade of IL-33R signaling impairs lung function and airway repair.
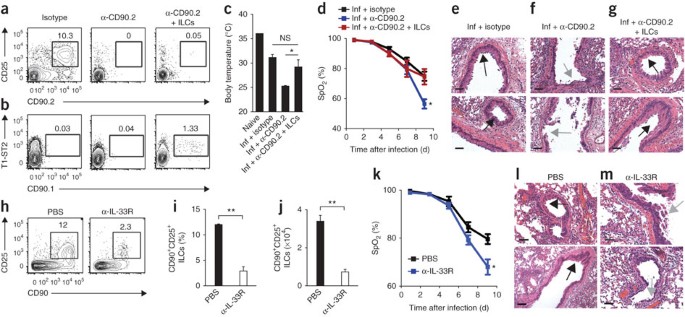
(a,b) Flow cytometry of endogenous Lin−CD90.2+CD25+ ILCs (a) and donor Lin−CD90.1+T1-ST2+ ILCs (b) in the lungs of _Rag1_−/− mice infected intranasally with 0.5 LD50 PR8 on day 0 and treated with 200 μg isotype or mAb to CD90.2 intraperitoneally the day before infection (day –1) and on days 2, 5 and 8 after infection, either alone (α-CD90.2) or with intravenous injection of 1 × 105 Lin−CD90.1+CD25+T1-ST2+ lung ILCs (sorted by flow cytometry) on days 0 and 5 after infection (α-CD90.2 + ILCs), assessed 10 d after infection. (c) Body temperature of naive mice and _Rag1_−/− mice infected as in a,b and treated with isotype (Inf + isotype) or mAb to CD90.2 without additional cells (Inf + α-CD90.2) or with the injection of ILCs (Inf + α-CD90.2 + ILCs) as in a,b, assessed 10 d after infection. (d) Blood oxygen saturation over the course of infection as in a,b. (e–g) Hematoxylin-and-eosin staining of lung tissue from _Rag1_−/− mice infected as in a,b and treated with isotype (e), anti-CD90.2 alone (f) or anti-CD90.2 plus ILCs (g) as in a,b, assessed 10 d after infection. Black arrows indicate epithelial cell hyperplasia; gray arrows indicate regions of epithelial shedding and/or necrosis in the bronchioles. Scale bars, 50 μm. (h–j) Flow cytometry (h), frequency (i) and number (j) of CD90+CD25+ ILCs in the lungs of C57BL/6 wild-type mice given PBS or 200 μg mAb to IL-33R (α-IL-33R) every 3 d after infection with 0.5 LD50 PR8, assessed 10 d after infection. (k) Blood oxygen saturation of mice infected and treated as in h–j. (l,m) Hematoxylin-and-eosin staining of lung tissue from mice infected with PR8 and treated with PBS (l) or anti-IL-33R (m) as in h–j, assessed 10 d after infection; arrows as in e–g. Scale bars, 100 μm. NS, not significant. *P < 0.05 and **P < 0.001 (unpaired Student's _t_-test). Data are representative of two independent experiments with three to four mice per group (a–g) or three independent experiments with three to four mice per group (h–m; mean and s.e.m. in c,d,i–k).
IL-33–IL-33R signaling is critical for lung ILC responses
Given the ability of lung ILCs to respond to stimulation with IL-33 (Fig. 1e,h) and published studies demonstrating that IL-33 is upregulated in the lung during infection with influenza virus15,30, we sought to test whether IL-33–IL-33R signaling was required for ILC-dependent maintenance of airway epithelial integrity. We infected wild-type mice with influenza virus and then administered mAb to T1-ST2 (to block IL-33–IL-33R signaling) or PBS. Blockade of IL-33–IL-33R signaling resulted in a significantly lower frequency and total number of CD90+ CD25+ ILCs in the lungs of mice treated with anti-IL-33R than in PBS-treated control mice (Fig. 5h–j). Additionally, lung function was severely impaired, with blood oxygen saturation reaching 68% in mice treated with anti-IL-33R, compared with 79% in PBS-treated control mice (Fig. 5k). Histological examination of lung parenchyma at day 10 after infection showed regions of epithelial cell necrosis and bronchial degeneration in the airways of mice treated with anti-IL-33R (Fig. 5l,m), indicative of severe damage to the airway epithelial barrier similar to the results obtained after depletion of ILCs with anti-CD90.2 (Fig. 4j). Together these results confirmed the results of the ILC-depletion studies and further demonstrated that a treatment that severely impaired lung ILCs compromised tissue repair and lung function in wild-type mice.
ILC-mediated repair is independent of IL-13 and IL-22
Published studies have demonstrated that IL-13 can promote the proliferation of epithelial cells and goblet cells as well as epithelial production of mucin in the context of airway inflammation and fibrosis16,31,32. To test the hypothesis that ILC-derived IL-13 may be needed to promote tissue homeostasis after infection with influenza virus, we administered recombinant IL-13 to _Rag1_−/− mice treated with anti-CD90.2. Although we detected IL-13 protein in BAL fluid (Supplementary Fig. 4a) and observed goblet cell hyperplasia in the bronchioles of mice treated with recombinant IL-13 (Supplementary Fig. 4b), which confirmed the biological activity of the recombinant IL-13 in the lung, mice treated with anti-CD90.2 that received recombinant IL-13 had much lower blood oxygen saturation, similar to that of mice that received anti-CD90.2 alone (59% and 62%, respectively; Supplementary Fig. 4c). Furthermore, mice that received anti-CD90.2 alone or in combination with recombinant IL-13 suffered severe influenza virus–induced morbidity, succumbing to infection 4 d earlier than did isotype-treated mice (day 9 versus day 13 after infection, respectively; data not shown). Together these results indicated that recombinant IL-13 was not sufficient to restore lung function in mice depleted of ILCs, which suggested that ILCs may use IL-13-independent mechanisms to promote lung tissue homeostasis.
Intestinal ILCs express IL-22, and interactions between IL-22 and its receptor regulate intestinal immunity and inflammation7,9,26. To determine whether IL-22 influenced airway epithelial barrier function after exposure to influenza virus, we treated infected wild-type mice with isotype or IL-22-neutralizing mAb. Neutralization of IL-22 in infected mice resulted in rates of weight loss and diminished blood oxygen saturation similar to those of isotype-treated control mice (Supplementary Fig. 5a,b), and examination of lung pathology showed no apparent defect in the ability of infected mice to mount a hyperproliferative epithelial remodeling response (Supplementary Fig. 5c). Consistent with a published report33, these data indicated that blockade of signaling via IL-22 and its receptor did not substantially influence the restoration of lung-tissue homeostasis after infection with influenza virus.
