Synergistic growth inhibition by Iressa and Rapamycin is modulated by VHL mutations in renal cell carcinoma (original) (raw)
Main
Renal cell carcinoma (RCC) afflicts annually over 30 000 individuals in North America with 12 000 deaths. Approximately 70–75% of RCCs have clear-cell morphology and one-half or more of these have mutations or epigenetic silencing of the von Hippel–Lindau (VHL) gene (Latif et al, 1993; Herman et al, 1994). As part of an E3 ubiquitin ligase (VBC), VHL targets specific substrates for polyubiquitylation and subsequent proteasome-mediated destruction with hypoxia inducible factor alpha subunits (HIF_α_) being the best characterized (Kaelin, 2003). Deregulation of HIF_α_ appears to be essential to the disease process (Kondo et al, 2002; Maranchie et al, 2002), although other VHL targets including fibronectin (Ohh et al, 1998), hnRNP molecules (Pioli and Rigby, 2001) and deubiquitinating enzymes (Li et al, 2002) may contribute.
Overexpression of the EGF receptor (EGFR) in RCC has been recognised for some time and EGFR signalling is mitogenic for malignant and normal renal tubular cells (Gomella et al, 1989; Humes et al, 1991; Uhlman et al, 1995; de Paulsen et al, 2001). TGF_α_, an EGFR ligand under transcriptional control by HIF, is constitutively expressed in VHL mutant cells (Knebelmann et al, 1998; de Paulsen et al, 2001; Gunaratnam et al, 2003). These observations have led to clinical trials of EGFR inhibitors in RCC (Drucker et al, 2003; Motzer et al, 2003; Foon et al, 2004; Rowinsky et al, 2004; Amato, 2005). However, as single –agents, the response rates have been low although stable disease may be prolonged. In contrast, the combination of EGFR and VEGF inhibitors may be more active than either agent alone (Hidalgo, 2003).
Downstream effectors of EGFR signalling include the Ras/Raf/MAP kinase and phosphatidyl inositol 3-OH kinase (PI3K) pathways. PI3K signalling leads to phosphorylation and activation of AKT, which blocks apoptosis (Thompson and Thompson, 2004), promotes cytoplasmic sequestration of FoxO transcription factors (Brunet et al, 1999), and activates protein translation initiation via the mammalian target of rapamycin (mTOR) (Hay and Sonenberg, 2004). Phosphatase and tensin homolog (PTEN) mutations, which activate AKT, have been described in RCC in association with more aggressive disease (Kondo et al, 2001; Shin Lee et al, 2003). Mammalian target of rapamycin (mTOR) is inhibited by the tuberous sclerosis (TSC) complex, which, in turn, is inhibited by AKT and patients with TSC mutations have an increased incidence of RCC (Al-Saleem et al, 1998). Biochemically, mouse embryo fibroblasts defective in TSC resemble RCCs with VHL mutations as they express elevated levels of HIF_α_, VEGF and glycolytic proteins (Brugarolas et al, 2003). Tuberous sclerosis mutant cells have an exaggerated HIF_α_ response to hypoxia, which is blocked by treatment with rapamycin. A rapamycin prodrug, CCI-779 (Wyeth), has been examined as a single agent in RCC (Atkins et al, 2004). The response rate was low (7%) but included one complete response. Rapamycin has shown additive or synergistic activity when combined with other tyrosine kinase inhibitors such as Gleevec and PKC412 (Mohi et al, 2004). Rapamycin also enhances chemotherapy responses in breast cancer cell lines (Mondesire et al, 2004) and in Myc-driven murine lymphomas (Ruggero et al, 2004). Tumours with loss of PTEN, loss of p53 or amplification of GLI may be particularly sensitive to rapamycin (Hosoi et al, 1999; Louro et al, 1999; Neshat et al, 2001).
Mammalian target of rapamycin, a serine/threonine kinase, positively affects protein translation initiation by phosphorylating two key regulators, 4E-BP1 and p70S6 kinase (S6K1) (Hay and Sonenberg, 2004). eIF4E binds the 5′-methyl cap (me7GpppN) modification of mRNAs and is essential for cap-dependent translation initiation. Unphosphorylated 4E-BP1 binds and inhibits eIF4E. Overexpression of eIF4E can transform NIH 3T3 cells (Lazaris-Karatzas et al, 1990) and can cause rapamycin-sensitive murine lymphomas to become rapamycin-resistant (Ruggero et al, 2004). In contrast to inhibiting 4E-BP1 function, S6K is activated by mTOR which leads to the phosphorylation of ribosomal protein S6 (RPS6) and increased translation of mRNAs, particularly those encoding components of the translation machinery itself (e.g. RPS19). Other signalling pathways also affect protein translation initiation. For instance, ERK1/2 activation affects multiple translation initiation components including S6K, p90 RSK, MNK and 4E-BP1, among others (Roux and Blenis, 2004). Also, in addition to eIF4E-mediated cap-dependent initiation, there are internal ribosome entry sites present in several mRNAs important in RCC such as MYC, VEGF and Cyclin D1 (Stoneley and Willis, 2004; Shi et al, 2005), which may allow translation initiation in a cap-independent manner.
We hypothesised that the combination of an EGFR inhibitor plus rapamycin might inhibit growth in RCC cell lines in a synergistic manner. This proved to be correct but synergy at low drug concentrations was only observed in cell lines with wild-type (wt) VHL. Mechanistically, loss of ERK1/2 and RPS6 phosphorylation following EGFR inhibition was confined to wt-VHL cells. Rapamycin more effectively inhibited phospho-4E-BP1 and this was VHL-independent. In contrast, neither agent inhibited phospho-AKT and similarly, neither agent nor combination blocked MYC translation initiation. However, blocking AKT with the PI3K inhibitor, LY-294002, substantially downregulated the association of MYC mRNA with ribosomes (i.e. polysome loading). Among seven parental RCC cell lines analysed biochemically, we found one with overexpression of eIF4E. With these results in mind, we examined a series of patient tumours. Extracellular signal-regulated kinase (ERK) and AKT pathways showed similar evidence for activation, although levels of phospho-ERK were relatively much greater. One tumour contained elevated eIF4E and several biochemical characteristics closely matched the one cell line with eIF4E overexpression. Most tumours, as expected, contained elevated EGFR protein compared to normal kidney, although these levels were substantially lower than in RCC cell lines. Interestingly, phospho-RPS6 was markedly reduced in tumours, suggesting that mTOR activity was lower than observed in cell lines. These preclinical findings in cell lines suggest that combined EGFR and mTOR inhibitors may benefit a particular subset of RCC patients but the lack of AKT inhibition potentially limits the response. Apparent differences between primary tumours and cell lines suggest that it would be particularly useful to examine tumours pre- and post-treatment with phospho-specific antibodies to ERK, AKT and RPS6 (or S6K).