Enrichment for genes encoding wound-healing molecules in ILCs
To investigate potential mechanisms by which lung ILCs influence the maintenance of airway epithelial integrity or restoration of lung tissue homeostasis, we did genome-wide transcriptional profiling of lung-resident ILCs. We sort-purified Lin−CD90+CD25+ lung ILCs and Lin−CD90+ CD4+ splenic LTi cells from the lungs and spleens of naive C57BL/6 mice and then hybridized mRNA isolated from these populations to microarray chips for genome-wide transcriptional profiling. Examination of the top 100 genes with the greatest difference in expression in lung ILCs versus splenic LTi cells showed substantial differences between these two ILC populations (Fig. 6a). Splenic LTi cells were characterized by high expression of genes encoding CD4, the chemokine receptor CCR6, IL-23R and RORγt, in agreement with published studies7,8, whereas the top transcripts in the lung ILCs included those encoding the T1-ST2 receptor IL-1RL1, the IL-25 receptor IL-17RB, and the cytokines IL-2 and IL-5 (Fig. 6b). In addition, in that group of 100 genes with high expression in lung ILCs, there was notable expression of many genes encoding molecules associated with tissue remodeling, including genes encoding the extracellular matrix proteins decorin, asporin and dermatopontin, as well as members of the epidermal growth factor family, such as amphiregulin (Fig. 6b). Analysis of the genes with the greatest difference in expression in lung ILC populations versus splenic LTi cell populations with the DAVID database of functional annotation tools identified a transcriptional signature of lung ILCs that was significantly enriched for genes encoding molecules linked to pathways that regulate wound healing, immune defense responses, inflammatory responses and cell proliferation (Fig. 6c and Supplementary Table 1). In contrast, splenic LTi cells were significantly enriched for genes encoding molecules linked to processes associated with hematopoietic and lymphoid organ development, immune responses and cell activation (Fig. 6c and Supplementary Table 1).
Figure 6: Global gene-expression profiling of lung-resident ILCs shows enrichment for genes encoding molecules that regulate wound-healing pathways.
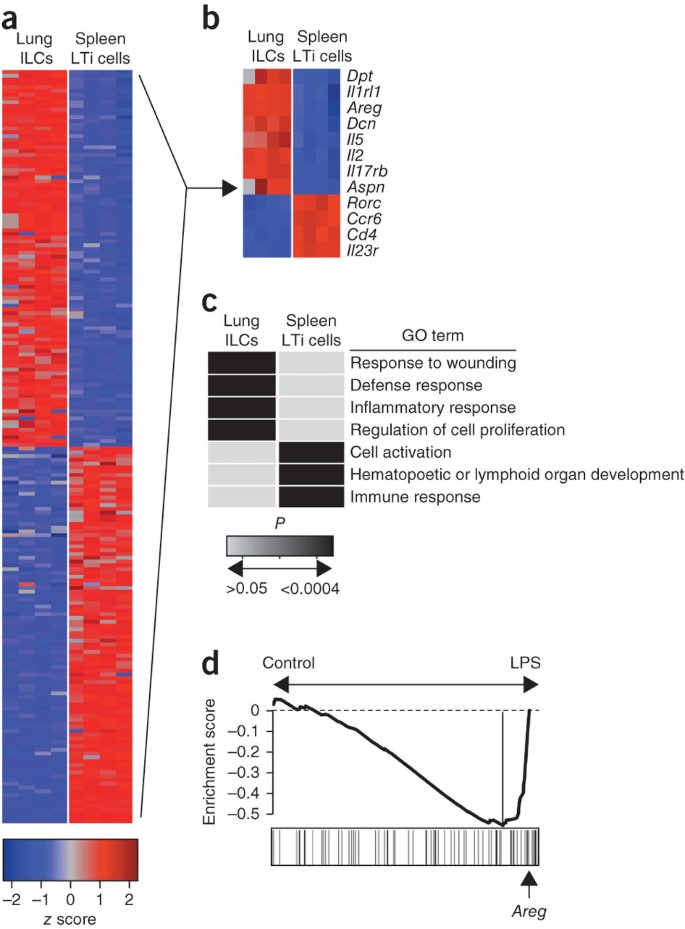
(a) Heat-map presentation of gene-expression profiles of the top 100 genes with the greatest differences in expression (red, high; blue, low) in Lin−CD90+CD25+ lung ILCs versus Lin−CD90+CD4+ splenic LTi cells sorted by flow cytometry from naive C57BL/6 wild-type mice. (b) Heat map of key genes from the profiling in a. (c) Functional classification of the gene-expression signatures of lung ILCs and splenic LTi cells (genes with a difference in expression of twofold or more) according to terms from the Gene Ontology project (GO term), analyzed with the DAVID database of functional annotation tools and presented as significance of enrichment (light gray, P > 0.05; black, P < 0.0004). (d) Gene-set enrichment analysis comparing lung ILC gene-expression signatures with those of a published data set examining the effects of LPS-induced acute lung injury34. Arrow indicates position of Areg in the analysis of the top transcripts in the LPS-treated group that define the 'leading edge' (long vertical line, right). Data are representative of one experiment with four biological replicates, each consisting of 1.5 × 104 to 2 × 104 ILCs (six mice per replicate) or LTi cells (ten mice per replicate).
We then used gene-set enrichment analysis to compare the gene-expression signature of lung ILCs with models of lung damage or inflammation reported before34. Direct comparison of the lung ILC gene set with a published data set examining the effects of lipopolysaccharide (LPS)-induced acute lung injury34 showed enrichment for the transcriptional signatures of lung ILCs in the LPS-treated group relative to their presence in the PBS-treated control group (Fig. 6d), consistent with a signature of tissue damage and wound repair. Further analysis of the transcripts that most contributed to the bias toward the LPS-treated group (right of the vertical line in Fig. 6d) identified the presence of the gene encoding amphiregulin (Areg; Fig. 6d and Supplementary Table 2). Thus, the gene encoding this epidermal growth factor, which had high expression in the lung ILC gene-expression profile, was one of the strong 'driver genes' in this enrichment pattern. Collectively, these results provided genome-wide transcriptional profiling of lung ILCs and demonstrated gene signatures of wound healing in the lung ILC population.