Methods
Cell lines, genotyping and patient tumours
786-O cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Stable 786-O transfectants, WT8 (containing wt-VHL) and PRC3 (vector control), were kindly provided by Dr William Kaelin. Additional wt-VHL transfectants, MPR6 and MEA2, were kindly provided by Dr Robert D Burk. 786-O cells were grown in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal calf serum (FCS). WT8, PRC3, MPR6 and MEA2 were maintained in the same medium plus 1000 _μ_g ml−1 G418. SKRC-02, SKRC-17, SKRC-39 and SKRC-45 were kindly provided by Dr Elisabeth Stockert (Tumour Cell Bank, Memorial Sloan-Kettering Cancer Center, New York, NY, USA) and maintained in RPMI-1640 with 10% FCS. ACHN was grown in McCoy's medium with 15% FCS and KRCY was grown in RPMI-1640 with 15% FCS. Cells were cultured under standard conditions in a humidified 5% CO2 atmosphere. Rapamycin and LY294002 were obtained from Sigma, St. Louis, MO, USA. EGF from Chemicon, Int., Temecula, CA, USA, while ZD-1839 was obtained from Astra-Zeneca, Inc., Wilmington, DE, USA, through the auspices of Dr Paul Bunn. Cell lines were confirmed to be of independent origin by either microsatellite genotyping analysis or VHL mutation determination using DNA sequencing. Genotyping analysis was performed by the University of Colorado Cancer Center DNA Sequencing & Analysis Core using the Applied Biosystems (Foster City, CA, USA). AmpF_l_STR Profiler Plus PCR amplification kit (No. 4303326). The microsatellite genotyping analysis was performed as indicated by the manufacturer. The following 10 loci were examined: D3S1358, vWA, FGA, Amelogenin, D8S1179, D21S11, D18S551, D5S818, D13S317 and D7S820. If a second allele (or Y chromosome in the case of Amelogenin) was not detected, it is listed as ‘nd’. The results (i.e. allele 1/allele 2) for each locus listed above in order are: SKRC2: 17/nd, 15/17, 20/nd, X/nd, 8/nd, 28/30, 12/19, 12/nd, 11/nd, 9/11; SKRC12: 15/nd, 14/16, 20/22, X/nd, 13/14, 29/nd, 12/16, 11/12, 11/13, 8/9; SKRC17: 15/nd, 14/16, 22/nd, X/nd, 13/14, 30/32.2, 15/18, 11/12, 11/12, 8/12; SKRC39: 16/nd, 17/nd, 24/nd, X/nd, 13/nd, 29/31.2, 14/15, 13/nd, 11/nd, 8/11; SKRC45: 15/nd, 15/16, 22/23, X/nd, 12/14, 27/28, 15/nd, 12/nd, 11/12, 12/nd; ACHN: 17/nd, 16/17, 22/nd, X/nd, 12/nd, 30/nd, 16/nd, 12/nd, 12/nd, 9/11; A498: 15/nd, 18/nd, 18/20, X/nd, 13/15, 28/32, 17/nd, 11/nd, 12/nd, 10/11; CAKI2: 14/nd, 16/17, 22/nd, X/Y, 10/nd, 27/31, 17/nd, 11/nd, 10/nd, 12/nd; KRCY: 15/nd, 17/18, 22/nd, X/nd, 14/15, 29/30.2, 14/nd, 12/13, 12/nd, 8/12; KV6: 14/nd, 16/nd, 21/22, X/nd, 12/15, 29/nd, 16/17, 12/nd, 9/13, 9/10.
Frozen sections from 12 RCC tumours and adjacent normal-appearing kidney were processed for Western blot, genomic DNA isolation and quantitative RT–PCR by pulverisation in liquid N2 and storage in aliquots at −80°C. Pathological parameters for this tumour cohort are provided in Table 1.
Table 1 Clinical characteristics and von Hippel–Lindau (VHL) status of 12 RCC tumours
MTT assays
Microtitre plates were seeded with 2000 cells per well on day 1. Agents and vehicle were added on day 2 and growth continued for an additional 5 days. MTT reagent (3- (4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide, Sigma) was then added to 400 _μ_g ml−1 and incorporated for 2–4 h. Deposited dye was solubilised with 75% isopropanol, 240 mM HCl and the absorbance measured at 490 nm in a 96-well plate reader (Molecular Devices, Inc., Sunnyvale, CA, USA). The combination index was calculated by CALCUSYN (Chou and Talalay, 1984).
Primers for real-time RT–PCR and VHL sequencing
RNA isolation, cDNA synthesis and quantitative RT–PCR assays were performed as previously described (Drabkin et al, 2002). Primer sequences (5′–3′) were: EGFR (for) AGATGGAGGAAGACGGCGTC, (rev) GGAGTCACCCCTAAATGCCAC; ErbB-2 (for) ACCTACCTGCCCACCAATGC, (rev) GGTGGTATTGTTCAGCGGGTC; ErbB-3 (for) TCCGCTTGACTCAGCTCACC, (rev) CCCTTGCAAACCTCATGACAG; ErbB-4 (for) CCAGCCCAGCGATTCTCAG, (rev) AAGGAGAGGTCCCGGTTGTG; RPS19 (for) CCTCAAAAAGTCCGGGAAGC, (rev) TGGAAGCAGCTCGCGTGTAG; _β_-actin (for) ACCGCGAGAAGATGACCCAG, (rev) AGGTCCAGACGCAGGATGG; MYC (for), ACACCCTTCTCCCTTCGGG, (rev) AGCCGCTCCACATACAGTCC; VEGF (for) CAAGACAAGAAAATCCCTGTGG, (rev) CCTCGGCTTGTCACATCTG; GAPDH (for) TGCACCACCAACTGCTTAGC, (rev) GGCATGGACTGTGGTCATGAG; VHL (for) GGACACACGATGGGCTTCTG, (rev) CAACCTGGAGGCATCGCTC.
Primers used for genomic amplification of VHL were: either PF1 (upstream of promoter): ACTTTATAAGCGTGATGATTGGGTG or Exon 1 (for): CGAAGACTACGGAGGTCGACTC, (rev) CGTGCTATCGTCCCTGCT; Exon 2 (for) GGATTTAGAGCTTTAAGTACGCGCTC, (rev) TGGATACCGTGCCTGACATCA; Exon 3 (for): GTTGTTGGCAAAGCCTCTTGTTC, (rev) CCATCAAAAGCTGAGATGAAAC. Amplification products were treated with ExoSAP-IT (USB Corporation, Cleveland, OH, USA, 1 _μ_l/10 _μ_l PCR reaction for 30 min at37°C followed by 45 min at 80°C) to remove excess primers and dNTPs. The products were sequenced using ABI Prism BigDye terminators version 1.1 on an ABI 3730 automated DNA sequencer. Sequencing of VHL Exon 2 utilised the primer CAGGACGGTCTTGATCTCCTG together with VHL Exon 2 (rev).
Western blots
Protein lysates were prepared in ice-cold buffer containing 25 mM Tris-HCl (pH 7.2), 150 mM NaCl, 5 mM DTT, 2 mM MgCl2, 0.5% NP-40, 1 mM PMSF, 5 _μ_g ml−1 aprotinin, 5 _μ_g ml−1 leupeptin, 1 _μ_g ml−1 pepstatin A, 1 mM 1,10-phenanthroline, 10 mM _N_-ethylmaleimide, 1 mM activated Na-orthovanadate and 1 mM NaF, clarified in a microcentrifuge (5 min at 10K RPM), and protein concentrations measured by the Bradford assay. Samples were denatured in Laemmli buffer, resolved by SDS–PAGE and transferred to PVDF membranes. Membranes were blocked in PBS/0.1% Tween/10% nonfat dry milk (NFDM) and incubated with primary antibodies for 1 h to overnight in PBS/0.1% Tween/1% NFDM. Filters were washed, incubated with HRP-conjugated secondary antibodies, then rewashed extensively in PBS/0.1% Tween. Detection used the ECL Lightning Plus reagent from Amersham, Inc., Piscataway, NJ, USA. The following antibodies were obtained from Cell Signalling, Beverly, MA, USA, and used according to the manufacturer's directions; EGFR, ErbB2, phospho-EGFR-Y845, phospho-EGFR-Y1068, phospho-ERK1/2, phospho-AKT, AKT, phospho-RPS6, RPS6, 4EBP1 and eIF4E. Anti-ERK antibody was obtained from Promega, Inc., Madison, WI, USA, while the anti-HA (Y11), ErbB3 and ErbB4 antibodies were from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA. The mouse monoclonal 11E12, directed against the VHL C-terminus, was a kind gift from Dr RD Burk.
Polysome fractionation on sucrose gradients
Cultured cells treated for 2 h with inhibitors or vehicle were incubated for 5 min in medium containing 0.1 mg ml−1 cycloheximide (CHX). Adherent cells were washed three times with ice-cold PBS/0.1 mg ml−1 CHX, and harvested with trypsin/0.1 mg ml−1 CHX. The cell pellet was lysed for 10 min on ice in 500 _μ_l of 0.3 M NaCl/15 mM Tris-HCl pH 7.5/15 mM MgCl2/1% Triton X-100/0.1 mg ml−1 CHX and 1 mg ml−1 heparin. Following centrifugation (10K, 5 min at 4°C), the clarified supernatant was layered onto a 10–50% sucrose step-gradient prepared the night before containing 300 mM NaCl, 15 mM Tris-HCl, pH 7.5, 15 mM MgCl2, 1 mg ml−1 heparin and 0.1 mg ml−1 CHX. Gradients were centrifuged at 37K RPM for 2.5 h in an SW41 rotor, collected from the top using an ISCO tube piercing device (ISCO, Inc., Lincoln, NE, USA) and fractions monitored by absorbance at 280 nm. RNA was recovered using sequential guanidine-HCl extraction and ethanol precipitation followed by further purification on RNeasy columns (Qiagen, Inc., Alameda, CA, USA) and on-column digestion with RNase-free DNase using conditions recommended by the manufacturer.