Amphiregulin restores lung function in mice depleted of ILCs
Amphiregulin is a member of the epidermal growth factor family that has been linked to the regulation of tissue remodeling and repair in the context of acute epithelial injury and asthma35,36,37. Quantitative PCR analysis confirmed the microarray results, showing higher expression of Areg mRNA by lung ILCs than by splenic LTi cells (Fig. 7a). Further, stimulation of sort-purified CD90+CD25+T1-ST2+ lung ILCs with IL-2 and IL-7 in combination with IL-33 resulted in more production of amphiregulin protein than that of cultures stimulated with IL-2 and IL-7 alone (Fig. 7b). Analysis of lung parenchyma tissue showed that at 10 d after infection with influenza virus, _Rag1_−/− mice had significantly higher expression of Areg mRNA (Fig. 7c) and amphiregulin protein (Fig. 7d) than did naive control mice, which indicated that amphiregulin expression was upregulated in the lung after infection with influenza virus and subsequent airway damage. Analysis of lung tissue from mice treated with anti-CD90.2 showed less Areg mRNA in the absence of CD90.2+ ILCs (Fig. 7e), which indicated that lung ILCs were a source of amphiregulin in vivo and that secretion of this protein may be one mechanism by which ILCs promote airway epithelial integrity and restore lung function.
Figure 7: Amphiregulin is produced by lung ILCs and can restore lung function, barrier integrity and remodeling of respiratory tissues after influenza virus–induced damage.
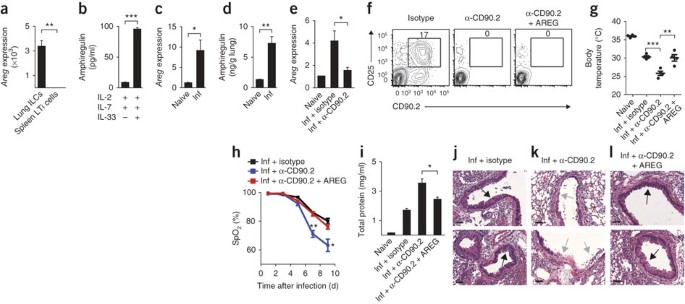
(a) Expression of Areg mRNA in sort-purified Lin−CD90+CD25+T1-ST2+ lung ILCs and Lin−CD90+CD4+ splenic LTi cells, presented relative to expression in splenic LTi cells. (b) Enzyme-linked immunosorbent assay of amphiregulin in sort-purified lung ILCs stimulated for 4 d with IL-2 and IL-7, with or without IL-33. (c,d) Expression of Areg mRNA (c) and amphiregulin protein (d) in the lungs of naive or PR8-infected _Rag1_−/− mice at 10 d after infection. Areg mRNA results are presented relative to those of naive lung tissue. (e) Expression of Areg mRNA in the lungs of naive _Rag1_−/− mice (Naive) or PR8-infected _Rag1_−/− mice that received isotype (Inf + istoype) or mAb to CD90.2 (Inf + α-CD90.2), assessed 10 d after infection; results are presented relative to expression in naive lung tissue. (f) Flow cytometry of lung ILCs from _Rag1_−/− mice infected intranasally with 0.5 LD50 PR8 and treated with isotype (Isotype), mAb to CD90.2 alone (α-CD90.2) or mAb CD90.2 plus 5–10 μg recombinant mouse amphiregulin administered intraperitoneally every 2 d (α-CD90.2 + AREG). (g) Body temperature of naive _Rag1_−/− mice (Naive) or PR8-infected _Rag1_−/− mice that received isotype (Inf + istoype), mAb to CD90.2 alone (Inf + α-CD90.2) or mAb to CD90.2 plus amphiregulin (Inf + α-CD90.2 + AREG) as in f, assessed 10 d after infection (presented as in Fig. 4d,e). (h) Blood oxygen saturation in mice infected and treated as in f. (i) Protein in BAL fluid of mice infected and treated as in f, assessed 10 d after infection. (j–l) Hematoxylin-and-eosin staining of lung tissue from _Rag1_−/− mice infected as in f and treated with isotype (j), mAb to CD90.2 (k) or mAb to CD90.2 plus amphiregulin (l) as in f, assessed 10 d after infection (arrows as in Fig. 5e–g). Scale bars, 50 μm. *P < 0.05, **P < 0.01 and ***P < 0.001 (unpaired Student's _t_-test). Data are representative of two independent experiments with three to four mice per group (mean and s.e.m. in a–e,h,i).
To directly determine whether delivery of amphiregulin could promote airway epithelial integrity and restore lung function after influenza virus–induced damage, we administered recombinant amphiregulin to PR8-infected _Rag1_−/− mice treated with anti-CD90.2 (Fig. 7f). Although the expression of IL-5 and IL-13 in the lung was not altered substantially after treatment with amphiregulin (Supplementary Fig. 6a,b), mice that received recombinant amphiregulin had higher mean body temperatures (Fig. 7g) and significantly improved lung function relative to that of control mice treated with anti-CD90.2, reaching blood oxygen saturation similar to that of isotype-treated mice at day 9 after infection (78% and 80%, respectively; Fig. 7h). Additionally, mice treated with recombinant amphiregulin had less total protein present in the BAL fluid than did control mice treated with anti-CD90.2 (Fig. 7i), and histological analysis of lungs from mice treated with anti-CD90.2 that received recombinant amphiregulin showed regions of epithelial cell hyperplasia and goblet cell proliferation in the bronchial airways (Fig. 7j–l), which indicated that treatment with recombinant amphiregulin promoted the restoration of airway epithelial integrity and lung tissue homeostasis. Collectively, these data provided both in vitro and in vivo evidence that amphiregulin is one mechanism by which ILCs can promote tissue homeostasis after infection with influenza virus.
Discussion
Many studies have identified heterogeneous ILC populations in mouse and human intestine and have indicated a role for these cell populations in regulating immunity and inflammation in the gut4,5. The data we have presented here have identified a population of ILCs constitutively present in the respiratory tract of humans and mice. Our results have also demonstrated a previously unknown function for mouse lung ILCs in regulating airway epithelial barrier integrity and tissue homeostasis after influenza virus–induced pulmonary damage.