Results
Epidermal growth factor receptor expression and synergistic growth inhibition by Iressa plus rapamycin
Previous reports have shown that EGFR expression is elevated in RCC although there is less information concerning other ErbB family members. To address this question, we used real-time RT–PCR to measure EGFR, ErbB-2, ErbB-3 and ErbB-4 mRNA levels in 14 RCC cell lines (Table 2), genetically verified to be of independent origin (see Methods). Expression was normalised to the housekeeping gene, GAPDH, although equivalent results were obtained using _β_-actin (not shown). The results demonstrate that EGFR is the predominantly expressed ErbB family member. Although ErbB-3 was next most highly and frequently expressed, these levels were low in comparison to EGFR. In addition, we determined the VHL mutant status for most of the cell lines and the level of expression for lines with wt-VHL (Table 2). For comparison, VHL mRNA levels were 6.7% of GAPDH in the immortalised renal epithelial cell line, HEK293 (not shown). It is apparent that most lines have either mutant VHL or express this gene at low levels making any potential correlations between VHL and EGFR problematic. Corresponding EGFR protein levels for some of these cell lines are shown in Figure 1A. There was no consistent relationship between EGFR mRNA and protein levels (e.g. SKRC-45 and KRCY). These results suggest that there must be post-transcriptional differences affecting EGFR in RCC. The other three ErbB family members were not detectable by Western analysis.
Table 2 Expressiona of ErbB family genes and von Hippel–Lindau (VHL) mutant status in renal cell carcinoma (RCC) cell lines
Figure 1
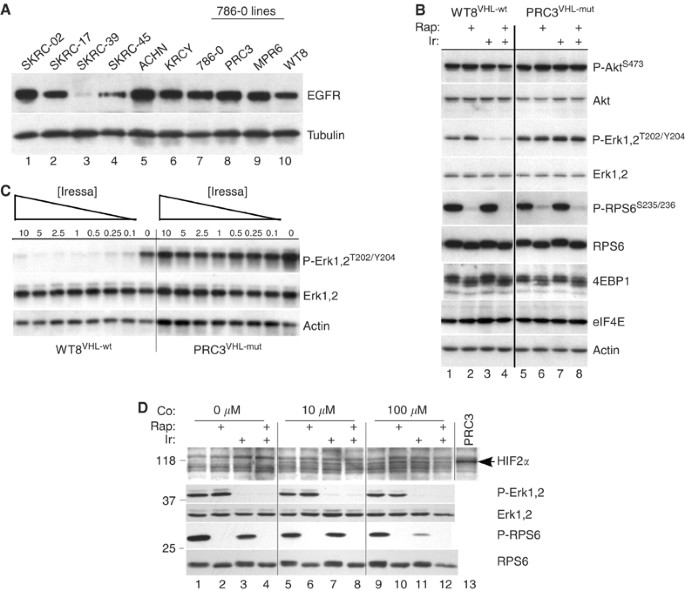
Differential EGFR levels and ERK phosphorylation in RCC cells. (A) Aliquots (10 μ_g) of cell lysates were analysed by Western blot for levels of EGFR protein. PRC3, MPR6 and WT8 are derived from 786-O cells by transfection of empty vector (PRC3) or independent wild-type VHL expression constructs (MPR6 and WT8). Tubulin served as a loading control. (B) RCC cell lines WT8VHL-wt and PRC3VHL-mut were grown until 50% confluent, then treated for 2 h with DMSO (lanes 1 and 5), rapamycin (10 nM; lanes 2 and 6), Iressa (10 μ M; lanes 3 and 7) or both (lanes 4 and 8). Aliquots were analysed for the indicated total and phospho-proteins. (C) WT8VHL-wt and PRC3VHL-mut cells at 50% confluency were treated for 2 h with decreasing doses of Iressa (10–0.1 μ M) and analysed as above. (D) WT8VHL-wt cells at 50% confluency were treated for 20 h with 10 or 100 μ M CoCl2, or not, as indicated (Co:). For the last 2 h, selected cultures were treated with rapamycin (10 nM), Iressa (10 μ M) or both; control cells were treated with DMSO. Cells were washed twice with PBS, harvested and analysed with the indicated antibodies. One lane of PRC3VHL-mut lysate was included as a positive control for the HIF2_α antibody (arrow). This lane was not analysed with the other four antibodies.
The quantitative EGFR results suggested that a selective inhibitor, such as Iressa (Gefitinib, ZD1839), might inhibit growth especially when combined with rapamycin. We tested this hypothesis initially using the paired cell lines, WT8 and PRC3. These lines were derived from 786-O, a clear-cell carcinoma that contains a deletion and frameshift of the VHL gene (Iliopoulos et al, 1995). WT8 cells contain stably transfected, HA-tagged VHL (p24 form), while PRC3 cells contain the vector-only control. Low-density cultures of WT8 and PRC3 cells were treated with varying concentrations of Iressa, rapamycin or the combination. After 5 days, growth was measured by MTT assays and the results converted to a combination index using CALCUSYN (Chou and Talalay, 1984). Values much less than 1.0 indicate synergy while values much greater than 1.0 are indicative of antagonism. As shown in Figure 2, Iressa plus rapamycin at various doses synergistically inhibited growth of WT8 cells. Nearly identical results were obtained using an independent stable VHL transfectant, MPR6 (Schoenfeld et al, 1998). These results include Iressa concentrations within the reported IC50 range for EGFR (i.e. 30–50 nM). At high Iressa and low rapamycin concentrations, a combination index close to 1.0 was obtained (indicating no interaction). In contrast, in the VHL-mutant cell lines 786-O and PRC3, low concentrations of Iressa plus rapamycin were antagonistic. Thus, the introduction of a wt-VHL gene into 786-O cells confers sensitivity (synergy) to the combination of EGFR plus mTOR inhibition at low drug concentrations. Of note, the levels of rapamycin used are readily achievable in patients.
Figure 2
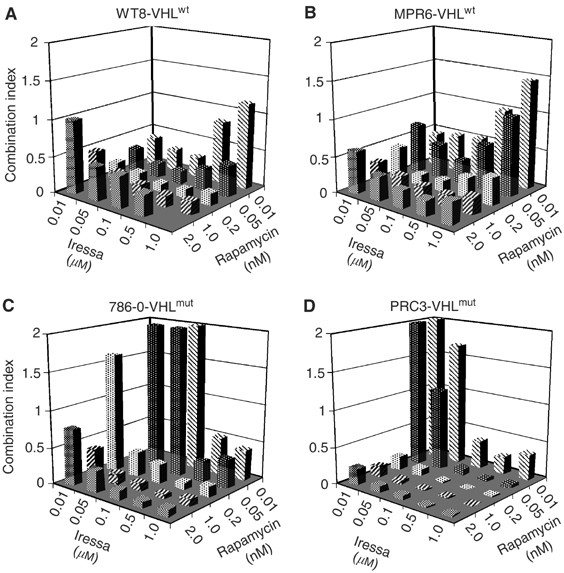
Growth inhibition by Iressa and rapamycin in derivatives of 786-O cells. The parental RCC line 786-O and three stably transfected derivatives were tested for the effects of varying doses of Iressa and rapamycin on growth using MTT assays. MTT absorbance values from single and combined agents were converted to the combination index (see Methods).
Biochemical correlates of Iressa and rapamycin exposure
To explore the difference in growth inhibition, we examined selected biochemical targets of EGFR and mTOR signalling in the PRC3 and WT8 cell lines. Short-term (2 h) treatment with Iressa inhibited phosphorylation of ERK1/2 in WT8 but not in PRC3 cells (Figure 1B). Iressa had no effect on AKT or 4E-BP1 phosphorylation in either cell line. Rapamycin, on the other hand, inhibited phosphorylation of both RPS6 and 4E-BP1 in a VHL-independent manner. Thus, in these paired cell lines, we identified biochemical changes that correlated with the observed synergistic growth inhibition in the MTT assays. Since higher concentrations of Iressa (10 μ M) were used in these short-term studies, we performed a dose–response analysis of ERK1/2 phosphorylation (Figure 1C). Inhibition was seen in WT8 cells at the lowest Iressa dose used (100 nM) while no inhibition was detected in PRC3.