The ILC population in mice is heterogeneous and includes cell populations found in lymphoid and intestinal tissues with distinct developmental requirements and patterns of effector cytokine expression4,5. In this report, we have shown that the development of lung-resident ILCs in mice was Id2 dependent and that these cells expressed a panel of cell surface markers characteristic of other ILC populations, including CD90, CD25, CD127 and T1-ST2. Furthermore, lung-resident ILCs produced IL-5 and IL-13 in response to stimulation with IL-33, but notably did not make large amounts of IL-22 or IL-17A after culture with IL-23. The cell-surface phenotype and cytokine profile of these lung-resident ILCs indicated that they most closely resembled NHCs or nuocytes, cell types previously thought to be restricted to sites in the gut-associated lymphoid tissue and fat-associated lymphoid clusters4,5,12,13. It remains to be determined whether ILCs in the mouse respiratory tract constitute a defined subset of ILCs distinct from nuocytes or NHCs or are developmentally related cells that exist in multiple anatomical locations.
In humans, published studies have identified RORγt+ ILC populations in the tonsils and Peyer's patches that share a phenotype and cytokine profile similar to that of mouse LTi cells and NKp46+ ILCs23,24,25,26,27. In this report, we identified ILCs in the human respiratory tract and lung parenchyma. Human lung ILCs expressed CD25, CD127 and IL-33R, which indicated they were nuocyte- or NHC-like ILCs. Therefore, like mouse ILCs, human ILC populations showed heterogeneity in tissue distribution, phenotype and functional ability. The examination of other lymphoid and mucosa-associated tissues may lead to the identification of additional ILC-like cells and provide insight into the regulation of human ILC populations.
Although published studies have indicated the involvement of NHCs and nuocytes in promoting TH2 cytokine–dependent antihelminth immunity4,5,12,13, we identified a role for ILCs in repairing airway epithelial integrity and maintaining lung tissue homeostasis. Lung injury is a key feature of many diseases, including viral infections such as infection with influenza virus, but also chronic obstructive pulmonary disease, acute respiratory distress syndrome, sarcoidosis, asthma, allergy and others16,17. However, the mechanisms involved in effective versus ineffective lung repair and tissue remodeling are poorly understood. The identification of lung ILCs as an important cell type in the regulation of tissue homeostasis not only indicates the involvement of an additional hematopoietic cell type that orchestrates the repair and/or regeneration of nonhematopoietic cells but also demonstrates previously unappreciated functions for the ILC lineage.
Genome-wide transcriptional profiling showed the expression by lung ILCs of many genes encoding molecules associated with wound healing and tissue repair, including the extracellular matrix proteins decorin, asporin and dermatopontin as well as members of the epidermal growth factor family, such as amphiregulin. Amphiregulin is expressed in many organs, including the lung, and has been shown to have critical roles in wound repair and tissue remodeling through the promotion of epithelial cell and fibroblast proliferation in settings of cancer, acute epithelial injury and asthma35,36,38,39. The demonstration that lung ILCs had high expression of amphiregulin, that depletion of ILCs resulted in loss of amphiregulin expression and that administration of amphiregulin to mice depleted of ILCs after infection with influenza virus restored epithelial integrity and tissue repair indicated that ILC-derived amphiregulin is one means by which ILCs regulate lung homeostasis.
The TH2 cell cytokines IL-5 and IL-13 have also been shown to mediate hyperplasia of epithelial cells and goblet cells in the lung31,32,40. Although lung-resident ILCs were able to produce large amounts of IL-5 and IL-13 in response to stimulation with IL-33, the administration of IL-13 failed to restore lung function or promote respiratory tissue remodeling in influenza virus–infected mice depleted of ILCs, which distinguishes the tissue-repair functions of lung ILCs from those of other nuocyte and NHC populations that are reported to act mainly by IL-13-dependent mechanisms12,13,14,15. It is unclear at present whether this difference indicates a minor role for IL-13 in the disease context examined here or additional heterogeneity in the functional diversity of ILC populations in the lungs. ILC-derived IL-13 might have a more prominent role in other airway-disease contexts. For example, a published report has identified a role for NHC-derived IL-13 through the use of a different model of infection with H3N1 influenza virus that causes airway hyper-reactivity early during infection15. In those studies, however, the NHC-produced IL-13 contributed to airway hyper-reactivity, which suggests that in some settings, the type 2 cytokines produced by lung ILC populations might have detrimental effects.
The effect of lung ILC-derived cytokines and tissue-remodeling proteins might depend on the pattern of lung injury (that is, a distinct pathogenesis of infectious or inflammatory disease) or other environmental factors. Nonetheless, the identification of a major pathway of tissue repair coordinated by lung ILCs suggests that these cells and their products could be targets for therapeutic manipulation in settings of lung infection, injury and chronic inflammation. Further studies are needed to fully characterize the various subtypes of ILCs that might exist in the respiratory tract. Comparison of the transcriptional profiles of the diverse ILC populations such as those presented here will be an important tool for elucidating the lineage relationships between RORγt-dependent LTi cells and RORγt-independent ILCs as well as highlighting biological features unique to these different cell types. In the context of lung inflammation and damage, it will be important to determine the extent of potential crosstalk between the epithelial repair pathways induced by IL-13 and the tissue-repair and/or tissue-remodeling proteins, such as amphiregulin, expressed by lung ILCs. It is notable that IL-33, as a key product of epithelial lineages and other cells of the immune system, induced the production of both IL-13 and amphiregulin by lung ILCs. This indicates that a degree of crosstalk exists between the damaged tissue and the ILC population that initiates the repair process. In summary, the results presented in our report here have identified a previously unrecognized role for lung ILCs in promoting airway epithelial integrity and lung tissue homeostasis through the production of amphiregulin. Given our findings, targeting ILC responses in the airway and other tissues may offer new therapeutic potential in the clinical management of tissue damage or chronic inflammation.
Methods
Mice.
C57BL/6 wild-type and _Rag1_−/− mice were from the Jackson Laboratory. All mice were maintained in specific pathogen–free facilities at the University of Pennsylvania. All protocols were approved by and all experiments were done according to the guidelines of the University of Pennsylvania Institutional Animal Care and Use Committee. Germ-free mice were maintained in plastic isolator units, were fed autoclaved feed and water and were routinely monitored to ensure the absence of microbial contamination. The generation of Id2-deficient mice and fetal liver chimeras has been described41,42. For the generation of Id2-deficient bone marrow chimeras, 10 × 106 to 20 × 106 bone marrow cells from Id2+/+ or _Id2_−/− fetal liver chimeras were transferred intravenously into irradiated hosts (900 rads) of a different congenic marker. All chimeras were provided drinking water with antibiotics (sulfamethoxazole and trimethoprim; Hi-Tech Pharmacal) for 2 weeks after irradiation and were allowed to reconstitute for 10 weeks before analysis.
Flow cytometry.
Single-cell suspensions were stained with a combination of the following fluorescence-conjugated mAbs (all from eBioscience unless specified otherwise): fluorescein isothiocyanate–conjugated anti-T1-ST2 (DJ8; MD Bioproducts); phycoerythrin-conjugated anti-ICOS (7E.17G9), anti-Sca-1 (D7), anti-c-Kit (2B8), anti-NKp46 (29AI.4) and anti–Siglec F (E50-2440; BD Pharmigen); peridinin chlorophyll protein–cyanine 5.5–conjugated anti-CD3 (17A2), anti-CD5 (53-73), anti-CD27 (LG.7F9), anti-NK1.1 (PK136), anti-TCRβ (H57-597), anti-CD11b (MI/70), anti-CD11c (N418) and anti-B220 (RA3-6B2); allophycocyanin-conjugated anti-CD44 (IM7), anti-c-Kit (2B8) and anti-CD90.1 (HIS51); eFluor 450–conjugated anti-CD127 (A7R34) and anti-CD45.1 (A20); phycoerythrin-indotricarbocyanine–conjugated CD25 (PC61.5); Alexa Fluor 700–conjugated anti-CD90.2 (30-H12) and anti-Ly6G (1A8; Biolegend); eFluor 780–conjugated anti-CD11b (MI/70), anti-CD11c (N418), anti-TCRβ (H57-597), anti-B220 (RA3-6B2) and anti-CD45.2 (104); and phycoerythrin–Texas red–conjugated anti-CD4 (GK1.5; Abcam). For measurement of intracellular cytokine expression, surfaces of stimulated cells were stained with a combination of the antibodies listed above, then cells were fixed and made permeable with a commercially available kit (eBioscience) and were stained with allophycocyanin–anti-IL-5 (TRFK5; BD Pharmigen), phycoerythrin–anti-IL-13 (eBio13A; eBioscience) or phycoerythrin–anti-IL-17A (TC11-18H10; BD Pharmigen), or with anti-IL-22 (IL22-02; Pfizer) conjugated to Alexa Fluor 647 according to the manufacturer's instructions (Molecular Probes). For measurement of intracellular RORγt, cells were fixed and made permeable and were stained with phycoerythrin–anti-RORγt (AFKJS-9; eBioscience). For all staining, dead cells were excluded from analysis through the use of a LIVE/DEAD Fixable Aqua Dead Cell Stain kit (Invitrogen). Samples were acquired on an LSR II (BD Biosciences) and were analyzed with FlowJo software (v9.2; TreeStar).
Isolation of cells from mouse lung tissues.
Lungs were perfused with 10 ml PBS through the right ventricle of the heart before removal. Lungs were then cut into small pieces with scissors and digested for 30–45 min at 37 °C with collagenase D (1 mg/ml in PBS; Roche) with vortexing every 10 min. Samples were mashed through 70-μm cell strainers and washed with DMEM (supplemented with 10% (vol/vol) FBS, 1% (vol/vol) L-glutamine (Gibco) and 1% (vol/vol) pencillin-streptomycin (Gibco)), and any remaining red blood cells were lysed. Single-cell suspensions were used for subsequent flow cytometry staining.
Analysis of human BAL fluid and lungs.
Human BAL samples were obtained as residual material from patients undergoing routine surveillance bronchoscopy during the first year after lung transplantation. Patients were on a regimen of immunomodulatory drugs after lung transplantation, including tacrolimus and prednisone, in addition to either mycophenolate or azathioprine. Use of human BAL samples for research purposes was approved by the University of Pennsylvania institutional review board and carried out in accordance with protocols for use of residual clinical material with a waiver of consent.
Human lung tissue was obtained from cadaver organ donors in collaboration with the New York Organ Donor Network. Consent for use of lung tissue from human cadavers for research purposes was obtained from the donors' families. Lung tissues were cut into small pieces with curved scissors and then were incubated for 1 h at 37 °C with shaking in collagenase solution (collagenase D (1 mg/ml), trypsin inhibitor (1 mg/ml) and DNAse I (25 μg/ml)). Tissues were then mechanically dissociated with a gentleMACS Dissociator (Miltenyi Biotech) and were strained through a wire mesh tissue sieve. The remaining connective tissue was then removed by passage of the cell suspension through a syringe column fitted with glass wool. Lymphocytes were subsequently separated by centrifugation through Ficoll (LSM; Invitrogen).
For analysis of ILC populations by flow cytometry, single-cell suspensions of BAL fluid and lung parenchyma were stained with the following monoclonal antibodies: fluorescein isothiocyanate–conjugated anti-ST2 (B4E6; MD Bioproducts), peridinin chlorophyll protein–cyanine 5.5–conjugated anti-CD3 (intracellular staining; SK7; BD Pharmigen), allophycocyanin-conjugated anti-CD11b (MI/70; eBioscience), eFluor 450–conjugated anti-CD19 (HIB19; eBioscience), phycoerythrin-indotricarbocyanine–conjugated anti-CD25 (M-A251; BD Pharmigen), Alexa Fluor 700–conjugated anti-CD56 (CMSSB; BD Pharmigen), eFluor 780–conjugated anti-CD127 (eBioRDR5; eBioscience), phycoerythrin-indodicarbocyanine–conjugated anti-CD11c (B-ly6; BD Pharmigen), phycoerythrin-indodicarbocyanine–conjugated anti-TCRαβ (IP26; eBioscience) and a LIVE/DEAD Aqua Fixable Dead Cell Stain kit (Invitrogen) Samples were acquired on an LSR II (BD Biosciences) and analyzed with FlowJo software (v9.2; Tree Star).
RNA isolation and real-time quantitative PCR.
Purified populations of Lin−CD90+CD25+ lung ILCs, Lin−CD90+CD4+ splenic LTi cells or B220+ splenic B cells were sorted on a FACSAria (BD Biosciences). RNA was isolated from those cells with an RNeasy mini kit according to the manufacturer's instructions (Qiagen), then cDNA was generated with SuperScript reverse transcriptase (Invitrogen). SYBR Green Master Mix (Applied Biosystems), commercially available primer sets (Qiagen) and an ABI7500 real-time PCR system (Applied Biosystems) were used for real-time quantitative PCR amplification of cDNA. Results were normalized to those of β-actin and are presented relative to expression in purified B cells (Id2 and Rorc) or naive lung tissue (Areg, Il5 and Il13).