Differential sensitivity to Iressa in WT8 cells could result from the absence of HIF_α_, a consequence of re-expressing wild-type VHL. As an initial assessment of HIF dependence, we treated WT8 cells with cobalt chloride to chemically induce an hypoxic state and thus upregulate HIF. Cobalt-treated cells were then tested for changes in response to Iressa (Figure 1D). Treatment with 10 and 100 μ M CoCl2 induced similar levels of the 118 kDa HIF2_α_, the only HIF_α_ isoform expressed in 786-O cells (Iliopoulos et al, 1995). Induction of HIF2_α_ had no effect on the ability of Iressa to inhibit Erk phosphorylation (compare lanes 3, 7 and 11), suggesting that under these conditions, differential sensitivity was not HIF dependent.
We expanded the biochemical analysis to six additional RCC cell lines (Figure 3). In the wt-VHL lines, ACHN and KRCY, Iressa inhibited both ERK1/2 and RPS6 phosphorylation while phospho-AKT levels were unaffected. In SKRC-39, which expressed the highest level of wt-VHL, the basal protein patterns were strikingly different. These cells overexpressed eIF4E and had low to undetectable levels of phospho-ERK1/2 (Figure 3). Epidermal growth factor receptor levels were also substantially reduced (Figure 1A). Among the three mutant VHL cell lines, only SKRC-45 showed any response to Iressa, consisting of a partial reduction of ERK1/2 and RPS6 phosphorylation. Rapamycin uniformly inhibited phospho-RPS6 regardless of the VHL status. In summary, Iressa was substantially more effective at inhibiting ERK and RPS6 phosphorylation in RCC cell lines with wt-VHL. Using a Wilcoxon Rank Sum Test, this approached but did not reach statistical significance (_P_=0.08) because of the limited number of cell lines.
Figure 3
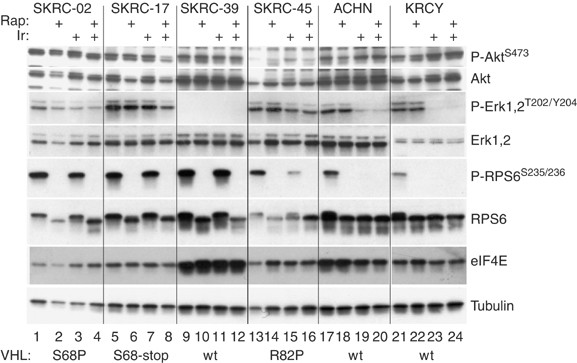
Phospho-protein analysis in additional RCC cell lines. Six RCC cell lines were grown to 50% confluency, then treated for 2 h with DMSO (lanes 1, 5, 9, 13, 17 and 21), rapamycin (10 nM; lanes 2, 6, 10, 14, 18 and 22), Iressa (10 μ M; lanes 3, 7, 11, 15, 19 and 23) or both (lanes 4, 8, 12, 16, 20 and 24). Lysates were analysed as in Figure 1. VHL mutational status is indicated along the bottom.
EGFR protein levels, phosphorylation and polysome loading of mRNA
Based on the differential responses to EGFR blockade, we examined levels of EGFR protein in PRC3 and WT8 cells following treatment with Iressa or rapamycin (Figure 4A). Epidermal growth factor receptor levels were higher in the VHL mutant, PRC3, although no changes were induced by treatment. These differences were more apparent at shorter exposures (compare Figures 1A, 2A, 3 and 4A). To examine the phosphorylation dynamics of EGFR, we serum-starved WT8 and PRC3 cells for 2 h, then added back serum plus EGF. Protein lysates were examined at multiple time points using phospho-specific EGFR antibodies (Figure 4B). In these experiments, more protein was loaded for WT8 to compensate for the lower levels of total EGFR. For tyrosine 845, maximum phosphorylation was seen at 10 min in WT8 cells compared to 60 min in PRC3. For tyrosine 1068, the onset of maximum phosphorylation was similar. For both sites, the duration of phosphorylation was prolonged in PRC3. Similar results were obtained in an independent experiment carried out to 180 min after EGF addition with evidence of persistent phosphorylation in PRC3 (not shown). Despite these differences, prior exposure of both cell lines to Iressa for 15 min blocked tyrosine phosphorylation (not shown). Thus, while the presence of wt-VHL in these paired cell lines appears to affect EGFR protein levels and activation, the results do not explain the differential sensitivity to Iressa. One likely possibility is that VHL mutations upregulate signalling from other kinases.
Figure 4
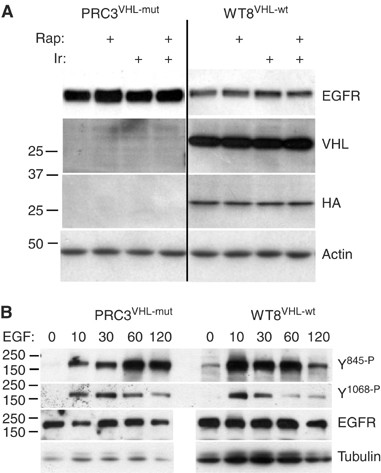
Total and phospho-EGFR levels in PRC3 and WT8. (A) Protein lysates, as described in Figure 1, were analysed for total EGFR protein. The blot was stripped and reprobed with anti-VHL and anti-HA antibodies. (B) PRC3 and WT8 cultures were grown to 50% confluency, washed with PBS, then starved in 0.1% serum for 2 h. Cells were harvested at the indicated time points (min) after the addition of 10% serum and 50 ng ml−1 EGF. More total protein was loaded in the WT8 lanes to compensate for the lower amounts of EGFR in this cell line. Otherwise, equal protein aliquots were analysed for the phosphorylation levels of tyrosine 845 and 1068 using phospho-specific antibodies.
To determine whether the elevated EGFR protein in PRC3 was the result of mRNA differences, possibly due to clonal variation, we examined message levels by real-time RT–PCR in parental 786-O, PRC3 and WT8 cells. The analyses, performed in triplicate, demonstrated that there were no differences among the cell lines (Table 3). Like the original series of RCCs examined, ErbB-2, ErbB-3 and ErbB-4 mRNA levels were barely detectable and corresponding proteins were undetectable by Western analysis. In contrast, VEGF expression, as expected, was higher in the VHL mutants 786-O and PRC3 than WT8. These results indicate that the observed differences in EGFR protein levels are post-transcriptional, possibly involving protein translation initiation or degradation. To examine the question of protein translation initiation, we isolated polysome fractions from WT8 and PRC3 cells and analysed these by quantitative real-time RT–PCR (Figure 5). No reproducible differences were identified in ribosome-bound EGFR mRNA between these cell lines. Furthermore, short-term treatment with rapamycin or Iressa had no consistent effects on polysome-associated EGFR message (not shown). These results suggest that the difference in EGFR protein levels (and phosphorylation duration) between PRC3 and WT8 involves internalisation or degradation.
Table 3 Expressiona of ErbB family genes in 786-O cell lines
Figure 5
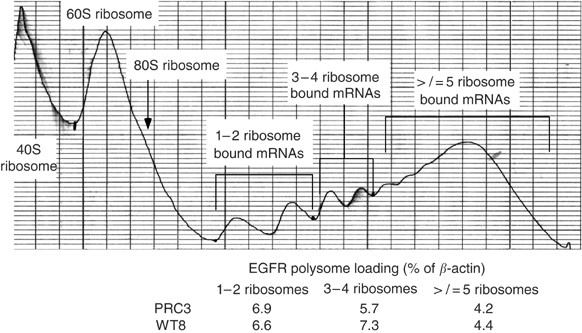
Polysome analysis of WT8 cells. WT8 cells were grown to 70% confluency, then treated for 2 h with DMSO, rapamycin (10 nM), Iressa (10 μ M) or both, and harvested for polysome fractionation. A representative A280 fractionation profile is shown along with the fractions pooled and the results from quantitative RT–PCR analysis.
Downregulation of MYC polysome loading by PI3K inhibition
Activated AKT is known to play an important antiapoptotic role and also positively regulates protein translation initiation. As noted above, EGFR and mTOR inhibitors did not affect phospho-AKT levels and did not have a major effect on EGFR polysome loading. We wished to determine, therefore, whether short-term blockade of AKT (using the PI3K inhibitor LY-294002) would have a demonstrable effect. We chose to focus on MYC translation initiation (polysome loading) in WT8 cells, since this gene is upregulated in RCC and because MYC mRNA contains an internal ribosome entry site, which provides a potential eIF4E-independent means of translation initiation (Shi et al, 2005). Also, since WT8 cells were more responsive to Iressa and rapamycin, we combined LY-294002 with these agents. As shown in Table 4, neither Iressa, rapamycin nor the combination substantially reduced MYC mRNA ribosome association and these agents may even have increased polysome loading in some fractions. As a control, we included RPS19 that contains a 5′-untranslated polypyrimidine stretch known to be sensitive to mTOR inhibition (Meyuhas, 2000). RPS19 loading was substantially reduced in all ribosome fractions by both agents. In contrast, MYC polysome loading was reduced only by LY-294002. This effect appeared to be somewhat greater when LY-294002 was combined with rapamycin or rapamycin plus Iressa.
Table 4 mRNAa loading on polysomes in WT8 cells
Analysis of primary RCCs
Cell lines differ from primary tumours in terms of growth rates, altered microenvironment (nutrients, oxygen tension and extracellular matrix) and the absence of interacting stroma. To test for differences in phosphoprotein levels, we examined 12 clear-cell carcinomas and their corresponding uninvolved kidney (Figure 6). Table 1 (Methods) provides the clinical characteristics of these tumours including mutational analysis of VHL, which is summarised in Figure 6. Phospho-ERK1/2 was readily detectable and upregulated in nine of the 12 tumours (Figure 6A). Of note, most of these same tumour samples contained elevated levels of phospho-AKT. Total AKT, ERK and Coomassie staining are shown as controls. Compared to the cell lines, longer radiographic exposures were required to detect phospho-AKT. The difference in phospho-AKT was confirmed in side-by-side comparisons with selected tumours and cell lines (Figure 6C), while comparable levels of phospho-ERK were detected in both cell lines and tumours.
Figure 6
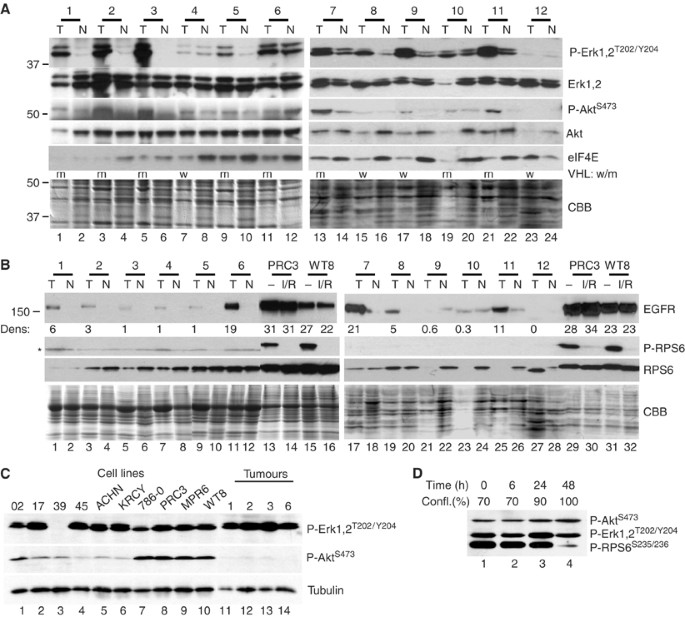
Analysis of primary tumours. (A) Protein (10 _μ_g) from 12 matched pairs of renal tumours (T) and normal kidney (N) were analysed with the indicated antibodies on two separate sets of blots. Equivalent gels were stained with Coomassie blue (CBB) for loading. The low protein content of tumour #10 indicates that relative Erk phosphorylation was even higher than the band intensity suggests. VHL mutational status is summarised by m=mutant; w=wild type. (B) In total, 30 _μ_g of tumour and normal lysates were analysed for levels of EGFR, phospho-RPS6 and total RPS6. The four cell line lysates were treated with Iressa and rapamycin (I/R) for 2 h or not (−). Densitometric analysis for the left panel was normalised to the tumour with the lowest EGFR signal. Densitometry for the right panel utilised a shorter exposure to remain within the linear response range. Even the longest exposures failed to detect EGFR in T-12. The asterisk denotes a background band. (C) Direct comparisons of phospho-Erk and phospho-Akt levels in a subset of tumours and cell lines on the same filter. (D) WT8VHL-wt cultures starting at 70% confluency (time zero) were grown under standard culture conditions over 48 h and periodically sampled. Phospho-RPS6 levels were downregulated at 100% confluency.
Levels of eIF4E were generally higher in the matched normal samples than in the tumours, with the exception of tumour #12 that had substantially increased eIF4E (compare lanes 23 and 24). Tumour 12 had sarcomatoid features (Table 1), indicating a more aggressive tumour and correspondingly poorer prognosis (Cheville et al, 2004). This tumour showed no detectable Erk phosphorylation despite abundant Erk protein and no detectable EGFR (Figure 6B). The biochemical characteristics of tumour 12 thus closely match the cell line SKRC-39 and verify that a subset of RCCs appear to be activated at this downstream translation regulatory point.
Most of the tumours expressed more EGFR protein than the corresponding normal tissue, yet EGFR levels were up to 30-fold higher in cell lines despite comparable levels of mRNA (Figure 6B, Tables 2 and 5). Among the tumours, only nos. 6 and 7 had EGFR protein levels that were as high as the cell lines and at least T-6 also expressed elevated EGFR message. In contrast, ErbB-3 mRNA was much higher in the primary tumours (and matched normal kidney) than in RCC cell lines, while ErbB-4 message was markedly reduced in tumours compared to normal (Table 5).
Table 5 Expressiona of mRNA for ErbB Genes in renal cell carcinoma (RCC) tumours
An additional major difference between tumours and cell lines involved phospho-RPS6, an indicator of mTOR activity. Phosphorylated RPS6 was undetectable in both tumours and normal kidney (Figure 6B), despite usually abundant levels of total RPS6 protein. Cell–cell contacts in solid tumours may result in alterations akin to contact inhibition in tissue culture. We therefore examined the effect of confluency on the phosphorylation state of RPS6 in WT8 cells (Figure 6D). While phospho-Akt and phospho-Erk were unaffected, phosphorylation of RPS6 on Ser235/236 was dramatically downregulated at 100% confluency. Thus, differences in RPS6 phosphorylation between tumours and cell lines may be a result of different growth rates or of cell–cell interaction.
Discussion
The results reported here demonstrate that combined inhibition of EGFR and mTOR provides a more effective growth blockade in RCC cell lines than either agent alone. Moreover, this effect was influenced by the mutational state of VHL. While we observed synergistic growth inhibition in both mutant and wt-VHL cell lines at high drug concentrations, synergy at Iressa doses near the IC50 for EGFR were limited to wt-VHL cells. Western blot analysis of phospho-proteins demonstrated that EGFR inhibition selectively blocked phosphorylation of ERK1/2 and RPS6 in cells with wt-VHL. The inhibition of RPS6 phosphorylation by Iressa, however, was not universal, which may relate to the presence of a PTEN mutation in 786-O and its derivatives (Kau et al, 2003). Mutant VHL lines showed little response to Iressa in terms of ERK and RPS6 phosphorylation and AKT phosphorylation was unaffected by either agent irrespective of VHL. These results indicate that wt-VHL cells are more sensitive to EGFR inhibition (or more dependent upon EGFR signalling for growth) than their mutant counterparts. Induction of HIF2_α_ using cobalt treatments failed to prevent Iressa inhibition of Erk phosphorylation in WT8VHL-wt cells, suggesting that this effect is independent of HIF.
Perera et al (2000) also noted that wt-VHL conveyed sensitivity to the EGFR blocking antibody, C225. However, changes in phospho-protein signalling were not described. Interestingly, we observed that the combination of low-dose Iressa and rapamycin was antagonistic in cells with mutant-VHL. This raises the possibility that certain drug targets might be regulated in an opposite manner depending on the state of VHL. Similar AKT-dependent results have been reported for single-agent rapamycin (Gera et al, 2004). However, since we did not detect changes in AKT phosphorylation, another target(s) that is VHL-dependent presumably would be responsible. Our studies suggest that there are at least three biochemical patterns in RCC cell lines: those responsive to EGFR inhibition in terms of ERK and RPS6 phosphorylation, those that are resistant and a third subset (likely resistant) with overexpression of eIF4E and downregulation of upstream signalling components. Of note, in cells where the eIF4E-binding protein 4E-BP1 is limiting, eIF4E overexpression also drives translation initiation in a rapamycin-resistant manner (Dilling et al, 2002).