Enzyme-linked immunosorbent assay of cytokines.
Lin−CD90+CD25+T1-ST2+ ILCs were sort-purified from the lungs of C57BL/6 mice with a FACSAria (BD). Lung ILCs (2 × 104) were cultured for 4 d in DMEM supplemented with 10% (vol/vol) heat-inactivated FBS, 2 mM glutamine, 100 μg/ml of streptomycin, 25 mM HEPES, pH 7.4, and 50 μM β-mercaptoethanol, in the presence of recombinant mouse IL-7 (10 ng/ml), recombinant IL-2 (10 ng/ml) and/or recombinant mouse IL-33 (30 ng/ml; for measurement of IL-5, IL-13 and amphiregulin) or recombinant mouse IL-23 (50 ng/ml; for measurement of IL-17A; all recombinant cytokines from R&D Systems). Supernatants were collected after 4 d of culture. For measurement of amphiregulin in whole tissue, lung tissues from naive or PR8-infected mice were homogenized in sterile PBS and centrifuged to remove cell debris. IL-5, IL-13 and IL-17A were measured with commercially available enzyme-linked immunosorbent assay kits (eBioscience). Amphiregulin was measured with a DuoSet ELISA kit (R&D Systems).
Influenza infection, antibody treatment and cytokine administration.
For infection with PR8, C57BL/6 or _Rag1_−/− mice were anesthetized intraperitoneally with 2.5% Avertin and infected intransally with 0.5 LD50 PR8 in a volume of 25 μl. PR8 was grown and titers were measured as described43. The replication and pathogenicity of this recombinant PR8 strain were not substantially different from those of nonrecombinant virus39.
Monoclonal anti-CD90.2 (30H12) and anti-NK1.1 (PK136) were from BioXCell; anti-IL-33R (245707) was from R&D Systems; and anti-IL-22 (IL22-01) was from Pfizer. All mAbs were administered intraperitoneally every 3 d at a dose of 200 μg per mouse starting on the day before infection.
For cytokine treatment, recombinant mouse IL-13 or amphiregulin (R&D Systems) was administered intraperitoneally at a dose of 5–10 μg every 2 d starting on the day of infection.
Adoptive transfer of ILCs.
Lin−CD90+CD25+T1-ST2+ ILCs (1 × 105) were sort-purified from the lungs of naive CD90.1 wild-type mice and were transferred intravenously on day 0 and day 5 after infection into _Rag1_−/− mice treated with mAb to CD90.2 and infected with influenza virus (0.5 LD50 PR8).
Lung histological sections.
The left lobes of lungs were perfused and fixed with 4% (vol/vol) paraformaldehyde and embedded in paraffin, and sections 5 μm in thickness were stained with hematoxylin and eosin. For analysis of bronchial epithelial degeneration, lung tissue sections stained with hematoxylin and eosin were graded (by researchers 'blinded' to sample identity) on a scale of 0–10 for the degree of bronchial and/or bronchiolar epithelial degeneration and necrosis.
Pulse oximetry.
A MouseOx pulse oximeter (Starr Life Sciences) was used for measurement of blood oxygen saturation (SpO2) in PR8-infected mice during the course of infection. A depilatory agent (Nair; Church & Dwight) was applied to the necks of anesthetized mice 2 d before influenza infection to remove hair and delay future hair growth. For measurements, the oximeter clip was placed on the neck and SpO2 was measured each second over several minutes; results are presented as the average of SpO2 measurements recorded over 3–5 min in each mouse.
Measurement of protein in BAL fluid.
BAL fluid was collected from naive or PR8-infected mice by the following method. A small cut was made in the trachea and a thin tube was inserted. Lungs were lavaged with 500 μl PBS and fluid was immediately placed on ice. BAL fluid was centrifuged to pellet cells and cell-free supernatants were collected. Total protein was measured with a Micro BCA Protein Assay according to the manufacturer's instructions (Pierce Thermo Scientific).
Quantification of influenza virus.
Lung tissues were homogenized and RNA was isolated with an RNeasy mini kit according to the manufacturer's instructions (Qiagen), then cDNA was generated by MultiScribe reverse transcription (Invitrogen). An ABI7500 real-time PCR system (Applied Biosystems) was used for real-time quantitative PCR analysis of cDNA with primers and carboxyfluorescein-labeled TaqMan probes generated to be specific for sequence encoding the PA protein of PR8. The viral RNA concentration (nanograms per gram of lung tissue) in each sample was determined by comparison to a standard curve of PR8 RNA concentrations and was converted to microliters of virus per gram of lung tissue. The TCID50 of PR8 (per microliter of virus) was determined by plaque assay on Madin-Darby canine kidney cell monolayers as described39, then the result was multiplied by the microliters of virus present in the lung tissue to determine the TCID50 equivalent.
Microarray gene-expression profiling.
Lin−CD90+CD25+ ILCs and Lin−CD90+CD4+ LTi cells were sorted from the lungs (ILCs) and spleens (LTi cells) of naive C57BL/6 wild-type mice. Four biological replicates were collected, each containing 1.5 × 104 to 2 × 104 cells sorted to a purity of >97% from six pooled lungs (ILCs) or ten pooled spleens (LTi cells). Cells were sorted directly into TRIzol LS (Invitrogen), then mRNA was isolated, amplified and hybridized to an Affymetrix GeneChip (Mouse Gene 1.0ST).
Data normalization and gene-expression profiling.
Affymetrix Power Tools software was used for processing and quantile normalization of fluorescence hybridization signals by the robust multichip averaging method44. Transcripts were log2-normalized and average values for technical replicates were obtained for analysis of change in expression (fold). With the ClassNeighbors module of the GenePattern genomic analysis platform, genes were ranked by signal-to-noise ratio, and class-biased genes were identified by permutation testing45. Genes were considered to have a difference in expression if the change in expression was over twofold (P < 0.05).
Gene Ontology enrichment analysis.
Genes with a difference of twofold or more in expression in both the naive ILC and naive LTi cell transcriptional profiles were uploaded to the DAVID database (Database for Annotation, Visualization and Integrated Discovery of the National Institute of Allergy and Infectious Diseases)46, and Fischer's exact test was used for the identification of genes with significant enrichment, with terms from the Gene Ontology project46 (full list of terms in Fig. 6c is in Supplementary Table 1).
Gene set enrichment analysis.