During these investigations, we identified higher levels of EGFR protein in PRC3 cells (mutant VHL) compared to WT8. In agreement, EGFR phosphorylation following EGF stimulation was also prolonged. However, this appears not to be the explanation for differential Iressa sensitivity since pretreatment of cells with Iressa blocked EGFR phosphorylation equally. Also, EGFR protein levels were not affected by Iressa or rapamycin. We also noted that EGFR protein was generally higher in cell lines than primary tumours despite comparable mRNA levels. Whether this represents an in vitro selection phenomenon with preferential growth of these cells is unknown. Previous investigators have not reported suppression of EGFR protein after re-expression of wt-VHL (Knebelmann et al, 1998; Gunaratnam et al, 2003). However, in the report by Gunaratnam et al, (2003), adenovirus-mediated introduction of wt-VHL into 786-O cells visibly reduced EGFR levels (see their Figure 2A), although this difference was either discounted or not further clarified (Gunaratnam et al, 2003). In the report of Knebelmann et al, (1998), the Western blot appears too overexposed for optimal quantitation (Knebelmann et al, 1998). On the other hand, we have not detected reduced EGFR protein in MPR6, an independent wt-VHL derivative of 786-O. However, MPR6 expresses two anti-VHL reactive bands at considerably lower levels than the single band detected (with either anti-HA or anti-VHL antibodies) in WT8. Thus, we cannot conclude that the changes in EGFR are a consequence of VHL. This difference could, for example, be explained by an acquired clonal variation affecting some aspect of EGFR protein processing or degradation. In any case, since EGFR mRNA levels in 786-O, PRC3 and WT8 were essentially identical, and since we were unable to detect consistent differences in EGFR ribosome association between PRC3 and WT8, we suspect that the difference involves EGFR degradation, which would be consistent with the observed differences in the duration of receptor phosphorylation. This would also be consistent with the absence of EGFR mRNA among a set of messages whose translation initiation was affected by VHL mutation (Galban et al, 2003).
EGFR overexpression in RCC has previously been well documented (Gomella et al, 1989; Uhlman et al, 1995). The finding that TGF_α_ is constitutively expressed as a consequence of VHL mutations (de Paulsen et al, 2001), and that TGF_α_ is a mitogen for renal epithelial cells, considerably strengthened the hypothesis that EGFR signalling is important in RCC development. However, in contrast to lung cancer, activating mutations in exons 19 and 21 of EGFR were not detected in 16 kidney tumours (Lynch et al, 2004) and, clinically, tumour regressions following EGFR inhibition have been minimal (Amato, 2005). If EGFR signalling is deregulated in RCC, then why are clinical responses so limited? Possible explanations include involvement of additional ErbB family members not affected by selective EGFR inhibitors, activation of other growth factor pathways such that EGFR blockade is insufficient to induce tumour regression, and changes in apoptotic components that prevent cell death in response to EGFR blockade.
Regarding other ErbB family members, in cell lines we found that EGFR was by far the predominant component among mRNAs and the only ErbB protein detectable by Western analysis. For ErbB-2, previous reports have been inconsistent. Stumm et al (1996) reported that p185erbB-2 was overexpressed in RCC while Freeman et al, (1989) found ErbB-2 to be reduced (Freeman et al, 1989; Stumm et al, 1996). We found ErbB-2 mRNA was expressed at low levels in both cell lines and primary tumours. For ErbB-3 and ErbB-4, Thomasson et al (2004) reported that both receptors were downregulated. Our results are in agreement for ErbB-4, although ErbB-3 was discordant, being downregulated in cell lines but maintained at substantial levels in primary tumours. Potentially, this is an important difference although the biological consequences are unclear. Although ErbB-3 lacks kinase activity (Burgess et al, 2003), it couples efficiently to phosphatidylinositol 3-kinase (Fedi et al, 1994; Burgess et al, 2003), has distinct preferred ligands (i.e. NDF, HB-EGF and BTC)(Beerli and Hynes, 1996) and is endocytosed less readily than EGFR (Baulida et al, 1996). Interestingly, in mice one of these ligands, NDF, is most highly expressed in the kidney and during development correlates with urogenital organ formation (Castagnino et al, 2000). At least in keratinocytes, NDF is induced by hepatocyte growth factor (HGF) (Castagnino et al, 2000). If this occurred in the kidney, then RCCs with VHL mutations, which are known to upregulate MET and be hyper-responsive to HGF (Oh et al, 2002; Koochekpour et al, 1999), might behave differently in terms of ErbB signalling and EGFR inhibition than cell lines.
Regarding other growth factor pathways that might render EGFR blockade insufficient, we note that phospho-AKT was not affected by short-term Iressa, rapamycin or the combination. Similarly, phospho-ERK1/2 showed no response to Iressa in mutant VHL cell lines. These results strongly suggest that other growth factor signalling pathways are activated. The inability of Iressa and rapamycin to affect AKT led us to ask whether blocking AKT activation with the PI3K, LY294002, would alter protein translation initiation of MYC. We chose MYC because of its overexpression in RCC (Yao et al, 1988) and because it contains an internal ribosome entry site that may allow initiation in an eIF4E-independent manner. MYC mRNA polysome loading was not affected by Iressa or rapamycin while the control RPS19 was substantially downregulated. In contrast, a strong reduction in MYC polysome loading was achieved by 2 h PI3K inhibition.
Lastly, how relevant are the phospho-protein analyses to primary tumours? Levels of phospho-ERK1/2 were similar between cell lines and primary tumours and these same tumours also had elevated phospho-AKT, although less so than in cell lines. Thus, phospho-ERK and AKT may be useful biomarkers in RCC. While most tumours overexpressed EGFR compared to matched normal controls, only two of 12 contained EGFR levels comparable to those observed in RCC cell lines. Another striking difference was the absence of RPS6 phosphorylation in any of the tumour/normal samples despite easily detectable total RPS6 protein. RPS6 is phosphorylated by p70S6K, which in turn is activated by mTOR (Hay and Sonenberg, 2004). Similar results were suggested by Kenerson et al (2002) who found increased phospho-RPS6 in RCCs derived from patients with tuberous sclerosis but not in sporadic RCCs (Kenerson et al, 2002). Other studies of non-RCC primary tumours have frequently identified mTOR activation. A broad study of phosphorylated S6K1 in eight common tumour types found nearly all with some reactivity while intense staining (3+) was present in up to 60% of colon adenocarcinomas and nearly 50% of lymphomas, ovarian, melanoma, lung and brain tumours (Xu et al, 2004).
Our results suggest that mTOR may be relatively inactive in primary RCCs. However, there are two important caveats. The first is that mTOR activity could be confined to areas of growth or invasion that would have been missed by our examination of bulk tumour protein. This interpretation is strengthened by our observation that confluency dramatically affected phospho-RPS6 levels in WT8 cells. Alternatively, mTOR may be activated in only a subset of RCCs in agreement with the observed 7% response rate to CCI-779 (Atkins et al, 2004).
One cell line (SKRC-39) expressed high levels of eIF4E in the absence of upstream signalling, and one tumour (#12) showed a very similar biochemical pattern, with high eIF4E but no detectable EGFR or phospho-Erk. Interestingly, this tumour had sarcomatoid differentiation, an indicator of more aggressive behaviour and poor prognosis (Cheville et al, 2004). Thus, eIF4E overexpression appears to define a subset of RCCs that are likely to be resistant to rapamycin and/or Iressa, consistent with the aggressive nature of sarcomatoid RCCs.