The Broad Institute GSEA software was used for gene-set enrichment analysis as described47. Published gene-expression arrays of whole lungs categorized as being from either control or LPS-treated groups were downloaded from Gene Expression Omnibus34. The normalized enrichment score and q value were calculated for enrichment of the top 100 genes with differences in expression in the naive lung ILC data sets (as defined before by the ClassNeighbors signal-to-noise ratio) and publicly accessible gene arrays (described above; 'leading-edge' genes, Supplementary Table 2.)
Statistical analysis.
Statistical significance was determined with the unpaired Student's _t_-test. Prism GraphPad software v5.0 was used for statistical analyses.
References
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809 (2009).
Article CAS PubMed Google Scholar - Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 8, 411–420 (2008).
Article CAS PubMed Google Scholar - Sonnenberg, G.F., Fouser, L.A. & Artis, D. Functional biology of the IL-22-IL-22R pathway in regulating immunity and inflammation at barrier surfaces. Adv. Immunol. 107, 1–29 (2010).
Article CAS PubMed Google Scholar - Spits, H. & Di Santo, J.P. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 12, 21–27 (2011).
Article CAS PubMed Google Scholar - Saenz, S.A., Noti, M. & Artis, D. Innate immune cell populations function as initiators and effectors in Th2 cytokine responses. Trends Immunol. 31, 407–413 (17 March 2010).
Article CAS PubMed Google Scholar - Rankin, L. & Belz, G.T. Diverse roles of inhibitor of differentiation 2 in adaptive immunity. Clin. Dev. Immunol. published online, doi:10.1155/2011/281569 (2011).
- Sonnenberg, G.F., Monticelli, L.A., Elloso, M.M., Fouser, L.A. & Artis, D. CD4+ lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34, 122–134 (2011).
Article CAS PubMed Google Scholar - Takatori, H. et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206, 35–41 (2009).
Article CAS PubMed PubMed Central Google Scholar - Satoh-Takayama, N. et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29, 958–970 (2008).
Article CAS PubMed Google Scholar - Buonocore, S. et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375 (2010).
Article CAS PubMed PubMed Central Google Scholar - Saenz, S.A. et al. IL25 elicits a multipotent progenitor cell population that promotes TH2 cytokine responses. Nature 464, 1362–1366 (2010).
Article CAS PubMed PubMed Central Google Scholar - Moro, K. et al. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature 463, 540–544 (2010).
Article CAS PubMed Google Scholar - Neill, D.R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Article CAS PubMed PubMed Central Google Scholar - Price, A.E. et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 107, 11489–11494 (2010).
Article CAS PubMed PubMed Central Google Scholar - Chang, Y.J. et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat. Immunol. 12, 631–638 (2011).
Article CAS PubMed PubMed Central Google Scholar - Crosby, L.M. & Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L715–L731 (2010).
Article CAS PubMed PubMed Central Google Scholar - Rock, J.R. & Hogan, B.L. Epithelial progenitor cells in lung development, maintenance, repair, and disease. Annu. Rev. Cell Dev. Biol. published online, doi:10.1146/annurev-cellbio-100109–104040 (31 May 2011).
- Yokota, Y. et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature 397, 702–706 (1999).
CAS PubMed Google Scholar - Satoh-Takayama, N. et al. IL-7 and IL-15 independently program the differentiation of intestinal CD3−NKp46+ cell subsets from Id2-dependent precursors. J. Exp. Med. 207, 273–280 (2010).
Article CAS PubMed PubMed Central Google Scholar - Boos, M.D., Yokota, Y., Eberl, G. & Kee, B.L. Mature natural killer cell and lymphoid tissue-inducing cell development requires Id2-mediated suppression of E protein activity. J. Exp. Med. 204, 1119–1130 (2007).
Article CAS PubMed PubMed Central Google Scholar - Sanos, S.L. et al. RORγt and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol. 10, 83–91 (2009).
Article CAS PubMed Google Scholar - Sawa, S. et al. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 12, 320–326 (2011).
Article CAS PubMed Google Scholar - Crellin, N.K. et al. Regulation of cytokine secretion in human CD127+ LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity 33, 752–764 (2010).
Article CAS PubMed Google Scholar - Cupedo, T. et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+CD127+ natural killer-like cells. Nat. Immunol. 10, 66–74 (2009).
Article CAS PubMed Google Scholar - Hughes, T. et al. Interleukin-1β selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity 32, 803–814 (2010).
Article CAS PubMed PubMed Central Google Scholar - Cella, M. et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457, 722–725 (2009).
Article CAS PubMed Google Scholar - Cella, M., Otero, K. & Colonna, M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1β reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. USA 107, 10961–10966 (2010).
Article CAS PubMed PubMed Central Google Scholar - Kohlmeier, J.E. & Woodland, D.L. Immunity to respiratory viruses. Annu. Rev. Immunol. 27, 61–82 (2009).
Article CAS PubMed Google Scholar - Bradley, J.E., Ramirez, G. & Hagood, J.S. Roles and regulation of Thy-1, a context-dependent modulator of cell phenotype. Biofactors 35, 258–265 (2009).
Article CAS PubMed PubMed Central Google Scholar - Le Goffic, R. et al. Influenza virus infection induces IL-33 in mouse lungs. Am. J. Respir. Cell Mol. Biol. published online, doi:10.1165/rcmb.2010–0516OC. (3 June 2011).
- Doherty, T. & Broide, D. Cytokines and growth factors in airway remodeling in asthma. Curr. Opin. Immunol. 19, 676–680 (2007).
Article CAS PubMed Google Scholar - Wilson, M.S. & Wynn, T.A. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2, 103–121 (2009).
Article CAS PubMed PubMed Central Google Scholar - Guo, H. & Topham, D.J. Interleukin-22 (IL-22) production by pulmonary natural killer cells and the potential role of IL-22 during primary influenza virus infection. J. Virol. 84, 7750–7759 (2010).
Article CAS PubMed PubMed Central Google Scholar - Altemeier, W.A. et al. Modulation of lipopolysaccharide-induced gene transcription and promotion of lung injury by mechanical ventilation. J. Immunol. 175, 3369–3376 (2005).
Article CAS PubMed Google Scholar - Enomoto, Y. et al. Tissue remodeling induced by hypersecreted epidermal growth factor and amphiregulin in the airway after an acute asthma attack. J. Allergy Clin. Immunol. 124, 913–920 (2009).
Article CAS PubMed Google Scholar - Fukumoto, J. et al. Amphiregulin attenuates bleomycin-induced pneumopathy in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L131–L138 (2010).