In summary, our results provide rationale for the testing of combined EGFR and mTOR inhibitors, at least in the subset of RCC patients with wt-VHL. It will be important to determine whether mTOR activation is heterogeneous or activated in only certain subsets of tumours and whether in vivo phospho-AKT is affected by these treatments.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
- Al-Saleem T, Wessner LL, Scheithauer BW, Patterson K, Roach ES, Dreyer SJ, Fujikawa K, Bjornsson J, Bernstein J, Henske EP (1998) Malignant tumors of the kidney, brain, and soft tissues in children and young adults with the tuberous sclerosis complex. Cancer 83: 2208–2216
Article CAS PubMed Google Scholar - Amato RJ (2005) Renal cell carcinoma: review of novel single-agent therapeutics and combination regimens. Ann Oncol 16: 7–15
Article CAS PubMed Google Scholar - Atkins MB, Hidalgo M, Stadler WM, Logan TF, Dutcher JP, Hudes GR, Park Y, Liou SH, Marshall B, Boni JP, Dukart G, Sherman ML (2004) Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol 22: 909–918
Article CAS PubMed Google Scholar - Baulida J, Kraus MH, Alimandi M, Di Fiore PP, Carpenter G (1996) All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J Biol Chem 271: 525–5257
Google Scholar - Beerli RR, Hynes NE (1996) Epidermal growth factor-related peptides activate distinct subsets of ErbB receptors and differ in their biological activities. J Biol Chem 271: 6071–6076
Article CAS PubMed Google Scholar - Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin Jr WG (2003) TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 4: 147–158
Article CAS PubMed Google Scholar - Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868
Article CAS PubMed Google Scholar - Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S (2003) An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell 12: 541–552
Article CAS PubMed Google Scholar - Castagnino P, Lorenzi MV, Yeh J, Breckenridge D, Sakata H, Munz B, Werner S, Bottaro DP (2000) Neu differentiation factor/heregulin induction by hepatocyte and keratinocyte growth factors. Oncogene 19: 640–648
Article CAS PubMed Google Scholar - Cheville JC, Lohse CM, Zincke H, Weaver AL, Leibovich BC, Frank I, Blute ML (2004) Sarcomatoid renal cell carcinoma: an examination of underlying histologic subtype and an analysis of associations with patient outcome. Am J Surg Pathol 28: 435–441
Article PubMed Google Scholar - Chou TC, Talalay P (1984) Quantitative analysis of dose–effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regulat 22: 27–55
Article CAS Google Scholar - de Paulsen N, Brychzy A, Fournier MC, Klausner RD, Gnarra JR, Pause A, Lee S (2001) Role of transforming growth factor-alpha in von Hippel–Lindau (VHL)(−/−) clear cell renal carcinoma cell proliferation: a possible mechanism coupling VHL tumor suppressor inactivation and tumorigenesis. Proc Natl Acad Sci USA 98: 1387–1392
CAS PubMed PubMed Central Google Scholar - Dilling MB, Germain GS, Dudkin L, Jayaraman AL, Zhang X, Harwood FC, Houghton PJ (2002) 4E-binding proteins, the suppressors of eukaryotic initiation factor 4E, are down-regulated in cells with acquired or intrinsic resistance to rapamycin. J Biol Chem 277: 13907–13917
Article CAS PubMed Google Scholar - Drabkin HA, Parsy C, Ferguson K, Guilhot F, Lacotte L, Roy L, Zeng C, Baron A, Hunger SP, Varella-Garcia M, Gemmill R, Brizard F, Brizard A, Roche J (2002) Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia 16: 186–195
Article CAS PubMed Google Scholar - Drucker B, Bacik J, Ginsberg M, Marion S, Russo P, Mazumdar M, Motzer R (2003) Phase II trial of ZD1839 (IRESSA) in patients with advanced renal cell carcinoma. Invest New Drugs 21: 341–345
Article CAS PubMed Google Scholar - Fedi P, Pierce JH, di Fiore PP, Kraus MH (1994) Efficient coupling with phosphatidylinositol 3-kinase, but not phospholipase C gamma or GTPase-activating protein, distinguishes ErbB-3 signaling from that of other ErbB/EGFR family members. Mol Cell Biol 14: 492–500
Article CAS PubMed PubMed Central Google Scholar - Foon KA, Yang XD, Weiner LM, Belldegrun AS, Figlin RA, Crawford J, Rowinsky EK, Dutcher JP, Vogelzang NJ, Gollub J, Thompson JA, Schwartz G, Bukowski RM, Roskos LK, Schwab GM (2004) Preclinical and clinical evaluations of ABX-EGF, a fully human anti-epidermal growth factor receptor antibody. Int J Radiat Oncol Biol Phys 58: 984–990
Article CAS PubMed Google Scholar - Freeman MR, Washecka R, Chung LW (1989) Aberrant expression of epidermal growth factor receptor and HER-2 (erbB-2) messenger RNAs in human renal cancers. Cancer Res 49: 6221–6225
CAS PubMed Google Scholar - Galban S, Fan J, Martindale JL, Cheadle C, Hoffman B, Woods MP, Temeles G, Brieger J, Decker J, Gorospe M (2003) von Hippel–Lindau protein-mediated repression of tumor necrosis factor alpha translation revealed through use of cDNA arrays. Mol Cell Biol 23: 2316–2328
Article CAS PubMed PubMed Central Google Scholar - Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, Lichtenstein AK (2004) AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem 279: 2737–2746
Article CAS PubMed Google Scholar - Gomella LG, Sargent ER, Wade TP, Anglard P, Linehan WM, Kasid A (1989) Expression of transforming growth factor alpha in normal human adult kidney and enhanced expression of transforming growth factors alpha and beta 1 in renal cell carcinoma. Cancer Res 49: 6972–6975
CAS PubMed Google Scholar - Gunaratnam L, Morley M, Franovic A, de Paulsen N, Mekhail K, Parolin DA, Nakamura E, Lorimer IA, Lee S (2003) Hypoxia inducible factor activates the transforming growth factor-alpha/epidermal growth factor receptor growth stimulatory pathway in VHL(−/−) renal cell carcinoma cells. J Biol Chem 278: 44966–44974
Article CAS PubMed Google Scholar - Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18: 1926–1945
Article CAS PubMed Google Scholar - Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM (1994) Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA 91: 9700–9704
Article CAS PubMed PubMed Central Google Scholar - Hidalgo M (2003) Erlotinib: preclinical investigations. Oncology (Huntingt) 17 (11 Suppl 12): 11–16
Google Scholar - Hosoi H, Dilling MB, Shikata T, Liu LN, Shu L, Ashmun RA, Germain GS, Abraham RT, Houghton PJ (1999) Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res 59: 886–894
CAS PubMed Google Scholar - Humes HD, Beals TF, Cieslinski DA, Sanchez IO, Page TP (1991) Effects of transforming growth factor-beta, transforming growth factor-alpha, and other growth factors on renal proximal tubule cells. Lab Invest 64: 538–545
CAS PubMed Google Scholar - Iliopoulos O, Kibel A, Gray S, Kaelin Jr WG (1995) Tumour suppression by the human von Hippel–Lindau gene product. Nat Med 1: 822–826
Article CAS PubMed Google Scholar - Kaelin Jr WG (2003) The von Hippel–Lindau gene, kidney cancer, and oxygen sensing. J Am Soc Nephrol 14: 2703–2711
Article PubMed Google Scholar - Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR, Silver PA (2003) A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell 4: 463–476
Article CAS PubMed Google Scholar - Kenerson HL, Aicher LD, True LD, Yeung RS (2002) Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res 62: 5645–5650
CAS PubMed Google Scholar - Knebelmann B, Ananth S, Cohen HT, Sukhatme VP (1998) Transforming growth factor alpha is a target for the von Hippel–Lindau tumor suppressor. Cancer Res 58: 226–231
CAS PubMed Google Scholar - Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin Jr WG (2002) Inhibition of HIF is necessary for tumor suppression by the von Hippel–Lindau protein. Cancer Cell 1: 237–246
Article CAS PubMed Google Scholar - Kondo K, Yao M, Kobayashi K, Ota S, Yoshida M, Kaneko S, Baba M, Sakai N, Kishida T, Kawakami S, Uemura H, Nagashima Y, Nakatani Y, Hosaka M (2001) PTEN/MMAC1/TEP1 mutations in human primary renal-cell carcinomas and renal carcinoma cell lines. Int J Cancer 91: 219–224