Article CAS PubMed Google Scholar - Dolinay, T. et al. Gene expression profiling of target genes in ventilator-induced lung injury. Physiol. Genomics 26, 68–75 (2006).
Article CAS PubMed Google Scholar - Wang, S.W. et al. Amphiregulin expression in human mast cells and its effect on the primary human lung fibroblasts. J. Allergy Clin. Immunol. 115, 287–294 (2005).
Article CAS PubMed Google Scholar - Okumura, S., Sagara, H., Fukuda, T., Saito, H. & Okayama, Y. FcɛRI-mediated amphiregulin production by human mast cells increases mucin gene expression in epithelial cells. J. Allergy Clin. Immunol. 115, 272–279 (2005).
Article CAS PubMed Google Scholar - Broide, D.H. Immunologic and inflammatory mechanisms that drive asthma progression to remodeling. J. Allergy Clin. Immunol. 121, 560–570 (2008).
Article CAS PubMed PubMed Central Google Scholar - Cannarile, M.A. et al. Transcriptional regulator Id2 mediates CD8+ T cell immunity. Nat. Immunol. 7, 1317–1325 (2006).
Article CAS PubMed Google Scholar - Monticelli, L.A. et al. Transcriptional regulator Id2 controls survival of hepatic NKT cells. Proc. Natl. Acad. Sci. USA 106, 19461–19466 (2009).
Article CAS PubMed PubMed Central Google Scholar - Decman, V. et al. Cell-intrinsic defects in the proliferative response of antiviral memory CD8 T cells in aged mice upon secondary infection. J. Immunol. 184, 5151–5159 (2010).
Article CAS PubMed Google Scholar - Irizarry, R.A. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003).
Article PubMed Google Scholar - Reich, M. et al. GenePattern 2.0. Nat. Genet. 38, 500–501 (2006).
Article CAS PubMed Google Scholar - Huang da, W. et al. in Current Protocols in Bioinformatics Ch. 13 (ed. Baxevanis, A.D.) Unit 13 11, 1–13 (John Wiley & Sons, 2009).
- Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Article CAS PubMed PubMed Central Google Scholar
Acknowledgements
We thank all members of the Artis and Wherry laboratories; M. Siracusa, S. Saenz, L. Osborne, E. Tait Wojno, M. Noti, M. Nair and A. Crawford for discussions and critical reading of the manuscript; D. Kobuley and D. Hill for care of the germ-free mouse facility; Yadav and A. Fitzgerald for assistance with the human BAL samples; and L. Fouser, S. Olland, R. Zollner, K. Lam and A. Root (Pfizer) for the preparation of anti-IL-22. Supported by US National Institutes of Health (U19AI083022, AI071309 and HHSN266200500030C to E.J.W.; HL098957 to R.G.C. for human pulmonary BAL studies; AI061570, AI074878, AI087990, AI095608, AI091759, AI095466 and U01AI095608 to D.A.; T32AI007532 to L.A.M. and G.F.S.; and T32AI05528 to M.C.A.), the Burroughs Wellcome Fund (D.A.), the National Institute of Diabetes and Digestive and Kidney Diseases Center for the Molecular Studies in Digestive and Liver Diseases Molecular Pathology and Imaging Core (DK50306) and the University of Pennsylvania (D.A.).
Author information
Authors and Affiliations
- Department of Microbiology and Institute for Immunology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA
Laurel A Monticelli, Gregory F Sonnenberg, Michael C Abt, Theresa Alenghat, Carly G K Ziegler, Travis A Doering, Jill M Angelosanto, Brian J Laidlaw, E John Wherry & David Artis - Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA
Laurel A Monticelli, Gregory F Sonnenberg, Michael C Abt, Theresa Alenghat & David Artis - Department of Biology, University of California San Diego, La Jolla, California, USA
Cliff Y Yang & Ananda W Goldrath - Department of Surgery and the Columbia Center for Translational Immunology, Columbia University Medical Center, New York, New York, USA
Taheri Sathaliyawala, Masaru Kubota, Damian Turner & Donna L Farber - Department of Medicine, School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA
Joshua M Diamond & Ronald G Collman
Authors
- Laurel A Monticelli
You can also search for this author inPubMed Google Scholar - Gregory F Sonnenberg
You can also search for this author inPubMed Google Scholar - Michael C Abt
You can also search for this author inPubMed Google Scholar - Theresa Alenghat
You can also search for this author inPubMed Google Scholar - Carly G K Ziegler
You can also search for this author inPubMed Google Scholar - Travis A Doering
You can also search for this author inPubMed Google Scholar - Jill M Angelosanto
You can also search for this author inPubMed Google Scholar - Brian J Laidlaw
You can also search for this author inPubMed Google Scholar - Cliff Y Yang
You can also search for this author inPubMed Google Scholar - Taheri Sathaliyawala
You can also search for this author inPubMed Google Scholar - Masaru Kubota
You can also search for this author inPubMed Google Scholar - Damian Turner
You can also search for this author inPubMed Google Scholar - Joshua M Diamond
You can also search for this author inPubMed Google Scholar - Ananda W Goldrath
You can also search for this author inPubMed Google Scholar - Donna L Farber
You can also search for this author inPubMed Google Scholar - Ronald G Collman
You can also search for this author inPubMed Google Scholar - E John Wherry
You can also search for this author inPubMed Google Scholar - David Artis
You can also search for this author inPubMed Google Scholar
Contributions
L.A.M., G.F.S. and M.C.A. did experiments and analyzed data; T.A. analyzed lung histological specimens; C.G.K.Z. and T.A.D. did microarray analysis; J.M.A. and B.J.L. designed the method for quantifying influenza virus by quantitative RT-PCR; C.Y.Y. and A.W.G. generated and provided Id2-deficient fetal liver chimeras; T.S., M.K., D.T. and D.L.F. collected and processed human lung tissues; J.M.D. and R.G.C. collected and processed human BAL fluid; and L.A.M., E.J.W. and D.A. designed the study, analyzed experiments and wrote the manuscript.
Corresponding authors
Correspondence toE John Wherry or David Artis.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Monticelli, L., Sonnenberg, G., Abt, M. et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus.Nat Immunol 12, 1045–1054 (2011). https://doi.org/10.1038/ni.2131
- Received: 02 August 2011
- Accepted: 01 September 2011
- Published: 25 September 2011
- Issue Date: November 2011
- DOI: https://doi.org/10.1038/ni.2131