Article CAS PubMed Google Scholar - Koochekpour S, Jeffers M, Wang PH, Gong C, Taylor GA, Roessler LM, Stearman R, Vasselli JR, Stetler-Stevenson WG, Kaelin Jr WG, Linehan WM, Klausner RD, Gnarra JR, Vande Woude GF (1999) The von Hippel–Lindau tumor suppressor gene inhibits hepatocyte growth factor/scatter factor-induced invasion and branching morphogenesis in renal carcinoma cells. Mol Cell Biol 19: 5902–5912
Article CAS PubMed PubMed Central Google Scholar - Latif F, Tory K, Gnarra J (1993) Identification of the von Hippel–Lindau disease tumor suppressor gene. Science 260: 1317–1320
Article CAS PubMed Google Scholar - Lazaris-Karatzas A, Montine KS, Sonenberg N (1990) Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature 345: 544–547
Article CAS PubMed Google Scholar - Li Z, Wang D, Na X, Schoen SR, Messing EM, Wu G (2002) Identification of a deubiquitinating enzyme subfamily as substrates of the von Hippel–Lindau tumor suppressor. Biochem Biophys Res Commun 294: 700–709
Article CAS PubMed Google Scholar - Louro ID, McKie-Bell P, Gosnell H, Brindley BC, Bucy RP, Ruppert JM (1999) The zinc finger protein GLI induces cellular sensitivity to the mTOR inhibitor rapamycin. Cell Growth Differ 10: 503–516
CAS PubMed Google Scholar - Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350: 2129–2139
Article CAS PubMed Google Scholar - Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD (2002) The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 1: 247–255
Article CAS PubMed Google Scholar - Meyuhas O (2000) Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem 267: 6321–6330
Article CAS PubMed Google Scholar - Mohi MG, Boulton C, Gu TL, Sternberg DW, Neuberg D, Griffin JD, Gilliland DG, Neel BG (2004) Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci USA 101: 3130–3135
Article CAS PubMed PubMed Central Google Scholar - Mondesire WH, Jian W, Zhang H, Ensor J, Hung MC, Mills GB, Meric-Bernstam F (2004) Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res 10: 7031–7042
Article CAS PubMed Google Scholar - Motzer RJ, Amato R, Todd M, Hwu WJ, Cohen R, Baselga J, Muss H, Cooper M, Yu R, Ginsberg MS, Needle M (2003) Phase II trial of antiepidermal growth factor receptor antibody C225 in patients with advanced renal cell carcinoma. Invest New Drugs 21: 99–101
Article CAS PubMed Google Scholar - Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL (2001) Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA 98: 10314–10319
Article CAS PubMed PubMed Central Google Scholar - Oh RR, Park JY, Lee JH, Shin MS, Kim HS, Lee SK, Kim YS, Lee SH, Lee SN, Yang YM, Yoo NJ, Lee JY, Park WS (2002) Expression of HGF/SF and Met protein is associated with genetic alterations of VHL gene in primary renal cell carcinomas. APMIS 110: 229–238
Article CAS PubMed Google Scholar - Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, Gavin BJ, Kley N, Kaelin Jr WG, Iliopoulos O (1998) The von Hippel–Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell 1: 959–968
Article CAS PubMed Google Scholar - Perera AD, Kleymenova EV, Walker CL (2000) Requirement for the von Hippel–Lindau tumor suppressor gene for functional epidermal growth factor receptor blockade by monoclonal antibody C225 in renal cell carcinoma. Clin Cancer Res 6: 1518–1523
CAS PubMed Google Scholar - Pioli PA, Rigby WF (2001) The von Hippel–Lindau protein interacts with hnRNP A2 and regulates its expression. J Biol Chem 276: 40346–40352
Article CAS PubMed Google Scholar - Roux PP, Blenis J (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68: 320–344
Article CAS PubMed PubMed Central Google Scholar - Rowinsky EK, Schwartz GH, Gollob JA, Thompson JA, Vogelzang NJ, Figlin R, Bukowski R, Haas N, Lockbaum P, Li YP, Arends R, Foon KA, Schwab G, Dutcher J (2004) Safety pharmacokinetics and activity of ABX-EGF a fully human anti-epidermal growth factor receptor monoclonal antibody in patients with metastatic renal cell cancer. J Clin Oncol 22: 3003–3015
Article CAS PubMed Google Scholar - Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, Pandolfi PP (2004) The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med 10: 484–486
Article CAS PubMed Google Scholar - Schoenfeld A, Davidowitz EJ, Burk RD (1998) A second major native von Hippel–Lindau gene product initiated from an internal translation start site functions as a tumor suppressor. Proc Natl Acad Sci USA 95: 8817–8822
Article CAS PubMed PubMed Central Google Scholar - Shi Y, Sharma A, Wu H, Lichtenstein A, Gera J (2005) Cyclin D1 and c-Myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK and ERK-dependent pathway. J Biol Chem 280: 10964–10973
Article CAS PubMed Google Scholar - Shin Lee J, Seok Kim H, Bok Kim Y, Cheol Lee M, Soo Park C (2003) Expression of PTEN in renal cell carcinoma and its relation to tumor behavior and growth. J Surg Oncol 84: 166–172
Article PubMed Google Scholar - Stoneley M, Willis AE (2004) Cellular internal ribosome entry segments: structures trans-acting factors and regulation of gene expression. Oncogene 23: 3200–3207
Article CAS PubMed Google Scholar - Stumm G, Eberwein S, Rostock-Wolf S, Stein H, Pomer S, Schlegel J, Waldherr R (1996) Concomitant overexpression of the EGFR and erbB-2 genes in renal cell carcinoma (RCC) is correlated with dedifferentiation and metastasis. Int J Cancer 69: 17–22
Article CAS PubMed Google Scholar - Thomasson M, Hedman H, Junttila TT, Elenius K, Ljungberg B, Henriksson R (2004) ErbB4 is downregulated in renal cell carcinoma – a quantitative RT–PCR and immunohistochemical analysis of the epidermal growth factor receptor family. Acta Oncol 43: 453–459
Article CAS PubMed Google Scholar - Thompson JE, Thompson CB (2004) Putting the rap on Akt. J Clin Oncol 22: 4217–4226
Article CAS PubMed Google Scholar - Uhlman DL, Nguyen P, Manivel JC, Zhang G, Hagen K, Fraley E, Aeppli D, Niehans GA (1995) Epidermal growth factor receptor and transforming growth factor alpha expression in papillary and nonpapillary renal cell carcinoma: correlation with metastatic behavior and prognosis. Clin Cancer Res 1: 913–920
CAS PubMed Google Scholar - Xu G, Zhang W, Bertram P, Zheng XF, McLeod H (2004) Pharmacogenomic profiling of the PI3K/PTEN-AKT-mTOR pathway in common human tumors. Int J Oncol 24: 893–900
CAS PubMed Google Scholar - Yao M, Shuin T, Misaki H, Kubota Y (1988) Enhanced expression of c-myc and epidermal growth factor receptor (C-erbB-1) genes in primary human renal cancer. Cancer Res 48: 6753–6757
CAS PubMed Google Scholar
Acknowledgements
We thank Dr William Kaelin for providing cell lines PRC3 and WT8, Dr Robert D Burk for providing MPR6, MEA2 and the anti-VHL antibody and Dr Paul Bunn for providing ZD-1839. Statistical analysis was performed by Drs Anna Baron and Chan Zeng of the University of Colorado Cancer Center Biostatistics Core. The Biostatistics Core and the DNA Sequencing & Analysis Core are supported by an NIH/NCI grant, CA046934. We thank B Helfrich for helpful discussions during the course of this work. These studies were supported by NCI grant CA76035 to HD and RG.
Author information
Authors and Affiliations
- Division of Medical Oncology, University of Colorado at Denver and Health Sciences and Cancer Centers, PO Box 6511, Mail Stop 8117, Aurora, 80045-0511, CO, USA
R M Gemmill, L Costa, C Korch & H A Drabkin - Cleveland Clinic Taussig Cancer Center, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, 44195, OH, USA
M Zhou & R M Bukowski
Authors
- R M Gemmill
- M Zhou
- L Costa
- C Korch
- R M Bukowski
- H A Drabkin
Corresponding author
Correspondence toR M Gemmill.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Gemmill, R., Zhou, M., Costa, L. et al. Synergistic growth inhibition by Iressa and Rapamycin is modulated by VHL mutations in renal cell carcinoma.Br J Cancer 92, 2266–2277 (2005). https://doi.org/10.1038/sj.bjc.6602646
- Received: 31 January 2005
- Revised: 21 April 2005
- Accepted: 28 April 2005
- Published: 14 June 2005
- Issue Date: 20 June 2005
- DOI: https://doi.org/10.1038/sj.bjc.6602646